

Abstract
In this work, we explored the molecular framework of the known CB1R allosteric modulator PSNCBAM-1 with the aim to generate new bioactive analogs and to deepen the structure-activity relationships of this type of compounds. In particular, the introduction of a NH group between the pyridine ring and the phenyl nucleus generated the amino-phenyl-urea derivative SN15b that behaved as a positive allosteric modulator (PAM), increasing the CB1R binding affinity of the orthosteric ligand CP55,940. The functional activity was evaluated using serum response element (SRE) assay, which assesses the CB1R-dependent activation of the MAPK/ERK signaling pathway. SN15b and the biphenyl-urea analog SC4a significantly inhibited the response produced by CP55,940 in the low μM range, thus behaving as negative allosteric modulators (NAMs). The new derivatives presented here provide further insights about the modulation of CB1R binding and functional activity by allosteric ligands.
Keywords: Allosteric modulator, CB1 receptor, Endocannabinoid system, MAPK/ERK signaling pathway, Serum response element
Graphical Abstract
1. Introduction
The endocannabinoid system (ECS) is abundantly expressed throughout the body and it consists of cannabinoid receptors, their endogenous ligands (endocannabinoids), and the enzymes involved in the metabolism of endocannabinoids.1, 2 Since the discovery of the cannabinoid receptors (CB1R and CB2R), several studies have been extensively devoted to their synthetic ligands. In fact, the direct activation of these receptors results in several beneficial effects, in the brain and in the periphery.3–7 CB1R is the most expressed G-protein coupled receptor (GPCR) in brain, where it transduces the endocannabinoid signaling and thereby acts as a modulator of neurotransmitter release.8 In the periphery, CB1R regulates metabolic processes including substrate storage and mobilization.9, 10 On the other hand, CB2R is mainly localised in peripheral immune cells and tissues, though recent studies showed its presence in CNS, and it has been investigated for treating pain and inflammation.11–14
CB1R is shown to be involved in regulation of several physiological functions, such as food intake and energy balance, cardiovascular and reproductive functions, immune modulation and cell apoptosis.15–17 Therefore, CB1R has been suggested as an important target of small-molecule pharmacotherapeutics, for ameliorating some pathological conditions such as pain, nausea, obesity, type 2 diabetes, glaucoma and substance abuse.9, 10
Several CB1R agonist and antagonists/inverse agonists have been reported in literature with different potencies and selectivities, showing effects in several preclinical disease models.3, 18–20 However, the orthosteric action of CB1R agonists and antagonists/inverse agonists induces important side effects.21, 22 In particular, CB1R orthosteric agonists produce psychotropic effects, addiction and cognitive impairment.3, 23 On the other hand, CB1R orthosteric antagonists/inverse agonists are associated with undesirable side effects, such as nausea, anxiety, depression, and in some cases, suicidal tendencies.21, 24
Recently, it has been suggested that CB1R contains (an) allosteric binding site(s) topologically distinct from the orthosteric site.25–29 Compared to orthosteric ligands, allosteric modulators offer several advantages. In fact, they show greater subtype selectivity due to the higher sequence divergence at extracellular allosteric binding sites, respect to the conserved orthosteric domains; furthermore, allosteric modulators show tissue selectivity, because they exert effects only where endocannabinoids are present; finally, the effect of allosteric modulators is saturable because of their dependence on endogenous ligands.30, 31 For these reasons, allosteric modulators might offer an alternative strategy to pharmacologically modulate CB1R without producing unwanted side effects associated with CB1R orthosteric agonists and antagonists/inverse agonists.
Allosteric modulators can be classified as positive allosteric modulators (PAMs) or negative allosteric modulators (NAMs), depending on the type of modulation on the affinity and/or efficacy of the orthosteric agonists.32 Various classes of compounds have been reported as CB1R allosteric modulators, such as the indole derivatives (e.g., the NAM Org27569),25 the urea derivatives (e.g., the NAM PSNCBAM-1),27 and other molecules (e.g the PAMs RTI-37133 and ZCZ01134) (Figure 1).
Figure 1.
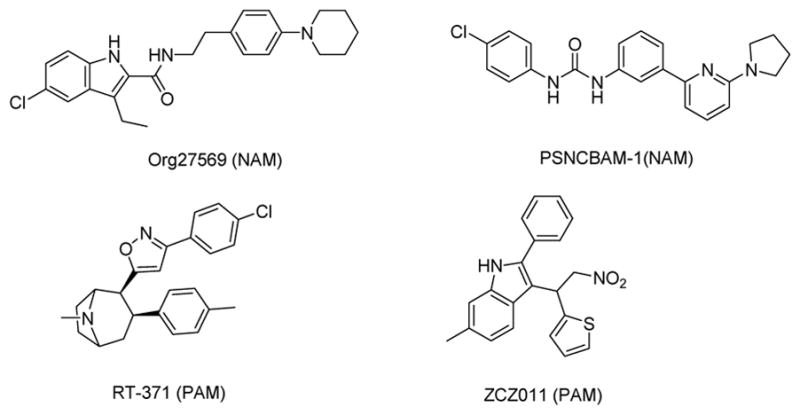
Chemical structures of some CB1R allosteric modulators.
Furthermore, recent works reported natural PAMs and NAMs of CB1R, such as the lipoxin A4, the peptide pepcan-12 (RVD-hemopessin), and pregnenolone (Figure 2).26, 28, 29
Figure 2.
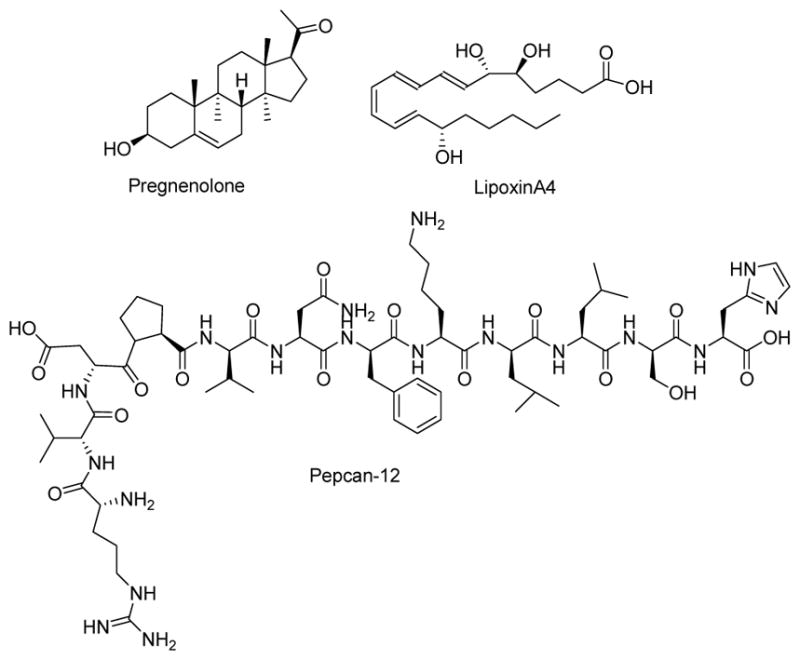
Chemical structures of endogenous CB1R allosteric modulators.
