

Abstract
The heat shock transcription factors (HSFs) were discovered over 30 years ago as direct transcriptional activators of genes regulated by thermal stress, encoding heat shock proteins. The accepted paradigm posited that HSFs exclusively activate the expression of protein chaperones in response to conditions that cause protein misfolding by recognizing a simple promoter binding site referred to as a heat shock element. However, we now realize that the mammalian family of HSFs comprises proteins that independently or in concert drive combinatorial gene regulation events that activate or repress transcription in different contexts. Advances in our understanding of HSF structure, post-translational modifications and the breadth of HSF-regulated target genes have revealed exciting new mechanisms that modulate HSFs and shed new light on their roles in physiology and pathology. For example, the ability of HSF1 to protect cells from proteotoxicity and cell death is impaired in neurodegenerative diseases but can be exploited by cancer cells to support their growth, survival and metastasis. These new insights into HSF structure, function and regulation should facilitate the development tof new disease therapeutics to manipulate this transcription factor family.
Heat shock transcription factors (HSFs) are a family of DNA-binding proteins, largely conserved from fungi to humans, that regulate gene expression at the level of transcription1,2. They were originally described to recognize a consensus heat shock element (HSE) DNA binding site and activate genes encoding protein chaperones in response to elevated temperatures. However, it has become apparent that mammalian HSFs and in particular the two most-studied members of the family — HSF1 and HSF2 — exhibit unanticipated complexity in their structure, DNA-binding selectivity, post-translational modifications (PTMs), interacting partners and regulation and that they have various roles in response to a wide range of stresses3–6.
In light of this complexity, it is perhaps not surprising that deregulation of HSF activity has been linked to human disease. For example, the pathology of neurodegenerative diseases that arise from protein misfolding, such as Huntington disease and Parkinson disease, has been shown to be associated with compromised activation of HSF1, which further exacerbates protein misfolding3,7. By contrast, various cancers show elevated levels of activated HSF1, which allow cancer cells to cope with the biosynthetic demands and stress resulting from rapid proliferation and promote invasion and metastasis4,5. To further complicate matters, HSF1 and HSF2 bind to distinct and overlapping sites in the genome and, in some instances, form hetero-oligomers, thereby enhancing the breadth of the regulatory control mechanisms imposed on each HSF isoform in a combinatorial manner6.
In this Review, we focus on HSF1 and provide an updated overview of our understanding of how HSFs are regulated and contribute to a range of functions in normal cells and in disease states. We emphasize advances based on structural insights, unanticipated roles in normal physiology and development and detail how HSF deregulation supports pathology. We highlight the role of HSFs in neurodegenerative diseases and cancer cells, as these pathological states provide good examples of the diversity of HSF functions and their dysfunction in disease.
Overview of the HSF activation cycle
In the human genome, there are several HSF isoforms, which are encoded by separate genes; their functions and differences in sequence and domain organization are described in BOX 1. Studies have largely focused on understanding the roles and mechanisms of action and regulation of the HSF1 and HSF2 family members because of their established role in the expression of stress-responsive genes and their link to disease1. The function of HSF1 and HSF2 is modulated at multiple stages, ranging from non-coding RNAs that regulate their expression levels (see Supplementary information S1 (box)) to the generation of splicing isoforms (see Supplementary information S2 (box)) and changes in oligomerization, subcellular compartmentalization, PTMs (TABLE 1; see Supplementary information S3 (box)), target gene activation and protein stability in normal and disease conditions. Deciphering these regulatory steps serves as an important basis for understanding HSF function and for the eventual development of pharmacological activators and inhibitors of HSFs for therapeutic approaches.
Box 1. Domain organization, family members and isoforms of human HSFs.
The human genome encodes six heat shock transcription factor (HSF) proteins: HSF1, HSF2, HSF4, HSF5, HSFX and HSFY. HSF1, HSF2 and HSF4 contain an amino-terminal, winged helix–turn–helix (wHTH) DNA-binding domain (DBD), which has yet to be validated in the other isoforms1. The leucine zipper oligomerization domain contains two heptad repeats, HR-A and HR-B, composed of hydrophobic and charged residues that are predicted to form inter-molecular leucine zippers when aligned upon oligomerization12. LZ1-3 is required for oligomerization, and its deletion produces a constitutively monomeric HSF16. The intrinsically disordered regulatory domain (RD) is post- translationally modified and regulates HSF1 activity and stability. Encompassed within LZ4 is HR C, which interacts with LZ1 3 and represses oligomerization. Some HSFs also contain an activation domain (AD). The figure shows HSF family members with their conserved domains and their splicing isoforms. The various splice isoforms are discussed in more detail in Supplementary information S2 (box).
HSF1 is the master regulator of protein quality-control machinery expression in response to proteotoxic stress conditions1 and further regulates gene expression to support cell survival4,116. HSF2 is highly expressed during early development117 and in the testis118, and has been described as an activator of protein chaperone genes in the febrile range of temperatures and as a tumour suppressor and activator79. HSF4 is required for growth and differentiation during eye lens development, and HSF4 mutations cause cataracts119,120. HSF4 lacks an LZ4 domain, resulting in constitutive trimerization and DNA-binding activity121. HSF4 is also expressed in the heart, brain, skeletal muscle and pancreas121. HSF5 has only been validated at the transcript level in humans, whereas HSF3 has been identified in mice but not in humans122. HSFX is located on the X chromosome, and its function has not been extensively explored117. HSFY is located on the Y chromosome; HSFY is primarily expressed in the testis, and HSFY deletion contributes to male infertility123.
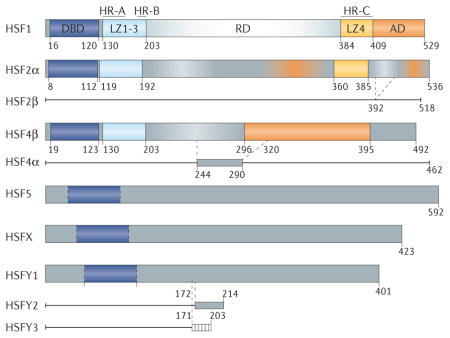
Table 1.
Post-translational modifications described for HSF1
| PTM | Enzyme | Effect | Physiological context | Refs |
|---|---|---|---|---|
| Phosphorylation | ||||
| Ser121 | MK2, AMPK | HSP90 binding and repression | Cancer (↓), metabolic stress (↑) | 41,126,127 |
| Ser127 | – | – | 128 | |
| Thr142 | CK2 | DNA binding activation | Heat shock (↑) | 129 |
| Ser195 | – | LZ1–3 and LZ4 dissociation and transactivation | Heat shock (↑) | 130 |
| Ser216 | PLK1 | Protein degradation, mitotic progression | Mitosis (↑) | 25 |
| Ser230 | CaMKII | Activation | Heat shock (↑) | 126,131 |
| Ser292 | – | – | 39,126 | |
| Ser303 | GSK3β, ERK1, MEK1, CK2 | Attenuation and degradation | Cancer (↓), HD (↑) | 3,40,126,128, 132 134 |
| Ser307 | GSK3β, ERK1, MEK1, CK2 | Attenuation and degradation | Cancer (↓), HD (↑) | 3,40,126 128 |
| Ser314 | – | – | Cancer (↓) | 126,127 |
| Ser319 | – | – | – | 126 |
| Ser320 | PKAcα | Activation | Heat shock (↑) | 135 |
| Thr323 | – | Repression | Heat shock (↑) | 127 |
| Ser326 | mTOR, ERK1/2, MEK1 | Activation | Heat shock (↑), cancer (↓) | 106,126,136 |
| Ser333 | PKCθ | HSP90 dissociation and activation | Cancer (↑) | 137 |
| Ser344 | – | – | 126 | |
| Ser363 | PKCα, PKCζ, JNK | Repression | Heat shock (↓) | 126,128,134 |
| Thr367 | – | Repression | Cancer (↓) | 127 |
| Ser419 | PLK1 | Nuclear localization and activation | Heat shock (↓) | 126 |
| Ser444 | – | – | 126 | |
| Acetylation | ||||
| Lys80 | GCN5, p300 | Inhibition, DNA binding dissociation | Heat shock (↓) | 31,37,38 |
| SIRT1, HDAC7, HDAC9 | ||||
| Lys116 | – | – | 31 | |
| Lys118 | p300 | Inhibition | Heat shock (↑) | 31,32 |
| Lys126, Lys148 | – | – | 31 | |
| Lys157, Lys188 | – | – | 31,138 | |
| Lys208 | p300 | Protein stabilization | Basal conditions (↑) | 31,32 |
| Lys224 | – | – | 31 | |
| Lys298 | p300 | Protein stabilization | Basal conditions (↑) | 31,32 |
| Lys524 | – | Heat shock response | Heat shock (↑) | 32 |
| Sumoylation | ||||
| Lys62, Lys91, Lys116, Lys118, Lys126, Lys131, Lys139, Lys148, Lys157, Lys162, Lys184, Lys208 | – | – | 43 | |
| Lys298 | UBC9 | Repression | Heat shock (↓) | 43,132 |
| Lys372 | – | – | 43 | |
Arrows indicate stress and/or pathological conditions in which alterations in particular modifications have been reported. ‘–’ indicates that the function of this post-translational modification is not known or has not been assessed yet. AMPK, 5′-AMP-activated protein kinase; CaMKII, calcium/calmodulin-dependent protein kinase type II; CK2, casein kinase 2; ERK1, extracellular signal-regulated kinase; GSK3β, glycogen synthase kinase 3β; HD, Huntington disease; HDAC, histone deacetylase; HSF1, heat shock factor 1; HSP90, heat shock protein 90; JNK, JUN N-terminal kinase; LZ, leucine zipper; MEK1 (mitogen-activated protein kinase kinase 1; MK2, MAP kinase-activated protein kinase 2; mTOR, mechanistic target of rapamycin; PKAcα, cAMP-dependent protein kinase catalytic subunit; PKC, protein kinase C; PLK1, polo-like kinase 1; SIRT1, NAD-dependent protein deacetylase sirtuin 1.
Activation and oligomerization of HSF1
The HSF1 activation cycle is a multistep and highly regulated process incorporating elements that tune every step of the cycle. Under standard cell culture growth conditions, mammalian HSF1 exists predominantly in an inactive form, probably as a monomer. In response to a wide range of stresses such as heat shock, exposure to heavy metals, oxidants and proteotoxic agents, HSF1 is converted into a DNA-binding-competent, active form, thought to be a homotrimer2 (FIG. 1). Activation of HSF1 results in its accumulation in the nucleus due to a potent bipartite nuclear localization signal8. What maintains HSF1 in the monomeric, inactive state and how HSF1 senses stress are currently unclear. However, it is likely that HSF1 is activated through different mechanisms depending on the type of stress, as it is able to differentiate between thermal stress and other stresses at normo-temperature conditions.
Figure 1. Heat shock transcription factor 1 activation cycle.

