

Abstract
Aims
Analysis of the stability and safety of Sulfolobus monocaudavirus 1 (SMV1) during passage through the mammalian GI tract.
Methods and Results
A major challenge of using nano-vectors to target gut microbiome is their survival during passage through the extremely acidic and proteolytic environment of the mammalian GI tract. Here, we investigated the thermo-acidophilic archaeal virus SMV1 as a candidate therapeutic nano-vector for the distal mammalian GI tract microbiome. We investigated the anatomical distribution, vector stability and immunogenicity of this virus following oral ingestion in mice and compared these traits to the more classically used Inovirus vector M13KE. We found that SMV1 particles were highly stable under both simulated GI tract conditions (in vitro) and in mice (in vivo). Moreover, SMV1 could not be detected in tissues outside the GI tract and it elicited a nearly undetectable inflammatory response. Finally, we used human intestinal organoids (HIOs) to show that labelled SMV1 did not invade or otherwise perturb the human GI tract epithelium.
Conclusion
Sulfolobus monocaudavirus 1 appeared stable and safe during passage though the mammalian GI tract.
Significance and Impact of the Study
This is the first study evaluating an archaeal virus as a potential therapeutic nanoparticle delivery system and it opens new possibilities for future development of novel nanoplatforms.
Keywords: archaeal virus, bacteriophage, mammalian GI tract, nanoparticle, thermophilic and acid-resistant virus
Introduction
The importance of the gut microbiome for human health and during certain diseases has been established, and the diversity of the microbial community in the human GI tract is vast (averaging 1013–1014 cells with an estimated 5 000 species (Turnbaugh et al. 2007)) and is largely individual-specific. Therefore, the gut microbiome is a potential new target for personalized drug therapy (Jia et al. 2008; Vinje et al. 2014). Optimal personalized therapies may take the form of drug- or vaccine-delivery vectors on the same size scale as micro-organisms (nano-vectors), which could be noninvasively administered. However, development of oral nano-vectors for the human gut remains challenging. In particular, nano-vectors must be stable in diverse chemical environments and remain intact when they reach the desired body site, which for orally administered vectors mean survival in extremely acidic and proteolytic environments (e.g. pepsins, proteases and other degradative enzymes of the GI tract). The intestinal mucus layer is another obstacle; many nanoparticles are trapped in the mucus and are rapidly removed (Ensign et al. 2012). Moreover, nano-vectors used specifically for microbiome therapy should not cross the epithelial gut barrier and enter into systemic circulation so as to prevent therapeutic complications due to immunogenicity and off-target effects (Shaji and Patole 2008). Further, when developing therapeutic nano-vectors, the ease of production, scale up and chemical/biochemical modification are all important factors to consider.
Viruses are naturally occurring nanoparticle platforms and despite their disease-causing ability in some contexts, they have many desirable attributes for novel nanobiotechnological applications. In the field of biomedicine, plant and bacterial viruses (bacteriophages) are favoured as they are nonpathogenic to animals, which limits safety concerns (Soto and Ratna 2010). Cowpea mosaic virus and other virus-based protein cage architectures have been engineered for viral delivery of tissue- or cell-specific fluorescent reporters and drugs (Robertson and Liu 2012; Niikura et al. 2013). In some instances, chemical modifications were added to the virus to increase evasion of the host immune system, for increasing circulation time, and for increasing tissue distribution (Kim et al. 2008). Often, there is a significant disconnect between the in vitro development of novel viral nano-vectors and their translation into the clinic because promising in vitro attributes are not maintained in vivo (Yildiz et al. 2011). Thus, evaluating potential nano-vectors in vivo is a critical step in the development of these novel therapeutics.
To our knowledge, no thermostable archaeal virus (i.e. viruses infecting cells of the domain Archaea) has been examined as a potential nanoparticle vector in mammalian systems. This study addresses the use of Sulfolobus monocaudavirus 1 (SMV1) as a candidate therapeutic nano-vector for targeting microbial cells of the mammalian gut. SMV1 infects strains of the hyperthermophilic archaeal genus Sulfolobus, and was originally isolated from an acidic hot spring (pH 2–3 and 75–80°C) in Yellowstone National Park (Erdmann et al. 2013). The lemon-shaped SMV1 particles are enveloped and have a diameter of about 120 nm and tail fibres protruding from one end (see Fig. 1 for TEM image). Owing to their extremophile nature (hyperthermostable and acid resistant) (Uldahl et al. 2016a), SMV1 particles are promising candidates for surviving passage through a mammalian gut. As with plant viruses and bacteriophages, SMV1 should not be pathogenic to humans or animals. To our knowledge, an archaeal virus has never been evaluated in vivo, but the potential for their use as nano-vector platforms has been proposed (Steinmetz et al. 2008; Evans 2009; Uldahl and Peng 2013) and recently, an in vitro study investigating SMV1 and its interaction with mammalian cells provided promising results (Uldahl et al. 2016b). In this study, SMV1 was shown to be efficiently internalized by both human endothelial cells of brain and umbelical vein origin without any detrimental effect on cell morphology, plasma membrane and mitochondrial functionality (Uldahl, Wu, Hall, Papathanasiou, Peng, and Moghimi 2016b), thus indicating SMV1 as a promising therapeutic nano-vector.
Figure 1.

Characteristics of the archaeal virus Sulfolobus monocaudavirus 1 (SMV1) and the bacteriophage M13KE.
In this study, we first compared the in vitro stability of SMV1 in simulated gastric conditions against the more classically used Inovirus vector, M13KE. We then quantified viral faecal shedding in mice following oral challenge as well as viral infectivity after passage through the GI tract. In parallel, we examined pro-inflammatory cytokine levels in serum of orally challenged mice to quantify possible host immune response. Finally, to generate more human-relevant data, we used human intestinal organoids (HIOs) and fluorescently labelled SMV1 particles to determine whether the virus readily crossed the human gut epithelium. Our results provide strong evidence that SMV1 is extremely stable in the mammalian GI tract, it does not stimulate the murine immune system, and it does not readily cross the human intestinal epithelium.
