

To the Editor
Mechanisms underlying exacerbations of allergic asthma, although poorly defined, are clearly linked with viral infections.1 Rhinoviruses are the most common viruses associated with asthma exacerbations.E1–E3 Influenza virus infections have a similar association,E4–E6 resulting in increased disease severity in this populationE7,E8 (see Table E1 in this article’s Online Repository at www.jacionline.org). Defects in innate cell antiviral responses represent one potential mechanism underlying the link between viral infections and atopic disease exacerbations. For example, high-affinity IgE receptor (FcεRI) stimulation on plasmacytoid dendritic cells inhibits influenza-induced IFN-α secretion2 and suppresses Toll-like receptor 72 and Toll-like receptor 9 expression.3 Decreasing serum IgE with omalizumab in allergic patients with asthma restores ex vivo antiviral IFN-α responses, and correlates with improved clinical outcomes.4 Monocytes are also recruited to respiratory sites during viral infectionsE17 and allergen exposure,5, E18 and implicated in food allergy development.6 In addition, allergic stimulation via IgE cross-linking impacts monocyte functions including apoptosis,E19 cytokine secretion, and phagocytosis.7 Defining IgE-mediated effects on monocyte antiviral responses could reveal additional pathways involved in atopic disease pathogenesis.
Antigen presentation, which drives T-cell activation and differentiation, represents a critical monocyte antiviral function. Influenza exposure increased monocyte-driven TH1 priming of naive CD4+ lymphocytes in primary human monocyte-T-cell cocultures, but did not significantly affect TH2 differentiation (Fig 1, A; see Fig E1 in this article’s Online Repository at www.jacionline.org). Influenza-driven TH1 differentiation inversely correlated with monocyte surface FcεRI expression (Fig 1, B), a surrogate for serum IgE,E20 suggesting that IgE-mediated stimulation may impact monocyte-directed TH1 priming during viral infection. Confirming this concept, IgE cross-linking of influenza-exposed monocytes significantly disrupted TH1 differentiation (Fig 1, C, and Fig E1), a finding consistent across all donors (Fig 1, D). Similarly, IgE cross-linking decreased IFN-γ secretion (Fig 1, E) and T-cell proliferation (Fig 1, F, and Fig E1), but had no effect on TH2 development (see Fig E2 in this article’s Online Repository at www.jacionline.org). The decrease in TH1 cell stimulation was not due to IgE-mediated inhibition of influenza virus infection of monocytes because IgE cross-linking did not affect percentages of monocytes expressing the influenza proteins hemagglutinin and nucleoprotein (Fig 1, G and H). These data suggest that allergen-mediated IgE cross-linking on monocytes during concomitant viral infection could result in impaired development of virus-specific TH1 cells in sensitized individuals in vivo.
FIG 1.
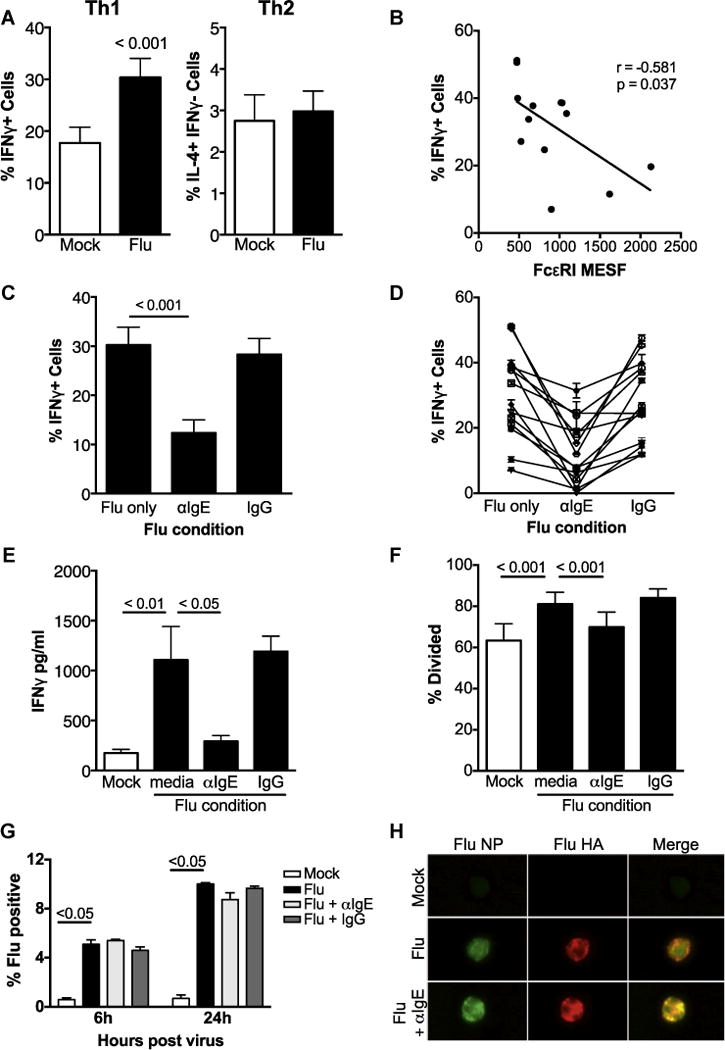