In response to proteotoxic stress conditions, heat shock transcription factor 1 (HSF1) is subject to a multistep activation and attenuation cycle. Inactive HSF1 monomers are retained in the cytoplasm in complex with regulatory proteins such as heat shock proteins (HSPs) 40, 70 and 90, as well as the cytosolic chaperonin TCP1 ring complex (TRiC). Upon stress sensing, HSF1 is activated, causing the dissociation of inhibitory proteins, HSF1 oligomerization and nuclear retention. HSF1 is modified by several activating post-translational modifications (PTMs) that promote DNA binding and transcriptional activation of target genes in concert with cofactor recruitment. HSF1 is then modified by different inhibitory PTMs, and by p23, causing DNA dissociation, HSF1 inactivation and HSF1 degradation (TABLE 1; see Supplementary information S3 (box)). It is currently unknown where HSF1 degradation occurs and to what extent HSF1 is newly synthesized or recycled into the cytoplasm. Ultimately, after attenuation, HSF1 is maintained in the cytoplasm by an inhibitory protein complex in a negative-feedback mechanism. Colour code: DNA-binding domain (dark blue), leucine zipper oligomerization domain LZ1 3 (light blue), regulatory domain (grey), LZ4 (yellow) and activation domain (orange).
Several intra-molecular and inter-molecular inter-actions have been proposed to repress HSF1 oligomerization, but the precise details of how the monomer-to-multimer inter-conversion occurs are not well understood. Evidence has demonstrated that increased temperature causes intrinsic structural changes in HSF1 that may facilitate oligomerization and activation9. This study closely examined the structural changes in heptad repeat A (HR-A) and HR-B and the repressive properties of HR-C, which is proposed to intra-molecularly repress HSF1 oligomerization via hydrophobic and ionic interactions with HR-A and HR-B (BOX 1)10. The mutagenesis of key residues predicted to be required for this interaction results in constitutively oligomeric species, both in vivo and in vitro10–12. Although many extrinsic factors could regulate HSF1 oligomerization in vivo, including PTMs and interacting proteins, the propensity of HSF1 to oligomerize in vitro in response to elevated temperature13 raises the question of whether HSF1 is an intrinsic ‘thermosensor’ (REF. 12). In vitro, the temperature-induced conformational dynamics of HSF1 were analysed using hydrogen–deuterium exchange mass spectrometry (HDX-MS)9, which relies on the difference in the exchange rate of hydrogen and deuterium between the solution and the more structured (less accessible to the solvent) and less structured (solvent-exposed) regions of a protein. This approach has the advantage of probing the dynamics of different regions of HSF1 at different temperatures, offering dynamic insights rather than a static crystal structure. Interestingly, only a few regions of HSF1 were structured at 20°C, including parts of the DNA-binding domain (DBD), oligomerization domain (LZ1–3) and HR-C. The HDX-MS studies found that dramatic structural changes occur in two regions at elevated temperatures; a temperature-dependent unfolding of the HR-C domain and a temperature-dependent stabilization of HR-A. This is consistent with the structural changes in the inactive monomer predicted to accompany oligomerization, including the interaction of HR-C with HR-A and HR-B (FIG. 1). The temperature-dependent stabilization of HR-A and unfolding of HR-C demonstrate that HSF1 possesses an intrinsic capacity to ‘sense’ temperature changes. However, because activation of HSF1 can occur at different temperatures in different tissues and differential temperature sensitivity is observed in different organisms with identical primary sequences of the protein14, HSF1 oligomerization is likely to also be regulated by other factors such as PTMs (TABLE 1; see Supplementary information S3 (box)) and other protein–protein interactions14.
Regulatory role of chaperone proteins
Although oligomerization has been linked to HSF1 transcriptional activity, treating human cells with anti-inflammatory agents such as sodium salicylate induced HSF1 oligomerization and DNA binding to the promoter of one of the canonical HSF1 target genes, heat shock protein 70 (HSP70), but did not activate its transcription15. Therefore, oligomerization of HSF1 is necessary but not sufficient for transcriptional activity16. Activation of HSF1 is regulated by a multi-molecular chaperone complex composed of HSP40, HSP70 and HSP90 (FIG. 1), as well as other proteins such as 14-3-3, which participates in the repression of HSF1 (REFS 1,2,17,18). The activation–inactivation mechanism of HSF1 follows the parsimonious chaperone titration model in which, upon stress conditions, HSF1 is liberated to oligomerize and then activates its corresponding targets, including HSPs. This results in increased levels of free HSPs, leading to HSF1 inactivation in a feedback response19. Whereas HSP90 is thought to inhibit HSF1 oligomerization and DNA binding20, HSP70 is proposed to inhibit the transactivation capacity of HSF1 (REFS 1,17). However, the direct role of these chaperones in repressing HSF1 activation is not completely understood, and some results seem to be contradictory. The use of HSP90 inhibitors such as geldanamycin results in HSF1 activation20, whereas recent in vitro studies suggest that HSP90 facilitates HSF1 trimerization during thermal stress9. Furthermore, overexpression of HSP70 is insufficient to suppress DNA-binding activity in vivo, but it seems to play a part in deactivating HSF1 after prolonged heat stress or during recovery from stress17,18. In addition to HSPs, a novel regulatory interaction between HSF1 and the cytosolic chaperonin TCP1 ring complex (TRiC) has been demonstrated (FIG. 1), expanding the pool of protein folding machinery that regulates HSF1. This is similar to the regulation of the bacterial thermal stress-responsive transcription factor σ32 by the bacterial chaperonin GroEL (also known as 60 kDa chaperonin)1,18. Interestingly, all eight genes encoding the TRiC subunits are also targets of HSF1, providing an integrated feedback regulatory loop. In addition, valosin-containing protein (VCP; also known as transitional endoplasmic reticulum ATPase), histone deacetylase 6 (HDAC6) and other proteins are thought to contribute to the formation of one or more repressive hetero-complexes that control HSF1 activity21.
Regulatory role of HSF degradation
Proteasomal degradation is also an important mechanism that regulates HSF activity, and HSF protein levels are often abnormal in the context of disease (see also below). For example, in cancer cells, HSF1 protein levels are increased4,22, thereby driving the transcriptome signature of cancer cells, whereas, in neurodegenerative diseases such as Alzheimer, Parkinson and Huntington diseases, HSF1 is considerably depleted, and the expression of HSF1 target genes is blunted3,7,23,24. In addition, HSF protein levels change during mitosis. HSF1 is degraded in a phosphorylation-dependent manner by polo-like kinase 1 (PLK1) and the S-phase kinase-associated protein (SKP)–cullin–F-box–βTrCP (SCFβTrCP) E3 ligase complex to promote progression through mitosis25. Failure to drive HSF1 degradation during mitosis results in aneuploidy and genomic instability25. HSF2 protein levels also decline during mitosis, and, although this has been attributed to decreased transcription, degradation of HSF2 by the anaphase-promoting complex (APC/C; also known as the cyclosome) has also been suggested26. The mechanisms that regulate HSF protein turnover differ in cancer and in neurodegenerative disease, but both events involve key enzymes and PTMs as rate-limiting steps in HSF degradation (TABLE 1).
HSF binding to target genes
HSF1 and HSF2 bind to canonical HSEs consisting of alternating inverted repeats with the sequence nGAAn, where n denotes any nucleotide1. Structural studies of the HSF DBD from the yeast Kluyveromyces lactis previously demonstrated that a recognition helix, containing a conserved Ser-Phe-Val-Arg-Gln amino acid sequence, inserts into the major groove of the HSE27. A conserved Arg in this sequence forms hydrogen bonds with the guanine of nGAAn and is essential for DNA binding27.
Two crystallographic studies of the human HSF1 and HSF2 DBDs were recently solved in complex with DNA11,28. These structures demonstrated similar HSF–DNA interactions as those observed for the K. lactis HSF, but they also provided new understanding of the architecture of HSFs in their DNA-bound form (FIG. 2a–c). Previous models predicted that the LZ1–3 of each monomer is positioned directly above the DBD, effectively occluding the portion of the DBD not in contact with the DNA from additional interactions (FIG. 2c). The new studies revealed that a carboxy-terminal helix of the DBD instead wraps around the DNA and directs LZ1–3 to the opposite side of the DNA (FIG. 2a, b, red helix; FIG. 2c). In both HSF1 and HSF2, this topology is stabilized by the favourable packing of a conserved amphipathic helix into a hydrophobic pocket and through additional DNA backbone contacts with conserved Arg and Lys residues11,28. These studies also revealed that, when bound to DNA, the different HSF isoforms expose biochemically distinct surfaces (FIG. 2d, e), and these isoforms can thus be subject to differential regulation by protein–protein interactions and PTMs (TABLE 1; see Supplementary information S3 (box)).
Figure 2. Structural insights into heat shock transcription factor–DNA interaction topology.

a, b | Crystal structure of the DNA-binding domain (DBD) of heat shock transcription factor 1 (HSF1)28 (Protein Data Bank (PDB) accession number: 3HTS) and crystal structure of HSF2 (REF. 11) bound to a two-site heat shock element (HSE) as a dimer (PDB: 5D8K). These independently solved structures revealed that a previously unknown carboxy-terminal (C term) helix (red) that is conserved in both HSF1 and HSF2 directs these HSFs to wrap around the HSE DNA, resulting in a topology where the DBD and the remainder of the HSF protein (not present in the crystal structure) occupy opposite sides of the DNA. c | A new model for the HSF–DNA interaction. Structural studies support a model that is in contrast to the previous model for the topology of DNA-bound HSF oligomers. In the old model (left), the oligomerization domains (light blue) were positioned on top of the DBD, such that the free surface of the DBD (shown in green, in contrast to the DNA-bound portion of the DBD shown in blue) was buried by the rest of the protein. In the new model (right), this free surface of the DBD is solvent exposed, which makes it available for interactions with regulatory proteins and to accept post-translational modifications. d | Surface representations of HSF1, HSF2 and HSF4 in their DNA-bound state with identical amino acids shared by all three family members shown in blue and divergent residues in green, cyan and orange, respectively. The surfaces that contact DNA are highly conserved, whereas the solvent-exposed surfaces are divergent. e | Sequence alignment of the DBDs of HSF1, HSF2 and HSF4. Residues conserved between isoforms are highlighted in black. Residues that contribute to the formation of α-helices, β-sheets and wing domains of HSF1 and HSF2 (from crystal structures11,28) are underlined (as no structural data for HSF4 are currently available, residues contributing to secondary structures are not designated). f | The HSF2 DBD structure (PDB: 5D8K) with a fully resolved wing domain and a line showing the location of Lys82, which is subject to regulatory sumoylation11. Unlike other winged helix–turn–helix DBDs, the wing domain of HSF2 does not contact the DNA backbone. Although the entire wing domain was not resolved for HSF1, a similar conformation was seen for the areas of the wing that were resolved27,28.