PSNCBAM-1 was discovered by Prosidion Limited in 2007 through an high throughput screening of a small library.27 This compound showed a pharmacological profile similar to Org27569, increasing the binding of CB1R agonists but decreasing their functional responses in several in vitro assays. In an in vivo study of acute feeding model, PSNCBAM-1 showed to decrease food intake and body weight.27 Several analogs of PSNCBAM-135–37 have been reported and structure-activity relationship studies showed that alkyl substitution at the 2-aminopyridine moiety is important for CB1R allosteric modulation; in particular, tertiary amine substitution is more favorable than secondary. Furthermore, the 4-position on the phenyl group tolerates structural modifications, but electron-withdrawing groups such as cyano or CF3 are preferred,35 and the substitution of the urea group with other spacers such as carbamate, methylated ureas or cyclic ureas, reduces the activity.36
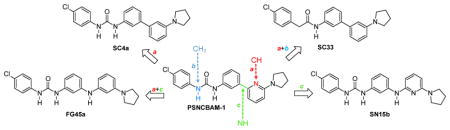
In an effort to deepen the knowledge on structural requirements for CB1R allosteric modulation within the PSNCBAM-1 template, we decided to introduce further modifications on the structure of PSNCBAM-1 obtaining compounds SC4a, SC33, SN15b and FG45a (Figure 3).
Figure 3.
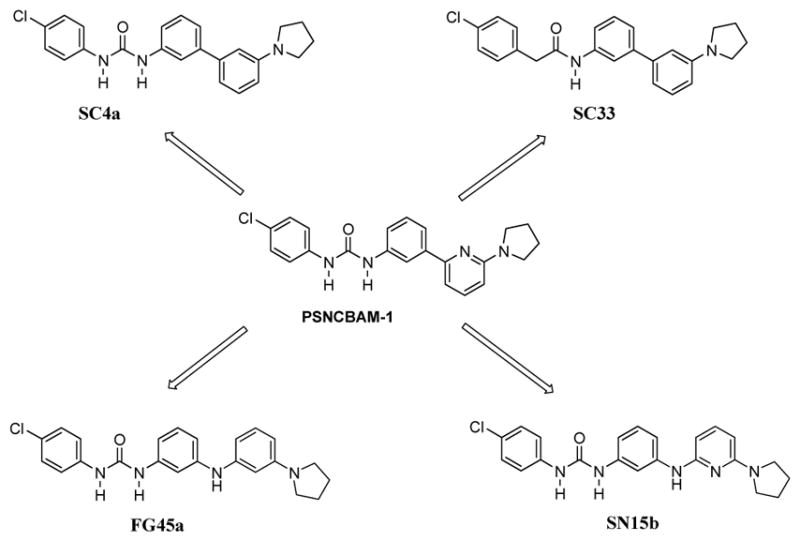
Compounds SC4a, SC33, SN15b and FG45a structurally derived from urea derivative PSNCBAM-1.
In particular, we investigated the role of the pyridine nitrogen atom by replacing it with a carbon atom (SC4a, FG45a). The derivative SC4a was previously reported36 and its pharmacological activity was determined by cAMP Hunter assay and CNR1 PathHunter assay (β-arrestin), but competitive radioligand experiments were not conducted. SC4a was prepared in our laboratory following a different synthetic route respect to that reported in Ref. 36, with an overall comparable yield. Moreover, we verified the importance of the urea group by replacing one NH group with a methylene group, thus obtaining the carboxamide derivative SC33. Finally, we evaluated the influence due to the introduction of a NH group between the pyridine ring and phenyl nucleus (SN15b, FG45a). For all these compounds, the 4-chlorophenyl group and the 2-pyrrolydyl substituent have been selected based on previous results obtained for PSNCBAM-1 analogs.35
In this work we reported the synthesis of compounds SC4a, SC33, SN15b and FG45a and their biological evaluation on the cannabinoid receptors, on the major metabolic enzymes of endocannabinoids, fatty acid amide hydrolase (FAAH), and monoacylglycerol lipase (MAGL) and on anandamide (AEA) uptake. Furthermore, the activity of the target compounds at CB1R was assessed in serum response element (SRE) assay.
2. Methods
2.1. Chemical synthesis
The synthesis of compound SC4a and SC33 is described in Scheme 1. Commercial 1,3-dibromobenzene 1 was subjected to a monoamination with pyrrolidine in the presence of a catalytic system constituted by tris(dibenzylideneacetone)dipalladium(0) (Pd2dba3), 2,2′-bis(diphenylphosphino)-1,1′-binaphtalene (BINAP) and t-BuONa (mol ratio: 0.05:0.15:2) in toluene, affording the intermediate 2. The subsequent Suzuki cross-coupling reaction with 3-nitrophenylboronic acid, in the presence of Pd(OAc)2, P(Ph)3 and acqueous Na2CO3 in 1,2-dimethoxyethane (DME), afforded compound 3. Then the nitro group was reduced with Raney nickel and hydrazine hydrate in EtOH and the amino derivative 4 thus obtained was treated with 4-chloro-phenylisocyanate in anhydrous CHCl3, affording the desired urea derivative SC4a. The treatment of 4 with (4-chlorophenyl)acetyl chloride in the presence of Et3N and DMAP in anhydrous CH2Cl2 afforded the final amide derivative SC33.
Scheme 1.
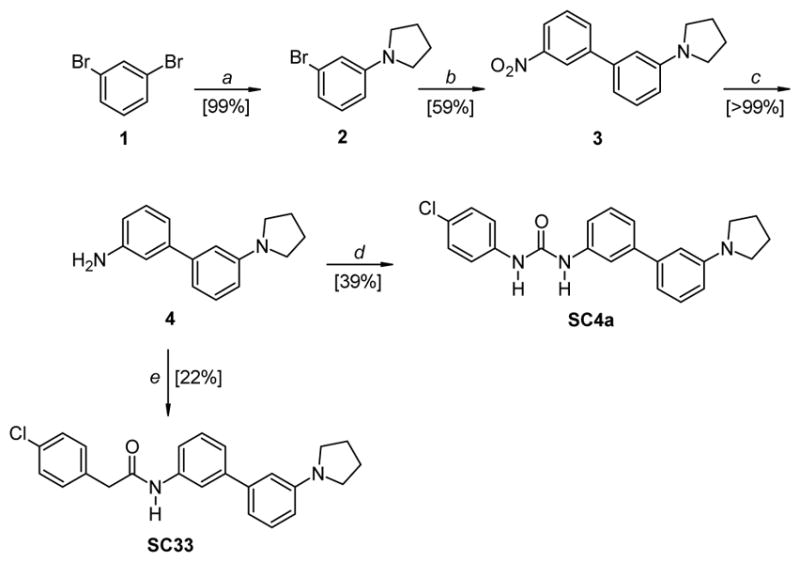
Reagents and conditions: a) pyrrolidine, Pd2(dba)3: BINAP: t-BuONa (mol ratio: 0.05:0.15:2), toluene, 80 °C, 4.5 h; b) 3-nitrophenylboronic acid, Pd(OAc)2, P(Ph)3, aq. NaHCO3, DME, 95 °C, ovenight; c) Raney nickel, NH2NH2·H2O, EtOH, 50 °C, 40 min – 1 h; d) 4-chloro-phenylisocyanate, anhydrous CHCl3, RT, 12 h; e) (4-chlorophenyl)acetyl chloride, Et3N, DMAP, anhydrous CHCl3, RT, 24 h.
The synthesis of compound FG45a is described in Scheme 2. The intermediate 5 was obtained by treating 2 with commercial 1,3-diaminobenzene 6, using the same reaction conditions described for the first step of Scheme 1. The subsequent reaction of 5 with 4-chloro-phenylisocyanate in anhydrous CHCl3 afforded the desired urea derivative FG45a.
Scheme 2.