IgE cross-linking inhibits virus-driven monocyte priming of TH1 cells. A, Naive CD4 T cells polarized with monocytes exposed to no virus (mock) or influenza A virus (Flu). B, Linear regression analysis was used to determine correlation between baseline monocyte FcεRI expression (x-axis) and TH1 priming capacity (%IFN-γ+ cells, y-axis) of influenza-exposed monocytes with P values and r2 values noted. N = 15. C and D, % IFN-γ+ T cells in monocyte-T-cell cocultures under IgE cross-linking conditions: (Fig 1, C) mean and (Fig 1, D) individual donor pairs. N = 15. E, Mean IFN-γ concentration in T-cell supernatants (N = 5). F, IgE cross-linking diminishes T-cell proliferation. Naive CD4 T cells labeled with VPD450 cellular dye before coculture. % divided defined as cells with lower VPD450 fluorescence compared with unstimulated T-cell controls, N = 3. Error bars represent the SEM. Pertinent P values are shown and were obtained with 2-way ANOVA analysis. G and H, Influenza A virus protein expression in monocytes at 6 and 24 hours postinfection by (Fig 1, G) flow cytometry of Flu-positive cells (NP/HA double positive) and (Fig 1, H) flow cytometry imaging at 6 hours postinfection (NP, green; HA, red). Shown is representative data from N = 3 experiments. HA, Influenza hemagglutinin; MESF, mean equivalent soluble fluorochrome; NP, influenza nucleoprotein. Error bars represent SEM, and P values represent results of 1-way ANOVA. All Material and Methods can be found in this article’s Online Repository at www.jacionline.org.
We evaluated potential roles for multiple cytokines in IgE-mediated inhibition of monocyte CD4+ T lymphocyte development, including IL-12, a critical factor in TH1 differentiation (see Figs E3 and E4 in this article’s Online Repository at www.jacionline.org). Our results indicate that IL-12, TNF-α, IL-6, and IL-10 do not regulate IgE-mediated suppression of monocyte-driven antiviral TH1 priming.
Upregulation of MHC and costimulatory molecules is required for engagement and activation of the T-cell receptor complex. These signals direct CD4+ T-cell maturation; stronger antigen signal strength favors TH1 differentiation, whereas weaker signals promote TH2 or regulatory phenotypes.E21 We evaluated the effect of IgE cross-linking on influenza-induced monocyte maturation. Influenza exposure significantly upregulated surface CD80, CD86, and MHC class I (HLA-ABC) and II (HLA-DR) expression, with levels significantly diminished by IgE-mediated stimulation (Fig 2, A–C). We hypothesized that IgE-mediated downregulation of HLA-DR and CD86 may disrupt monocyte-T-cell interactions. Visualization and quantitation of monocyte-T-cell interactions revealed that monocyte-T-cell clusters were significantly increased by influenza exposure, but impaired by IgE cross-linking (Fig 2, D and E). Because cell proliferation directly reflects T-cell activation, the IgE-mediated decrease in cell division (Fig 1, F) provides additional evidence that IgE cross-linking prevents monocytes from forming high-affinity interactions needed to activate multiple T cells simultaneously. These data support the model that allergic stimulation significantly impairs upregulation of proteins essential for antigen presentation, resulting in weaker T lymphocyte stimulation and diminished antiviral TH1 responses (see Fig E5 in this article’s Online Repository at www.jacionline.org).
FIG 2.