These crystal structures also offer a mechanistic explanation for the inhibitory role of Lys80 acetylation in regulating HSF1 binding to DNA. Lys acetylation removes the positive charge of this residue; therefore, it will neutralize DNA phosphate backbone interactions and compromise HSF1–DNA interactions11. The HSF2 structure revealed an identical interaction of Lys72 (analogous to Lys80 of HSF1) with the DNA backbone, although it is currently unclear whether HSF2 experiences a similar acetylation-mediated inhibition of DNA binding. These structures also revealed important insights into the role of the HSF wing domain (FIG. 2f), which is conserved in the family of winged helix–turn–helix (wHTH) DNA-binding proteins. Unlike the wing domains of other mammalian wHTH proteins, the wing domains of HSF1 and HSF2 do not make contacts with the DNA. Instead, the HSF1 and HSF2 wing domains are solvent exposed, leaving them accessible to various, often isoform-specific PTMs and protein–protein interactions and oligomerization. For example, the HSF1 wing domain interacts with replication factor A protein 1 (RPA1; also known as replication protein A 70 kDa DNA-binding subunit), which does not interact with HSF2 (REF. 29). In addition, the wing domains of HSFs are differentially regulated by sumoylation11 (see below). These data indicate that HSF wing domains provide additional and specific regulation of HSF activity. Collectively, these structural studies have substantially refined the model of HSF–DNA topology. The structures suggest that the presentation of biochemically distinct surfaces on HSFs drives unique regulatory interactions and allows different HSF proteins to occupy and regulate different genomic loci in vivo, despite having almost identical DNA-binding sequences.
PTMs and their role in HSF regulation
As noted above, PTMs serve an important function in regulating HSF activity, and the analysis of HSF1 and HSF2 under normal cell growth and stress conditions revealed the presence of numerous PTMs, primarily on HSF1 (REF. 30). These modifications include acetylation, phosphorylation and sumoylation and are thought to influence essentially every step of the HSF1 activation cycle (TABLE 1), having both activating and inhibiting effects. Some notable examples will be briefly discussed below (for further discussion, see Supplementary information S3 (box)).
Regulation of HSF1 activity and stability by acetylases
Recent discoveries have shown that reversible, site-specific acetylation modulates HSF1 DNA-binding persistence and/or protein stability, which has been confirmed by structural studies11,31,32. In the absence of stress, HSF1 steady-state levels are controlled by the histone acetyltransferase p300 (also known as EP300), which acetylates specific lysine residues (Lys208 and Lys298) to promote HSF1 stability by preventing its proteasomal degradation32. p300 also mediates acetylation of Lys80 of HSF1, which (as discussed above) inhibits the ability of HSF1 to directly interact with the DNA phosphate backbone, thereby inhibiting the binding of HSF1 to DNA. These effects can be counteracted by the NAD-dependent protein deacetylase sirtuin 1 (SIRT1), which deacetylates HSF1 (REFS 2,31). Silencing of p300 in HeLa cells results in reduced HSF1 protein levels and activity owing to increased proteasomal degradation32. Similarly, during cell differentiation, ageing and in neurodegeneration, HSF1 activity is attenuated, contributing to increased stress susceptibility, exacerbated protein misfolding and aggregation, and cell death. An age-related decline in stress response pathways has been widely documented and correlates with decreased HSF1 levels. For example, older mice have reduced HSF1 protein levels in heart and muscle compared with young mice, without changes in HSF1 mRNA levels33. Interestingly, p300 activity is attenuated in ageing mice, which could contribute to HSF1 depletion34. Although the mechanism underlying decreased HSF1 levels in ageing is unknown, it has been suggested that decreased SIRT1 protein levels contribute to decreased HSF1 abundance, perhaps resulting in increased Lys80 acetylation-dependent proteasomal degradation. Indeed, decreased SIRT1 levels during neuronal differentiation result in decreased HSF1 target gene expression35,36. Conversely, increased expression of HDAC1 in ageing cells contributes to inhibition of HSF1 activity by recruiting the histone acetyltransferase GCN5 (also known as KAT2A), with the involvement of the p23 co-chaperone37,38.
Phosphorylation regulates HSF1 activity and degradation
HSF1 is subjected to a plethora of phosphorylation events that modulate different steps of the activation cycle. For many years, HSF1 hyper-phosphorylation mediated by various kinases has been used as a surrogate for its activation during thermal stress (TABLE 1; see Supplementary information S3 (box)), but a comprehensive mutagenesis study revealed that there is no clear requirement for heat stress-induced phosphorylation in target gene activation39. However, these phosphorylation events seem to regulate HSF1 activity and stability under normo-temperature conditions and in different disease states (TABLE 1). Interestingly, the same residues can be phosphorylated by distinct protein kinases in different contexts. For example, Ser303 and Ser307 can be phosphorylated by glycogen synthase kinase 3 beta (GSK3β), casein kinase 2 (CK2), and the mitogen-activated protein (MAP) kinases MEK1 and ERK1, and these phosphorylation events promote HSF1 degradation and attenuate HSF1 activity (TABLE 1). In cancer cells, inhibition of GSK3β decreases Ser303 and Ser307 phosphorylation, resulting in increased HSF1 protein levels3,40. However, in Huntington disease, these residues are preferentially phosphorylated by CK2, which drives HSF1 inactivation and degradation3. This phosphorylation-regulated degradation of HSF1 seems to be mediated by its ubiquitylation in melanoma and in neurons affected by Huntington disease. Phosphorylated HSF1 is a substrate for FBXW7, a component of the E3 ubiquitin ligase complex, and its depletion dampens HSF1 ubiquitylation and increases total HSF1 protein levels3,40.
HSF1 phosphorylation is also strongly linked to metabolism. For example, 5′-AMP-activated protein kinase (AMPK), which plays a crucial part in cellular energy metabolism, directly inactivates HSF1 in both metabolic stress and under the unique metabolic conditions of cancer41,42 (FIG. 3; TABLE 1; see also below).
Figure 3. Heat shock transcription factor 1 at the forefront of metabolic regulation.

Heat shock transcription factor 1 (HSF1) acts as a core component of metabolic regulation through its ability to respond to metabolic and environmental stresses in key organs such as the liver and skeletal muscle. In this regard, HSF1 directly activates expression of the transcription factor peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α). Moreover, through direct protein protein interactions, PGC1α cooperates with HSF1 to activate the expression of chaperones and proteins that function in mitochondrial metabolism and biogenesis to prevent oxidative damage and increase oxidative phosphorylation48,51,53,55. The HSF1 and PGC1α network also functions in white adipose tissue browning, conferring heat production and beneficial effects on adiposity, insulin resistance and dyslipidaemia54. Intriguingly, this intricate network is simultaneously inhibited and activated by the metabolic stress sensor 5′-AMP-activated protein kinase (AMPK). AMPK phosphorylates and represses HSF1 function during nutrient deprivation, causing a proteotoxic stress response41. However, AMPK also activates PGC1α expression and transcriptional activity in adipose tissue124. This regulation focuses on modulating energy expenditure to improve metabolic fitness and stress protection. HSP40, heat shock protein 40; TRiC, TCP1 ring complex.
Sumoylation of HSFs regulates their activity in an isoform-specific manner
Sumoylation is an important PTM of HSFs, and both HSF1 and HSF2 are sumoylated on many different Lys residues43. The wing domains of HSFs are differentially sumoylated, as mentioned above. Lys82 in the HSF2 wing domain is sumoylated in vivo, which inhibits HSF2 DNA-binding activity44. Notably, the equivalent residue in HSF1 is not sumoylated, but enforced sumoylation of HSF1 by creating a chimeric HSF1 protein harbouring the wing domain from HSF2 compromised HSF1 activation in vivo, which led to diminished induction of HSP70 expression11. In contrast to HSF1 and HSF2, HSF4 does not contain a lys in its predicted wing domain that could be sumoylated (FIG. 2). This provides evidence for isoform-specific regulation of HSFs by SUMO.
HSF1 and metabolism
Previous work aimed at identifying genomic loci bound by HSF1 in Saccharomyces cerevisiae showed that HSF1 regulates a wide range of targets well beyond those encoding chaperones; the identified targets included transcription factors, proteins that maintain the integrity of the cell wall and cytoskeleton, and metabolic enzymes45. Indeed, mammalian HSF1 plays an important part in metabolic control, including in sensing metabolic stress and in regulating energy metabolism and white fat browning. These exciting findings will be briefly discussed below.
Cross talk between metabolic stress and the HSF1-mediated heat shock response
Metabolic stresses such as nutrient deprivation result in an increased intra-cellular AMP-to-ATP ratio that triggers a stress response to maintain cellular energy homeostasis. This response is largely coordinated by the kinase AMPK, the principal function of which is to enhance ATP generation and reduce ATP expenditure to ensure cell survival46. Concomitantly, metabolic stress decreases expression of the protein quality-control machinery, sensitizing cells to proteotoxic stress41. Intriguingly, this effect is mediated by the direct repression of HSF1 by AMPK (FIG. 3), which phosphorylates HSF1 Ser121. Accordingly, loss of AMPK elevated basal HSF1 activity and reversed the HSF1 repression imposed by glucose starvation41. In an analogous way, the yeast homologue of AMPK, Snf1, regulates yeast HSF47. Interestingly, metformin, used to treat type II diabetes mellitus, is also an AMPK agonist, and its application recapitulates the HSF1 repression observed under nutrient deprivation41. In support of the idea that HSF1 functions at the intersection between the proteotoxic stress response and metabolic stress, it has been demonstrated that HSF1−/− mice exhibit increased insulin signalling and increased AMPK activation in response to low glucose levels, indicating that HSF1 could potentially be targeted to control insulin resistance and diabetes42.
HSF1 and the PGC1α network in mitochondrial function
Previous studies elegantly demonstrated a crucial role for HSF1 in mitochondrial function and protection against oxidative stress48. Neurons have an extremely high demand for continued production of high-energy phosphate bonds such as those found in ATP, and reduced mitochondrial function is associated with neurodegeneration49. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α; which is encoded by PPARGC1A) is a central transcription regulator of mitochondrial (and peroxisomal) remodelling and biogenesis50,51. On one hand, PGC1α dysfunction contributes to neurodegenerative diseases, such as Huntington disease49,52. On the other hand, invasive cancer cells, which — similar to neurons — require high ATP production, exhibit a direct correlation between increased PGC1α expression and the formation of distant metastases. Silencing of PGC1α in cancer cells reduced invasive potential and attenuated metastasis53.
HSF1 directly activates PGC1α transcription54 (FIG. 3). Accordingly, increasing HSF1 protein levels in a mouse model of Huntington disease elevated the expression of PGC1α and its downstream targets such as cytochrome c and the mitochondrial transcription factor TFAM3.
HSF1 in systemic energy metabolism and white fat browning
In addition to the parts played by HSF1 and PGC1α in regulating mitochondrial metabolism, these two transcription factors function in thermogenesis and the browning of white fat54,55 (FIG. 3). An imbalance between energy input and output results in the accumulation of excessive fat tissue, leading to obesity, diabetes and cardiovascular dysfunction. White adipose tissue is the main site of energy storage, whereas brown fat is principally involved in energy expenditure. Evidence has demonstrated that HSF1 activates Ppargc1a expression and increased the energy expenditure of mice on a high-fat diet by inducing the browning of white adipose tissue and activating the expression of mitochondrial genes in muscle54. HSF1−/− mice exhibit increased lipid deposition and decreased brown fat markers, features associated with decreased PGC1α expression. These results demonstrate that, via the activation of PGC1α, HSF1 modulates mitochondrial and brown fat gene programmes that have an impact on thermogenic function and energy expenditure54.
