

Abstract
Aims
Myocardial fibrosis alters the cardiac architecture favouring the development of cardiac dysfunction, including arrhythmias and heart failure. Reducing myocardial fibrosis may improve outcomes through the targeted diagnosis and treatment of emerging fibrotic pathways. The European‐Commission‐funded ‘FIBROTARGETS’ is a multinational academic and industrial consortium with the main aims of (i) characterizing novel key mechanistic pathways involved in the metabolism of fibrillary collagen that may serve as biotargets, (ii) evaluating the potential anti‐fibrotic properties of novel or repurposed molecules interfering with the newly identified biotargets, and (iii) characterizing bioprofiles based on distinct mechanistic phenotypes involving the aforementioned biotargets. These pathways will be explored by performing a systematic and collaborative search for mechanisms and targets of myocardial fibrosis. These mechanisms will then be translated into individualized diagnostic tools and specific therapeutic pharmacological options for heart failure.
Methods and results
The FIBROTARGETS consortium has merged data from 12 patient cohorts in a common database available to individual consortium partners. The database consists of >12 000 patients with a large spectrum of cardiovascular clinical phenotypes. It integrates community‐based population cohorts, cardiovascular risk cohorts, and heart failure cohorts.
Conclusions
The FIBROTARGETS biomarker programme is aimed at exploring fibrotic pathways allowing the bioprofiling of patients into specific ‘fibrotic’ phenotypes and identifying new therapeutic targets that will potentially enable the development of novel and tailored anti‐fibrotic therapies for heart failure.
Keywords: Myocardial fibrosis, Fibrotic bioprofiles, Heart failure
Introduction
FIBROTARGETS is a multinational academic–industrial consortium funded by the European Commission Seventh Framework Programme. The consortium was built to create synergies and collaborative efforts between clinical investigators, basic scientists, and small‐ and medium‐sized enterprises, combining complementary approaches, disciplines, technologies, resources, and expertise. It aims at bridging pre‐clinical findings related to myocardial interstitial fibrosis (MIF) to the clinical setting with the objective of changing and improving outcomes through the advent of tailored anti‐fibrotic therapies.
The permanent cellular constituents of the heart mainly include fibroblasts, myocytes, endothelial cells, and vascular smooth muscle cells.1 Cardiac fibroblasts are the most prevalent cell type in the heart and are essential not only for normal cardiac function, being involved in the synthesis and deposition of the extracellular matrix (ECM), cell–cell communication with myocytes, and cell–cell signalling with other fibroblasts and with endothelial cells but also for post‐aggression repair processes (e.g. myocardial scar formation after infarction). Myocardial fibroblasts respond to several mediators, including pro‐inflammatory cytokines (e.g. tumour necrosis factor, interleukins 1 and 6, and transforming growth factor beta), vasoactive peptides (e.g. angiotensin II, endothelin 1, and natriuretic peptides), hormones (e.g. cortisol, renin, aldosterone, norepinephrine, and mechanical stretch), and changes in oxygen availability (e.g. ischaemia–reperfusion). Thus, any dysregulation at the level of these mediators may lead to dysfunctional excessive fibrogenesis affecting the electrophysiological pathways, mechanical properties, and signalling functions of the heart. Insults to the heart such as pressure or volume overload, ischaemic injury, valvular diseases, and genetic predispositions favour the development and influence the amount of myocardial fibrosis. The latter develops into two distinct patterns: as a scar to replace the loss of cardiomyocytes (referred to as focal reparative fibrosis) and as pervasive fibrosis diffusively invading the myocardial interstitium and the perivascular space (referred to as diffuse fibrosis). While both types of myocardial fibrosis result from a dysregulated collagen turnover (i.e. synthesis exceeds degradation, facilitating the deposition of highly cross‐linked, stiff, and degradation‐resistant collagen fibres)2, 3 impairing cardiac funtion,4 the excessive accumulation of collagen in the ECM may result from various altered signalling pathways.5 While fibrosis is essential for cardiac repair, its excessive and aberrant accumulation is deleterious; hence, there is a thin line separating ‘good’ from ‘bad’ fibrosis. The identification and validation of circulating biomarkers showing good correlation with the ‘intracardiac’ mechanistic pathways involved in interstitial fibrosis are essential for developing bioprofiles that could help in identifying patients with ‘bad’ fibrosis and who are likely to benefit from targeted therapeutic agents. However, it is also plausible that different pathological phenotypes result from the interweaving of several altered molecular pathways. Hence, an integrative and comprehensive approach is essential to better understand these fibrotic disease processes. Moreover, specific therapeutic options targeting myocardial fibrosis have failed to demonstrate undisputable efficacy, hence the need for novel therapeutic options.6, 7
By design, the FIBROTARGETS project integrates experimental and clinical research with the aim of (i) characterizing novel key mechanistic pathways involved in the metabolism of fibrillary collagen that may serve as biotargets, (ii) evaluating the potential anti‐fibrotic properties of novel or repurposed molecules interfering with the newly identified biotargets, and (iii) characterizing bioprofiles based on distinct mechanistic phenotypes involving the aforementioned biotargets targeting a tailored treatment approach. The purpose of this article is to describe the design of the clinical studies addressing the latter objective.
Study population
Taking advantage of its multinational structure, the FIBROTARGETS consortium has merged data from 12 patient cohorts in a common database available to individual consortium partners. The database consists of >12 000 patients with a large spectrum of cardiovascular clinical phenotypes.8 It integrates community‐based population cohorts (Stanislas9 and Health ABC10), cardiovascular risk cohorts (REVE‐1 and REVE‐2,11 ADELAHYDE,12 R2C2, HVC, REMI,13 and STOP‐HF14), and heart failure (HF) cohorts (Leitzaran, MEDIA‐DHF, and TIME‐CHF15). Cohort details are summarized in Table 1 and the Supporting Information, Tables S1 , S2 , and S3 .
Table 1.