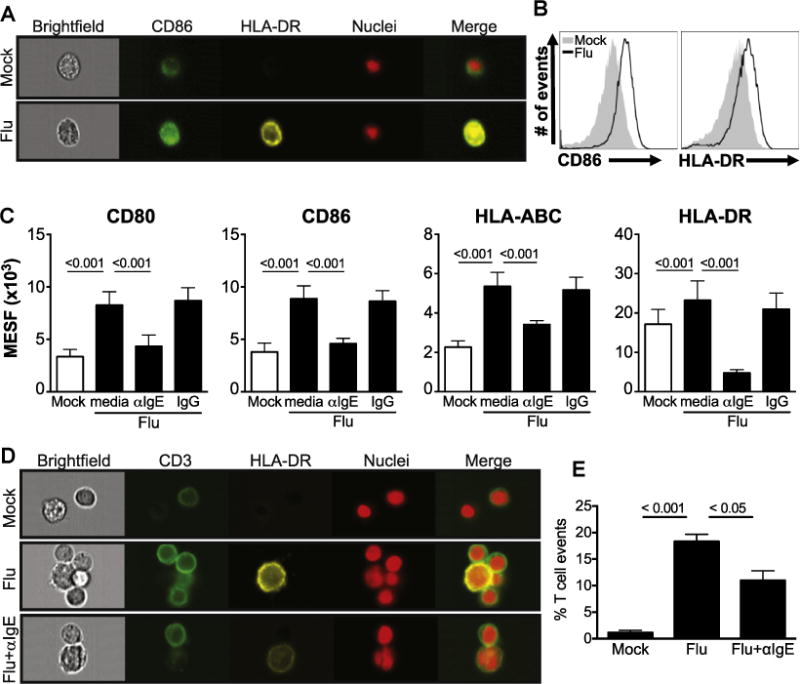
IgE cross-linking inhibits virus-induced monocyte maturation and monocyte-T-cell interactions. A–C, Monocytes treated with media alone (mock) or influenza A virus (Flu) ± IgE cross-linking or IgG control antibody and stained for various cell surface markers. Representative flow cytometry (Fig 2, A) imaging analysis and (Fig 2, B) histograms for CD86 and HLA-DR staining are shown for mock and Flu only (Fig 2, C). Mean cell surface marker expression by flow cytometry analysis of the mean equivalent soluble fluorochrome (MESF, expressed as ×103 units) value was determined under the various conditions, N = 3. D, Flow cytometry imaging analysis of monocyte-T-cell interactions. Cell clusters from monocyte-T-cell cocultures stained with anti-CD3 (green), anti–HLA-DR (yellow), and nuclei (red) were visualized, 60× magnification on the Amnis ImageStream flow cytometer (Amnis, Seattle, Wash). E, Quantitation of total monocyte-T-cell clusters was performed using the IDEAS Software (Amnis) (N = 3). Error bars represent SEM, and P values represent results of 1-way ANOVA. All Material and Methods can be found in this article’s Online Repository at www.jacionline.org.
IgE-mediated inhibition of virus-induced TH1 priming was remarkably conserved across all donors, and may reflect an evolutionarily maintained mechanism. Parasite-produced proteins have effects on antigen-presenting cells similar to IgE cross-linking on monocytes—downregulation of antigen presenting molecules, altered CD4 T-cell activation, and decreased TH1 responses.E22,E23 Parasite infection can even alter viral immune responses in coinfection models.E24–E27 Parasite-specific IgE is protective during parasitic infectionsE28,E29; diminished TH1 responses induced by IgE cross-linking could thus be immunomodulatory in the setting of either parasitic or atopic diseases. Along these lines, many epidemiological studies reveal that children with parasitic infections have decreased atopy.E30–E32 Parasitic infections are characterized by TH2-mediated immunity. Activation of IgE-mediated pathways by parasites, or allergen-specific IgE, could thus negatively regulate TH1 development, creating a TH2-predominant, more optimal antiparasitic response. Decreased TH1 differentiation could then dampen proinflammatory responses during parasitic (or even viral) infections, resulting in decreased inflammation and tissue damage,E33–E36 and providing potential benefit in allergic airway disease.
Alternatively, IgE-mediated disruption of antiviral TH1 responses could promote more severe viral disease via dysregulation of normal TH1/TH2 balance and contribute to exacerbations of allergic disease. The samples in our study were predominantly from unknown donors obtained via a local blood bank; no information regarding atopic status or serum IgE levels was available. We can thus only extrapolate our findings to those with defined atopic disease. However, the range of surface FcεRI expression suggests a spectrum of IgE levels in our study participants, and the inverse correlation with influenza-induced TH1 differentiation supports the concept that elevated IgE may contribute to defective monocyte-directed antiviral TH1 responses in vivo. Consistent with our findings, others have reported blunted TH1 responses to viral infections associated with atopy. Following rhinovirus infection of patients with asthma, blood and airway CD4 T cells secreted decreased IFN-γ.8 Similarly, patients with atopic dermatitis had decreased virus-specific TH1 cells after transcutaneous live virus vaccination, which inversely correlated with serum IgE levels.9 In future studies, it will be important to evaluate effects of IgE cross-linking on T-cell priming by monocytes exposed to other viruses, including rhinoviruses, and compare responses in atopic (high IgE) versus nonatopic individuals.