Although HSF1 is considered the master activator of chaperone expression and directly activates PGC1α, it is worth noting that HSF1 and PGC1α also physically interact and colocalize on several HSF1 target promoters55,56. Interestingly, cells and mice lacking Ppargc1a are more sensitive to thermal challenge, whereas ectopic expression of PGC1α activates ~30% of the genes commonly known to be involved in heat shock responses55. In addition, PGC1α functions in the HSF1-dependent induction of HSP70 in hyperthermia. However, it has also been reported that PGC1α functions as a repressor of HSF1-mediated transcriptional programmes in hepatocytes, in muscle and in cancer cell lines56. Although the regulatory mechanisms that control PGC1α and HSF1 interactions are not fully deciphered, it is clear that specific transcriptional programmes are controlled via cooperation between these two transcription factors at multiple levels. This finding suggests that other transcription factors regulate HSF1 target gene expression in different cell types or disease conditions.
HSF1 and neurodegenerative diseases
HSFs have essential roles in brain development and function through their engagement in regulating genetic programmes involved in the modulation of neuronal migration, the formation and maintenance of neuronal synapses and the resistance to proteotoxic stress57–61 (FIG. 4a; see Supplementary information S4 (box)). The impairment of HSF1 activity with ageing and in age- related diseases such as neurodegeneration has been widely documented2. At the same time, somewhat paradoxically, protein misfolding-based neurodegenerative diseases are associated with reduced expression of the protein quality-control machinery3,24,62 (FIG. 4b, c). Whereas impaired HSF1 activation does not cause neurodegenerative disease, it does seem to exacerbate protein misfolding and aggregation, at least in part by decreasing chaperone expression, thereby contributing to decreased protein quality control, neuronal dysfunction, neuronal cell death and disease progression. Studies in cell culture, fruitfly, worm and mouse models of neurodegenerative disease clearly demonstrate that enhancing protein-folding capacity and pro-survival functions via elevated expression of HSF1, as well as chaperone proteins and other HSF1 targets, has therapeutic potential62.
Figure 4. Heat shock transcription factor 1 inactivation or depletion is a common defect in neurodegenerative disease.

a | Healthy neuronal cells can cope with misfolded protein stress by activating heat shock transcription factor 1 (HSF1) in response to stress sensing, which then activates the transcription of target genes that promote neuronal function and survival. Target genes include protein chaperones, peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α), postsynaptic density protein 95 (PSD95), synapsin and brain-derived neurotrophic factor (BDNF). b | In Huntington disease, pathogenic mutant huntingtin protein (mHTT) with an expanded polyglutamine (polyQ) tract increases the levels of casein kinase 2 subunit-α′ (CK2α′) and an F box component of an E3 ubiquitin ligase, FBXW7, which drive HSF1 phosphorylation and ubiquitin-dependent proteolysis, respectively. This decreases HSF1 levels and impairs the resolution of protein aggregates, thereby contributing to increased proteotoxicity, neuronal dysfunction and death. c | In Parkinson disease, Alzheimer disease and amyotrophic lateral sclerosis, reduced HSF1 protein levels and/or activity has been reported and is associated with decreased expression of chaperones, exacerbating the aggregation of disease-relevant proteins, including α-synuclein, amyloid-β (Aβ), phosphorylated Tau (Tau-P), TAR DNA-binding protein 43 (TDP43), mutant superoxide dismutase 1 (mSOD1) and chromosome 9 open reading frame 72 (C9ORF72). Although it is unknown whether CK2 has an impact on HSF1 abundance in these neurodegenerative diseases, increased CK2 levels have been observed. Two E3 ubiquitin ligases, NEDD4 and FBXW7, have been implicated in HSF1 degradation in Parkinson disease and Huntington disease, respectively.
Huntington disease and other polyglutamine expansion diseases
The abnormal expansion of polyglutamine (polyQ)-encoding regions underlies 14 neurodegenerative diseases, the most common of which include Huntington disease, spinal and bulbar muscular atrophy (SBMA) and spinocerebellar ataxia. In general, polyQ expansions cause the misfolding and aggregation of proteins, leading to cellular dysfunction and death63.
Similar to what has been observed in neurons from older animals2,35, the expression of several HSF1 target genes encoding protein chaperones and anti-apoptotic proteins is decreased in polyQ diseases, thus exacerbating the progression of protein misfolding and neurodegeneration64–66,67. Selective neuronal death is a crucial feature of neurodegenerative disease; therefore, the elucidation of the mechanisms that underlie the cell-specific defects in HSF1 activity may lead to a better understanding of how neurodegeneration could be slowed.
Overexpression of individual protein chaperones has shown beneficial effects in protein-misfolding disease models by enhancing the refolding and solubilization of pathogenic polyQ proteins68–70. However, given that multiple individual chaperones work in obligatory molecular complexes65, coordinated activation of multiple chaperones via increased HSF1 activity or stability could lead to a more profound impact on the amelioration of protein aggregation in Huntington disease62. Consistent with HSF1 activating the expression of protein chaperones and stress-protective pathways, Hsf1 knockout in a Huntington disease mouse model led to increased brain aggregation of mutant huntingtin (mHTT) — a protein that harbours expanded polyQ tracts in Huntington disease — and a shortened lifespan71, whereas expression of a constitutively active form of HSF1 alleviated mHTT aggregation and prolonged lifespan72. Moreover, an Hsf+/− mouse model of SBMA, which contains a pathogenic polyQ repeat expansion in the androgen receptor, exhibited increased androgen receptor aggregation in neurons and non-neuronal tissues and increased neurodegeneration compared with Hsf1+/+ SBMA mice73.
Genome-wide HSF1 chromatin immunoprecipitation and sequencing analysis in striatum cells from wild-type mice and mice modelling Huntington disease revealed that expression of mHTT dramatically alters HSF1 binding to DNA74. After heat shock, HSF1 binding to target promoters was reduced by ~40% in mutant cells compared with control cells. Intriguingly, genes related to cytoskeletal organization, focal adhesions and the activity of GTPases were the most affected, whereas HSF1 binding to chaperone genes was not substantially altered despite evidence for decreased chaperone expression in Huntington disease. This finding could be explained by the fact that HSF1 can bind to its target HSE-containing promoters without inducing transcription75, suggesting that multiple inhibitory mechanisms control HSF1 activity in Huntington disease.
Whereas pharmacological activation of HSF1 has therapeutic potential in Huntington disease, an approach to activate HSF1 by HSP90 inhibition conferred only transient benefits in a mouse model65. Accumulating reports suggest that HSF1 protein levels are reduced in neurodegenerative disease3,66,73,76. Mouse models of SBMA also showed decreased levels of HSF1 in motor neurons and decreased chaperone expression73. Similarly, a cell model of spinocerebellar ataxia 6 harbouring polyQ expansions within the Cav2.1 calcium channel gene exhibits decreased HSF1 and HSP70 expression77. What causes HSF1 levels to decrease in the context of neurodegenerative diseases? In Huntington disease, this decrease is associated with the abnormal degradation of HSF1 during disease progression3. HSF1 is phosphorylated at Ser303 and Ser307 within the regulatory domain, and these two phosphorylation events are associated with HSF1 inactivation (see discussion above and TABLE 1). This modification in Huntington disease is mediated by CK2; in particular, the CK2α′ catalytic subunit is dramatically elevated in this disease. Phosphorylated HSF1 then recruits the FBXW7 F box component of an E3 ligase, which is also elevated, driving HSF1 ubiquitin-proteasome dependent degradation (FIG. 4b). Mutating Ser303 to Ala is sufficient to increase HSF1 levels in yeast3,78 and in polyQ-expressing mammalian cells, and the expression of HSF1 lacking the Ser303–Ser307 region in a Huntington disease mouse model ameliorates polyQ aggregation and associated phenotypes72. Similarly, pharmacological or genetic inhibition of CK2α′ restored HSF1 protein levels, increased chaperone expression, decreased mHTT aggregation in the striatum and increased the abundance of medium spiny neurons in a Huntington disease mouse model. Increasing HSF1 levels also had a positive impact on the expression of PGC1α54, the decreased expression of which is partly responsible for mitochondrial dysfunction in Huntington disease49. Therefore, restoring HSF1 levels increases protein folding capacity and the abundance of a key regulator of energy metabolism3. CK2-mediated degradation of HSF1 is a phenomenon that may also apply to other neurodegenerative diseases (see below).
HSF2 also modulates polyQ aggregation and Huntington disease progression, as mice modelling Huntington disease and harbouring an Hsf2 deletion exhibit a reduced lifespan and an increase in protein aggregation in the striatum79. HSF2 may function through cooperative interactions with HSF1 and increased expression of alpha-crystallin B chain (α(B)-crystallin), a small chaperone that binds to misfolded proteins and prevents aggregation79. In the absence of both HSF1 and HSF2, overexpression of α(B)-crystallin and other HSF targets such as the scaffolding protein PDZK3 can alleviate polyQ aggregation; the expression of these targets depends on cooperation between HSF1 and nuclear factor of activated T-cells, cytoplasmic 2 (NFATC2)71,79. Therefore, interactions between HSF1, HSF2 and other transcription factors modulate the expression of proteins that prevent protein aggregation. Differences in these interactions may determine the vulnerabilities of different neuronal cell types to the aggregation of proteins with polyQ expansions.
Parkinson disease
Parkinson disease is characterized by a progressive loss of dopaminergic neurons mainly due to α-synuclein (α-syn) aggregation and the formation of Lewy bodies in the substantia nigra. Early studies overexpressing HSP70 in a fly model of Parkinson disease demonstrated that increased levels of HSP70 prevent α-syn toxicity and dopaminergic neuronal loss80. Expression of constitutively active HSF1 in human cells modelling Parkinson disease increased HSP70 protein levels and decreased α-syn inclusions and toxicity81. Overexpression of α-syn in human cells results in HSF1 depletion, and this is further exacerbated in mutant cells modelling early onset Parkinson disease7. HSF1 is also depleted in the midbrain of mice expressing α-syn and in patients with diffuse Lewy body disease7. This depletion of HSF1 was ascribed to an aberrant degradation mechanism that involves the E3 ligase NEDD4, the levels of which are increased in Parkinson disease (FIG. 4c). Interestingly, SIRT1-mediated deacetylation was found to be crucial for NEDD4-mediated HSF1 degradation7. Although it is currently unclear whether HSF1 phosphorylation is also required for NEDD4-mediated degradation, several kinases that phosphorylate HSF1, such as CK2, have been reported to exhibit increased expression and activity in Parkinson disease82,83.