Materials and methods
SMV1 preparation
Sulfolobus monocaudavirus 1 was propagated in a highly susceptible strain, Sulfolobus islandicus CRISPR deletion mutant delta C1C2 (Gudbergsdottir et al. 2011). The host culture was grown in Sulfolobus medium supplemented with 0·2% tryptone, 0·1% yeast extract, 0·2% sucrose and 0·002% uracil (TYS + U medium) (Zillig et al. 1994). Cultures were started from −80°C stock; cells were transferred to 50 ml TYS + U medium and incubated at 78°C. After 24 h of incubation, the 50 ml cell culture was transferred to 950 ml of preheated (78°C) TYS + U medium. The culture was grown to an OD600 of 0·2–0·3 (typically 24 h) at which time point the host culture was infected with SMV1 suspension. The virion-containing supernatant was collected 48–72 h postinfection and concentrated by ultrafiltration using VivaSpin 1 000 kDa centrifugal concentrators (Sartorius, Aubagne Cedex, France). Additionally, the virus concentrate was purified by ultracentrifugation through a 10–40% continuous Iodixanol gradient. Continuous gradients were prepared by sequentially layering 10, 20, 30 and 40% iodixanol solution (OptiPrep™, Axis-Shield PoC AS, Oslo, Norway) in 10 mm Tris-acetate, pH 6 into 14 ml centrifuge tubes (Beckman Coulter UK Ltd., High Whickham, Bucks, UK), the gradients were allowed to diffuse overnight at 4°C in the dark. Ultrafiltrated virus sample was layered over the continuous gradient (1 ml) and centrifuged in a SW-41 rotor (Beckman Coulter) for 6 h at 35 000 rpm at 4°C. The opaque band was recovered and an additional ultrafiltration step was performed; the virus fraction was washed three times with 10 ml of 10 mmol l−1 Tris-acetate, pH 6 to remove the Iodixanol from the solution to prevent interference with downstream processes. TEM was used to confirm the presence of virus particles in the recovered fraction. The virus sample was then stored at 4°C until used.
Fluorescent labelling of SMV1 particles
Immediately before labelling, 1 μl of PKH67 dye (Sigma, St. Louis, MO, USA) was diluted in 1 ml of Diluent C (Sigma, St. Louis, MO, USA). Two volumes of diluted PKH67 was added to 1 volume of virus suspension (250 μl) and mixed by pipetting. After 3 min, the labelling was stopped by adding 3 volumes of 1% BSA, then incubated for 1 min to allow binding of excess dye. The virus particles were centrifuged at 17 000 g for 20 min at 4°C, then the supernatant was carefully removed. A tiny yellow pellet was observed. The pellet was re-suspended in 3 ml of 10 mmol l−1 Tris-acetate, pH 6 and centrifuged at 17 000 g for 20 min. The washing step was repeated twice. After the final wash, the virus pellet was re-suspended in 250 μl 10 mmol l−1 Tris-acetate, pH 6. The virus titre after labelling was determined by plaque assay. SMV1 PKH67 particles were stored at 4°C in the dark until use.
Determination of SMV1 plaque forming units
The virus titre of unlabelled and fluorescently labelled SMV1 particles was determined by plaque assays. Serial dilutions of a virus suspension (10 μl) were mixed with 2 ml of the exponentially growing host culture (2 × 108 cells). The mixtures were incubated for 30 min at 78°C to allow the adsorption of the virus to the host cells. Immediately following the addition of 2 ml of TYS + U medium containing 0·25% Gelrite (78°C), the sample was layered over a premade 0·8% Gelrite plate (78°C). The plates were incubated for 2 days at 78°C. Sulfolobus monocaudavirus 1 plaques appeared as small clear halos, easy to enumerate. Sulfolobus monocaudavirus 1 titres averaged between 109 and 1011 plaque forming units (PFU) per ml.
Bacteriophage M13KE preparation
Ph.D.-11 phage display peptide library kit was purchased from New England Bioloabs (Ipswich, MA, USA). The kit contained Escherichia coli (E. coli) host strain, ER2738 and bacteriophage M13KE. The host strain and phage were propagated according to the detailed manufacturer’s protocol. To ensure high virus titre, the phage amplification was performed in 2 × 20 ml host cultures. The final eluted phage solutions were serially diluted and quantified by plating on lawns of ER 2738 grown on agar containing 5-bromo-4-chloro-3indolyl-β-D-galactopyranoside (X-Gal) and isopropyl-β-D-thiogalactopyranoside (IPTG) (LB/IPTG/X-Gal plates), which show blue plaques after incubation at 37°C overnight. The plaques were then counted and the phage titre averaged between 1011 and 1013 PFU per ml.
Stability of SMV1 particles
A 10 μl suspension of SMV1 (3 × 107 PFU) was added to 990 μl of the tested buffers. Suspensions of ethanol/water and DMSO/water were mixed in proportions 1:1 for a final concentration of 50%. Simulated gastric fluid (SGF) pH 1·6 and simulated intestinal fluid (SIF) pH 6·5 were prepared according to Klein (2010). Both solutions were used within 48 h. The tube contents were mixed by mild vortexing and the tubes were immediately placed in a 37°C incubator simulating the temperature of the human body. As a reference, 10 μl of SMV1 (3 × 107 PFU) was added to 990 μl of 10 mmol l−1 Tris-acetate buffer, pH 6. One suspension was stored at 37°C, the other at 4°C (normal long-term storage temp.). Duplicates of each of the six different storage conditions were stored for 7 days. The suspensions were monitored by plaque assays 1 and 8 h after mixing and then once a day for the following 6 days.
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the care and use of laboratory animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of Montana State University. All mice were euthanized by osoflurane inhalation, and all efforts were made to minimize suffering.
Experimental animals
Nine-week-old, male C57BL/6 mice were used for these experiments. Experimental groups of mice were housed in separate cages. The bacteriophage M13KE, unlabelled archaeal virus SMV1, fluorescently labelled SMV1 (SMV1 PKH97) and Tris buffer controls were prepared as described above and introduced individually to three mice per tested sample via oral gavage. Each mouse received a 200 μl volume of either viral suspension or buffer. After gavage, mice were observed and faecal pellets were collected for 5 days. The dose of each virus was as follows: SMV1 = 3·2 × 1011 PFU, M13KE = 7 × 1011 PFU, and SMV1 PKH67 = 4 × 1010 PFU. Faecal pellets were collected from each mouse at 0, 1, 3, 5, 7, 11, 24, 48, 72, 96 and 120 h after oral gavage. Immediately after collection, pellets were stored at −20°C. Mice were euthanized after 5 days and tissue samples (stomach, small intestine and colon) were removed for a histopathological analysis. An identical experiment was conducted (two mice per group) for cytokine analysis and one mouse from each group was euthanized at 6 and 12 h postgavage. Blood was collected immediately after euthanasia by perfusing the carotid artery with physiological saline.
Monitoring for signs of disease/distress
Each mouse, in all three groups, was examined for lack of movement, ataxia, hunched posture, ruffled fur, hyperthermia, dehydration, diarrhoea, seizure and death. The mice were examined five times in the first 12 h after the oral administration, then once a day for the following 4 days. Body weights were recorded on a daily basis.