Clinical characteristics of the merged FIBROTARGETS cohort
| Parameter | Available data | Merged FIBROTARGETS database (12 922) |
|---|---|---|
| n | (Baseline) | |
| Demographics | ||
| Women, n (%) | 12 922 | 6365 (49) |
| Age, years | 12 921 | 54 ± 23 |
| BMI, kg/m2 | 12 419 | 25.7 ± 5.6 |
| Current smoker, n (%) | 11 318 | 2078 (18) |
| Current alcohol consumer, n (%) | 8859 | 4461 (50) |
| Medical history | ||
| Hypertension, n (%) | 7443 | 4906 (66) |
| Diabetes mellitus, n (%) | 8445 | 1624 (19) |
| Hypercholesterolaemia, n (%) | 4366 | 1565 (36) |
| Myocardial infarction, n (%) | 4742 | 1231 (26) |
| Heart failure, n (%) | 6158 | 1763 (29) |
| Atrial fibrillation, n (%) | 3695 | 655 (18) |
| Haemodynamics | ||
| Systolic blood pressure, mmHg | 12 486 | 129 ± 21 |
| Diastolic blood pressure, mmHg | 12 484 | 72 ± 14 |
| Heart rate, b.p.m. | 12 734 | 70 ± 13 |
| Blood biology | ||
| Total cholesterol (mmol/L) | 11 681 | 5.2 ± 1.3 |
| Glucose, mmol/L | 11 528 | 5.70 ± 1.91 |
| Haemoglobin, g/dL | 7286 | 14.0 ± 1.4 |
| Serum creatinine, μmol/L | 10 566 | 86.5 ± 35.6 |
| Blood biomarkers | ||
| BNP, pg/mL | 1441 | 36 (13–116) |
| NT‐proBNP, pg/mL | 1542 | 2502 (1074–6115) |
| Echocardiographic parameters | ||
| LVmass, g | 2823 | 167 ± 77 |
| LVEF, % | 4291 | 57 ± 14 |
| LVEF < 40% | 4291 | 586 (14%) |
| 40% ≤ LVEF ≤ 50% | 551 (13%) | |
| LVEF > 50% | 3154 (73%) | |
| E/A ratio | 3112 | 1.12 ± 1.60 |
BMI, body mass index; BNP, B‐type natriuretic peptide; LVEF, left ventricular ejection fraction; LVmass, left ventricular mass; NA, not applicable; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide. Continuous variables are described as mean ± standard deviation or, for non‐normal variables, as median (first quartile to third quartile). Dichotomous variables are described as number of events (% of available data).
Patients from the merged database were clustered into four main phenotypic groups: (i) ‘healthy subjects’, including (but not exclusively consisting of) participants without cardiovascular risk factors or renal dysfunction, (ii) participants at ‘risk’ of developing HF as evidenced by the presence of diabetes and/or hypertension or other cardiovascular risk factors and no overt cardiac dysfunction, (iii) patients with prevalent HF with reduced left ventricular ejection fraction regardless of HF symptoms, and (iv) patients with prevalent HF with preserved left ventricular ejection fraction regardless of HF symptoms.
Selection of biomarkers of interest
The biomarkers to be studied in the FIBROTARGETS consortium are related to the novel targets studied within the project and reflect various aspects related to myocardial fibrosis and are summarized in Table 2.
Table 2.
Examples of biomarkers to be studied in the FIBROTARGETS consortium
| Biomarkers | Examples |
|---|---|
| Collagen metabolism‐derived peptides | C‐terminal propeptide of procollagen type I |
| N‐terminal propeptide of procollagen type III | |
| C‐terminal telopeptide of collagen type I | |
| Matrix metalloproteinase 1 | |
| Cellular matrix proteins | Mimecan (osteoglycin) |
| SPARC (osteonectin) | |
| Biglycan | |
| Thrombospondin 2 | |
| Osteopontin | |
| Lumican | |
| Biomarkers related to the main pro‐fibrotic mediators | Aldosterone |
| Transforming growth factor β | |
| Neutrophil gelatinase‐associated lipocalin | |
| Galectin 3 | |
| Cardiotrophin 1 | |
| Apelin | |
| Inflammatory molecules | Growth differentiation factor 15 |
| Soluble ST‐2 | |
| CD40L | |
| Agouti‐related protein | |
| Circulating microRNAs and their targets | miR‐1 |
| miR‐19b | |
| miR‐21 | |
| miR‐29 | |
| miR‐122 | |
| miR‐133a | |
| miR‐208a | |
| miR‐499‐5p | |
| lncRNAs (e.g. LipCar) | |
| SOD2 (miR‐21 target) | |
| Fibulin 2 (miR‐1 target) | |
| SDF‐1/CXCR12 (progenitor cell recruitment) |
A number of molecules involved in the extracellular formation and degradation of collagen type I that is secreted from the heart to the blood stream, and thus detectable in either the serum or plasma by using immunoassay methods, have recently been proposed as biomarkers of diffuse interstitial fibrosis.16 For instance, serum C‐terminal propeptide of procollagen type I (PICP) has a good correlation with histologically proven severe myocardial interstitial deposition of collagen type I fibres and collagen volume fraction (r ≈ 0.7) in hypertensive patients with stage C HF.17 The molecular basis of this biomarker is that the cleavage of PICP from the procollagen type I precursor will determine the ability of the resulting collagen type I molecule to form fibrils.18 Excessive myocardial collagen cross‐linking is associated with HF hospitalization in hypertensive patients with HF, and the serum C‐terminal telopeptide of collagen type I (CITP) : matrix metalloproteinase 1 (MMP‐1) ratio allows identification of patients with increased collagen cross‐linking and high risk for HF hospitalization.19 The molecular basis of this biomarker relies on the consideration that a diminished circulating level of CITP (corrected by circulating MMP‐1) reflects reduced collagen type I fibre degradation by MMP‐1 given that high lysyl‐oxidase‐mediated cross‐linking increases the resistance of the fibre to MMP‐1 proteolysis.20 On the other hand, recent clinical data suggest that cardiotrophin 1 (CT‐1), a member of the interleukin 6 superfamily, behaves as a profibrotic cytokine. In fact, CT‐1 stimulates the differentiation of human cardiac fibroblasts to myofibroblasts and the expression of procollagen type I and III messenger RNAs in these patients.21 In addition, CT‐1 overexpression has been found to be associated with increased expression of collagen types I and III in the myocardium of HF patients.21 Moreover, an excess of plasma CT‐1 has been associated with both increased serum PICP and increased serum N‐terminal propeptide of procollagen type III, a molecule formed during the conversion of procollagen type III into collagen type III by the enzyme procollagen type III aminoterminal proteinase, which is correlated with the myocardial deposition of collagen type III fibres,22 in these same HF patients.21
Conversely, apelin, a small anti‐collagen production peptide, has been found to be decreased in the plasma of patients with advanced HF. In addition, in rodent cardiac fibroblasts, apelin has been shown to reduce the production of collagen and the activation of fibroblasts induced by transforming growth factor beta.23 Interestingly, exogenous administration of apelin was able to reduce myocardial fibrosis and improve cardiac function in a rodent model of HF.24