Alzheimer disease
Cognitive deficits in Alzheimer disease are believed to largely result from the accumulation of amyloid-β, a toxic peptide released after proteolysis of the β-amyloid precursor protein that misfolds and aggregates, contributing to neuronal loss in the hippocampus and cerebellum84. Pharmacological activation of HSF1 by HSP90 inhibition in a mouse model of Alzheimer disease ameliorated the synapse and memory loss induced by amyloid-β, owing in part to the increased expression of HSP70 and HSP25, as well as of various presynaptic and postsynaptic proteins85. Furthermore, intra-nasal administration of HSP70 to mice increased hippocampal and cortical neuron density, diminished amyloid-β accumulation and improved spatial memory86. Cerebellar Purkinje cells, the numbers of which are reduced in patients with Alzheimer disease and in mouse models, show a depletion of HSF1 and chaperones23, and overexpression of HSF1 in rat or mouse models rescues Purkinje cell numbers, lowers amyloid-β levels and ameliorates the cognitive deficits associated with Alzheimer disease23,87. Although it is not known if the decreased levels of HSF1 in the context of Alzheimer disease result from increased degradation88,89 (FIG. 4c), it is curious that CK2 is elevated in both mouse models and patients and that CK2 high level is correlated with disease progression88,89. CK2 elevation is associated with the inflammatory response driven by astrocytes, which influences amyloid formation88,90, although the connection between CK2, HSF1 and inflammation has yet to be explored.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects motor neurons in the brain and spinal cord, ultimately leading to paralysis. Motor neurons affected by ALS accumulate protein aggregates, such as aggregates of superoxide dismutase 1 (SOD1), which are associated with familial ALS91. HSF1 is thought to protect motor neurons by activating chaperones and taurine transporter (TauT) expression. Increased expression of TauT enhances taurine accumulation in motor neurons, and taurine in turn acts as an antioxidant by reducing the motor neuron loss caused by the oxidative stress resulting from SOD1 dysfunction92. The increased susceptibility of ALS motor neurons to protein aggregation and oxidative stress has been associated with diminished HSF1 activity93,94 (FIG. 4c). Mice modelling ALS and overexpressing SIRT1 showed increased HSF1 deacetylation and activation in the spinal cord, increased HSP70 levels and a prolonged lifespan95. In cultured motor neurons from model mice and patients with ALS, HSP70 expression is decreased, suggesting that HSF1 is compromised.
Apart from SOD1, ALS is also associated with the accumulation of misfolded, detergent-resistant (insoluble), ubiquitylated and hyper-phosphorylated TAR DNA-binding protein 43 (TDP43; also known as TARDBP), which is observed in more than 95% of individuals with familial ALS (and in ~25–50% of Alzheimer disease cases). HSF1 overexpression maintains the solubility of nuclear TDP43, decreases its hyper-phosphorylation and inhibits its cytotoxicity91,94. Accordingly, HSF1-deficient mice display increased TDP43 insolubility and ALS-associated phenotypes. Importantly, in mice showing increased levels of TDP43, HSF1 levels are reduced in motor neurons, and this reduction correlates with decreased HSP40 and HSP70 levels94. Similar to other neurodegenerative diseases, CK2 levels are also increased in ALS, but the role of CK2-mediated phosphorylation in regulating HSF1 abundance has not been reported in this context96,97. In summary, decreased HSF1 levels seem to contribute to the pathology of neurodegenerative diseases by reducing stress tolerance and neuronal cell survival (FIG. 4).
HSFs in cell proliferation and cancer
Elevated chaperone expression has been observed in many cancers and correlates with poor prognosis, increased metastatic potential and resistance to therapy98. The expression of both HSF1 and HSF2 has been shown to be altered in cancer, with evidence indicating distinct roles for these HSFs in tumorigenesis4,99,100.
HSF1 and HSF2 have different capacities for regulating gene expression in mitosis
Phosphorylation-dependent regulation of HSFs is important for resistance to stress during mitosis and for genomic integrity and stability25, as discussed above. A genome-wide study investigated how the genomic occupancy of HSF1 and HSF2 is altered in mitotic cells6. The results revealed that HSF1 has a dramatically reduced ability to bind to DNA during mitosis. This reduced binding is accompanied by the diminished transcription of HSF1-bound genes in mitotic cells compared with asynchronous cells, showing that HSF1 function is dampened in mitotic cells. By contrast, HSF2 exhibited only modestly reduced binding in mitotic cells, which was partly explained by a higher capacity to bind to condensed chromatin (which prevails in mitosis) compared with HSF1. These studies revealed both overlapping and distinct HSF binding patterns and suggested unique roles for HSF1 and HSF2 in the cell cycle and in mitosis (for further details, see Supplementary information S5 (box)).
HSF1 in cancer initiation and progression
The importance of HSF1 in cancer has been revealed by Hsf1-knockout mice, which have a decreased propensity to form tumours driven by oncogenic RAS, loss of p53 or chemical carcinogens22,42,101–103. One clear role that HSF1 has in cancer is to promote adaptation and survival in the rapidly changing and stressful conditions encountered by cancer cells, including hypoxia, acidosis and alterations in nutrient availability22. High levels of HSF1 maintain proteomes during stress and support the high biosynthetic demand of cancer cells. In a diverse array of cancers, HSF1 abundance and nuclear localization are strongly increased4. Cancer cells are described as having a ‘non-oncogene’ addiction to HSF1, meaning that these cells are more highly dependent on HSF1 function than are normal cells. However, it is important to note that increased levels of active HSF1 per se do not cause cancer initiation104.
High HSF1 levels correlate with poor prognosis, although the mechanisms by which HSF1 expression and activity are elevated in cancer cells may vary98. For example, increased HSF1 activity and protein levels are observed in ERBB2-overexpressing cancers in which HSF1 translation is increased105. Cancer cells also increase HSF1 levels by preventing its degradation through either reduced expression or mutagenesis of the E3 ligase FBXW7 (REF. 40).
Alterations in HSF1 activity in cancer cells also occur via PTMs. For example, HSF1 is activated by MEK- mediated Ser326 phosphorylation106 (TABLE 1). It has been shown that MEK, the upstream activator of ERK, forms a ternary complex with ERK and HSF1 in which ERK phosphorylates and inhibits MEK, resulting in decreased phosphorylation of HSF1 at Ser326, ultimately inactivating HSF1 and sensitizing cells to proteotoxic stress. Accordingly, MEK blockade decreased HSF1 Ser326 phosphorylation and induced proteotoxic stress in cancer cell lines, whereas ERK blockade increased this phosphorylation and prevented the formation of toxic amyloids106. In tumours driven by the loss of the tumour suppressor protein neurofibromin (NF1), MEK is over-active, resulting in constitutively active HSF1. Such high HSF1 protein levels and activity promote cell survival and further increase MAPK signalling, which supports cancer growth and proliferation103. Given that ~30% of human cancers contain mutations that deregulate MAPK signalling, this pathway seems to have an important role in activating HSF1 in cancer cells, preventing amyloidogenesis and subsequent apoptosis98.
Because key cancer-related proteins such as p53, AKT, RAF1, BCR–ABL1 fusion, cyclin-dependent kinase 4 (CDK4), Cyclin D, ERBB2, hormone receptors and hypoxia-inducible factor 1α (HIF1α) are highly dependent on chaperones for their activity and stability, it is not surprising that changes in HSF1 have an impact on oncoprotein abundance and function98. In addition to promoting the activity of oncoproteins, chaperones also drive cancer-specific signalling pathways to facilitate oncogenesis and inhibit apoptosis107 (FIG. 5). For example, HSP70 prevents stress-induced apoptosis through the JUN N-terminal kinase (JNK)-stress-activated protein kinase pathway, and HSP90 inhibits apoptosis through AKT, tumour necrosis factor receptors, and the function of nuclear factor-κB (NF-κB)107. In addition, chaperones buffer the folding of proteins destabilized by mutations, which are more frequent in cancer cells owing to their genomic instability98,107.
Figure 5. Distinct regulation of heat shock transcription factor 1 in cancer and neurodegenerative disease.

a | In cancer cells, heat shock transcription factor 1 (HSF1) is increased and drives a cancer-specific gene signature that supports cancer cell growth and survival. After establishment, tumours recruit and reprogramme cancer-associated fibroblasts (CAFs) from the surrounding stromal tissue, resulting in activation of pathways in CAFs that enhance cancer proliferation, metastasis and angiogenesis. The stromal-specific HSF1-dependent gene signature, which is distinct from that of cancer and healthy cells, includes activation of transforming growth factor-β (TGFβ) and stromal cell-derived factor 1 (SDF1) expression, leading to the secretion of cancer-supportive soluble proteins. By contrast, in neurodegenerative diseases such as Huntington disease, HSF1 is abnormally degraded, and its promoter occupancy is altered. b | HSF1 functions are distinct in neurodegenerative diseases, such as Huntington disease, and in cancer, contributing to gene expression signatures that are characteristic for each disease in Huntington disease, HSF1 levels decrease, which impairs the expression of genes with functions in processes that are crucial for striatal neuronal function74,125; in cancer, increased HSF1 levels are associated with inhibition of apoptotic genes and activation of genes that drive processes supporting cancer cell metabolism, proliferation, translation and genomic instability4,98. The decreased levels of HSF1 in Huntington disease result from increased degradation via a mechanism involving phosphorylation by the casein kinase 2 subunit-α′ (CK2α′), which is overexpressed, and subsequent ubiquitylation associated with increased FBXW7 (see FIG. 4b). In melanoma, HSF1 levels are elevated owing to decreased expression or mutation of FBXW7 and decreased ubiquitylation40. Two known mechanisms for activation of HSF1 in cancers involve increased phosphorylation of HSF1 at Ser326 by mitogen-activated protein kinase kinase (MEK) through oncogenic RAS signalling106 and decreased activity of 5′-AMP-activated protein kinase (AMPK), which ultimately lowers inhibitory Ser121 phosphorylation41. Full list of gene names is detailed in Supplementary Information S6 (box).
The role of HSF1 in regulating a cancer gene expression signature is clear, and it is well established that HSF1 drives oncogenesis in many ways beyond inducing the expression of chaperone proteins. HSF1 coordinates the activation of genes that support the initiation and maintenance of cancer cells through changes in processes including metabolism, protein translation, cell cycle control, signalling and proliferation108 (FIG. 5). Interestingly, HSF1 regulates these programmes through both gene activation and repression108. For example, HSF1 negatively regulates genes related to apoptosis, including those encoding microtubule-associated protein tau (MAPT) and the apoptosis regulator BAX4. In addition, chromatin immunoprecipitation followed by deep sequencing experiments revealed that the constellation of gene targets that are bound and regulated by HSF1 is quite unique in cancer — a staggering 60% of the genes bound in cancer cell lines were not bound in non-transformed cell lines, even under heat shock conditions. Other studies have also demonstrated the varied roles of HSF1 in cancer. For example, in mammary carcinogenesis, HSF1 increases the levels of Huantigen R (HuR; also known as ELAVL1), an RNA-binding protein that stabilizes mRNAs to promote translation. HuR targets include mRNAs encoding β-catenin, HIF1α, Cyclin D and HSF1 itself, which results in a positive regulatory feedback loop109,110. Translation of β-catenin, a transcription factor that stimulates proliferation, differentiation, migration, angiogenesis and survival, is further increased by the ability of HSF1 to suppress the synthesis of lincRNA-p21 (see Supplementary information S1 (box)), which negatively regulates β-catenin translation109. Studies have also highlighted the link between HSF1 and translational capacity, as depletion of HSF1 resulted in decreased levels of the ribosomal large subunit proteins RPL13 and RPL17, whereas hyperactive HSF1 increased the basal levels of these proteins106. HSF1 also activates translation by derepressing a major regulator of cellular growth, mechanistic target of rapamycin complex 1 (mTORC1). This derepression occurs through HSF1-mediated suppression of JNK, which normally inhibits mTORC1 (REFS 111,112).