Extraction of virus particles from faecal pellets following virus administration
Prior to extraction, each individual faecal pellet was weighed for later normalization. The individual pellets averaged 0·03 g and were homogenized in 20 ml of SM buffer (50 mmol l−1 Tris-HCl, pH 7·5, 100 mmol l−1 NaCl and 8 mmol l−1 MgSO4). Particulate matter was removed by centrifugation at 6 000 g for 10 min and the supernatant was filtered through 0·32 μm Supor membrane filters (Acrodisc® Syringe Filters, Pall Life Sciences, Corp., East Hills, NY, USA). The virus present in the filtrate was concentrated by ultrafiltration using 100K MWCO Spin-x centrifugal concentrators (Corning Incorporated). The final volume was adjusted to 500 μl. The viral nucleic acid (vDNA) was extracted from 200 μl concentrate using the PureLink viral RNA/DNA mini kit (Invitrogen, USA), according to the manufacturer’s protocol. The isolated vDNA was quantified by qPCR. Additionally, the concentrates of extracted viral particles were tested for infectivity using plaque assays.
Isolation of total DNA from mouse tissues
Total DNA was extracted from 25 mg brain, liver, spleen (10 mg) and caecal tip tissues, based on the DNeasy blood and tissue kit (Qiagen, Valencia, CA). We examined tissues from a total of six mice, groups 1–3 (from the cytokine experiment), euthanized at 6 or 12 h post-virus or buffer administration. All samples were processed according to the manufacturer’s protocol; lysing overnight in 180 μl ATL lysis buffer as outlined in the DNeasy user manual (“Purification of Total DNA from Animal Tissues” as found in DNeasy Blood & Tissue Handbook, Qiagen, Valencia, CA; July 2006). DNA was recovered in a single elution step with 100 μl AE solution from the kit.
qPCR assays
After isolating vDNA or total DNA from faecal samples, tissues and serum, qPCR was used to estimate the approximate amount of SMV1 or M13KE particles in each specific sample. Primer sets specific to the two viruses were designed for the assays. M13KE primers and conditions were previously published (Jaye et al. 2003). Sulfolobus monocaudavirus 1 primers were designed using SnapGene software. The primer pair was as follows: SMV1F(5′-CCTATTCAACGTATCAAATCCG-3′) and SMV1R(5′-CTTATCGCCTCAACTACTTTAT-GAAG-3′). The qPCR reaction mixtures (20 μl) consisted of 10 μl of SsoFast™ EvaGreen® Supermix (Bio-Rad), 0·8 μl forward primer (10 μmol l−1), 0·8 μl reverse primer (10 μmol l−1), template DNA preparation (2 μl) and sterile MilliQ water (6·4 μl). Amplification was performed with Rotor-Gene Q real-time PCR cycler (Qiagen). Sulfolobus monocaudavirus 1 qPCR cycling conditions were 95°C (5 min), followed by 30 3-step cycles at 95°C (5 s), 61°C (5 s) and 72°C (20 s) and a final cycle at 72°C (5 min). For M13KE cycling conditions, we followed Jaye, Nolte, Mazzucchelli, Geigerman, Akyildiz and Parkos (2003) (Jaye, Nolte, Mazzucchelli, Geigerman, Akyildiz and Parkos 2003). Samples from the control mice gavaged with Tris buffer were used as negative controls and all samples were run in triplicate. Plasmids containing the relevant template DNA were constructed to establish qPCR standard curves. Each amplicon was cloned into the pCR2·1-TOPO® vector (TOPO TA-cloning kit, Invitrogen), plated and subsequently, individual clones were picked and grown overnight in 3 ml LB medium containing ampicillin. The plasmids were purified using PureYield™ plasmid miniprep (Promega) and were sequenced to check for potential errors. Larger batches of correct plasmid DNA were prepared and the concentrations were determined by spectrophotometry. To generate standard curves, the plasmids were serially diluted. A standard curve consisted of at least five different dilutions ranging between 10 ng μl−1 and 100 fg μl−1, and was included on every run in duplicate. Real-time data capture was performed with Qiagen’s Rotor-Gene Q operating software. The faecal samples with signal above detection limit were screened by TEM for viral particles (see below).
Cytokine ELISA
Blood was collected from the heart using a syringe, loaded in a serum separator tube and immediately centrifuged at 10 000 g for 5 min. The separated serum was stored at −20°C until tested for 12 different pro-inflammatory cytokines using a Multi-Analyte ELISArray Kit (Qiagen, CA, USA) according to manufacturer’s protocols.
Histopathological analysis
After completion of the experimental time course, three mice were randomly picked for histopathological analysis, one from each treatment group and anaesthetized using isoflurane. The stomach, small intestine and colon were collected, fixed in 4% PBS paraformaldehyde and stored at 4°C. After 48 h, the organs were transferred to 70% ethanol. Tissue sections were cut into 5 μmol l−1 thick sections, mounted on glass slides, stained with haematoxylin and eosin (H&E) and examined by a pathologist, screening for apoptotic cells in sections from stomach, small intestine and colon.
Incubation of SMV1 with cultured human intestinal organoids
Human intestinal organoids (HIOs) were obtained from the laboratory of Jason R. Spence, University of Michigan. Generation, growth and maintenance of HIOs was carried out as previously described (McCracken et al. 2011). HIOs were grown to 2·5–3 mm in diameter and inoculated with virus suspension using thin-walled glass capillaries, pulled to fine-point needles. c. 3 μl SMV1 PKH67 virus suspension (2 × 109 PFU) or mock (10 mmol l−1 Tris-acetate buffer, pH 6) was injected using separate needles for each HIO. Immediately after inoculation, HIOs were suspended in fresh intestinal growth medium. Inoculated HIOs were incubated at 37°C and 5% CO2 for 8 h. At 8 h postinoculation, HIOs were washed three times in PBS and fixed with 2·5% gluteraldehyde in 0·1 mol l−1 sodium phosphate buffer overnight at 4°C. After fixation, HIOs were washed (PBS) and embedded in optimal-cutting-temperature freezing compound (Tissue-Tek® O.C.T. compound, Sakura® Finetek, USA), and frozen at −20°C to make blocks for sectioning. Sections (10 μm) of HIOs were cut using a cryostat and stained for immunofluorescence analysis. DAPI (4′,6-diamidino-2-phenylindole) was used as nuclear counterstain, 10−6: 1 Phalloidin-Tetramethylrhodamine B isothiocyanate (TRITC)/PBS was used to identify filamentous actin, and the alcian blue/PAS stain kit (Newcomer supply) was used to stain epithelial mucins.