Multiple studies have highlighted the association of inflammation with myocardial fibrosis, and several inflammation‐related biomarkers have been identified as predictors of clinical outcome in HF patients.25 For instance, serum galectin 3 has been reported to be a good predictor of dismal events and mortality in HF patients.26 Similarly, soluble ST2 has been reported to be independently associated with cardiovascular mortality in HF patients,27 and growth differentiation factor 15 with mortality and non‐fatal events in HF with preserved or reduced ejection fraction.28
Neutrophil gelatinase‐associated lipocalin (NGAL), also known as lipocalin 2 or oncogene 24p3, is considered as a biomarker of kidney injury because it is rapidly released in response to kidney tubular damage.29 However, increased systemic levels have also been reported in patients with HF and myocardial infarction, with these levels being associated with adverse cardiac events.30 For example, NGAL is up‐regulated in the cardiovascular system in various pathological models such as atherosclerosis,31 aortic abdominal aneurysm,32 and after myocardial infarction.33
Non‐coding RNAs
Many studies have shown that non‐coding RNAs such as microRNAs (miRNAs) and long non‐coding RNAs (lncRNAs) are released into the circulation where they exhibit specific profiles that differ between healthy subjects and diseased patients.34 These molecules are increasingly recognized as non‐invasive and readily accessible biomarkers for risk stratification, diagnosis, and prognosis of cardiac injury and multiple forms of cardiovascular disease. Several miRNAs have recently been linked to the diagnosis of HF.34, 35 Based on our screening results as well as literature findings,36, 37 several ‘cardiac remodelling’‐associated miRNAs and lncRNAs have been chosen to be tested in the FIBROTARGETS patient populations to determine their use for improving diagnosis of HF, risk and patient stratification, and prediction of individual patient prognosis. Standardized miRNA‐ or lncRNA‐specific PCR‐based protocols and normalization procedures are currently being used to achieve this goal.
Data management
The FIBROTARGETS database was elaborated and is maintained at the Inserm Clinical Investigation Center (CIC1433), Nancy, France. In order to be included in FIBROTARGETS, participating institutions and investigators must comply with the latest local, national, and international rules on data protection, biomedical research regulations, good clinical practices, and the Helsinki Declaration. All participants from each cohort must have provided written informed consent. Informative data relevant to the FIBROTARGETS consortium have been extracted from each cohort dataset. Thus, the merged dataset retains a maximum of information retrieved from each contributing cohort in order to include baseline characteristics of each participant (anthropometrics, medical history and medication use, routine haematological and biochemical measurements, electrocardiographic and echocardiographic variables) as well as follow‐up information, whenever available. A data dictionary has been elaborated based on agreed common definitions and criteria enabling classification of clinical events and phenotypes with a particular focus on HF events, left ventricular dysfunction, hypertension, diabetes, atrial fibrillation, myocardial infarction, and chronic kidney disease (see Table 1 and Supporting Information, Tables S1 , S2 , and S3 ).
The investigators of each study have also collected follow‐up data.
By ensuring a large diversity of phenotypes within the FIBROTARGETS population (in terms of age and presence or absence of co‐morbidities), the merged database provides a unique tool to evaluate the determinants and utility of profiling patients according to specific fibrotic pathways using candidate biomarkers.
Statistical and bioinformatics considerations
Predictive models (of the aforementioned phenotypic clusters) using biomarker variables will be developed in three ways: (i) using classical statistical methods (as described above) including selection procedures in the second phase, (ii) using machine‐learning tools such as regression trees and random forests, and (iii) using system biological or computational models based on available knowledge of myocardial fibrosis.
In addition to the prediction of phenotypic clusters, associations between biomarkers and clinical outcomes will be assessed, including all‐cause and cardiovascular mortality and HF hospitalizations. Cox models will be used to assess the associations between biomarkers and outcome.
Further bioinformatics analysis will integrate the obtained FIBROTARGETS biomarker data in the context of previous knowledge.38, 39 The biomarkers will first be analysed for their interaction with diseases and phenotypes (such as cardiovascular diseases, immune disorders, and cancer, among others), drug interactions, protein–protein interactions, and their predicted targeting by miRNAs. Next, correlation network modules will further refine the measured biomarkers in clusters that correlate with specific clinical phenotypes and pre‐measured metabolites (such as lipid metabolism). Finally, this cluster analysis will evolve in cluster enrichment for specific biological processes—such as leukocyte migration and fibroblast proliferation—using Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathways. In addition to classical biostatistics, this interaction, network, and clustering analysis will place the biomarkers in their biological context for improving selection of biomarkers, understanding their disease context, and refining patient stratification.
A matched case–control design will be chosen to assess the associations between candidate biomarkers and the four phenotypic clusters (as defined above).
Cases from the three patient clusters will be matched with a control from the ‘healthy subjects’ cluster. The distribution of biomarker levels in ‘healthy’ subjects and in patients will be described, and reference values established for each biomarker, with influence of age and gender. The association between candidate biomarkers, disease clinical phenotypes, and outcome will be assessed with appropriate statistical methods, using continuous or categorical variables, including conditional logistic regression adjusted for a set of confounders. The influence of common co‐morbid conditions (e.g. diabetes, hypertension, and obesity) will be taken into account in multivariate analyses. In each instance, sensitivity and specificity measurements of the predictions will be quantified.
Power calculations will be performed solely based on the comparison of biomarkers across the four clusters. In the first phase of the analysis, to account for the multiple comparisons and biomarkers, a two‐sided alpha of 0.01 will be assumed for sample size calculation. In order to achieve a statistical power of 90% to detect a minimum difference of an odds ratio of 1.35 per 1 SD of the biomarker, the minimum sample size would be 750 consisting of 250 cases and 500 controls.