A distinct reprogramming of HSF1 target genes is also observed in cells that surround cancer cells, called stromal cells, which are non-malignant and genetically stable but support cancer cell malignancy5 (FIG. 5). Stromal cells include immune cells, endothelial cells and fibroblasts, which comprise the tumour microenvironment and are essential for tumour formation and progression5. The most abundant stromal cells are cancer-associated fibroblasts (CAFs). HSF1 is activated to drive a CAF transcription programme that is complementary to but distinct from that observed in cancer cells4,5. For example, HSF1 in CAFs drives the expression of transforming growth factor-β and its cognate receptors, as well as of stromal cell-derived factor 1 (SDF1; also known as CXCL12), leading to the secretion of proteins that enhance the survival and proliferation of cancer cells in a non-cell-autonomous manner. Furthermore, high levels of nuclear, activated HSF1 found in the stroma correlate with higher tumour grades, reduced overall survival and poorer patient outcomes. Activated HSF1 in stromal cells may even be more predictive of patient survival than HSF1 levels in the tumour itself, indicating the crucial importance of stromal HSF1 activation in cancer progression4,5.
HSF2 as a potential tumour suppressor
Additional roles of HSF2 in cancer are emerging and indicate that HSF2 may act as a tumour suppressor26,99. A reduction in HSF2 levels increased the invasiveness of cancer organoids, further supporting the notion that low levels of HSF2 promote malignant invasiveness. What are the underlying mechanisms?
Transcriptome analyses in prostate cancer organoids showed that HSF2 silencing substantially affected gene expression related to translation, energy metabolism, membrane transport, RNA metabolism, invasion and chromatin assembly and disassembly99. HSF2 silencing supports invasive transformation via the regulation of actin cytoskeleton pathways and GTPase signalling. In addition, HSF2 silencing promotes epithelial–mesenchymal transition (EMT), whereby cells lose polarity and adhesion properties while gaining invasive properties, contributing to tumour progression. This transition is accomplished via reduced levels of E-cadherin, a hallmark of EMT. Although this compelling study demonstrates that HSF2 acts as a tumour suppressor in prostate cancer and HSF2 mRNA is reduced in many other cancers, another study demonstrated that HSF2 was increased in 38 out of 50 samples isolated from patients with lung cancer113. In addition, changes in HSF2 could alter HSF1 function, as HSF2 can hetero-oligomerize and modulate the activity of HSF1 (REFS 114,115). Further investigation is needed to clarify how HSF2 functions in different types of cancer, both in concert with HSF1 and independently.
Conclusions and perspective
Over the past few years, our understanding of the structure, regulation and function of HSFs has rapidly increased, providing new insights into the roles of these transcription factors in protein misfolding, metabolism, neurological disease and cancer. However, many unanswered questions remain. It will be important to gain a systematic understanding of the full network of HSF isoforms encoded in the human genome, their complete structures and how they interact within this family and with other proteins in unique combinations to regulate gene expression. although HSFs, particularly HSF1, are subject to a plethora of PTMs, we still have a poor understanding of their specific and context-dependent roles in HSF regulation under normal or disease situations. Furthermore, the mechanisms by which HSF1 and HSF2 are regulated in the broad context of metabolism, particularly by protein misfolding in neurodegeneration or by cancer signalling pathways, remain to be elucidated. A comprehensive understanding of this family of transcription factors could provide a basis for optimizing selective small molecule agonists or antagonists of HSF function, which may provide possibilities for new therapeutic interventions.
Supplementary Material
Acknowledgments
E.T.B. is supported by a Predoctoral Fellowship from the US National Institutes of Health (F31 GM119375-02). R.G.-P. is supported by a Postdoctoral Fellowship from the Huntington’s Disease Society of America Human Biology Project. The authors acknowledge Alex Jaeger for assistance with Figure 2 and the Reviewers for excellent comments and suggestions.
Glossary
- Winged helix–turn–helix (wHTH)
Structural feature of a protein containing α-helices, β-sheets and loops arranged to form a helix–turn–helix DNA-binding motif with a wing domain.
- Transactivation
Activation of gene expression by transcription factors.
- Chaperonin
Multi-subunit protein folding machines found from bacteria to humans that fold proteins in an ATP-dependent manner.
- Valosin-containing protein (p97/VCP)
A multitasking, chaperone-like AAA ATPase involved in protein ubiquitylation.
- Aneuploidy
An abnormal number of chromosomes.
- Genomic instability
The acquisition of insertions, deletions or rearrangements in chromosomes or loss of chromosomes.
- Wing domain
A structural feature in a winged helix–turn–helix domain typically composed of loops that come together to form a butterfly-like, ‘wing’ protrusion, typically in contact with the DNA backbone.
- Focal adhesions
Functional points of contact that facilitate signalling in response to stimuli such as force.
- GTPases
Guanine nucleotide-hydrolysing proteins that function in cellular signalling, protein translation, vesicular trafficking and other processes.
- Lewy bodies
Protein aggregates in Parkinson disease and other dementias.
- Substantia nigra
Midbrain structure of basal ganglia that has an important role in movement and reward function.
- Purkinje cells
Large GABAergic neurons in the cerebellar cortex with a large number of dendritic spines, which are small protrusions that receive axonal input.
- Astrocytes
Supporting glial cells in the brain and spinal cord that contribute to the function and health of other cells in the central nervous system.
- Oncogenic RAS
RAS is a family of related small GTPases involved in signal transduction pathways. RAS mutations are the most common cancer-associated mutations.
- Nuclear factor-κB (NF-κB)
A protein complex that controls the expression of genes involved in inflammation and a range of other functions.
- Mechanistic target of rapamycin complex 1 (mTORC1)
Controls protein synthesis and senses the cellular energy, nutrient and redox balance status.
Footnotes
Author contributions
R.G.-P., E.T.B. and D.J.T. contributed equally to researching data for the article, discussion of content and writing and reviewing the manuscript before submission.
Competing interests statement
The authors declare competing interests: see Web version for details.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol. 2010;11:545–555. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 3.Gomez-Pastor R, et al. Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington’s disease. Nat Commun. 2017;8:14405. doi: 10.1038/ncomms14405. This publication reported a mechanism for the dampened expression of chaperones in polyQ expansion disease through the targeted degradation of HSF1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendillo ML, et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. This work identifies the HSF1 cancer gene signature, a set of genes that are largely distinct from those activated by heat shock stress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scherz-Shouval R, et al. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell. 2014;158:564–578. doi: 10.1016/j.cell.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vihervaara A, et al. Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc Natl Acad Sci USA. 2013;110:E3388–E3397. doi: 10.1073/pnas.1305275110. This work describes the comprehensive genomic binding profiles of HSF1 and HSF2 during stress and in mitotically arrested cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim E, et al. NEDD4-mediated HSF1 degradation underlies α-synucleinopathy. Hum Mol Genet. 2015;25:211–222. doi: 10.1093/hmg/ddv445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mercier PA, Winegarden NA, Westwood JT. Human heat shock factor 1 is predominantly a nuclear protein before and after heat stress. J Cell Sci. 1999;112:2765–2774. doi: 10.1242/jcs.112.16.2765. [DOI] [PubMed] [Google Scholar]
- 9.Hentze N, Le Breton L, Wiesner J, Kempf G, Mayer MP. Molecular mechanism of thermosensory function of human heat shock transcription factor Hsf1. eLife. 2016;5:e11576. doi: 10.7554/eLife.11576. This work identifies mechanisms for intrinsic thermosensing by HSF1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neef DW, Jaeger AM, Thiele DJ. Genetic selection for constitutively trimerized human HSF1 mutants identifies a role for coiled-coil motifs in DNA binding. G3 (Bethesda) 2013;3:1315–1324. doi: 10.1534/g3.113.006692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaeger AM, Pemble CW, Sistonen L, Thiele DJ. Structures of HSF2 reveal mechanisms for differential regulation of human heat-shock factors. Nat Struct Mol Biol. 2016;23:147–154. doi: 10.1038/nsmb.3150. Structural biology studies demonstrated a new model for HSF2 binding to DNA and elucidated key regulatory distinctions between HSF1 and HSF2 via the DBD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabindran SK, Haroun RI, Clos J, Wisniewski J, Wu C. Regulation of heat shock factor trimer formation: role of a conserved leucine zipper. Science. 1993;259:230–234. doi: 10.1126/science.8421783. [DOI] [PubMed] [Google Scholar]
- 13.Ahn SG, Thiele DJ. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17:516–528. doi: 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gothard LQ, Ruffner ME, Woodward JG, Park-Sarge OK, Sarge KD. Lowered temperature set point for activation of the cellular stress response in T-lymphocytes. J Biol Chem. 2003;278:9322–9326. doi: 10.1074/jbc.M209412200. [DOI] [PubMed] [Google Scholar]
- 15.Jurivich DA, Pachetti C, Qiu L, Welk JF. Salicylate triggers heat shock factor differently than heat. J Biol Chem. 1995;270:24489–24495. doi: 10.1074/jbc.270.41.24489. [DOI] [PubMed] [Google Scholar]
- 16.Nakai A, editor. Heat Shock Factor. Springer; 2016. [Google Scholar]
- 17.Shi Y, Mosser DD, Morimoto RI. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998;12:654–666. doi: 10.1101/gad.12.5.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neef DW, et al. A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep. 2014;9:955–966. doi: 10.1016/j.celrep.2014.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sivéry A, Courtade E, Thommen Q. A minimal titration model of the mammalian dynamical heat shock response. Phys Biol. 2016;13:066008. doi: 10.1088/1478-3975/13/6/066008. [DOI] [PubMed] [Google Scholar]
- 20.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 21.Pernet L, et al. HDAC6–ubiquitin interaction controls the duration of HSF1 activation after heat-shock. Mol Biol Cell. 2014;25:4187–4194. doi: 10.1091/mbc.E14-06-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. This early work described a key function for HSF1 in cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang YQ, et al. Increased heat shock transcription factor 1 in the cerebellum reverses the deficiency of Purkinje cells in Alzheimer’s disease. Brain Res. 2013;1519:105–111. doi: 10.1016/j.brainres.2013.04.059. [DOI] [PubMed] [Google Scholar]
- 24.Goetzl EJ, et al. Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann Clin Transl Neurol. 2015;2:769–773. doi: 10.1002/acn3.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee YJ, et al. HSF1 as a mitotic regulator: phosphorylation of HSF1 by Plk1 is essential for mitotic progression. Cancer Res. 2008;68:7550–7560. doi: 10.1158/0008-5472.CAN-08-0129. [DOI] [PubMed] [Google Scholar]
- 26.Elsing AN, et al. Expression of HSF2 decreases in mitosis to enable stress-inducible transcription and cell survival. J Cell Biol. 2014;206:735–749. doi: 10.1083/jcb.201402002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Littlefield O, Nelson HC. A new use for the ‘wing’ of the ‘winged’ helix–turn–helix motif in the HSF–DNA cocrystal. Nat Struct Biol. 1999;6:464–470. doi: 10.1038/8269. [DOI] [PubMed] [Google Scholar]