To our knowledge, this is the first study demonstrating IgE-mediated impairment of human monocyte antiviral responses, via suppression of virus-induced monocyte maturation and TH1 differentiation. This extends our understanding of how allergen-mediated pathways alter host antiviral responses, highlighting a potential role for monocytes in virus-associated exacerbations of allergic disease. Further studies investigating the mechanisms underlying IgE-mediated suppression of virus-induced monocyte responses will enhance our comprehension of allergic disease pathogenesis and potentially uncover novel targets for therapeutic development.
Supplementary Material
Acknowledgments
We thank the participants for donating blood, Dr Angela Mobley and the UT Southwestern Flow Cytometry Facility for assistance with flow cytometry– based assays, Dr David Farrar for providing select reagents and assistance with experimental design, and the following funding sources for supporting our research: the National Institutes of Health: the National Institute of Allergy and Infectious Diseases (grant no. R01-AI098077 to M.A.G.), Ruth L. Kirschstein National Research Service Award (grant no. T32-AI005284 to D.M.P.), the National Institute of General Medical Sciences (grant no. T32-GM008014 to D.M.P.), and the National Heart, Lung, and Blood Institute (grant no. T32-HL098040 to R.K.R. and A.R.T.); Children’s Clinical Research Advisory Council (grant award to M.A.G.); and the UT Southwestern William A. and Joyce M. Sellars Distinguished Chair in Allergy and Immunology.
This study was supported by the National Institutes of Health: the National Institute of Allergy and Infectious Diseases (grant no. R01-AI098077 to M.A.G.), Ruth L. Kirschstein National Research Service Award (grant no. T32-AI005284 to D.M.P.), the National Institute of General Medical Sciences (grant no. T32-GM008014 to D.M.P.), and the National Heart, Lung, and Blood Institute (grant no. T32-HL098040 to R.K.R. and A.R.T.); Children’s Clinical Research Advisory Council (grant award to M.A.G.); and the UT Southwestern William A. and Joyce M. Sellars Distinguished Chair in Allergy and Immunology.
Disclosure of potential conflict of interest: M. A. Gill receives grant support from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH) and travel support from American Thoracic Society Meeting. R. K. Rowe receives grant support from the National Heart, Lung, and Blood Institute (NHLBI) and travel support from the Society for Pediatric Research. D. M. Pyle receives grant support from NIH/NRSA and NIH/National Institute of General Medical Sciences. A. R. Tomlinson receives grant support from NIH/NHLBI. Z. Hu and T. Lv receive grant support from the NIH.
References
- 1.Rowe RK, Gill MA. Asthma: the interplay between viral infections and allergic diseases. Immunol Allergy Clin North Am. 2015;35:115–27. doi: 10.1016/j.iac.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 2.Gill MA, Bajwa G, George TA, Dong CC, Dougherty II, Jiang N, et al. Counter-regulation between the FcepsilonRI pathway and antiviral responses in human plasmacytoid dendritic cells. J Immunol. 2010;184:5999–6006. doi: 10.4049/jimmunol.0901194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schroeder JT, Bieneman AP, Xiao H, Chichester KL, Vasagar K, Saini S, et al. TLR9- and FcepsilonRI-mediated responses oppose one another in plasmacytoid dendritic cells by down-regulating receptor expression. J Immunol. 2005;175:5724–31. doi: 10.4049/jimmunol.175.9.5724. [DOI] [PubMed] [Google Scholar]
- 4.Teach SJ, Gill MA, Togias A, Sorkness CA, Arbes SJ, Jr, Calatroni A, et al. Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. J Allergy Clin Immunol. 2015;136:1476–85. doi: 10.1016/j.jaci.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee YG, Jeong JJ, Nyenhuis S, Berdyshev E, Chung S, Ranjan R, et al. Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemo-attractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol. 2015;52:772–84. doi: 10.1165/rcmb.2014-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y, Collier F, Naselli G, Saffery R, Tang ML, Allen KJ, et al. Cord blood monocyte-derived inflammatory cytokines suppress IL-2 and induce nonclassic “TH2-type” immunity associated with development of food allergy. Sci Transl Med. 2016;8:321ra8. doi: 10.1126/scitranslmed.aad4322. [DOI] [PubMed] [Google Scholar]
- 7.Pyle DM, Yang VS, Gruchalla RS, Farrar JD, Gill MA. IgE cross-linking critically impairs human monocyte function by blocking phagocytosis. J Allergy Clin Immunol. 2013;131:491–500e5. doi: 10.1016/j.jaci.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, et al. Rhino-virus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc Natl Acad Sci U S A. 2008;105:13562–7. doi: 10.1073/pnas.0804181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slifka MK, Leung DY, Hammarlund E, Raue HP, Simpson EL, Tofte S, et al. Transcutaneous yellow fever vaccination of subjects with or without atopic dermatitis. J Allergy Clin Immunol. 2014;133:439–47. doi: 10.1016/j.jaci.2013.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.