In the second phase of the analysis, the most promising candidate biomarkers will be tested using a two‐sided alpha of 0.001, including 600 patients (200 cases and 400 controls) to provide a 90% power to identify a difference of 1.50 odds ratio per 1 SD of the biomarker. In total, for the first and second phases of the analysis, 450 cases and 900 controls would be required for a total sample size of 1350 patients.
Baseline characteristics of the study population
The characteristics of the main cohorts are described in Table 1 (‘merged’ dataset) and Supporting Information, Tables S1 (population/‘healthy’ cohorts), S2 (cardiovascular risk cohorts), and S3 (HF cohorts).
The FIBROTARGETS population consists of 12 922 individuals, 49% of whom are of female sex. The mean ± standard deviation (SD) age is 54 ± 23 years with a mean ± SD body mass index of 25.7 ± 5.6 kg/m2. Sixty‐six per cent of the individuals present a diagnosis of hypertension, 19% had diabetes mellitus, and 29% had HF.
Additionally, individuals were classified based on the four selected risk factors (hypertension, obesity, diabetes, and history of HF), allowing the establishment of subgroups based on disease/risk factors, as represented in Figure 1 .
Figure 1.
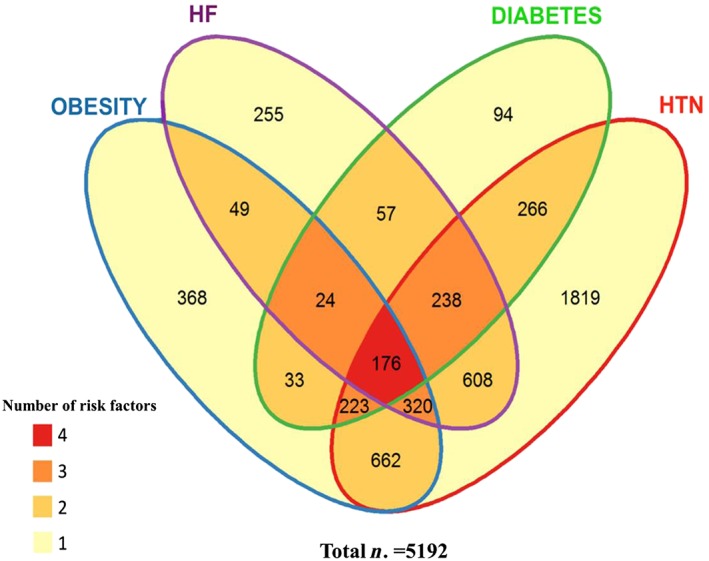
Venn diagram of the FIBROTARGETS merged cohort, with clustering of individuals with at least one of the listed risk factors. Legend: the merged FIBROTARGETS cohort has been stratified according to co‐morbidities [obesity, diabetes, hypertension (HTN), and heart failure (HF)]. Number of individuals displaying from 1 to 4 of these conditions is given within the intersection of corresponding ellipses.
Outcomes
The available outcomes for each study population are described in Table 3. Outcomes in each study cohort were adjudicated by independent committees.
Table 3.
Outcomes and follow‐up of the FIBROTARGETS study cohorts
| Population cohort | Cardiovascular risk patients | HF patients | Total baseline population | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stanislas | Health ABC | ADELAHYDE | R2C2 | HVC | REMI | REVE‐1 | REVE‐2 | STOP‐HF | Leitzaran | MEDIA‐DHF | TIME‐CHF | ||
| Death | 131 (3.8%) | 1896 (61.7%) | NA | 0 (0%) | 90 (12.4%) | 0 (0%) | NA | NA | NA | 70 (34.5%) | 35 (6.0%) | 247 (39.7%) | 2399 (28%) |
| CVD | NA | NA | NA | 0 (0%) | 6 (2.6%) | 0 (0%) | NA | NA | NA | 37 (18.2%) | 15 (2.6%) | 214 (34.4%) | 235 (3.6%) |
| FU until CVD or censoring | 7973 (7839–8135) | 4691 (2927–4848) | NA | 2353 (1928–2562) | 335 (88–985) | 182 (174–193) | NA | NA | NA | 1938 (1478–2117) | 364 (189–368) | 794 (423–1226) | 4819 (1463–7902) |
| HHF | NA | 656 (21.3%) | NA | NA | 25 (3.4%) | NA | NA | NA | NA | 92 (45.3%) | 48 (8.3%) | 317 (51.0%) | 1046 (21%) |
| FU until HHF or censoring | NA | 4294 (2439–4807) | NA | NA | 308 (85–923) | NA | NA | NA | NA | 1551 (562–2022) | 364 (183–367) | 596 (177–1078) | 1928 (390–4716) |
CVD, cardiovascular death; FU, follow‐up; HHF, hospitalization for heart failure; NA, not applicable.
Discussion
Therapeutic developments conducted in the last decades have led to a substantial decrease in mortality in HF with reduced ejection fraction. Therefore, demonstrating a therapeutic benefit on top of existing therapies in the logic of ‘one‐size‐fits‐all’ trials has become increasingly difficult and cost demanding. As a consequence, in the last three decades, landmark HF trial sizes have progressed from a few hundred to many thousand patients.40, 41 Operational obstacles and unbearable costs are major challenges for the successful implementation and completion of such large trials.42, 43 Even with pre‐specified subgroup analysis, regulators do not accept testing for responder profiles due to the risk of bias. Substantial warnings and approval deferrals have already been performed on the basis of subgroup results.44, 45 Precision medicine has emerged as one possible solution, aiming at identifying the different strata within a disease based on a deeper understanding of the mechanisms underpinning these strata. Accordingly, biomarker‐based stratification will likely enable the development of disease‐specific diagnostic tools and treatments. The FIBROTARGETS programme is based on the hypothesis that intervention on novel fibrosis‐related targets involved in the processes of fibroblast differentiation into myofibroblasts, on collagen synthesis over degradation balance, and/or on collagen maturation may allow for interstitial repair, thus providing a new strategy for the prevention and treatment of MIF, a key mechanism of cardiac remodelling involved in both the transition to and progression of HF. Multi‐panel circulating markers, descriptive of mechanisms involving the proposed novel targets, will be used to screen large‐scale population of patients and to stratify the latter based on specific ‘fibrotic’ profiles.