- 28.Neudegger T, Verghese J, Hayer-Hartl M, Hartl FU, Bracher A. Structure of human heat-shock transcription factor 1 in complex with DNA. Nat Struct Mol Biol. 2016;23:140–146. doi: 10.1038/nsmb.3149. This study deciphered the structure of the human HSF1 DBD, elucidating key features of HSF1 topology and a new model for the HSF1–DNA interaction. [DOI] [PubMed] [Google Scholar]
- 29.Fujimoto M, et al. RPA assists HSF1 access to nucleosomal DNA by recruiting histone chaperone FACT. Mol Cell. 2012;48:182–194. doi: 10.1016/j.molcel.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 30.Xu YM, Huang DY, Chiu JF, Lau AT. Post-translational modification of human heat shock factors and their functions: a recent update by proteomic approach. J Proteome Res. 2012;11:2625–2634. doi: 10.1021/pr201151a. [DOI] [PubMed] [Google Scholar]
- 31.Westerheide SD, et al. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raychaudhuri S, et al. Interplay of acetyltransferase EP300 and the proteasome system in regulating heat shock transcription factor 1. Cell. 2014;156:975–985. doi: 10.1016/j.cell.2014.01.055. A key role for acetylation in regulating HSF1 degradation. [DOI] [PubMed] [Google Scholar]
- 33.Carnemolla A, et al. Contesting the dogma of an age-related heat shock response impairment: implications for cardiac-specific age-related disorders. Hum Mol Genet. 2014;23:3641–3656. doi: 10.1093/hmg/ddu073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Q, Xiao H, Isobe K. Histone acetyltransferase activities of cAMP-regulated enhancer-binding protein and p300 in tissues of fetal, young, and old mice. J Gerontol A Biol Sci Med Sci. 2002;57:B93–98. doi: 10.1093/gerona/57.3.b93. [DOI] [PubMed] [Google Scholar]
- 35.Yang J, Oza J, Bridges K, Chen KY, Liu AY. Neural differentiation and the attenuated heat shock response. Brain Res. 2008;1203:39–50. doi: 10.1016/j.brainres.2008.01.082. [DOI] [PubMed] [Google Scholar]
- 36.Liu DJ, et al. SIRT1 knockdown promotes neural differentiation and attenuates the heat shock response. J Cell Physiol. 2014;229:1224–1235. doi: 10.1002/jcp.24556. [DOI] [PubMed] [Google Scholar]
- 37.Zelin E, Zhang Y, Toogun OA, Zhong S, Freeman BC. The p23 molecular chaperone and GCN5 acetylase jointly modulate protein–DNA dynamics and open chromatin status. Mol Cell. 2012;48:459–470. doi: 10.1016/j.molcel.2012.08.026. This study demonstrated how the chaperone p23 cooperates with the acetyltransferase GCN5 to modulate HSF1 genomic occupancy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zelin E, Freeman BC. Lysine deacetylases regulate the heat shock response including the age-associated impairment of HSF1. J Mol Biol. 2015;427:1644–1654. doi: 10.1016/j.jmb.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Budzynśki MA, Puustinen MC, Joutsen J, Sistonen L. Uncoupling stress-inducible phosphorylation of heat shock factor 1 from its activation. Mol Cell Biol. 2015;35:2530–2540. doi: 10.1128/MCB.00816-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kourtis N, et al. FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat Cell Biol. 2015;17:322–332. doi: 10.1038/ncb3121. High levels of HSF1 drive cancer survival and metastasis. This work demonstrated that, in some cancers, HSF1 levels are stabilized by loss of the FBXW7 F box protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai S, et al. Suppression of the HSF1-mediated proteotoxic stress response by the metabolic stress sensor AMPK. EMBO J. 2015;34:275–293. doi: 10.15252/embj.201489062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin X, Moskophidis D, Mivechi NF. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 2011;14:91–103. doi: 10.1016/j.cmet.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hendriks IA, et al. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat Struct Mol Biol. 2017;24:325–336. doi: 10.1038/nsmb.3366. [DOI] [PubMed] [Google Scholar]
- 44.Anckar J, et al. Inhibition of DNA binding by differential sumoylation of heat shock factors. Mol Cell Biol. 2006;26:955–964. doi: 10.1128/MCB.26.3.955-964.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol Cell Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hardie DG. AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem Soc Trans. 2011;39:1–13. doi: 10.1042/BST0390001. [DOI] [PubMed] [Google Scholar]
- 47.Hahn JS, Thiele DJ. Activation of the Saccharomyces cerevisiae heat shock transcription factor under glucose starvation conditions by Snf1 protein kinase. J Biol Chem. 2004;279:5169–5176. doi: 10.1074/jbc.M311005200. [DOI] [PubMed] [Google Scholar]
- 48.Yan LJ, et al. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J. 2002;21:5164–5172. doi: 10.1093/emboj/cdf528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsunemi T, et al. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra197. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu Z, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic co-activator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 51.Bagattin A, Hugendubler L, Mueller E. Transcriptional co-activator PGC-1α promotes peroxisomal remodelling and biogenesis. Proc Natl Acad Sci USA. 2010;107:20376–20381. doi: 10.1073/pnas.1009176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weydt P, et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1α in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 53.LeBleu VS, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16:992–1003. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma X, et al. Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1–PGC1α transcriptional axis. Cell Metab. 2015;22:695–708. doi: 10.1016/j.cmet.2015.08.005. This study identified a role for HSF1 in metabolic regulation through its activation of PGC1α. [DOI] [PubMed] [Google Scholar]
- 55.Xu L, Ma X, Bagattin A, Mueller E. The transcriptional coactivator PGC1α protects against hyperthermic stress via cooperation with the heat shock factor HSF1. Cell Death Dis. 2016;7:e2102. doi: 10.1038/cddis.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Minsky N, Roeder RG. Direct link between metabolic regulation and the heat-shock response through the transcriptional regulator PGC-1α. Proc Natl Acad Sci USA. 2015;112:E5669–5678. doi: 10.1073/pnas.1516219112. This work demonstrates a direct interaction between HSF1 and PGC1α in metabolic regulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.El Fatimy R, et al. Heat shock factor 2 is a stress-responsive mediator of neuronal migration defects in models of fetal alcohol syndrome. EMBO Mol Med. 2014;6:1043–1061. doi: 10.15252/emmm.201303311. This work identifies a role for HSF2 in neuronal migration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Homma S, et al. Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in hsf1-deficient mice. J Neurosci. 2007;27:7974–7986. doi: 10.1523/JNEUROSCI.0006-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ingenwerth M, Estrada V, Stahr A, Müller HW, von Gall C. HSF1-deficiency affects gait coordination and cerebellar calbindin levels. Behav Brain Res. 2016;310:103–108. doi: 10.1016/j.bbr.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 60.Uchida S, et al. Impaired hippocampal spinogenesis and neurogenesis and altered affective behavior in mice lacking heat shock factor 1. Proc Natl Acad Sci USA. 2011;108:1681–1686. doi: 10.1073/pnas.1016424108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hooper PL, Durham HD, Török Z, Crul T, Vígh L. The central role of heat shock factor 1 in synaptic fidelity and memory consolidation. Cell Stress Chaperones. 2016;21:745–753. doi: 10.1007/s12192-016-0709-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neef DW, Jaeger AM, Thiele DJ. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov. 2011;10:930–944. doi: 10.1038/nrd3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Havel LS, Li S, Li XJ. Nuclear accumulation of polyglutamine disease proteins and neuropathology. Mol Brain. 2009;2:21. doi: 10.1186/1756-6606-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hay DG, et al. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13:1389–1405. doi: 10.1093/hmg/ddh144. [DOI] [PubMed] [Google Scholar]
- 65.Labbadia J, et al. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest. 2011;121:3306–3319. doi: 10.1172/JCI57413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chafekar SM, Duennwald ML. Impaired heat shock response in cells expressing full-length polyglutamine-expanded huntingtin. PLoS ONE. 2012;7:e37929. doi: 10.1371/journal.pone.0037929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hodges A, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 68.Labbadia J, et al. Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain. 2012;135:1180–1196. doi: 10.1093/brain/aws022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kampinga HH, Bergink S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016;15:748–759. doi: 10.1016/S1474-4422(16)00099-5. [DOI] [PubMed] [Google Scholar]
- 70.Kakkar V, et al. The S/T-rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol Cell. 2016;62:272–283. doi: 10.1016/j.molcel.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 71.Hayashida N, et al. Heat shock factor 1 ameliorates proteotoxicity in cooperation with the transcription factor NFAT. EMBO J. 2010;29:3459–3469. doi: 10.1038/emboj.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fujimoto M, et al. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J Biol Chem. 2005;280:34908–34916. doi: 10.1074/jbc.M506288200. [DOI] [PubMed] [Google Scholar]
- 73.Kondo N, et al. Heat shock factor-1 influences pathological lesion distribution of polyglutamine-induced neurodegeneration. Nat Commun. 2013;4:1405. doi: 10.1038/ncomms2417. This work demonstrates that HSF1 has a key role in reducing the extent and tissue distribution of protein aggregation in a polyQ neurodegenerative disease mouse model. [DOI] [PubMed] [Google Scholar]
- 74.Riva L, et al. Poly-glutamine expanded huntingtin dramatically alters the genome wide binding of HSF1. J Huntingtons Dis. 2012;1:33–45. This work demonstrates the genome-wide dysregulation of HSF1 in Huntington disease. [PMC free article] [PubMed] [Google Scholar]
- 75.Trinklein ND, Murray JI, Hartman SJ, Botstein D, Myers RM. The role of heat shock transcription factor 1 in the genome-wide regulation of the mammalian heat shock response. Mol Biol Cell. 2004;15:1254–1261. doi: 10.1091/mbc.E03-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maheshwari M, et al. Dexamethasone induces heat shock response and slows down disease progression in mouse and fly models of Huntington’s disease. Hum Mol Genet. 2013;23:2737–2751. doi: 10.1093/hmg/ddt667. [DOI] [PubMed] [Google Scholar]
- 77.Li L, Saegusa H, Tanabe T. Deficit of heat shock transcription factor 1-heat shock 70 kDa protein 1A axis determines the cell death vulnerability in a model of spinocerebellar ataxia type 6. Genes Cells. 2009;14:1253–1269. doi: 10.1111/j.1365-2443.2009.01348.x. [DOI] [PubMed] [Google Scholar]
- 78.Batista-Nascimento L, Neef DW, Liu PC, Rodrigues-Pousada C, Thiele DJ. Deciphering human heat shock transcription factor 1 regulation via post-translational modification in yeast. PLoS ONE. 2011;6:e15976. doi: 10.1371/journal.pone.0015976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shinkawa T, et al. Heat shock factor 2 is required for maintaining proteostasis against febrile-range thermal stress and polyglutamine aggregation. Mol Biol Cell. 2011;22:3571–3583. doi: 10.1091/mbc.E11-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 81.Liangliang X, et al. Dominant-positive HSF1 decreases α-synuclein level and α-synuclein-induced toxicity. Mol Biol Rep. 2010;37:1875–1881. doi: 10.1007/s11033-009-9623-2. [DOI] [PubMed] [Google Scholar]
- 82.Lee G, et al. Casein kinase II-mediated phosphorylation regulates α-synuclein/synphilin-1 interaction and inclusion body formation. J Biol Chem. 2004;279:6834–6839. doi: 10.1074/jbc.M312760200. [DOI] [PubMed] [Google Scholar]
