

Table of content entry
A biomimetic, nanoparticulate anticancer vaccine is fabricated by coating the membrane derived from cancer cells onto a highly immunostimulatory core. The resulting nanoformulation is capable of promoting immunity against multiple tumor antigens. When the nanovaccine is combined with checkpoint blockade therapy, significant control of tumor growth is achieved. The reported approach may ultimately be adapted towards the design of potent autologous vaccines made from patient-derived tumor material.
Keywords: anticancer vaccine, immunotherapy, biomimetic nanoparticle, nanomedicine, personalized medicine
Recent successes in the field of immunotherapy have provided convincing evidence that, if properly stimulated, the immune system is capable of successfully battling a variety of cancer types.[1–3] Despite this fact, an effective anticancer vaccine that is widely applicable and facile to administer, while highly sought after, has continued to remain elusive.[4,5] Fundamentally, the challenge lies in the need to generate potent and specific immune responses that enable the body to successfully distinguish between healthy and diseased tissue.[6] By the time a neoplastic growth reaches the malignant stage, the most immunogenic tumor-specific antigens have generally been eliminated via negative selection.[7,8] Some promising strategies under clinical investigation have focused on common tumor-associated antigens, which are dysregulated wild-type proteins.[9,10] However, the applicability of such single-antigen approaches is dependent on tumor phenotype, and they may also be subject to some of the same limitations facing targeted monotherapies as tumors evolve mechanisms of escape.[11,12] On the other end of the spectrum, whole cell vaccine preparations are capable of delivering a wide range of autologous antigens,[13,14] but they have traditionally been ineffective. This may be a result of significant interference from a surplus of non-tumor-related antigenic material[15] or difficulties in direct administration, which have necessitated more complex cell-based strategies.[14,16,17] Additionally, the immunosuppressive microenvironment of established tumors is often hard to break,[18,19] leading to suboptimal efficacy despite effective training of the immune system.
Advances in genomics have enabled the elucidation of individual cancer mutanomes, which can be leveraged to identify multiple vaccine epitopes on a personalized level.[20,21] Other studies have demonstrated that high mutational burden can lead to neoantigen targets that are recognized by the immune system, and this correlates with clinical response to checkpoint blockade therapies.[22,23] While personalized epitope identification and vaccine manufacture may currently not be practical at large-scale, these findings confirm that, despite the challenges facing whole cell formulations, there is a wealth of relevant antigens to be found in autologous tumor material. Applying the principles of biomimetic nanotechnology,[24–26] we explored the presentation of cancer-derived membrane material in a context that could enable potent, multi-antigenic immune responses for anticancer vaccine design (Figure 1). It was demonstrated that nanoparticulate delivery of the membrane, along with an immunostimulatory adjuvant, could facilitate enhanced antigen presentation, leading to the activation of tumor-specific cellular responses. Further, when used in conjunction with checkpoint blockade therapy to help break tumor immunosuppresion,[27–29] the nanovaccine formulation was able to achieve significant control of tumor growth in a therapeutic setting.
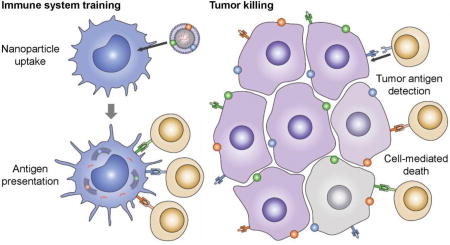
Figure 1.

Schematic of CpG-CCNPs for anticancer vaccination. Membrane derived from cancer cells (purple), along with the associated tumor antigens (small colored spheres), is coated onto adjuvant-loaded nanoparticle cores (CpG-NPs) to yield a nanoparticulate anticancer vaccine (CpG-CCNPs). Upon delivery to antigen presenting cells (blue), the vaccine formulation enables activation of T cells (tan) with multiple specificities. After detecting the antigens present on the tumor, the T cells are capable of initiating cancer cell death (gray).
CpG oligodeoxynucleotide 1826 (CpG), a nucleic acid-based immunological adjuvant known to trigger the maturation of antigen presenting cells, was encapsulated into biodegradable poly(lactic-co-glycolic acid) (PLGA) nanoparticle cores via a double emulsion process (Figure 2a). The amount of CpG that could be loaded started saturating at an initial input of 1 nmol per 1 mg of PLGA, and approximately 100 pmol of the adjuvant could be loaded at this ratio. To introduce tumor antigen material, the membrane derived from B16–F10 mouse melanoma cells was coated onto CpG-loaded PLGA cores (CpG-NPs). The process used for coating did not significantly alter the amount of adjuvant within the polymeric cores (Figure S1, Supporting Information). Dynamic light scattering (DLS) measurements showed an increase in nanoparticle size after coating, and the zeta potential of the adjuvant-loaded, cancer cell membrane-coated nanoparticles (CpG-CCNPs) increased to approximately that of pure membrane (Figure 2b,c). Successful coating was confirmed by transmission electron microscopy (TEM), which revealed a characteristic core-shell structure (Figure 2d). Over time, the CpG-CCNPs stayed stable in solution (Figure 2e). Importantly, the presence of known membrane-bound tumor-associated antigens,[30] including MART1, TRP2, and gp100, was confirmed by western blotting (Figure 2f). When normalized by total protein amount, significant antigen enrichment was observed on the derived membrane and CpG-CCNPs when compared with whole cell lysate.
Figure 2.