In syndromes that are clinically and pathophysiological heterogeneous and frequently associated with several co‐morbid conditions such as HF (and HF with preserved ejection fraction in particular), more appropriate biomarkers are needed to improve screening, diagnosis, monitoring, and treatment response. Mitigating or preventing the development of MIF is one of the therapeutic strategies warranting further evaluation. Mineralocorticoid receptor antagonists (MRAs) are the only treatments to date demonstrated to be capable of reducing cardiac fibrosis during HF‐induced remodelling.46, 47 In fact, post hoc evidence suggests that MRAs are likely to work best in subgroup of patients with elevated cardiac collagen markers46 or with visceral obesity, conditions known to display marked fibrosis. The discovery of more powerful and specific modulators of fibrosis could potentially provide the basis for new drug development and patient selection for future trials.
Previously assessed biomarkers can be classified as collagen quantity (e.g. PICP and N‐terminal propeptide of procollagen type III)16 and quality markers (e.g. CITP : MMP‐1).16 , 18 While these biomarker assessments in patients at risk of and with HF have been associated with poor prognosis, only two collagen‐derived serum peptides have been shown to be specifically associated with the extent of myocardial fibrosis: PICP and C‐terminal propeptide of procollagen type III.16 Beyond these end‐products of collagen turnover, FIBROTARGETS aims to explore upstream mechanistic pathways as potential discrete biotargets for novel therapies preventing and/or limiting the progression of MIF.
One of the many challenges is that fibrosis is a mechanism involved in a number of overlapping and/or coexisting risk and disease states and co‐morbidities48, 49 such as hypertension, chronic kidney disease, diabetes mellitus, obesity, atrial fibrillation, ischaemic heart disease, and prevalent HF, such that a number of circulating biomarkers can be present in these various conditions.26, 46, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61 Our hypothesis is that novel ‘mechanistic’ markers of cardiac fibrosis are differentially expressed in various risk and disease states and co‐morbidities.
Conclusions
While many biomarkers have been tested, reproducibility and external validity are not sufficiently robust however to derive firm conclusions as to which biomarker(s) can best assess cardiac fibrosis or assess treatment response. Moreover, these biomarkers have not proved to determine senescent cell accumulation in fibrotic myocardial tissue, especially given that myofibroblasts have been identified as the predominant cardiac cell population undergoing senescence and are critical regulators of cardiac fibrogenesis.62 The FIBROTARGETS programme will explore novel and mechanistic fibrotic targets that will enable the development of disease‐specific therapies. The programme includes the investigation of multi‐panel circulating markers descriptive of mechanisms involving the proposed novel targets. These will form the basis of the stratification of patients at risk into specific ‘fibrotic’ bioprofiles.
Conflict of interest
All authors are actively involved in the FIBROTARGETS consortium.
Funding
This work was funded through the European Commission Seventh Framework Programme (FIBROTARGETS project grant HEALTH‐2013‐602904) and the REBIRTH Excellence Cluster, Hannover Medical School (to T.T.).
Supporting information
Table S1. Details on the population cohorts.
Table S2. Details on patient cohorts at risk for CV.
Table S3. Details on HF patient cohorts.
Acknowledgements
The authors wish to express their gratitude to all the partners of the FIBROTARGETS consortium: Faiez Zannad and Frédéric Jaisser, Institut National de la Santé et de la Recherche Médicale, France; Catherine Clusel and Marie‐Alix Fauvel, Institut National de la Santé et de la Recherche Médicale‐Transfert SA, France; Javier Díez and Arantxa González, Centro de Investigación Médica Aplicada, Spain; Kenneth Mac Donald, University College Dublin, National University of Ireland, Ireland; Stephane Heymans and Anna Papageorgiou, Universiteit Maastricht, The Netherlands; Thomas Thum, Medizinische Hochschule Hannover, Germany; Mariann Gyöngyösi and Johannes Winkler, Medical University of Vienna, Austria, Austria; Quoc‐Tuan Do, Greenpharma S.A.S., France; Hueseyin Firat and Kaidre Bendjama, Firalis S.A.S., France; Isbaal Ramos and Clarisa Salado, Innovative Technologies in Biological Systems, Spain; Natalia López‐Andrés, Fundación Pública Miguel Servet, Spain.
The authors would also like to acknowledge Mr Pierre Pothier for editing of the manuscript.
Ferreira, J. P. , Machu, J.‐L. , Girerd, N. , Jaisser, F. , Thum, T. , Butler, J. , González, A. , Diez, J. , Heymans, S. , McDonald, K. , Gyöngyösi, M. , Firat, H. , Rossignol, P. , Pizard, A. , and Zannad, F. (2018) Rationale of the FIBROTARGETS study designed to identify novel biomarkers of myocardial fibrosis. ESC Heart Failure, 5: 139–148. doi: 10.1002/ehf2.12218.