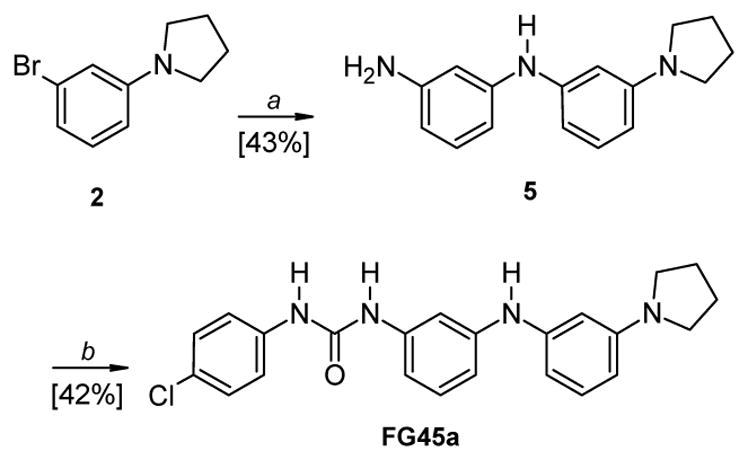
Reagents and conditions: a) 1,3-diaminobenzene, Pd2(dba)3: BINAP: t-BuONa (mol ratio: 0.05:0.15:2), toluene, 80 °C, 4.5 h; b) 4-chloro-phenylisocyanate, anhydrous CHCl3, RT, 12 h.
The synthesis of compound SN15b is described in Scheme 3. Commercially available 1,3-diaminobenzene 6 and 2,6-dibromopyridine were refluxed for 48 h in anhydrous toluene, in the presence of an excess of t-BuOK, obtaining the intermediate 7, which was refluxed in an excess of pyrrolidine for 12 h, affording compound 8. The subsequent reaction of 8 with 4-chloro-phenylisocyanate in anhydrous CHCl3 afforded the desired urea derivative SN15b.
Scheme 3.
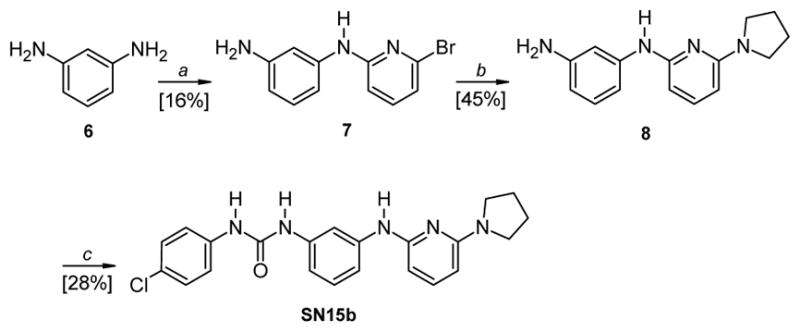
Reagents and conditions: a) 2,6-dibromopyridine, t-BuOK, anhydrous toluene, reflux, 48 h; b) pyrrolidine, reflux, 12 h; c) 4-chloro-phenylisocyanate, anhydrous CHCl3, RT, 12 h.
2.2. Biological assays
Cannabinoid receptors binding was evaluated by incubating the target compounds at different concentrations with membrane preparations obtained from CHO-K1 cells overexpressing hCB1R or hCB1R in presence of 0.5 nM of [3H]CP55,940.38
The inhibitory activity of all four compounds and of compound PSNCBAM-1 on FAAH, MAGL and on AEA cell uptake was evaluated using U937 cell homogenate for metabolic enzymes and intact U937 cells for AEA uptake, in the presence of AEA and of [ethanolamine-1-3H]AEA (0.5 nM) as tracer.39, 40 Finally the functional activity of the new compounds was determined by SRE assay using HEK293 cells transfected with CB1R.41
3. Results and Discussion
Since the discovery of PSNCBAM-1, some SAR studies35–37 showed that CB1R allosteric modulators with improved affinity, efficacy and potency, could be generated from PSNCBAM-1 template. With the aim to extend the knowledge about the structural requirements of PSNCBAM-1 template as CB1R allosteric modulator, in this work we studied some derivatives designed by introducing various modifications on PSNCBAM-1 structure.
Firstly, with the purpose of studying the role of the pyridine nitrogen atom, we synthesized compound SC4a bearing a phenyl ring in replacement of the pyridine nucleus. Pharmacological characterization of SC4a showed its behavior as positive allosteric modulator for the orthosteric ligand [3H]CP55,940 at CB1R, inducing a dose-dependently binding increase by approximately 15–20%. The estimated EC50 is 43 (5–520) nM (Fig. 4).
Figure 4.

CB1 receptor binding. Compounds SC4a, SN15b, SC33, FG45a and PSNCBAM-1 were incubated at different concentrations with membrane preparations obtained from CHO-K1 cells overexpressing hCB1R in the presence of 0.5 nM of [3H]CP55,940. Data show mean values ± SD of at least 3 independent experiments each performed in triplicates.
In the second step, with the aim to verify the importance of the urea group, we synthesized compound SC33 characterized by a carboxamide group. The results of pharmacological studies showed that SC33 does not influence [3H]CP55,940 binding to CB1R up to 10 μM, while only a minor binding (approx. 10%) seems to occur at 100 μM (Fig. 4).
Furthermore, we evaluated the influence of NH group placed between the pyridine ring and the phenyl nucleus, synthesizing compound SN15b. This derivative behaves as a positive allosteric modulator at CB1R for the orthosteric ligand [3H]CP55,940. The estimated EC50 is 1.3 (0.41–5.8) μM and the binding is increased by 35% (Fig. 4).
Finally, we synthesized compound FG45a as an analog of SC33 and characterized by the same spacer (NH) of SN15b between the two phenyl rings. FG45a behaves as a positive allosteric modulator by enhancing [3H]CP55,940 binding to CB1R with an EC50 value of 13.5 (5.1–35.3) μM and reaching 182% of maximal binding (Fig. 4).
We also tested the progenitor compound PSNCBAM-1 which increased [3H]CP55,940 binding at CB1R by 15–20% with an EC50 value of 270 (140–520) nM, thus showing a similar behavior as its analogs SC4a, SN15b and FG45a (Table 1 and Fig. 4)
Table 1.
Summary data for SC33, SN15b, SC4a and FG45a.a
| EC50 (μM) | IC50 (μM) | Ki value (μM) | IC50 (μM) | |||
|---|---|---|---|---|---|---|
|
| ||||||
| Compound | CB1RBinding | CB1RSRE assay | CB2RBinding | FAAH | MAGL | AEA uptake |
| PSNCBAM-1 | 0.27 (0.14–0.52) | 0.23 (0.16–0.34) | >10 | 2.1 (1.9–2.4) | >100 | >10 |
|
| ||||||
| SC33 | >10 | 1.9 (0.52–6.9) | >100 | >10 | >10 | >10 |
| SN15b | 1.3(0.41–5.8) | 0.18 (0.09–0.36) | 52.6(21.5–88.4) | >10 | >10 | >10 |
| SC4a | 0.043(0.005–0.52) | 0.29 (0.10–0.84) | 49.8(12.9–91.5) | >10 | >10 | >10 |
| FG45a | 13.5(5.1–35.3) | 9.6 (0.36 – 25) | >100 | >10 | >10 | >10 |
Data show mean and 95% CI; n = 3.
Functional activity was evaluated using a serum response element (SRE) reporter assay. HEK293 cells were transiently transfected with both pGL4.33 [luc2P/SRE/Hygro] and hCB1R. The next day cells were treated with ligands for 5 h, following which transcriptional activation of the SRE was detected using luminescence from the luciferase reporter (Ref 41 and experimental procedures). The SRE assay assesses the contribution of MAPK/ERK signalling pathway and thus encompasses both G-protein dependent and G-protein independent signalling.41 Recent findings showed the roles of MAPK/ERK signaling pathways in human neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis.42 Moreover the ERK signaling pathway plays a central role in several steps of cancer development.43 As shown in Figure 5, all the compounds inhibited the response produced by CP55,940 at 33 nM (concentration corresponding to EC80). SC4a and SN15b were the most potent, with IC50 values of 291 (101–848) nM and 182 (92–357) nM, respectively, which were comparable to that of PSNCBAM-1 (IC50 value of 234 (161–337) nM). SC33 and FG45 were less potent, with IC50 values of 1.9 (0.52–6.9) μM and 9.6 (0.36–25) μM, respectively.