Transmission electron microscopy
Virus containing samples were adsorbed onto carbon-coated copper grids for 5 min and stained with 2% uranyl acetate. Images were recorded using a Zeiss Leo 912-Omega transmission electron microscope (operating at 100 KV) with a ProScan 2048 × 2048-CCD camera.
Results
SMV1 retains stability and infectivity after exposure to simulated gastric conditions
Sulfolobus monocaudavirus 1 was selected as a potential nano-vector based on its extremophilic nature. A concentrated stock of SMV1 particles (107 PFU) were diluted into 990 μl of each of the storage solutions and incubated for 7 days at 37°C. The stability of the particles was monitored by using plaque assays to estimate any drop in infectivity over time. Overall, the infectivity of SMV1 particles in 1:1 DMSO/H2O and the two reference control groups (Tris-Acetate buffer, 4°C and 37°C) remained constant at 107 PFU per ml over the 7 days (Fig. 2). In contrast, the infectivity of SMV1 particles incubated in 1:1 EtOH/H2O dropped reproducibly after 1 h and was undetectable by 24 h. A difference in SMV1 infectivity was observed (Student’s t-test, P < 0·01) between incubation in SGF (pH 1·6) and SIF (pH 6·5). Sulfolobus monocaudavirus 1 particles incubated in SIF dropped two orders of magnitude in infectivity over the 7 days, though a significant difference was not observed until 72 h and onwards compared to the Tris 37°C control, whereas SMV1 particles incubated in SGF dropped three orders of magnitude in infectivity over the first 72 h and were undetectable after this point (Fig. 2). We next compared the stability of SMV1 to bacteriophage M13KE under similar conditions. Whereas SMV1 retained 80% of its starting infectivity after 1 h incubation in SGF, M13KE phage particles incubated in SGF at 37°C showed no infectivity at the three tested time points (1, 24 and 72 h) (Fig. S1), suggesting that strong acid is rapidly detrimental to M13KE. Moreover, the M13KE infectivity dropped about 100 times after 72 h incubation in SIF (Fig. S1). In accordance with its hyperthermophilic nature, we also found that SMV1 infectivity was unaffected by high heat (95°C for 5 min), whereas M13KE was completely inactivated (data not shown). Overall, these results indicate that SMV1 is more stable under simulated gastric conditions than M13KE.
Figure 2.
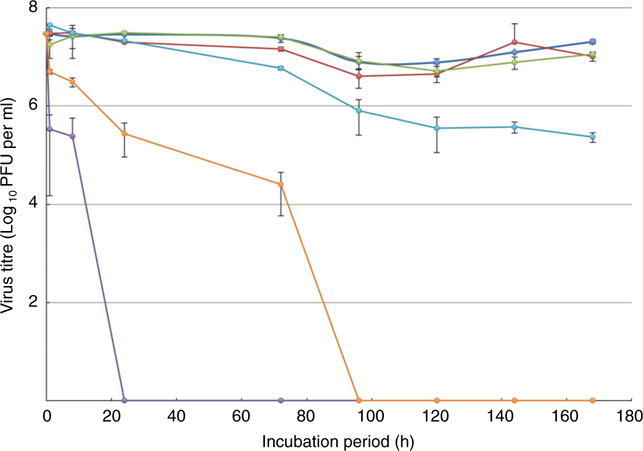
Survival of Sulfolobus monocaudavirus 1 (SMV1) particles. The infectivity of SMV1 after incubation at 37°C in 50% DMSO, 50% EtOH, simulated intestinal fluid (SIF) pH 6·5, and Simulated gastric fluid (SGF) pH 1·6. Incubation in 10 mmol l−1 Tris-acetate pH 6·0 was monitored as a reference at two temperatures; 4°C and 37°C. The number of plaque forming units per millilitre (plaque forming units (PFU) per ml) was determined by plaque assay and is shown as a function of time in hours. Three replicates were performed and the error bars represent standard deviation. (
 ) Tris buffer 4°C; (
) Tris buffer 4°C; (
 ) Tris buffer 37°C; (
) Tris buffer 37°C; (
 ) DMSO; (
) DMSO; (
 ) EtOH; (
) EtOH; (
 ) SIF pH 6·5; (
) SIF pH 6·5; (
 ) SGF pH 1·6. [Colour figure can be viewed at wileyonlinelibrary.com]
) SGF pH 1·6. [Colour figure can be viewed at wileyonlinelibrary.com]
Trafficking of SMV1 through the murine GI tract
The faecal shedding profiles for SMV1 were investigated in mice over a 5-day period. No indications of pain or distress were observed and mouse weights did not change significantly (Fig. S2). First, viral DNA in the faecal samples was quantified by qPCR using sequence-specific primers for SMV1. For inter-individual comparison, viral genome copy number per pellet was standardized to the average pellet weight (0·03 g) over the course of the experiment. Faecal SMV1 shedding was detected already at 3 h postoral gavage, peaked at 7 h and dropped below the detection limit by the 24 h time point, thus a shedding period between 3 and 11 h postoral gavage was observed (Fig. 3a). To test whether the recovered virus fractions were still infectious, plaque assays were performed. The plaque assays detected infectious viral particles at 5 and 7 h, corresponding to the highest virus shedding detected by qPCR (Fig. 3b). Passage through the GI tract appeared to have a significant influence on SMV1 infectivity, as the abundance of PFU per pellet was very low (103) compared to qPCR quantification of SMV1 genome equivalents per pellet (1010). Nevertheless, SMV1 particles appeared to be excreted in a stable form as TEM analysis of SMV1 particles recovered from faecal pellets revealed intact morphology (Fig. S3). Finally, to test whether modified SMV1 particles behave similarly to unlabelled SMV1 particles, we performed an identical GI tract passage experiment with fluorescently labelled SMV1 particles (SMV1 PKH67). SMV1 PKH67 particles had an identical shedding profile compared to the unmodified virus, and therefore, qPCR data from both virus groups were integrated (Fig. 3a).
Figure 3.