Preparation and characterization of CpG-CCNPs. a) CpG encapsulation into PLGA cores with increasing inputs, normalized by polymer weight (n = 3, mean ± SD). b) Size of CpG-NPs, B16–F10 membrane vesicles, and CpG-CCNPs (n = 3; mean ± SD). c) Surface zeta potential of CpG-NPs, B16–F10 membrane vesicles, and CpG-CCNPs (n = 3; mean ± SD). d) TEM image of CpG-CCNPs negatively stained with uranyl acetate. Scale bar = 100 nm. e) Size stability over time of CpG-CCNPs stored in 10% sucrose (n = 3; mean ± SD). f) Western blots for known melanoma-associated antigens MART1, TRP2, and gp100 on B16–F10 cells, B16–F10 membrane, and CpG-CCNPs.
To study the interaction of the nanoformulation with antigen presenting cells, bone marrow-derived dendritic cells (BMDCs) were employed. When incubated with dye-labeled CpG-CCNPs, quick uptake was observed until saturation was achieved at approximately 6 h (Figure 3a). CpG is known to activate pro-inflammatory responses in antigen presenting cells,[31] which is necessary for generating potent antitumor immunity. Using a fluorescently tagged CpG, the adjuvant was shown to much more readily be internalized by BMDCs when encapsulated within the membrane-coated nanoparticles, which are in the ideal size range for endocytosis[32,33] (Figure 3b). To test the implications of this enhanced internalization and confirm the integrity of CpG after encapsulation, the biological activity of CpG in free form versus nanoparticulate form was assessed (Figure 3c,d). Secretion of two representative pro-inflammatory cytokines, interleukin-6 (IL-6) and IL-12, was significantly enhanced for the CpG-CCNP formulation, which was approximately an order of magnitude more immunostimulatory than free CpG. This effect is likely due to the fact that nanoparticulate CpG more readily localizes to the endosomal compartment during uptake, where it can engage its endosomal recognition site on toll-like receptor 9 (TLR-9).[34] It should be noted that CCNPs without adjuvant induced significantly less cytokine secretion when incubated with BMDCs at equivalent nanoparticle concentrations (Figure S2, Supporting Information). While the CpG employed in the studies here was murine-specific, other variants could easily be substituted to promote immunity in humans.[35] Further, the integrity of the nanoparticle structure was assessed by fluorescent imaging using dye-labeled CpG and membrane protein, and significant colocalization of the two signals confirmed the ability of the CpG-CCNPs to co-deliver both adjuvant and antigen to the same BMDC (Figure 3e). Upon in vivo administration subcutaneously via the hock, the nanoformulation could easily be detected at the draining lymph node after 1 h, with some appearing at an adjacent node after 24 h (Figure S3, Supporting Information). Little signal was observed at the spleen given its considerable distance from the injection site. Within the draining lymph node, antigen presenting cells such as dendritic cells and macrophages exhibited the highest percentage of nanoparticle uptake; B cells and granulocytes also displayed some uptake, while the limited amount of signal observed for T cells was likely the result of nonspecific interactions with the nanoformulation (Figure 3f).
Figure 3.