Figure 5.
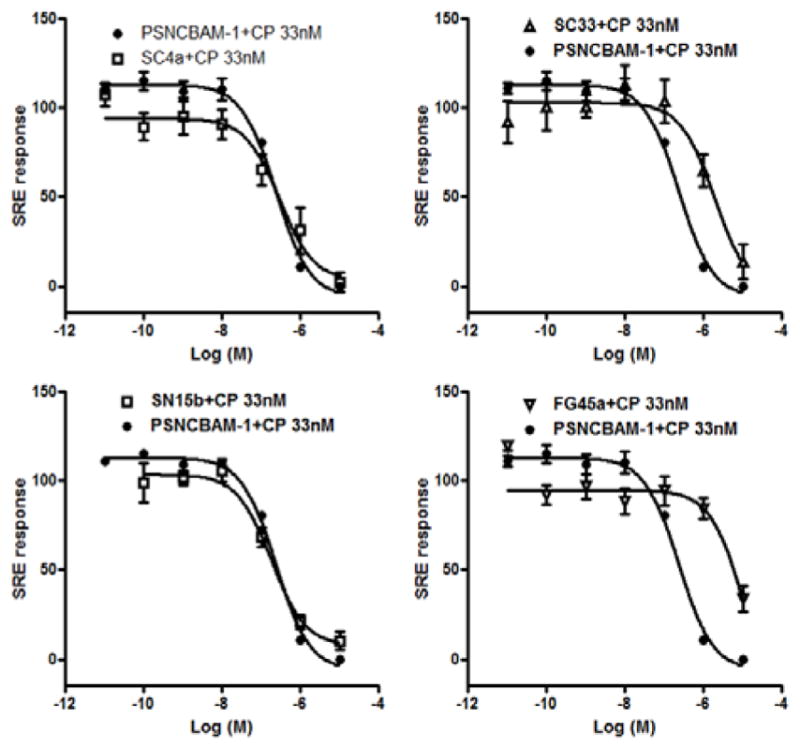
Compounds SC4a, SN15b, SC33 and FG45a were found to decrease the SRE response induced by CB1 agonist CP55,940 (33 nM). PSNCBAM-1 was used as a reference compound. Data show mean values ± SD of at least 3 independent experiments each performed in triplicates.
Compounds were also tested for binding to CB2R and for interaction with the other major targets of the ECS. As shown in Figure 6, compound SC4a and SC33 weakly bound to CB2R with Ki values of approximately 50 μM, while SN15b and FG45a did not significantly interact with the receptor up to 100 μM. The binding curves suggest that SC4a and SC33 behave as weak orthosteric CB2R ligands and do not interact with a distinct allosteric pocket. PSNCBAM-1 did not show any significant binding to CB2R up to 10 μM.
Figure 6.
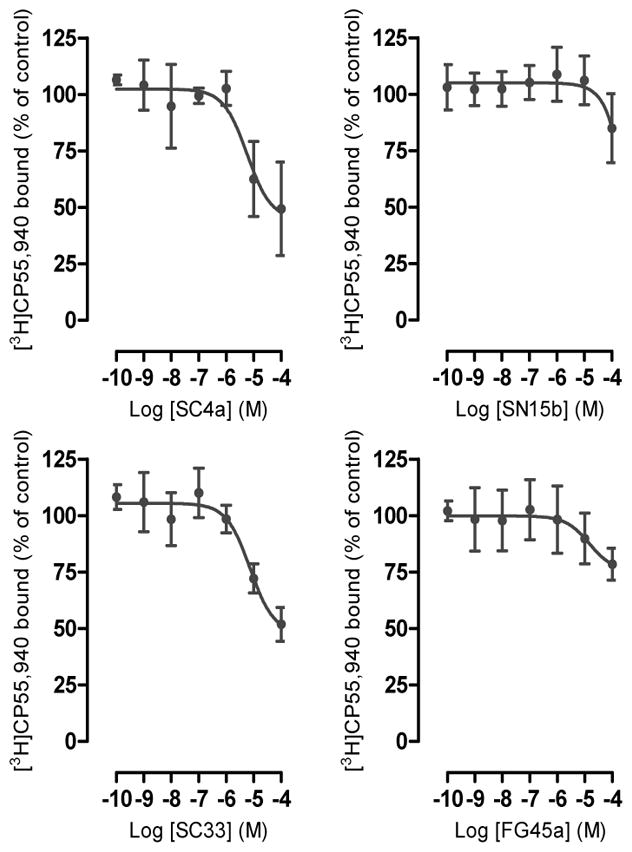
CB2 receptor binding. Compound SC4a, SN15b, SC33 and FG45a were incubated at different concentrations with membrane preparations obtained from CHO-K1 cells overexpressing hCB2 receptors in presence of 0.5 nM of [3H]CP55,940. Data show mean values ± SD of at least 3 independent experiments each performed in triplicates.
In further experiments, all four compounds did not exhibit any significant inhibition of FAAH, MAGL and AEA cell uptake activity up to 10 μM. The progenitor compound PSNCBAM-1 was negative for MAGL and AEA cell uptake, but inhibited FAAH activity with an IC50 value of 2.1 (1.9–2.4) μM (Table 1). Altogether, these data show that, analogously to PSNCBAM-1, SN15b and SC4a are potent PAMs at CB1R for the orthosteric ligand [3H]CP55,940 with high selectivity over CB2R and the other targets of the ECS (Table 1). Furthermore all new compounds decreased the functional response in SRE assay showing to be NAMs.
4. Conclusion
With the aim to deepen the knowledge on structural requirements for CB1R allosteric modulation, we have designed and synthesized some analogs of urea derivative PSNCBAM-1, a known allosteric modulator of CB1R. The new compounds showed to be PAMs on binding affinity of orthosteric ligand (CP55,940) but NAMs in functional assays. This behavior is in line with the complex receptor pharmacology previously described for PSNCBAM-1.27, 44, 45 The obtained results showed that the pyridine nitrogen atom in PSNCBAM-1 is not an important feature for CB1R modulating activity, indeed the compound SC4a, concentration-dependently increased [3H]CP,55940 binding to CB1R. Furthermore, a spacer (NH group) between the pyridine ring and phenyl nucleus (SN15b and FG45a) is allowed for the modulation of CB1R activity. These compounds showed also high selectivity over CB2R and the other targets of the ECS. Moreover, all compounds inhibited the response produced by CP55,940 in SRE assay, indicating a behaviour as NAMs. In particular, SC4a and SN15b were the most potent, reducing the MAPK/ERK signalling pathway with an activity similar to that of PSNCBAM-1. The new urea derivatives presented here provide further insights regarding the modulation of CB1R binding and offer new opportunities for modulating specific signalling pathway associated with CB1R, avoiding unwanted side effects usually caused by CB1R orthosteric modulation.
5. Experimental
5.1. Chemistry
Commercially available reagents were purchased from Sigma Aldrich or Alfa Aesar and used without purification. 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE III™ 400 spectrometer (operating at 400 MHz). Chemical shift (δ) are reported in parts per million related to the residual solvent signal, while coupling constants (J) are expressed in Hertz (Hz). All synthesized compounds were analyzed by HPLC, showing a purity ≥ 95%. A Bechman HPLC instrument equipped with a System Gold Solvent Delivery module (Pumps) 125, System Gold UV/VIS Detector 166, Detector set to 278 nm was employed. Analyses were performed on a reverse phase C18 column (Phenomenex 250 × 4.6 mm, 5 mm particle size, Gemini). The mobile phase was constituted by a mixture of H2O/AcOH (0.1% v/v) (eluent A) and ACN (eluent B). A gradient starting from 50% of B, changing to 100% of B over 20 min., and returning to the initial conditions over 10 min., was used for compounds SC4a, SC33b and FG45a. For compound SN15b was chosen an isocratic elution with 100% of B. The flow rate was 1.0 ml/min. Evaporation was carried out in vacuo using a rotating evaporator. Silica gel flash chromatography was performed using silica gel 60 Å (0.040–0.063 mm; MERK). Reactions was monitored by TLC on Merck aluminium silica gel (60 F254) plates that were visualized under a UV lamp (λ = 254 nm). Melting points were determined on a Kofler hot-stage apparatus and are uncorrected.