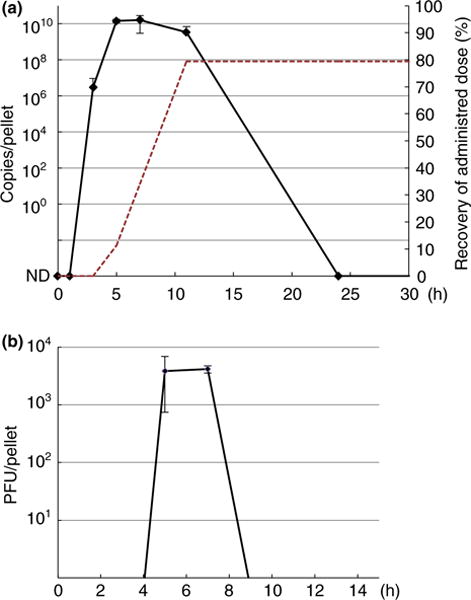
Sulfolobus monocaudavirus 1 (SMV1) shedding profiles. Faecal samples were collected at 0, 1, 3, 5, 7, 11, 24, 48, 72, 96 and 120 h, all were assayed by qPCR (a) and plaque assays (b) and results within the first 24 h (a) or 9 h (b) are depicted. The values (DNA copies/pellet in A and PFU per pellet in (b) were standardized to a pellet of 0·03 g. The Tris control group showed no positive signal at any time point. The red dotted line in (a) indicates the cumulative amount of viral DNA shedded over the peak period (5–11 h postoral gavage) as a percentage of the total administered dose. The cumulative amount is based on the average viral DNA concentration in the pellets from one mouse collected over the peak period with a stool frequency of 7 pellets per h (the cumulative curves were comparable for all mice in the group). Three replicates were performed and the error bars represent standard deviation (SD). ND, nondetectable. [Colour figure can be viewed at wileyonlinelibrary.com]
Based on the qPCR data, we estimated the cumulative amount of SMV1 particles shedded as a percentage of the original dose (Fig. 3a). This estimation was based on the average concentration of SMV1 DNA detected in the pellets collected during the peak shedding period and a published stool frequency estimate of 7 pellets per h for C57BL/6 mice (Li et al. 2006; Chandrasekharan et al. 2009; Ren et al. 2011). Based on this approach, a high percentage (c. 80%) of the original SMV1 travelled through the entire murine GI tract and was recovered in the faecal samples.
M13KE was also tested for the passage through murine GI tract (Fig. S2). Although the starting administered dose of M13KE particles (7 × 1011 PFU, see Materials and Methods) was similar to that of SMV1 (Table S1), no infectious M13KE was recovered from the faecal samples as revealed by plaque assays and we were not able to detect any excreted M13KE particles using TEM. qPCR analysis revealed an order of magnitude lower M13KE DNA (average 6·8 × 108 copies per pellet) in faecal samples compared to SMV1 (average 7·9 × 109 copies per pellet) (Fig. 3b, Table S1). Also, detection was limited to a narrower time span between 5 and 7 h after administration. Overall, these results indicate that SMV1 can traverse the mouse GI track more successfully than M13KE.
SMV1 is not detected in off-target tissues/organs
To determine whether SMV1 and M13KE particles trafficked to tissues outside the murine GI tract, we collected and analysed different tissues for the presence of vDNA by qPCR. Brain, liver, spleen, caecum, caecal tip and serum were analysed from one animal in each treatment group 6 and 12 h postoral gavage. Cardiac perfusion was used prior to tissue dissection to exclude virus that may have been present in the bloodstream. No SMV1 or M13KE vDNA was detected in the Tris-acetate controls and no SMV1 vDNA was detected in any tissue/organ. M13KE vDNA was detected in the caecal tip at both 6 and 12 h and the signal (Ct) was comparable between these time points (Table S2).
Inflammatory cytokine profiling
Serum levels of 12 different pro-inflammatory cytokines were quantified to determine whether oral administration of SMV1 triggered a pro-inflammatory host immune response (Table 1). Serum was collected from an individual in each of the treatment groups above at 6 and 12 h postoral gavage. All cytokines were below the limit of detection, with the exception of a slight increase in IL1β in the mouse that received M13KE and a slight increase in IFN-γ in the mouse that received SMV1. Both of these signals were above the limit of detection at 6 h postgavage, but back below the limit of detection at 12 h postgavage.
Table 1.
Inflammatory cytokine profiling in serum of mice inoculated orally with virus
| Conc. In 100 μl mouse serum (the ELISA was run on a 10E-1 dilution)
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cytokine | IL1 α | IL1 β | IL2 | IL4 | IL6 | IL10 | IL12 | IL17A | IFN-γ | TNF-α | G-CSF | GM-CSF |
| − Control | 0·07 | 0·089 | 0·054 | 0·049 | 0·108 | 0·061 | 0·086 | 0·052 | 0·06 | 0·058 | 0·064 | 0·119 |
| 6 h Tris | −0·08 | −0·01 | −0·06 | 0·02 | −0·22 | 0·13 | −0·21 | 0·28 | 0·03 | 0·02 | 0·07 | −0·12 |
| 6 h SMV1 | −0·02 | 0 | −0·12 | −0·07 | −0·37 | 0·12 | −0·2 | 0·05 | 0·87 | −0·02 | 0·09 | 0·07 |
| 6 h M13KE | 0·06 | 0·6 | 0·06 | 0·05 | −0·24 | 0·14 | −0·06 | 0·31 | 0·05 | 0·02 | −0·04 | 0·12 |
| 12 h Tris | −0·01 | −0·2 | 0 | 0·04 | −0·29 | 0·13 | −0·19 | 0·12 | 0·16 | 0·18 | −0·02 | −0·04 |
| 12 h SMV1 | 0·05 | −0·04 | 0·13 | 0·12 | −0·2 | 0·2 | −0·14 | 0·26 | 0·13 | 0·2 | 0·11 | 0·07 |
| 12 h M13KE | 0·04 | 0 | 0·04 | 0·06 | −0·29 | 0·16 | −0·15 | 0·2 | −0·11 | 0·21 | 0·01 | 0·11 |
| + Control | 1·65 | 1·05 | 1·83 | 1·87 | 1·13 | 0·83 | 1·41 | 2·45 | >2·5* | 1·70 | 0·29 | 1·82 |
Absorbance values >2·5 are not within the linear range of the assay. Lower limit of detection; values less than two times the negative control should not be interpreted. Absorbance read at 450 nm.
The serum levels of 12 pro-inflammatory cytokines measured under identical conditions in experimental animals at time points 6 h and 12 h postoral administration. Numbers in bold are significantly higher than detection limit.
SMV1 particle interaction with human intestinal epithelial cells
To test whether SMV1 particles were taken up by human epithelial cells, we inoculated concentrated SMV1 PKH67 particles (2 × 109 PFU) into the lumen of human intestinal organoids (HIOs). These structures are derived from embryonic stem cells and form 3-dimensional spheres, representing a single layer of columnar epithelium. Human intestinal organoids contain the five major cell types found in the human small intestine, including absorptive enterocytes, goblet cells, enteroendocrine cells, Paneth cells and self-renewing stem cells. This monolayer encloses an internal lumen and is surrounded by a layer of mesenchymal cells on the basal surface (Finkbeiner et al. 2012). HIOs were inoculated with either the concentrated SMV1 suspension or Tris buffer, incubated for 8 h, and prepared for confocal microscopy (see methods section). Very few green fluorescent SMV1 PKH67 particles were observed in the HIO sections, but some particles were visibly associated with the mucus layer on the surface of the HIO epithelium (Fig. 4). No particles were observed inside or between epithelial cells, suggesting that uptake, if present, was a rare event.