References
- 1. Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther 2009; 123: 255–278. [DOI] [PubMed] [Google Scholar]
- 2. Kong P, Christia P, Frangogiannis N. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 2014; 71: 549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li A‐H, Liu PP, Villarreal FJ, Garcia RA. Dynamic changes in myocardial matrix and relevance to disease: translational perspectives. Circ Res 2014; 114: 916–927. [DOI] [PubMed] [Google Scholar]
- 4. Weber KT, Pick R, Jalil JE, Janicki JS, Carroll EP. Patterns of myocardial fibrosis. J Mol Cell Cardiol 1989; 21: 121–131. [DOI] [PubMed] [Google Scholar]
- 5. Hansen NU, Genovese F, Leeming DJ, Karsdal MA. The importance of extracellular matrix for cell function and in vivo likeness. Exp Mol Pathol 2015; 98: 286–294. [DOI] [PubMed] [Google Scholar]
- 6. Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand JL, Cohen‐Tervaert JW, Drexler H, Filippatos G, Felix SB, Gullestad L, Hilfiker‐Kleiner D, Janssens S, Latini R, Neubauer G, Paulus WJ, Pieske B, Ponikowski P, Schroen B, Schultheiss HP, Tschope C, Van Bilsen M, Zannad F, McMurray J, Shah AM. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2009; 11: 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heymans S, Gonzalez A, Pizard A, Papageorgiou AP, Lopez‐Andres N, Jaisser F, Thum T, Zannad F, Diez J. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur J Heart Fail 2015; 17: 764–771. [DOI] [PubMed] [Google Scholar]
- 8. Jacobs L, Thijs L, Jin Y, Zannad F, Mebazaa A, Rouet P, Pinet F, Bauters C, Pieske B, Tomaschitz A, Mamas M, Diez J, McDonald K, Cleland JG, Rocca HP, Heymans S, Latini R, Masson S, Sever P, Delles C, Pocock S, Collier T, Kuznetsova T, Staessen JA. Heart ‘omics’ in AGEing (HOMAGE): design, research objectives and characteristics of the common database. J Biomed Res 2014; 28: 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ferreira JP, Girerd N, Bozec E, Machu JL, Boivin JM, London GM, Zannad F, Rossignol P. Intima‐media thickness is linearly and continuously associated with systolic blood pressure in a population‐based cohort (STANISLAS Cohort Study). J Am Heart Assoc 2016; 5: pii: e003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kalogeropoulos A, Psaty BM, Vasan RS, Georgiopoulou V, Smith AL, Smith NL, Kritchevsky SB, Wilson PW, Newman AB, Harris TB, Butler J. Validation of the health ABC heart failure model for incident heart failure risk prediction: the Cardiovascular Health Study. Circ Heart Fail 2010; 3: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lamblin N, Fertin M, de Groote P, Bauters C. Incidence, determinants and consequences of left atrial remodelling after a first anterior myocardial infarction. Arch Cardiovasc Dis 2012; 105: 18–23. [DOI] [PubMed] [Google Scholar]
- 12. Toupance S, Watfa G, Lakomy C, Kearney‐Schwartz A, Labat C, Rossignol P, Lacolley P, Zannad F, Benetos A. 2C.01: fixed ranking of leukocyte telomere length in elderly people: results from 8 year follow‐up of the Adelahyde cohort. J Hypertens 2015; 33: e25. [Google Scholar]
- 13. Lemarie J, Huttin O, Girerd N, Mandry D, Juilliere Y, Moulin F, Lemoine S, Beaumont M, Marie PY, Selton‐Suty C. Usefulness of speckle‐tracking imaging for right ventricular assessment after acute myocardial infarction: a magnetic resonance imaging/echocardiographic comparison within the relation between aldosterone and cardiac remodeling after myocardial infarction study. J Am Soc Echocardiogr 2015; 28: 818–827, e4. [DOI] [PubMed] [Google Scholar]
- 14. Ledwidge M, Gallagher J, Conlon C, Tallon E, O'Connell E, Dawkins I, Watson C, O'Hanlon R, Bermingham M, Patle A, Badabhagni MR, Murtagh G, Voon V, Tilson L, Barry M, McDonald L, Maurer B, McDonald K. Natriuretic peptide‐based screening and collaborative care for heart failure: the STOP‐HF randomized trial. JAMA 2013; 310: 66–74. [DOI] [PubMed] [Google Scholar]
- 15. Zurek M, Maeder MT, Rickli H, Muzzarelli S, Sanders‐van Wijk S, Abbuhl H, Handschin R, Jeker U, Pfisterer M, Brunner‐la Rocca HP. Differential prognostic impact of resting heart rate in older compared with younger patients with chronic heart failure—insights from TIME‐CHF. J Card Fail 2015; 21: 347–354. [DOI] [PubMed] [Google Scholar]
- 16. Lopez B, Gonzalez A, Ravassa S, Beaumont J, Moreno MU, San Jose G, Querejeta R, Diez J. Circulating biomarkers of myocardial fibrosis: the need for a reappraisal. J Am Coll Cardiol 2015; 65: 2449–2456. [DOI] [PubMed] [Google Scholar]
- 17. Lopez B, Querejeta R, Gonzalez A, Larman M, Diez J. Collagen cross‐linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: potential role of lysyl oxidase. Hypertension 2012; 60: 677–683. [DOI] [PubMed] [Google Scholar]
- 18. Prockop DJ, Sieron AL, Li SW. Procollagen N‐proteinase and procollagen C‐proteinase. Two unusual metalloproteinases that are essential for procollagen processing probably have important roles in development and cell signaling. Matrix Biol 1998; 16: 399–408. [DOI] [PubMed] [Google Scholar]
- 19. Lopez B, Ravassa S, Gonzalez A, Zubillaga E, Bonavila C, Berges M, Echegaray K, Beaumont J, Moreno MU, San Jose G, Larman M, Querejeta R, Diez J. Myocardial collagen cross‐linking is associated with heart failure hospitalization in patients with hypertensive heart failure. J Am Coll Cardiol 2016; 67: 251–260. [DOI] [PubMed] [Google Scholar]
- 20. Robins SP. Biochemistry and functional significance of collagen cross‐linking. Biochem Soc Trans 2007; 35: 849–852. [DOI] [PubMed] [Google Scholar]