5.1.1. 1-(3-Bromophenyl)pyrrolidine (2)
Commercial 1,3-dibromobenzene 1 (200.0 mg, 0.85 mmol), pyrrolidine (60.5 mg, 0.85 mmol) and 157.0 mg of a reagent constituted by tris(dibenzylideneacetone)dipalladium(0), BINAP and t-BuONa (mol ratio: 0.05:0.15:2) were suspended in toluene (2.5 mL) and allowed to react in a sealed tube, at 80 °C for 4.5 h, under magnetic stirring. After cooling to room temperature, the mixture was filtered under reduced pressure and the resulting solution evaporated in vacuo. The residue was dissolved in CHCl3 and washed with a 10% aqueous solution of NaOH. The organic phase was dried over anhydrous Na2SO4, filtered, evaporated in vacuo, and purified by flash column chromatography on silica gel (petroleum ether), obtaining the intermediate 2 as a transparent oil (190.5 mg, 0.84 mmol). Yield: 99%. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.05 (t, 1H, J = 8.0 Hz), 6.76–6.74 (m, 1H), 6.68 (m, 1H), 6.46 (m, 1H), 3.27–3.24 (m, 4H), 2.05–1.99 (m, 4H).
5.1.2. 1-(3′-Nitrobiphenyl-3-yl)pyrrolidine (3)
In a sealed tube, under a flux of nitrogen, were introduced DME (40.0 mL), Pd(OAc)2 (158.1 mg, 0.24 mmol) and P(Ph)3 (307.9 mg, 1.17 mmol). The mixture was left under magnetic stirring at room temperature for 15 min, allowing the in situ formation of the catalyst. Then, compound 2 (701.0 mg, 3.10 mmol), NaHCO3 (775.0 mg, 9.23 mmol), H2O (17.6 mL) and 3-nitrophenylboronic acid (571.5 mg, 3.42 mmol) were added. The mixture was allowed to react at 95 °C, overnight. After cooling to room temperature, the solvent was removed in vacuo and the residue partitioned between water and ethyl acetate. The organic phase was dried over anhydrous Na2SO4, filtered, evaporated in vacuo, and purified by flash column chromatography on silica gel (petroleum ether/CH2Cl2 7:3), obtaining the intermediate 3 as an orange solid (491.0 mg, 1.83 mmol). Yield: 59%. 1H NMR (CDCl3, 400 MHz) δ (ppm): 8.46–8.45 (m, 1H), 8.19–8.16 (m, 1H), 7.94–7.91 (m, 1H), 7.58 (t, 1H, J = 7.9 Hz), 7.33 (t, 1H, J = 7.8 Hz), 6.89 (d, 1H, J = 8.0 Hz), 6.75–6–74 (m, 1H), 6.65–6.63 (m, 1H), 3.38–3.35 (m, 4H), 2.07–2.03 (m, 4H).
5.1.3. 3′-(Pyrrolidin-1-yl)biphenyl-3-amine (4)
Hydrazine hydrate (0.2 mL, 66 mmol) was added to a solution of 3 (63.8 mg, 0.29 mmol) in ethanol (4.0 mL). The mixture was stirred at 50 °C for 15 min. Then, an excess of Raney nickel (∼ 60 mg) was added. After the bubbling ceased (the solution changed from orange to colorless), the mixture was cooled to room temperature and filtered through a celite pad. The filtrate was evaporated in vacuo, affording intermediate 4 (58.5 mg) as a white solid, which was used in the next step without further purification. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.28 (t, 1H, J = 7.8 Hz), 7.22 (t, 1H, J = 7.8 Hz), 7.03–7.01 (m, 1H), 6.95–6.92 (m, 1H), 6.88–6.85 (m, 1H), 6.75–6.73 (m, 1H), 6.69–6.65 (m, 1H), 6.59–6.55 (m, 1H), 3.36–3.33 (m, 4H), 2.05–1.99 (m, 4H).
5.1.4. N-[3-(pyrrolidin-1-yl)phenyl]-1,3-diamino-benzene (5)
Obtained by treating 2 (714.0 mg, 3.16 mol) with commercial 1,3-diaminobenzene 6 (410 mg, 3.79 mmol) and 628.0 mg of a reagent constituted by tris(dibenzylideneacetone)dipalladium(0), BINAP and t-BuONa (mol ratio: 0.05:0.15:2) in toluene (7.9 mL), following the same reaction and work-up conditions used for the synthesis of 2. The crude was purified by flash column chromatography on silica gel (petroleum ether/AcOEt 9:1), obtaining 5 as an yellow oil (342.0 mg, 1.35 mmol). Yield: 43%. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.11 (t, 1H, J = 8.0 Hz), 7.02 (t, 1H, J = 8.0 Hz), 6.50–6.42 (m, 3H), 6.32–6.31 (m, 1H), 6.25–6.20 (m, 2H), 5.58 (bs, 1H), 3.66 (bs, 2H), 3.28–3.25 (m, 4H), 2.00–1.97 (m, 4H).
5.1.5. N-(6-bromopyridin-2-yl)benzene-1,3-diamine (7)
A mixture of 1,3-diaminobenzene 6 (2.60 g, 24.0 mmol), 2,6-dibromopyridine (7.11 g, 30.0 mmol), t-BuOK (6.16 g, 55.2 mmol) and anhydrous toluene (130.0 mL) was refluxed for 48 h, under magnetic stirring. After cooling to room temperature, the solvent was removed in vacuo, the residue dissolved in AcOEt and washed with water. The organic phase was dried over anhydrous Na2SO4, filtered, evaporated in vacuo, and purified by flash column chromatography on silica gel (petroleum ether/AcOEt 6:4), obtaining the intermediate 7 as a brown oil (147.0 mg, 0.56 mmol). Yield: 16%. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.29 (td, 1H, J = 7.9 Hz, J = 0.3 Hz), 7.10 (td, 1H, J = 7.7 Hz, J = 0.8 Hz), 6.86 (dd, 1H, J = 7.5 Hz, J = 0.6 Hz), 6.81 (dd, 1H, J = 8.2 Hz, J = 0.6 Hz), 6.63–6.58 (m, 2H), 6.44–6.41 (m, 1H), 4.00 (bs, 2H).
5.1.6. N-[6-(pyrrolidin-1-yl)pyridin-2-yl]benzene-1,3-diamine (8)
A mixture of 7 (362.0 mg, 1.37 mmol) and pyrrolidine (2.17 mL) was refluxed for 12 h. After cooling to room temperature, pyrrolidine was removed in vacuo, obtaining a brown oil that was dissolved in CHCl3 and washed with water. The organic phase was dried over anhydrous Na2SO4, filtered, evaporated in vacuo, and purified by flash column chromatography on silica gel (petroleum ether/AcOEt 6:4), obtaining the intermediate 8 as a white solid (153.0 mg, 0.60 mmol). Yield: 45%. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.29 (t, 1H, J = 7.9 Hz), 7.06 (t, 1H, J = 7.9 Hz), 6.87–6.84 (m, 1H), 6.72 (dd, 1H, J = 8.0 Hz, J = 1.9 Hz), 6.43 (bs, 1H), 6.32 (dd, 1H, J = 7.8 Hz, J = 1.9 Hz), 6.12 (d, 1H, J = 7.8 Hz), 5.82 (d, 1H, J = 8.1 Hz), 3.47–3.42 (m, 4H), 3.20 (bs, 2H), 2.01–1.96 (m, 4H).