Delivery of antigen and adjuvant to immune cells. a) Uptake kinetics of dye-labeled CpG-CCNPs by BMDCs (n = 3; mean ± SD). b) Uptake kinetics of dye-conjugated CpG in free form or within CpG-CCNPs by BMDCs (n = 3; mean ± SD). c,d) Secretion of the pro-inflammatory cytokines IL-6 (c) and IL-12p40 (d) by BMDCs when incubated with either free CpG or CpG-CCNPs (n = 3; mean ± SD). e) Confocal microscopy colocalization of CpG and membrane proteins upon uptake of dual-labeled CpG-CCNPs by a BMDC. Green = CpG, red = membrane, blue = cell nucleus; scale bar = 10 µm. f) Uptake of dye-labeled CpG-CCNPs by different immune cell subsets in the draining lymph node after in vivo administration (n = 6; mean ± SD).
The effect of the nanoformulation on BMDC maturation in vitro was studied by looking at the upregulated expression of co-stimulatory markers CD40, CD80, and CD86, as well as MHC-II (Figure S4, Supporting Information). Consistent with the fact that the dendritic cell maturation process is largely driven by the detection of pathogen-associated molecular patterns such as CpG, it was observed that both CpG-CCNPs and CpG-NPs without any antigen were equally potent. Without CpG, the antigen-only CCNP formulation exhibited significantly decreased activity. A similar pattern was seen when assessing the secretion of IL-6 and IL-12 by the BMDCs (Figure S5, Supporting Information). When administered in vivo, the CpG-CCNP and CpG-NP formulations were likewise able to induce significant dendritic cell maturation at the draining lymph node after 24 hours (Figure 4a–d). They also outperformed additional controls, including CCNPs with free CpG and whole cell lysate with free CpG, highlighting the advantage of nanoparticulate formulations. The level of cytokine secretion at the draining lymph node was shown to be mostly dependent on the presence of CpG, with all adjuvanted formulations performing similarly (Figure 4e,f). This effect was localized, as analysis of cytokine levels in the serum did not yield anything significantly above baseline (Figure S6, Supporting Information).
Figure 4.

Characterization of in vivo dendritic cell maturation. a–d) Analysis of dendritic cell maturation markers CD40 (a), CD80 (b), CD86 (c), and MHC-II (d) in the draining lymph nodes after administration with CpG-CCNPs and various control formulations, including whole cell lysate with free CpG (WC + fCpG), CCNPs with free CpG (CCNP + fCpG), CCNPs, CpG-NPs, and blank solution (n = 4; mean ± SD). e,f) Concentration of pro-inflammatory cytokines IL-6 (e) and IL-12p40 (f) secreted by immune cells isolated from the draining lymph nodes after vaccination with CpG-CCNPs or various control formulations (n = 4; mean ± SEM). * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 (compared to CpG-CCNP); one-way ANOVA.
To confirm the utility of the CpG-CCNP formulation for antitumor vaccination, its ability to elicit antigen-specific immune responses was verified using T cell-based assays. First, pmel-1 CD8+ T cells, which specifically recognize a gp100 epitope, were adoptively transferred to recipient mice, which were subsequently vaccinated with the various formulations (Figure 5a). Treatment with CpG-CCNPs resulted in the highest degree of pmel-1 T cell proliferation, indicating that the formulation was able to effectively deliver the gp100 antigen for presentation under an immunostimulatory context. Additionally, after a set of vaccinations in naïve mice, the CpG-CCNPs were able to promote the native generation of T cells with multiple tumor antigen specificities (Figure 5b,c). T cells specific for both gp100 and TRP2 could be isolated and expanded from mice vaccinated with the CpG-CCNPs. Further, when cultured ex vivo, immune cell preparations from mice vaccinated with the formulation showed significantly enhanced production of IFNγ and IL-2 when stimulated with a gp100 peptide, a TRP2 peptide, or whole cell lysate, suggesting robust effector-level response against those targets (Figure S7, Supporting Information). While these studies were generally limited to probing for immunity against well characterized epitopes, it could be reasonably inferred that the CpG-CCNP formulation was concurrently generating additional responses against other tumor-relevant antigens.
Figure 5.

Characterization of in vivo T cell responses. a) Proliferation index of adoptively transferred pmel-1 CD8+ T cells after in vivo stimulation by CpG-CCNPs or various control formulations, including whole cell lysate with free CpG (WC + fCpG), CCNPs with free CpG (CCNP + fCpG), CCNPs, CpG-NPs, and blank solution (n = 3; mean ± SD). b,c) Tetramer staining analysis of T cells specific for gp100 (b) and TRP2 (c) after ex vivo restimulation of splenocytes from mice vaccinated with CpG-CCNPs or various control formulations (n = 3; mean ± SD). * p < 0.05, ** p < 0.01, **** p < 0.0001 (compared to CpG-CCNP); one-way ANOVA.
To assess if the enhanced cellular immunity afforded by the CpG-CCNP formulation could translate into functional rejection of tumor cells, a prophylactic study using the wild-type B16–F10 model, which is poorly immunogenic,[21,36,37] was carried out (Figure 6a–c). In mice vaccinated with CpG-CCNPs, there was significant activity, and tumor occurrence was prevented in 86% of mice 150 days after challenge with the tumor cells. Formulations consisting of either whole cell lysate with free CpG or CCNPs with free CpG both showed modest control of tumor growth, extending median survival from 20 days for the untreated group to 34 and 40 days, respectively. All but one of the mice in these groups reached the experimental endpoint by day 48 after challenge. CCNPs without adjuvant had minimal protective benefit, with the mice in these groups achieving a median survival of 28 days. Finally, mice vaccinated with CpG-NPs that had no antigenic material exhibited tumor growth kinetics identical to the blank control and displayed a median survival of 22 days. The results suggest that co-delivery of both tumor antigen material and the CpG adjuvant together in the same vehicle is necessary for eliciting maximal antitumor immunity. The fact that CpG-NPs alone had no effect is encouraging and demonstrates that the inclusion of cancer membrane material helped to provide appropriate cues for the specific detection and elimination of malignant cells by the immune system.
Figure 6.