Figure 4.
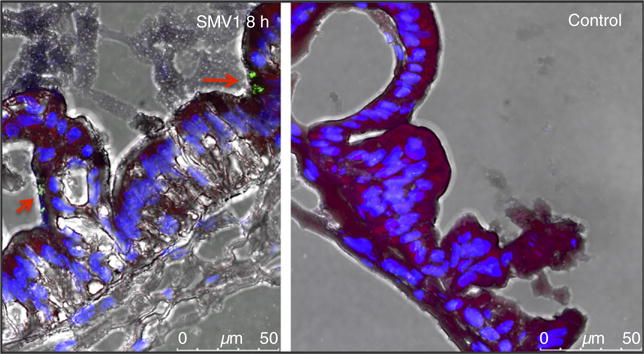
Sulfolobus monocaudavirus 1 (SMV1) particle interaction with human intestinal epithelial cells. The uptake of SMV1 particles in human intestinal organoids was evaluated by confocal microscopy. SMV1 were fluorescently labelled with the green dye PKH67, the cell nuclei were stained blue with DAPI, the actin filaments were red, and the mucus dye appear black. [Colour figure can be viewed at wileyonlinelibrary.com]
Histopathological analysis
A single mouse from each treatment group was examined for histologic signs of inflammation. As seen in Fig. 5, tissue from the stomach, small intestine and colon were similar in virus-treated and control mice. No differences were noted between these tissues by a trained pathologist.
Figure 5.
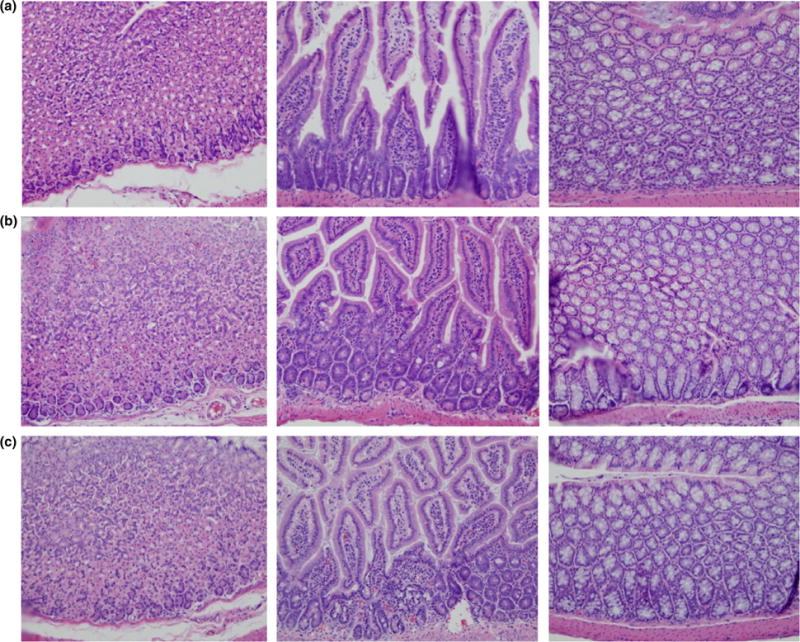
Histological examination of tissues from mice challenged with Sulfolobus monocaudavirus 1 (SMV1) and M13KE. Histopathological observation of tissues obtained from stomach, small intestine and colon, demonstrating no treatment-related microscopic effects. (a). Tris-acetate control; (b). Bacteriophage M13KE; (c). Archaeal virus SMV1. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
The main objective of this study was to evaluate the stability of the acido-thermophilic archaeal virus SMV1 in the GI tract as a first step towards developing this virus as nanoplatform for biomedical gut applications. Sulfolobus monocaudavirus 1 was chosen owing to its extreme thermal stability, lack of potential hosts in animals and its ease of large-scale production. Sulfolobus monocaudavirus 1 extremophilic nature suggests it can survive the harsh conditions present in the GI tract of humans and animals. In order to evaluate SMV1 against a commonly used viral nanoplatform, we compared it to the E. coli bacteriophage, M13KE, which is the most commonly used phage for in vivo phage display experiments (Tothova et al. 2012) and as an addressable nanoprotein cage architecture (Yi et al. 2012). Sulfolobus monocaudavirus 1 showed no decline in infectivity when incubated in 50% DMSO. Stability in DMSO is advantageous, as this chemical is often used as a solvent in bioconjugation protocols (Jin et al. 2009; Wen et al. 2012). Storage in 50% EtOH proved to be the most damaging condition for SMV1 particles with infectivity undetectable after 24 h. Ethanol is a useful solvent when working with silica, which is used for surface functionalization of nanoparticles with nanosilica in applied nanotechnology (Rahman et al. 2009). However, if a 10–100 fold drop in infectivity can be tolerated, short experiments (1–8 h) appear feasible (Fig. 1). We have shown that SMV1 particles lose infectivity under certain conditions but the virions may maintain structural integrity providing a noninfectious protein cage, which could prove useful in certain types of applications.
Viral stability in simulated GI conditions is valuable for prediction of in vivo behaviour of drug formulations (Klein 2010) and potential viral nanoplatform-based vectors, like phages (Tothova, Babickova, and Celec 2012). Two types of media were used to evaluate the stability of SMV1 particles: SIF (simulation of small intestinal conditions in the fasted state), and SGF (simulation of stomach conditions in the fasted state). The stability of SMV1 measured in our experiments (only a 2 log drop in infectivity in SIF over the 7 days and 3 log drop in infectivity in SGF over 72 h) indicates that this virus can easily survive passage through the GI tract. In particular, the stomach emptying of mice and humans as quantified by27Al nuclear magnetic resonance was 50 and 30 min (Schwarz et al. 2002), respectively, and the total GI transit time in mice and humans has been approximated to 6–10 h and 2–4 days respectively (Graff et al. 2001; Thompson et al. 2010; Padmanabhan et al. 2013). Within the corresponding emptying time frames, SMV1 infectivity dropped only 10 fold in SGF and less than 10 fold in SIF whereas M13KE lost completely its infectivity (c. 1010 fold drop) in SGF and dropped about 100 fold in infectivity in SIF. Thus, our in vitro results suggest that SMV1 is likely to remain stable and infectious in the GI tract of both of these mammals and would be more capable of oral delivery and passage through the GI tract than M13KE.