5.1.7. General procedure for the synthesis of urea derivatives SC4a, FG45a and SN15b
4-Chloro-phenylisocyanate (1 eq) was added to a solution of the suitable amine intermediate (1 eq) in the minimum amount of freshly distilled CHCl3. The resulting mixture was left at room temperature, under magnetic stirring, for 12 h. The solvent was removed by evaporation in vacuo or by filtration and the residue purified.
5.1.8. 1-(4-Chlorophenyl)-3-[3′-(pyrrolidin-1-yl)biphenyl-3-yl]urea (SC4a)
Prepared from 4-chloro-phenylisocyanate (39.0 mg, 0.25 mmol) and intermediate 4 (58.5 mg, 0.25 mmol) dissolved in 10.2 mL of freshly distilled CHCl3. Purification by flash column chromatography on silica gel (petroleum ether/AcOEt 7:3). SC4a (37.1 mg, 0.10 mmol). Yield: 39%. M. p.: 168–172 °C. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.84 (s, 1H), 8.79 (s, 1H), 7.72–7.69 (m, 1H), 7.49 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′XX′ = 1.6 Hz), 7.43–7.41 (m, 1H), 7.35–7.30 (m, 1H), 7.33 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′XX′ = 1.6 Hz), 7.26–7.22 (m, 2H), 6.81 (d, 1H, J = 7.3 Hz), 6.72–6.68 (m, 1H), 6.55 (dd, 1H, J = 8.0 Hz, J = 1.7 Hz), 3.30–3.27 (m, 4H), 1.99–1.96 (m, 4H). 13C NMR (DMSO-d6, 400 MHz) δ (ppm): 152.49, 148.07, 141.98, 141.15, 139.89, 138.71, 129.44, 129.16, 128.61, 125.32, 120.56, 119.75, 117.20, 116.71, 113.76, 110.96, 109.78, 41.33, 24.96. HPLC analysis: retention time = 20.62 min; peak area, 99% (280 nm).
5.1.9. 2-(4-Chlorophenyl)-N-[3′-(pyrrolidin-1-yl)biphenyl-3-yl]acetamide (SC33)
The reaction was conducted under nitrogen atmosphere. Et3N (29.3 mg, 0.29 mmol) and DMAP (35.4 mg, 0.29 mmol) were added to a solution of 4 (69.4 mg, 0.29 mmol) in anhydrous CH2Cl2 (2.0 mL). Then, the mixture was cooled to −5 °C (ice-NaCl bath) and (4-chlorophenyl)acetyl chloride (54.8 mg, 0.29 mmol) was added. The reaction mixture was left, under magnetic stirring, at room temperature for 24 h. The crude was washed twice with an aqueous saturated solution of NaHCO3 and purified by flash column chromatography on silica gel (petroleum ether/AcOEt 8:2), obtaining the desired product SC33 as a solid (24.8 mg, 0.06 mmol). Yield: 22%. M. p.: 138–140 °C. 1H NMR (CDCl3, 400 MHz) δ (ppm): 7.68 (bs, 1H), 7.64 (bs, 1H), 7.52–7.47 (m, 1H), 7.40–7.30 (m, 4H), 7.30–7.23 (m, 2H), 6.86 (d, 1H, J = 7.6 Hz), 6.75–6.71 (m, 1H), 6.59 (dd, 1H, J = 8.1 Hz, J = 1.9 Hz), 3.67 (s, 2H), 3.35–3.32 (m, 4H), 2.04–2.01 (m, 4H). 13C NMR (CDCl3, 400 MHz) δ (ppm): 168.66, 141.84, 137.90, 133.75, 133.02, 130.98, 129.67, 129.43, 129.36, 123.77, 118.85, 118.78, 44.21, 29.85, 25.56. HPLC analysis: retention time = 20.75 min; peak area, 98% (280 nm).
5.1.10. 1-(4-Chlorophenyl)-3-(3-{[3-(pyrrolidin-1-yl)phenyl]amino}phenyl)urea (FG45a)
Prepared from 4-chloro-phenylisocyanate (31.8 mg, 0.21 mmol) and intermediate 5 (52.5 mg, 0.21 mmol) dissolved in 8.5 mL of freshly distilled CHCl3. Purification by flash column chromatography on silica gel (CHCl3) and subsequent crystallization from EtOH. FG45a (34.7 mg, 0.09 mmol). Yield: 42%. M. p.: 168–171 °C. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.73 (s, 1H), 8.60 (s, 1H), 7.97 (s, 1H); 7.46 (AA′XX′, 2H, JAX = 9.0 Hz, JAA′XX′ = 2.6 Hz), 7.36–7.35 (m, 1H), 7.31 (AA′XX′, 2H, JAX = 8.9 Hz, JAA′XX′ = 2.6 Hz), 7.07 (t, 1H, J = 8.0 Hz), 7.00 (t, 1H, J = 8.0 Hz), 6.79–6.77 (m, 1H), 6.66–6.64 (m, 1H), 6.36–6.30 (m, 2H), 6.08–6.07 (m, 1 H), 3.22–3.19 (m, 4H), 1.94–1.92 (m, 4H). 13C NMR (DMSO-d6, 400 MHz) δ (ppm): 152.35, 148.57, 144.44, 143.78, 140.17, 138.80, 129.43, 129.25, 128.60, 125.20, 119.59, 110.64, 109.17, 106.04, 105.18, 104.29, 100.78, 47.24, 24.95. HPLC analysis: retention time = 16.53 min; peak area, 97% (280 nm).
5.1.11. 1-(4-Chlorophenyl)-3-{[6-(pyrrolidin-1-yl)pyridin-2-yl]amino}phenyl)urea (SN15b)
Prepared from 4-chloro-phenylisocyanate (20.3 mg, 0.13 mmol) and intermediate 8 (33.8 mg, 0.13 mmol) dissolved in 5.4 mL of freshly distilled CHCl3. Purification by crystallization from EtOH. SN15b (14.8 mg, 0.04 mmol). Yield: 28%. M. p.: dec. at 220 °C. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 8.81 (s, 1H), 8.67 (s, 1H), 8.54 (s, 1H), 7.88–7.82 (m, 1H), 7.50–7.47 (m, 2H), 7.44–7.42 (m, 1H), 7.33–7.31 (m, 2H), 7.28–7.24 (m, 1H), 7.11–7.07 (m, 1H), 6.81 (d, 1H, J = 8.1 Hz), 6.03 (d, 1H, J = 7.9 Hz), 5.77 (d, 1H, J = 7.6 Hz), 3.44–3.37 (m, 4H), 1.96–1.90 (m, 4H). 13C NMR (DMSO-d6, 400 MHz) δ (ppm): 155.99, 154.65, 152.31, 142.80, 139.52, 138.88, 138.03, 128.58, 125.11, 119.52, 111.76, 110.06, 107.86, 97.03, 95.56, 46.42, 25.01. HPLC analysis: retention time = 4.65 min; peak area, 95% (280 nm).