- 21. Lopez B, Gonzalez A, Querejeta R, Larman M, Rabago G, Diez J. Association of cardiotrophin‐1 with myocardial fibrosis in hypertensive patients with heart failure. Hypertension 2014; 63: 483–489. [DOI] [PubMed] [Google Scholar]
- 22. Klappacher G, Franzen P, Haab D, Mehrabi M, Binder M, Plesch K, Pacher R, Grimm M, Pribill I, Eichler HG et al. Measuring extracellular matrix turnover in the serum of patients with idiopathic or ischemic dilated cardiomyopathy and impact on diagnosis and prognosis. Am J Cardiol 1995; 75: 913–918. [DOI] [PubMed] [Google Scholar]
- 23. Koguchi W, Kobayashi N, Takeshima H, Ishikawa M, Sugiyama F, Ishimitsu T. Cardioprotective effect of apelin‐13 on cardiac performance and remodeling in end‐stage heart failure. Circ J 2012; 76: 137–144. [DOI] [PubMed] [Google Scholar]
- 24. Pchejetski D, Foussal C, Alfarano C, Lairez O, Calise D, Guilbeau‐Frugier C, Schaak S, Seguelas MH, Wanecq E, Valet P, Parini A, Kunduzova O. Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur Heart J 2012; 33: 2360–2369. [DOI] [PubMed] [Google Scholar]
- 25. Collinson P. The role of cardiac biomarkers in cardiovascular disease risk assessment. Curr Opin Cardiol 2014; 29: 366–371. [DOI] [PubMed] [Google Scholar]
- 26. Lopez B, Gonzalez A, Querejeta R, Zubillaga E, Larman M, Diez J. Galectin‐3 and histological, molecular and biochemical aspects of myocardial fibrosis in heart failure of hypertensive origin. Eur J Heart Fail 2015; 17: 385–392. [DOI] [PubMed] [Google Scholar]
- 27. Bayes‐Genis A, de Antonio M, Vila J, Penafiel J, Galan A, Barallat J, Zamora E, Urrutia A, Lupon J. Head‐to‐head comparison of 2 myocardial fibrosis biomarkers for long‐term heart failure risk stratification: ST2 versus galectin‐3. J Am Coll Cardiol 2014; 63: 158–166. [DOI] [PubMed] [Google Scholar]
- 28. Wollert KC, Kempf T, Wallentin L. Growth differentiation factor 15 as a biomarker in cardiovascular disease. Clin Chem 2017; 63: 140–151. [DOI] [PubMed] [Google Scholar]
- 29. Savoia C, Touyz RM, Amiri F, Schiffrin EL. Selective mineralocorticoid receptor blocker eplerenone reduces resistance artery stiffness in hypertensive patients. Hypertension 2008; 51: 432–439. [DOI] [PubMed] [Google Scholar]
- 30. van Deursen VM, Damman K, Voors AA, van der Wal MH, Jaarsma T, van Veldhuisen DJ, Hillege HL. Prognostic value of plasma neutrophil gelatinase‐associated lipocalin for mortality in patients with heart failure. Circ Heart Fail 2014; 7: 35–42. [DOI] [PubMed] [Google Scholar]
- 31. Eilenberg W, Stojkovic S, Piechota‐Polanczyk A, Kaun C, Rauscher S, Groger M, Klinger M, Wojta J, Neumayer C, Huk I, Demyanets S. Neutrophil gelatinase‐associated lipocalin (NGAL) is associated with symptomatic carotid atherosclerosis and drives pro‐inflammatory state in vitro. Eur J Vasc Endovasc Surg 2016; 51: 623–631. [DOI] [PubMed] [Google Scholar]
- 32. Tarin C, Fernandez‐Garcia CE, Burillo E, Pastor‐Vargas C, Llamas‐Granda P, Castejon B, Ramos‐Mozo P, Torres‐Fonseca MM, Berger T, Mak TW, Egido J, Blanco‐Colio LM, Martin‐Ventura JL. Lipocalin‐2 deficiency or blockade protects against aortic abdominal aneurysm development in mice. Cardiovasc Res 2016; 111: 262–273. [DOI] [PubMed] [Google Scholar]
- 33. Hemdahl AL, Gabrielsen A, Zhu C, Eriksson P, Hedin U, Kastrup J, Thoren P, Hansson GK. Expression of neutrophil gelatinase‐associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler Thromb Vasc Biol 2006; 26: 136–142. [DOI] [PubMed] [Google Scholar]
- 34. Viereck J, Thum T. Circulating noncoding RNAs as biomarkers of cardiovascular disease and injury. Circ Res 2017; 120: 381–399. [DOI] [PubMed] [Google Scholar]
- 35. Watson CJ, Gupta SK, O'Connell E, Thum S, Glezeva N, Fendrich J, Gallagher J, Ledwidge M, Grote‐Levi L, McDonald K, Thum T. MicroRNA signatures differentiate preserved from reduced ejection fraction heart failure. Eur J Heart Fail 2015; 17: 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Derda AA, Thum S, Lorenzen JM, Bavendiek U, Heineke J, Keyser B, Stuhrmann M, Givens RC, Kennel PJ, Schulze PC, Widder JD, Bauersachs J, Thum T. Blood‐based microRNA signatures differentiate various forms of cardiac hypertrophy. Int J Cardiol 2015; 196: 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Devaux Y, Zangrando J, Schroen B, Creemers EE, Pedrazzini T, Chang CP, Dorn GW 2nd, Thum T, Heymans S. Long noncoding RNAs in cardiac development and ageing. Nat Rev Cardiol 2015; 12: 415–425. [DOI] [PubMed] [Google Scholar]
- 38. Summer G, Kelder T, Radonjic M, van Bilsen M, Wopereis S, Heymans S. The Network Library: a framework to rapidly integrate network biology resources. Bioinformatics 2016; 32: i473–i478. [DOI] [PubMed] [Google Scholar]
- 39. Summer G, Kelder T, Ono K, Radonjic M, Heymans S, Demchak B. cyNeo4j: connecting Neo4j and Cytoscape. Bioinformatics 2015; 31: 3868–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR. Angiotensin–neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371: 993–1004. [DOI] [PubMed] [Google Scholar]
- 41. Swedberg K, Kjekshus J. Effects of enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). Am J Cardiol 1988; 62: 60A–66A. [DOI] [PubMed] [Google Scholar]
- 42. Ferreira JP, Girerd N, Rossignol P, Zannad F. Geographic differences in heart failure trials. Eur J Heart Fail 2015; 17: 893–905. [DOI] [PubMed] [Google Scholar]
- 43. Pfeffer MA, Claggett B, Assmann SF, Boineau R, Anand IS, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Heitner JF, Lewis EF, O'Meara E, Rouleau JL, Probstfield JL, Shaburishvili T, Shah SJ, Solomon SD, Sweitzer NK, McKinlay SM, Pitt B. Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) Trial. Circulation 2015; 131: 34–42. [DOI] [PubMed] [Google Scholar]