The in vivo stability of SMV1 was determined by quantifying both viral genomic DNA by qPCR and viral infectivity by plaque assays. For qPCR, we reasoned that viral DNA would be protected in vivo when packaged inside intact viral capsids thus providing a good indicator for intact viral particles. The faecal clearance interval of orally administered virus particles was comparable to previous noted GI transit times (see above) with peak viral shedding 7 h postsample gavage. c. 80% of the administered SMV1 was recovered in faecal samples. However, M13KE particles were present about 10 times less in excreted faecal pellets although the initial administered dose was similar, suggesting that this phage is easily degraded and may not be sufficiently stable to carry effector functions to distal GI tract targets. In addition to the stability of the unlabeled SMV1 virus, we also found that a fluorescently modified variant had similar passage dynamics, suggesting that chemical modifications of the viral envelope have minimal or no negative effect on the stability, providing encouragement that future cellular targeting ligands can be incorporated onto the surface of the SMV1 particles. Taken together with the observed stability in 50% DMSO, the modification potential of SMV1 particles without loss of stability appears promising.
We found no evidence that labelled SMV1 were taken up by epithelial cells of human intestinal organoids. These results suggest that SMV1 particles reaching the human intestines are likely to travel lumenally with minimal epithelial cell interaction. Further support for this hypothesis is that no SMV1 DNA was detected outside the GI tract, whereas M13KE DNA was detected in two caecal tip samples.
An important consideration when using a proteinaceous virus particle is the potential for a robust host immune response. As no archaeal viruses are known to be pathogenic to mammals, we expected to observe a minimal, if any, inflammatory response at the administered dose. We were encouraged to find that nearly all of the 12 pro-inflammatory cytokines were below our limit of detection, indicating that mice were highly tolerant of both SMV1 and M13KE. One limitation of our study is the low numbers of mice considered. However, multiple lines of evidence supported little or no murine response to viral challenge, making additional animal experiments difficult to justify. The further lack of histopathology (Fig. 5), signs of overt pain or distress or changes in weight all suggest that ingestion of SMV1 and M13KE was well tolerated by these mammalian hosts. Future studies are needed to develop SMV1 particles with the ability to attach to GI tract targets, such as absorptive enterocytes lining the GI mucosal surface.
To the best of our knowledge, this is the first study to evaluate an archaeal virus as a potential therapeutic nanoparticle delivery system for human GI tract. We demonstrated that SMV1 particles are exceptionally stable under simulated GI tract conditions. We also showed that SMV1 particles are stable in the GI tract of mice, elicited a nearly undetectable inflammatory response, and were well tolerated. Although several viral-based nanoplatforms are in development, SMV1 particles provide many favourable in vivo characteristics for bioengineering applications, such as drug and vaccine delivery.
Supplementary Material
Figure S1 Survival of M13KE particles. The infectivity of M13KE after incubation at 37°C in SIF pH 6·5 and SGF pH 1·6. Incubation in TBS buffer pH 7·5 was monitored as a control. The number of plaque forming units per millilitre (PFU ml−1) was determined by plaque assay and is shown as a function of time in hours.
Figure S2 Observations of mice body weights. Body weights were measured daily for 5 days. At the 0 h time point, a one-time administration of either 200 μl VLP solution or control Tris-acetate solution was carried out.
Figure S3 TEM images of recovered SMV1 particles from the faecal samples show intact morphology.
Table S1 Summary of virus titre. Estimated SMV1 titre in original inoculum and those detected in faecal pellets. Estimates based on qPCR and PFU assays are presented.
Table S2 Detection of SMV1 and M13KE DNA in tissues of mice inoculated orally with virus.
Acknowledgments
X.P. was supported by The Danish Council for Independent Research/Technology and production [grant number DFF – 7017-00060].
Footnotes
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Conflict of Interest
None.
References
- Chandrasekharan BP, Kolachala VL, Dalmasso G, Merlin D, Ravid K, Sitaraman SV, Srinivasan S. Adenosine 2B receptors (A(2B)AR) on enteric neurons regulate murine distal colonic motility. Faseb J. 2009;23:2727–2734. doi: 10.1096/fj.09-129544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensign LM, Cone R, Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv Drug Deliver Rev. 2012;64:557–570. doi: 10.1016/j.addr.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann S, Shah SA, Garrett RA. SMV1 virus-induced CRISPR spacer acquisition from the conjugative plasmid pMGB1 in Sulfolobus solfataricus P2. Biochem Soc Trans. 2013;41:1449–1458. doi: 10.1042/BST20130196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DJ. Exploitation of plant and archaeal viruses in bionanotechnology. Biochem Soc Trans. 2009;37:665–670. doi: 10.1042/BST0370665. [DOI] [PubMed] [Google Scholar]
- Finkbeiner SR, Zeng XL, Utama B, Atmar RL, Shroyer NF, Estes MK. Stem cell-derived human intestinal organoids as an infection model for rotaviruses. MBio. 2012;3:e00159–12. doi: 10.1128/mBio.00159-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Brinch K, Madsen JL. Gastrointestinal mean transit times in young and middle-aged healthy subjects. Clin Physiol. 2001;21:253–259. doi: 10.1046/j.1365-2281.2001.00308.x. [DOI] [PubMed] [Google Scholar]
- Gudbergsdottir S, Deng L, Chen ZJ, Jensen JVK, Jensen LR, She QX, Garrett RA. Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol Microbiol. 2011;79:35–49. doi: 10.1111/j.1365-2958.2010.07452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaye DL, Nolte FS, Mazzucchelli L, Geigerman C, Akyildiz A, Parkos CA. Use of real-time polymerase chain reaction to identify cell-and tissue-type-selective peptides by phage display. Am J Pathol. 2003;162:1419–1429. doi: 10.1016/S0002-9440(10)64275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W, Li HK, Zhao LP, Nicholson JK. Gut microbiota: a potential new territory for drug targeting. Nat Rev Drug Discov. 2008;7:123–129. doi: 10.1038/nrd2505. [DOI] [PubMed] [Google Scholar]