5.2. Biological assays
5.2.1. Radioligand binding assays on CB1R and CB2R
Cannabinoid receptors affinity was determined using membrane preparations obtained from CHO-K1 cells stably transfected with hCB1 or hCB2 as previously described.38 Briefly, 20 mg of membrane preparation were resuspended in 500 μL of binding buffer (50 mM Tris-HCl, 2.5 mM EDTA, 5 mM MgCl2, 0.5% fatty acid free BSA, pH 7.4) in silanized glass tubes. Membranes were incubated with the compounds at different concentrations (1 nM–100 μM) in presence of 0.5 nM of [3H]CP55,940 (168 Ci·mmol−1) for 1.5 h at 30 °C. Non-specific binding of the radioligand was determined in the presence of 10 μM of (R)-WIN55,512–2. Non-specific binding was around 10%. After the incubation time, membrane suspensions were rapidly filtered through a 0.05% polyethyleneimine presoaked 96-well microplate bonded with GF/B glass fiber filters (UniFilter-96GF/B, Perkin Elmer Life Sciences) under vacuum and washed 10 times with 167 μL of ice-cold washing buffer (50 mM Tris-HCl, 2.5 mM EDTA, 5 mM MgCl2, 0.5% fatty acid free BSA, pH 7.4). Filters were added to 3 mL of MicroScint 20 scintillation liquid and radioactivity was measured with the 1450 Micro Beta Trilux top counter. The non-specific binding was subtracted and results were expressed as [3H]CP55,940 bound as % of binding in vehicle-treated samples.
5.2.2. FAAH, MAGL and AEA uptake assays
FAAH and MAGL activity assay and AEA uptake assays were performed as previously described.39,40 Briefly, FAAH and MAGL activity assays were performed using U937 cell homogenate (100 μg) which were diluted in 200 μL of Tris–HCl 10 mM, EDTA 1 mM, pH 8 containing 0.1% fatty acid-free BSA. Compounds were added at the screening concentration of 10 μM and incubated for 30 min at 37 °C. Then, 100 nM of AEA containing 1 nM of [ethanolamine-1–3H]AEA as a tracer was added to the homogenates and incubated for 15 min at 37 °C. The reaction was stopped by the addition of 400 μL of ice-cold CHCl3:MeOH (1:1), samples were vortexed and rapidly centrifuged at 16’000 × g for 10 min. at 4 °C. The aqueous phases were collected and the radioactivity was measured for tritium content by liquid scintillation spectroscopy. For AEA uptake assay 0.5 × 106 of intact U937 cells were suspended in 500 μL of serum-free medium in silanized glass tubes and preincubated with a screening concentration (10 μM) of the compounds for 20 min at 37 °C. Then, the cells were incubated for 5 min at 37 °C with 100 nM of AEA and [ethanolamine-1-3H]AEA (0.5 nM) was added. The uptake process was stopped by transferring the tubes on ice and by rapid filtration over UniFilter®-96 GF/C filters pre-soaked with PBS supplemented with 0.2% BSA. Cells were washed three times with 100 μL ice-cold PBS supplemented with 1% fatty acid free BSA. After drying, 50 μL MicroScint 20 scintillation cocktail was added to the wells. The radioactivity was measured using a Trilux MicroBeta 1450.
5.2.3. Serum response element (SRE) assay
HEK293 cells were transiently transfected with hCB1R (5 ng/well) and pGL4.33 [luc2P/SRE/Hygro] vector (100 ng/well) reporter (Promega) plasmids using Lipofectamine 2000 as described by the manufacturer (Invitrogen).46 Transfected HEK293 cells were seeded (60,000 cells per well) in 96-well plates. Five hours later, medium was changed to 1% FBS/DMEM. Cells were incubated overnight. The next day cells were treated with ligands for 5 h in serum-free DMEM medium at 37 °C. After treatment, cells were lysed by 1X lysis buffer for 10 min. at room temperature. Plates were read to record bioluminescent after the injection of 40μl Luciferin (≥ 250 μM) per well. Luminescence was measured in an Envision 2104 Multilabel Reader (Perkin Elmer). Luminescence values are given as relative light units. Concentration-effect curves for agonist-mediated receptor activation were analyzed by nonlinear regression techniques using GraphPad Prism 5.0 software (GraphPad) and data were fitted to sigmoidal dose-response curves to obtain EC50 values.
Acknowledgments
This research was supported by grants from University of Pisa (Progetti di Ricerca di Ateneo, PRA_2017_51) and from the National Institute on Drug Abuse R01DA023204, R01DA035926 and P30DA013429.
References and notes
- 1.Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and their Ligands: Beyond CB(1) and CB(2) Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pertwee RG. Emerging Strategies for Exploiting Cannabinoid Receptor Agonists as Medicines. Br J Pharmacol. 2009;156:397–411. doi: 10.1111/j.1476-5381.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gowran A, Noonan J, Campbell VA. The multiplicity of action of cannabinoids: implications for treating neurodegeneration. CNS Neurosci Ther. 2011;17:637–644. doi: 10.1111/j.1755-5949.2010.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manera C, Arena C, Chicca A. Synthetic Cannabinoid Receptor Agonists and Antagonists: Implication in CNS Disorders. Recent Pat CNS Drug Discov. 2016;10:142–156. doi: 10.2174/1574889810666160519113853. [DOI] [PubMed] [Google Scholar]
- 6.Pisanti S, Picardi P, D’Alessandro A, Laezza C, Bifulco M. The endocannabinoid signaling system in cancer. Trends Pharmacol Sci. 2013;34:273–82. doi: 10.1016/j.tips.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Pertwee RG. Elevating endocannabinoid levels: pharmacological strategies and potential therapeutic applications. Proc Nutr Soc. 2014;73:96–105. doi: 10.1017/S0029665113003649. [DOI] [PubMed] [Google Scholar]
- 8.Elphick MR. The evolution and comparative neurobiology of endocannabinoid signalling. Philos Trans R Soc London, Ser B. 2012;367:3201–3215. doi: 10.1098/rstb.2011.0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pacher P, Kunos G. Modulating the endocannabinoid system in human health and disease successes and failures. FEBS J. 2013;280:1918–1943. doi: 10.1111/febs.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thakur GA, Tichkule R, Bajaj S, Makriyannis A. Latest advances in cannabinoid receptor agonists. Expert Opin Ther Pat. 2009;19:1647–1673. doi: 10.1517/13543770903436505. [DOI] [PubMed] [Google Scholar]
- 11.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 12.Wilkerson JL, Gentry KR, Dengler EC, Wallace JA, Kerwin AA, Armijo LM, Kuhn MN, Thakur GA, Makriyannis A, Milligan ED. Intrathecal cannabilactone CB(2)R agonist, AM1710, controls pathological pain and restores basal cytokine levels. Pain. 2012;153:1091–1106. doi: 10.1016/j.pain.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manera C, Malfitano AM, Parkkari T, Lucchesi V, Carpi S, Fogli S, Bertini S, Laezza C, Ligresti A, Saccomanni G, Savinainen JR, Ciaglia E, Pisanti S, Gazzerro P, Di Marzo V, Nieri P, Macchia M, Bifulco M. New quinolone- and 1,8-naphthyridine-3-carboxamides as selective CB2 receptor agonists with anticancer and immuno-modulatory activity. Eur J Med Chem. 2015;97:10–18. doi: 10.1016/j.ejmech.2015.04.034. [DOI] [PubMed] [Google Scholar]
- 14.Han S, Thatte J, Buzard DJ, Jones RM. Therapeutic Utility of Cannabinoid Receptor Type 2 (CB2) Selective Agonists. J Med Chem. 2013;56:8224–8256. doi: 10.1021/jm4005626. [DOI] [PubMed] [Google Scholar]