- 83.Dzamko N, Zhou J, Huang Y, Halliday GM. Parkinson’s disease-implicated kinases in the brain; insights into disease pathogenesis. Front Mol Neurosci. 2014;7:57. doi: 10.3389/fnmol.2014.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mavroudis IA, et al. Morphological changes of the human Purkinje cells and deposition of neuritic plaques and neurofibrillary tangles on the cerebellar cortex of Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2010;25:585–591. doi: 10.1177/1533317510382892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y, et al. Hsp90 chaperone inhibitor 17-AAG attenuates Aβ-induced synaptic toxicity and memory impairment. J Neurosci. 2014;34:2464–2470. doi: 10.1523/JNEUROSCI.0151-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bobkova NV, et al. Therapeutic effect of exogenous Hsp70 in mouse models of Alzheimer’s disease. J Alzheimers Dis. 2014;38:425–435. doi: 10.3233/JAD-130779. [DOI] [PubMed] [Google Scholar]
- 87.Pierce A, et al. Over-expression of heat shock factor 1 phenocopies the effect of chronic inhibition of TOR by rapamycin and is sufficient to ameliorate Alzheimer’s-like deficits in mice modeling the disease. J Neurochem. 2013;124:880–893. doi: 10.1111/jnc.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Masliah E, et al. Casein kinase II alteration precedes tau accumulation in tangle formation. Am J Pathol. 1992;140:263–268. [PMC free article] [PubMed] [Google Scholar]
- 89.Rosenberger AF, et al. Increased occurrence of protein kinase CK2 in astrocytes in Alzheimer’s disease pathology. J Neuroinflamm. 2016;13:4. doi: 10.1186/s12974-015-0470-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Greenwood JA, Scott CW, Spreen RC, Caputo CB, Johnson GV. Casein kinase II preferentially phosphorylates human tau isoforms containing an amino-terminal insert. Identification of threonine 39 as the primary phosphate acceptor. J Biol Chem. 1994;269:4373–4380. [PubMed] [Google Scholar]
- 91.Lin PY, et al. Heat shock factor 1 over-expression protects against exposure of hydrophobic residues on mutant SOD1 and early mortality in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegener. 2013;8:43. doi: 10.1186/1750-1326-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jung MK, et al. Expression of taurine transporter (TauT) is modulated by heat shock factor 1 (HSF1) in motor neurons of ALS. Mol Neurobiol. 2013;47:699–710. doi: 10.1007/s12035-012-8371-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Batulan Z, et al. High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci. 2003;23:5789–5798. doi: 10.1523/JNEUROSCI.23-13-05789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen HJ, et al. The heat shock response plays an important role in TDP-43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain. 2016;139:1417–1432. doi: 10.1093/brain/aww028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Watanabe S, et al. SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol Brain. 2014;7:62. doi: 10.1186/s13041-014-0062-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carlomagno Y, et al. Casein kinase II induced polymerization of soluble TDP-43 into filaments is inhibited by heat shock proteins. PLoS ONE. 2014;9:e90452. doi: 10.1371/journal.pone.0090452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Herman AM, Khandelwal PJ, Stanczyk BB, Rebeck GW, Moussa CE. β-Amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res. 2011;1386:191–199. doi: 10.1016/j.brainres.2011.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dai C, Sampson SB. HSF1: guardian of proteostasis in cancer. Trends Cell Biol. 2016;26:17–28. doi: 10.1016/j.tcb.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Björk JK, et al. Heat-shock factor 2 is a suppressor of prostate cancer invasion. Oncogene. 2016;35:1770–1784. doi: 10.1038/onc.2015.241. HSF2 has a role in tumour suppression by altering gene expression profiles associated with cell differentiation and invasion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Meng L, Gabai VL, Sherman MY. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene. 2010;29:5204–5213. doi: 10.1038/onc.2010.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Min JN, Huang L, Zimonjic DB, Moskophidis D, Mivechi NF. Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene. 2007;26:5086–5097. doi: 10.1038/sj.onc.1210317. [DOI] [PubMed] [Google Scholar]
- 102.Xi C, Hu Y, Buckhaults P, Moskophidis D, Mivechi NF. Heat shock factor Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary tumorigenesis and metastasis. J Biol Chem. 2012;287:35646–35657. doi: 10.1074/jbc.M112.377481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dai C, et al. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest. 2012;122:3742–3754. doi: 10.1172/JCI62727. This work places HSF1 downstream of cancer signalling pathways. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao YH, et al. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009;28:3689–3701. doi: 10.1038/onc.2009.229. [DOI] [PubMed] [Google Scholar]
- 106.Tang Z, et al. MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell. 2015;160:729–744. doi: 10.1016/j.cell.2015.01.028. This study demonstrates a role for HSF1 in protecting cancer cells from proteomic instability, suggesting that HSF1 is a key target for cancer therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 108.Ciocca DR, Arrigo AP, Calderwood SK. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol. 2013;87:19–48. doi: 10.1007/s00204-012-0918-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chou SD, Murshid A, Eguchi T, Gong J, Calderwood SK. HSF1 regulation of β-catenin in mammary cancer cells through control of HuR/elavL1 expression. Oncogene. 2015;34:2178–2188. doi: 10.1038/onc.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gabai VL, et al. Heat shock transcription factor Hsf1 is involved in tumor progression via regulation of hypoxia-inducible factor 1 and RNA-binding protein HuR. Mol Cell Biol. 2012;32:929–940. doi: 10.1128/MCB.05921-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Su KH, Dai C. Protein quantity–quality balance licenses growth. Cell Cycle. 2016;15:3155–3156. doi: 10.1080/15384101.2016.1220714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Su KH, et al. HSF1 critically attunes proteotoxic stress sensing by mTORC1 to combat stress and promote growth. Nat Cell Biol. 2016;18:527–539. doi: 10.1038/ncb3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhong YH, Cheng HZ, Peng H, Tang SC, Wang P. Heat shock factor 2 is associated with the occurrence of lung cancer by enhancing the expression of heat shock proteins. Oncol Lett. 2016;12:5106–5112. doi: 10.3892/ol.2016.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Loison F, et al. Up-regulation of the clusterin gene after proteotoxic stress: implication of HSF1–HSF2 heterocomplexes. Biochem J. 2006;395:223–231. doi: 10.1042/BJ20051190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sandqvist A, et al. Heterotrimerization of heat-shock factors 1 and 2 provides a transcriptional switch in response to distinct stimuli. Mol Biol Cell. 2009;20:1340–1347. doi: 10.1091/mbc.E08-08-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Santagata S, et al. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science. 2013;341:1238303. doi: 10.1126/science.1238303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fujimoto M, Nakai A. The heat shock factor family and adaptation to proteotoxic stress. FEBS J. 2010;277:4112–4125. doi: 10.1111/j.1742-4658.2010.07827.x. [DOI] [PubMed] [Google Scholar]
- 118.Goodson ML, Park-Sarge OK, Sarge KD. Tissue-dependent expression of heat shock factor 2 isoforms with distinct transcriptional activities. Mol Cell Biol. 1995;15:5288–5293. doi: 10.1128/mcb.15.10.5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fujimoto M, et al. Analysis of HSF4 binding regions reveals its necessity for gene regulation during development and heat shock response in mouse lenses. J Biol Chem. 2008;283:29961–29970. doi: 10.1074/jbc.M804629200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fujimoto M, et al. HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J. 2004;23:4297–4306. doi: 10.1038/sj.emboj.7600435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nakai A, et al. HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol Cell Biol. 1997;17:469–481. doi: 10.1128/mcb.17.1.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fujimoto M, et al. A novel mouse HSF3 has the potential to activate nonclassical heat-shock genes during heat shock. Mol Biol Cell. 2010;21:106–116. doi: 10.1091/mbc.E09-07-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tessari A, et al. Characterization of HSFY, a novel AZFb gene on the Y chromosome with a possible role in human spermatogenesis. Mol Hum Reprod. 2004;10:253–258. doi: 10.1093/molehr/gah036. [DOI] [PubMed] [Google Scholar]
- 124.Wan Z, et al. Evidence for the role of AMPK in regulating PGC-1α expression and mitochondrial proteins in mouse epididymal adipose tissue. Obes (Silver Spring) 2014;22:730–738. doi: 10.1002/oby.20605. [DOI] [PubMed] [Google Scholar]
- 125.Runne H, et al. Dysregulation of gene expression in primary neuron models of Huntington’s disease shows that polyglutamine-related effects on the striatal transcriptome may not be dependent on brain circuitry. J Neurosci. 2008;28:9723–9731. doi: 10.1523/JNEUROSCI.3044-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Guettouche T, Boellmann F, Lane WS, Voellmy R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005;6:4. doi: 10.1186/1471-2091-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Asano Y, et al. IER5 generates a novel hypo-phosphorylated active form of HSF1 and contributes to tumorigenesis. Sci Rep. 2016;6:19174. doi: 10.1038/srep19174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Olsen JV, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 129.Soncin F, et al. Transcriptional activity and DNA binding of heat shock factor-1 involve phosphorylation on threonine 142 by CK2. Biochem Biophys Res Commun. 2003;303:700–706. doi: 10.1016/s0006-291x(03)00398-x. [DOI] [PubMed] [Google Scholar]
- 130.Calderwood SK, et al. Signal transduction pathways leading to heat shock transcription. Sign Transduct Insights. 2010;2:13–24. doi: 10.4137/STI.S3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Holmberg CI, et al. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J. 2001;20:3800–3810. doi: 10.1093/emboj/20.14.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hietakangas V, et al. Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol Cell Biol. 2003;23:2953–2968. doi: 10.1128/MCB.23.8.2953-2968.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hietakangas V, et al. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc Natl Acad Sci USA. 2006;103:45–50. doi: 10.1073/pnas.0503698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chu B, Zhong R, Soncin F, Stevenson MA, Calderwood SK. Transcriptional activity of heat shock factor 1 at 37 °C is repressed through phosphorylation on two distinct serine residues by glycogen synthase kinase 3 and protein kinases Cα and Cζ. J Biol Chem. 1998;273:18640–18646. doi: 10.1074/jbc.273.29.18640. [DOI] [PubMed] [Google Scholar]
- 135.Murshid A, et al. Protein kinase A binds and activates heat shock factor 1. PLoS ONE. 2010;5:e13830. doi: 10.1371/journal.pone.0013830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chou SD, Prince T, Gong J, Calderwood SK. mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS ONE. 2012;7:e39679. doi: 10.1371/journal.pone.0039679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sourbier C, et al. Englerin A stimulates PKCθ to inhibit insulin signaling and to simultaneously activate HSF1: pharmacologically induced synthetic lethality. Cancer Cell. 2013;23:228–237. doi: 10.1016/j.ccr.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tan K, et al. Mitochondrial SSBP1 protects cells from proteotoxic stresses by potentiating stress-induced HSF1 transcriptional activity. Nat Commun. 2015;6:6580. doi: 10.1038/ncomms7580. The mitochondrial SSBP1 DNA replication factor is re-localized in response to stress to interact with nuclear HSF1, potentiating the expression of a network of stress protective genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.