- Jin X, Newton JR, Montgomery-Smith S, Smith G. A generalized kinetic model for amine modification of proteins with application to phage display. Biotechniques. 2009;46:175–182. doi: 10.2144/000113074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KP, Cha JD, Jang EH, Klumpp J, Hagens S, Hardt WD, Lee KY, Loessner MJ. PEGylation of bacteriophages increases blood circulation time and reduces T-helper type 1 immune response. Microb Biotechnol. 2008;1:247–257. doi: 10.1111/j.1751-7915.2008.00028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010;12:397–406. doi: 10.1208/s12248-010-9203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZS, Schmauss C, Cuenca A, Ratcliffe E, Gershon MD. Physiological modulation of intestinal motility by enteric dopaminergic neurons and the D-2 receptor: analysis of dopamine receptor expression, location, development, and function in wild-type and knock-out mice. J Neurosci. 2006;26:2798–2807. doi: 10.1523/JNEUROSCI.4720-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken KW, Howell JC, Wells JM, Spence JR. Generating human intestinal tissue from pluripotent stem cells in vitro. Nat Protoc. 2011;6:1920–1928. doi: 10.1038/nprot.2011.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikura K, Sugimura N, Musashi Y, Mikuni S, Matsuo Y, Kobayashi S, Nagakawa K, Takahara S, et al. Virus-like particles with removable cyclodextrins enable glutathione-triggered drug release in cells. Mol BioSyst. 2013;9:501–507. doi: 10.1039/c2mb25420d. [DOI] [PubMed] [Google Scholar]
- Padmanabhan P, Grosse J, Asad AB, Radda GK, Golay X. Gastrointestinal transit measurements in mice with 99mTc-DTPA-labeled activated charcoal using NanoSPECT-CT. EJNMMI Res. 2013;3:60. doi: 10.1186/2191-219X-3-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, Seth D, Mukhopadhyaya SK, Brahmachary RL, Ulrichs C, Goswami A. Surface functionalized amorphous nanosilica and microsilica with nanopores as promising tools in biomedicine. Die Naturwissenschaften. 2009;96:31–38. doi: 10.1007/s00114-008-0445-1. [DOI] [PubMed] [Google Scholar]
- Ren TH, Grants I, Alhaj M, McKiernan M, Jacobson M, Hassanain HH, Frankel W, Wunderlich J, et al. Impact of disrupting adenosine A(3) receptors (A(3)(−/−) AR) on colonic motility or progression of colitis in the mouse. Inflamm Bowel Dis. 2011;17:1698–1713. doi: 10.1002/ibd.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KL, Liu JL. Engineered viral nanoparticles for flow cytometry and fluorescence microscopy applications. Wires Nanomed Nanobi. 2012;4:511–524. doi: 10.1002/wnan.1177. [DOI] [PubMed] [Google Scholar]
- Schwarz R, Kaspar A, Seelig J, Kunnecke B. Gastrointestinal transit times in mice and humans measured with 27Al and 19F nuclear magnetic resonance. Magn Reson Med. 2002;48:255–261. doi: 10.1002/mrm.10207. [DOI] [PubMed] [Google Scholar]
- Shaji J, Patole V. Protein and peptide drug delivery: oral approaches. Indian J Pharm Sci. 2008;70:269–277. doi: 10.4103/0250-474X.42967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto CM, Ratna BR. Virus hybrids as nanomaterials for biotechnology. Curr Opin Biotech. 2010;21:426–438. doi: 10.1016/j.copbio.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Steinmetz NF, Bize A, Findlay KC, Lomonossoff GP, Manchester M, Evans DJ, Prangishvili D. Site-specific and spatially controlled addressability of a new viral nanobuilding block: sulfolobus islandicus rod-shaped virus 2. Adv Funct Mater. 2008;18:3478–3486. [Google Scholar]
- Thompson CL, Hofer MJ, Campbell IL, Holmes AJ. Community dynamics in the mouse gut microbiota: a possible role for IRF9-regulated genes in community homeostasis. PLoS ONE. 2010;5:e10335. doi: 10.1371/journal.pone.0010335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tothova L, Babickova J, Celec P. Phage survival: the biodegradability of M13 phage display library in vitro. Biotech Appl Biochem. 2012;59:490–494. doi: 10.1002/bab.1050. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldahl K, Peng X. Biology, biodiversity and application of thermophilic viruses. In: Satyanarayana T, Littlechild J, Kawarabayasi Y, editors. Thermophilic Microbes in Environmental and Industrial Biotechnology. Dordrecht: Springer Science+Business Media; 2013. pp. 271–304. [Google Scholar]
- Uldahl KB, Jensen SB, Bhoobalan-Chitty Y, Martinez-Alvarez L, Papathanasiou P, Peng X. Life cycle characterization of sulfolobus monocaudavirus 1, an extremophilic spindle-shaped virus with extracellular tail development. J Virol. 2016a;90:5693–5699. doi: 10.1128/JVI.00075-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldahl KB, Wu L, Hall A, Papathanasiou P, Peng X, Moghimi SM. Recognition of extremophilic archaeal viruses by eukaryotic cells: a promising nanoplatform from the third domain of life. Sci Rep. 2016b;6:37966. doi: 10.1038/srep37966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinje S, Stroes E, Nieuwdorp M, Hazen SL. The gut microbiome as novel cardio-metabolic target: the time has come! Eur Heart J. 2014;35:883–887. doi: 10.1093/eurheartj/eht467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen AM, Lee KL, Yildiz I, Bruckman MA, Shukla S, Steinmetz NF. Viral nanoparticles for in vivo tumor imaging. J visual exp. 2012;69:e4352. doi: 10.3791/4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Ghosh D, Ham MH, Qi J, Barone PW, Strano MS, Belcher AM. M13 phage-functionalized single-walled carbon nanotubes as nanoprobes for second near-infrared window fluorescence imaging of targeted tumors. Nano Lett. 2012;12:1176–1183. doi: 10.1021/nl2031663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz I, Shukla S, Steinmetz NF. Applications of viral nanoparticles in medicine. Curr Opin Biotech. 2011;22:901–908. doi: 10.1016/j.copbio.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillig W, Kletzin A, Schleper C, Holz I, Janekovic D, Hain J, Lanzendorfer M, Kristjansson JK. Screening for sulfolobales, their plasmids and their viruses in icelandic solfataras. Syst App Microbiol. 1994;16:609–628. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Survival of M13KE particles. The infectivity of M13KE after incubation at 37°C in SIF pH 6·5 and SGF pH 1·6. Incubation in TBS buffer pH 7·5 was monitored as a control. The number of plaque forming units per millilitre (PFU ml−1) was determined by plaque assay and is shown as a function of time in hours.
Figure S2 Observations of mice body weights. Body weights were measured daily for 5 days. At the 0 h time point, a one-time administration of either 200 μl VLP solution or control Tris-acetate solution was carried out.
Figure S3 TEM images of recovered SMV1 particles from the faecal samples show intact morphology.
Table S1 Summary of virus titre. Estimated SMV1 titre in original inoculum and those detected in faecal pellets. Estimates based on qPCR and PFU assays are presented.
Table S2 Detection of SMV1 and M13KE DNA in tissues of mice inoculated orally with virus.