- 15.Howlett AC, Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Porrino LJ. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology. 2004;47(Suppl 1):345–358. doi: 10.1016/j.neuropharm.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 16.Pertwee RG. Cannabinoid pharmacology: the first 66 years. Br J Pharmacol. 2006;147(Suppl 1):S163–S171. doi: 10.1038/sj.bjp.0706406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackie K. Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol. 2006;46:101–122. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- 18.Palmer SL, Thakur GA, Makriyannis A. Cannabinergic ligands. Chem Phys Lipids. 2002;121:3–19. doi: 10.1016/s0009-3084(02)00143-3. [DOI] [PubMed] [Google Scholar]
- 19.Thakur GA, Bajaj S, Paronis C, Peng Y, Bowman AL, Barak LS, Caron MG, Parrish D, Deschamps JR, Makriyannis A. Novel adamantyl cannabinoids as CB1 receptor probes. J Med Chem. 2013;56:3904–3921. doi: 10.1021/jm4000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon DD, Tius MA, Thakur GA, Zhou H, Bowman AL, Shukla VG, Peng Y, Makriyannis A. C3-heteroaroyl cannabinoids as photolabeling ligands for the CB2 cannabinoid receptor. Bioorg Med Chem Lett. 2012;22:5322–5325. doi: 10.1016/j.bmcl.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet. 2007;370:1706–1713. doi: 10.1016/S0140-6736(07)61721-8. [DOI] [PubMed] [Google Scholar]
- 22.Volkow ND, Baler RD, Compton WM. Adverse health effects of marijuana use. N Eng J Med. 2014;370:2219–2227. doi: 10.1056/NEJMra1402309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skosnik PD, D’Souza DC, Steinmetz AB, Edwards CR, Vollmer JM, Hetrick WP. The effect of chronic cannabinoids on broadband EEG neural oscillations in humans. Neuropsychopharmacology. 2012;37:2184–2193. doi: 10.1038/npp.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janero DR. Cannabinoid-1 receptor (CB1R) blockers as medicines: beyond obesity and cardiometabolic disorders to substance abuse/drug addiction with CB1R neutral antagonists. Expert Opin Emerg Drugs. 2012;17:17–29. doi: 10.1517/14728214.2012.660916. [DOI] [PubMed] [Google Scholar]
- 25.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, McLean A, McIntosh L, Goodwin G, Walker G, Westwood P, Marrs J, Thomson F, Cowley P, Christopoulos A, Pertwee RG, Ross RA. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 26.Pamplona FA, Ferreira J, Menezes de Lima O, Jr, Duarte FS, Bento AF, Forner S, Villarinho JG, Bellocchio L, Wotjak CT, Lerner R, Monory K, Lutz B, Canetti C, Matias I, Calixto JB, Marsicano G, Guimarães MZ, Takahashi RN. Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proc Natl Acad Sci USA. 2012;109:21134–9. doi: 10.1073/pnas.1202906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, Reynet C, Wong Kai P. PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol. 2007;152:805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vallée M, Vitiello S, Bellocchio L, Hébert-Chatelain E, Monlezun S, Martin-Garcia E, Kasanetz F, Baillie GL, Panin F, Cathala A, Roullot-Lacarrière V, Fabre S, Hurst DP, Lynch DL, Shore DM, Deroche-Gamonet V, Spampinato U, Revest JM, Maldonado R, Reggio PH, Ross RA, Marsicano G, Piazza PV. Pregnenolone can protect the brain from cannabis intoxication. Science. 2014;343:94–98. doi: 10.1126/science.1243985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauer M, Chicca A, Tamborrini M, Eisen D, Lerner R, Lutz B, Poetz O, Pluschke G, Gertsch J. Identification and quantification of a new family of peptide endocanabinoids (Pepcans) showing negative allosteric modulation at CB1 receptors. J Biol Chem. 2012;287:36944–36967. doi: 10.1074/jbc.M112.382481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Christopoulos A. Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nat Rev Drug Disc. 2002;1:198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- 31.Bridges TM, Lindsley CW. G-protein-coupled receptors: From classical modes of modulation to allosteric mechanisms. ACS Chem Biol. 2008;3:530–541. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]
- 32.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat Rev Drug Disc. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Navarro HA, Howard JL, Pollard GT, Carroll FI. Positive allosteric modulation of the human cannabinoid (CB) receptor by RTI-371, a selective inhibitor of the dopamine transporter. Br J Pharmacol. 2009;156:1178–1184. doi: 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ignatowska-Jankowska BM, Baillie GL, Kinsey S, Crowe M, Ghosh S, Owens RA, Damaj IM, Poklis J, Wiley JL, Zanda M, Zanato C, Greig IR, Lichtman AH, Ross RA. A cannabinoid CB1 receptor-positive allosteric modulator reduces neuropathic pain in the mouse with no psychoactive effects. Neuropsychopharmacology. 2015;40:2948–2959. doi: 10.1038/npp.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.German N, Decker AM, Gilmour BP, Gay EA, Wiley JL, Thomas BF, Zhang Y. Diarylureas as allosteric modulators of the cannabinoid CB1 receptor: structure–activity relationship studies on 1-(4- Chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl)pyridin-2-yl]phenyl}urea (PSNCBAM-1) J Med Chem. 2014;57:7758–7769. doi: 10.1021/jm501042u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thakur GA, Tichkule RB, Kulkarni PM, Kulkarni AR. Allosteric modulators of the cannabinoid 1 receptor. WO2015027160 Patent. 2015
- 37.Kulkarni PM, Kulkarni AR, Korde A, Tichkule RB, Laprairie RB, Denovan-Wright EM, Zhou H, Janero DR, Zvonok N, Makriyannis A, Cascio MG, Pertwee RG, Thakur GA. Novel Electrophilic and Photoaffinity Covalent Probes for Mapping the Cannabinoid 1 Receptor Allosteric Site(s) J Med Chem. 2016;14:44–60. doi: 10.1021/acs.jmedchem.5b01303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chicca A, Caprioglio D, Minassi A, Petrucci V, Appendino G, Taglialatela-Scafati O, Gertsch J. Functionalization of β-caryophyllene generates novel polypharmacology in the endocannabinoid system. ACS Chem Biol. 2014;9:1499–1507. doi: 10.1021/cb500177c. [DOI] [PubMed] [Google Scholar]
- 39.Nicolussi S, Chicca A, Rau M, Rihs S, Soeberdt M, Abels C, Gertsch J. Correlating FAAH and anandamide cellular uptake inhibition using N-alkylcarbamate inhibitors: from ultrapotent to hyperpotent. Biochem Pharmacol. 2014;92:669–689. doi: 10.1016/j.bcp.2014.09.020. [DOI] [PubMed] [Google Scholar]
- 40.Chicca A, Nicolussi S, Bartholomäus R, Blunder M, Rey AA, Petrucci V, del Carmen Reynoso-Moreno I, Viveros-Paredes JM, Dalghi Gens M, Lutz B, Schiöth HB, Soeberdt M, Abels C, Charles R-P, Altmann K-H, Gertsch J. Chemical probes to potently and selectively inhibit endocannabinoid cellular reuptake. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1704065114. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng Z, Garvin D, Paguio A, Stecha P, Wood K, Fan F. Luciferase Reporter Assay System for Deciphering GPCR Pathways. Curr Chem Genomics. 2010;4:84–91. doi: 10.2174/1875397301004010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 43.Beloueche-Babari M, Jackson LE, Al-Saffar NM, Workman P, Leach MO, Ronen SM. Magnetic resonance spectroscopy monitoring of mitogen-activated protein kinase signaling inhibition. Cancer Res. 2005;65:3356–3363. doi: 10.1158/10.1158/0008-5472.CAN-03-2981. [DOI] [PubMed] [Google Scholar]
- 44.Baillie GL, Horswill JG, Anavi-Goffer S, Reggio PH, Bolognini D, Abood ME, McAllister S, Strange PG, Stephens GJ, Pertwee RG, Ross RA. Mol Pharmacol. 2013;83:322–338. doi: 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Horswill JG, Whalley BJ, Stephens GJ. Mol Pharmacol. 2011;79:758–767. doi: 10.1124/mol.110.068197. [DOI] [PubMed] [Google Scholar]
- 46.Console-Bram LM, Zhao P, Abood ME. Protocols and Good Operating Practices in the Study of Cannabinoid Receptors. Methods Enzymol. 2017;593:23–42. doi: 10.1016/bs.mie.2017.06.027. [DOI] [PubMed] [Google Scholar]