- 44. Mahaffey KW, Wojdyla DM, Carroll K, Becker RC, Storey RF, Angiolillo DJ, Held C, Cannon CP, James S, Pieper KS, Horrow J, Harrington RA, Wallentin L. Ticagrelor compared with clopidogrel by geographic region in the Platelet Inhibition and Patient Outcomes (PLATO) trial. Circulation 2011; 124: 544–554. [DOI] [PubMed] [Google Scholar]
- 45. Pocock S, Calvo G, Marrugat J, Prasad K, Tavazzi L, Wallentin L, Zannad F, Alonso Garcia A. International differences in treatment effect: do they really exist and why? Eur Heart J 2013; 34: 1846–1852. [DOI] [PubMed] [Google Scholar]
- 46. Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation 2000; 102: 2700–2706. [DOI] [PubMed] [Google Scholar]
- 47. Condorelli G, Jotti GS, Pagiatakis C. Fibroblast senescence as a therapeutic target of myocardial fibrosis: beyond spironolactone? J Am Coll Cardiol 2016; 67: 2029–2031. [DOI] [PubMed] [Google Scholar]
- 48. Ho JE, Enserro D, Brouwers FP, Kizer JR, Shah SJ, Psaty BM, Bartz TM, Santhanakrishnan R, Lee DS, Chan C, Liu K, Blaha MJ, Hillege HL, van der Harst P, van Gilst WH, Kop WJ, Gansevoort RT, Vasan RS, Gardin JM, Levy D, Gottdiener JS, de Boer RA, Larson MG. Predicting heart failure with preserved and reduced ejection fraction: the International Collaboration on Heart Failure Subtypes. Circ Heart Fail 2016; 9: pii: e003116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ganz P, Heidecker B, Hveem K, Jonasson C, Kato S, Segal MR, Sterling DG, Williams SA. Development and validation of a protein‐based risk score for cardiovascular outcomes among patients with stable coronary heart disease. Jama 2016; 315: 2532–2541. [DOI] [PubMed] [Google Scholar]
- 50. Lopez B, Gonzalez A, Varo N, Laviades C, Querejeta R, Diez J. Biochemical assessment of myocardial fibrosis in hypertensive heart disease. Hypertension 2001; 38: 1222–1226. [DOI] [PubMed] [Google Scholar]
- 51. Morillas P, Quiles J, de Andrade H, Castillo J, Tarazon E, Rosello E, Portoles M, Rivera M, Bertomeu‐Martinez V. Circulating biomarkers of collagen metabolism in arterial hypertension: relevance of target organ damage. J Hypertens 2013; 31: 1611–1617. [DOI] [PubMed] [Google Scholar]
- 52. O'Meara E, Rouleau JL, White M, Roy K, Blondeau L, Ducharme A, Neagoe PE, Sirois MG, Lavoie J, Racine N, Liszkowski M, Madore F, Tardif JC, de Denus S. Heart failure with anemia: novel findings on the roles of renal disease, interleukins, and specific left ventricular remodeling processes. Circ Heart Fail 2014; 7: 773–781. [DOI] [PubMed] [Google Scholar]
- 53. Su CT, Liu YW, Lin JW, Chen SI, Yang CS, Chen JH, Hung KY, Tsai WC, Huang JW. Increased procollagen type I C‐terminal peptide levels indicate diastolic dysfunction in end‐stage renal disease patients undergoing maintenance dialysis therapy. J Am Soc Echocardiogr 2012; 25: 895–901. [DOI] [PubMed] [Google Scholar]
- 54. Jellis CL, Sacre JW, Wright J, Jenkins C, Haluska B, Jeffriess L, Martin J, Marwick TH. Biomarker and imaging responses to spironolactone in subclinical diabetic cardiomyopathy. Eur Heart J Cardiovasc Imaging 2014; 15: 776–786. [DOI] [PubMed] [Google Scholar]
- 55. Ihm SH, Youn HJ, Shin DI, Jang SW, Park CS, Kim PJ, Kim HY, Chang K, Seung KB, Kim JH, Choi KB. Serum carboxy‐terminal propeptide of type I procollagen (PIP) is a marker of diastolic dysfunction in patients with early type 2 diabetes mellitus. In Int J Cardiol, Netherlands 2007; 122: e36–e38. [DOI] [PubMed] [Google Scholar]
- 56. Kosmala W, Jedrzejuk D, Derzhko R, Przewlocka‐Kosmala M, Mysiak A, Bednarek‐Tupikowska G. Left ventricular function impairment in patients with normal‐weight obesity: contribution of abdominal fat deposition, profibrotic state, reduced insulin sensitivity, and proinflammatory activation. Circ Cardiovasc Imaging 2012; 5: 349–356. [DOI] [PubMed] [Google Scholar]
- 57. Kosmala W, Przewlocka‐Kosmala M, Szczepanik‐Osadnik H, Mysiak A, Marwick TH. Fibrosis and cardiac function in obesity: a randomised controlled trial of aldosterone blockade. Heart 2013; 99: 320–326. [DOI] [PubMed] [Google Scholar]
- 58. Zhao F, Zhang S, Chen Y, Gu W, Ni B, Shao Y, Wu Y, Qin J. Increased expression of NF‐AT3 and NF‐AT4 in the atria correlates with procollagen I carboxyl terminal peptide and TGF‐beta1 levels in serum of patients with atrial fibrillation. BMC Cardiovasc Disord 2014; 14: 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang SH, Wang J, Jin TR, Zhang LX, Shao J. The role of spironolactone in the metabolism of serum type I collagen in elderly patients with atrial fibrillation. Eur Rev Med Pharmacol Sci 2014; 18: 2903–2907. [PubMed] [Google Scholar]
- 60. Barthelemy O, Beygui F, Vicaut E, Rouanet S, Van Belle E, Baulac C, Degrandsart A, Dallongeville J, Montalescot G. Relation of high concentrations of plasma carboxy‐terminal telopeptide of collagen type I with outcome in acute myocardial infarction. Am J Cardiol 2009; 104: 904–909. [DOI] [PubMed] [Google Scholar]
- 61. Li MJ, Huang CX, Okello E, Yanhong T, Mohamed S. Treatment with spironolactone for 24 weeks decreases the level of matrix metalloproteinases and improves cardiac function in patients with chronic heart failure of ischemic etiology. Can J Cardiol 2009; 25: 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Meyer K, Hodwin B, Ramanujam D, Engelhardt S, Sarikas A. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J Am Coll Cardiol 2016; 67: 2018–2028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Details on the population cohorts.
Table S2. Details on patient cohorts at risk for CV.
Table S3. Details on HF patient cohorts.