Prophylactic efficacy. a–c) Mice immunized with CpG-CCNPs and various control formulations, including whole cell lysate with free CpG (WC + fCpG), CCNPs with free CpG (CCNP + fCpG), CCNPs, CpG-NPs, and blank solution, on days 0, 7, and 14 were challenged with B16–F10 cells on day 21. Average tumor sizes (a), survival (b), and individual tumor growth kinetics (c) were plotted over time (n = 7; mean ± SEM). Reporting of average tumor sizes was halted after the first mouse died in each respective group. * p < 0.05, *** p < 0.001 (compared to CpG-CCNP in survival plot); log-rank test.
The utility of the nanoparticulate vaccine formulation was further tested in a more clinically relevant therapeutic setting (Figure 7a–c). In this study, mice were challenged with B16–F10 cells and subsequently treated with the nanoformulation. Using this design, CpG-CCNPs alone displayed a modest ability to control tumor growth and extend survival. Given the aggressive nature of the B16–F10 tumor model, the results were not unexpected, especially given that vaccination largely focuses on the training phase of adaptive immunity. Despite adequately enabling the immune system to recognize the appropriate targets, vaccine formulations for boosting cellular immunity may not be particularly well-suited for potentiating effector functionality in the presence of strong immunosuppression.[38] As such, the CpG-CCNPs were combined with a checkpoint blockade cocktail consisting of anti-CTLA4 and anti-PD1, and treatment with the combination enabled significantly enhanced control of tumor growth. Median survival was extended from 18 days for the blank control to 32 days for the treated group, and 50% of tumors were still below the experimental endpoint threshold on day 48 post-challenge. In contrast, the checkpoint blockades, which have not shown significant efficacy in a related B16 model,[39] was about as effective as CpG-CCNPs. The results confirm that the nanoparticulate vaccine formulation can act synergistically with other immunotherapies, modulating different aspects of immunity to promote the strongest antitumor responses.
Figure 7.

Therapeutic efficacy. a–c) After challenge with B16–F10 cells on day 0, mice were treated using CpG-CCNPs combined with a checkpoint blockade cocktail of anti-CTLA4 plus anti-PD1 (αCTLA4/αPD1), CpG-CCNPs alone, or the checkpoint blockade cocktail alone on days 1, 2, 4 and 7. Average tumor sizes (a), survival (b), and individual tumor growth kinetics (c) were plotted over time (n = 6; mean ± SEM). Reporting of average tumor sizes was halted after the first mouse died in each respective group. * p < 0.05, *** p < 0.001 (compared to CpG-CCNP + αCTLA4/αPD1 in survival plot); log-rank test.
In conclusion, we have reported on a biomimetic nanoparticulate anticancer vaccine formulation capable of activating multi-antigenic immunity. The design leverages the unique advantages of recent nanoparticle technology, delivering both syngeneic cancer material along with a potent immunological adjuvant in a format that promotes effective antigen presentation. The final formulation is capable of generating strong antitumor responses in vivo and can work together with other immunotherapies such as checkpoint blockades to help control tumor growth. It is increasingly understood that presentation of tumor antigens alone, even in highly immunogenic contexts, may not be able to overcome the immunosuppressive tumor microenvironment.[38,40] As such, efforts have shifted towards the rational design of combinatorial approaches that leverage multiple modes of action,[41–43] including employing such strategies as adjuvant therapies to surgical resection.[44] In doing such, the potential adverse effects of immunomodulatory cocktails will also need to be considered.[45] The present nanoparticle-based cancer cell membrane coating strategy represents a generalizable and effective means of boosting endogenous immunity against autologous material, which may, in the future, be derived from a patient’s own resected primary tumor as a means to prevent relapse. All of this is accomplished in a manner that is unique when compared to current strategies and can possibly pave the way for enhanced personalized anticancer vaccines.
Experimental Section
B16–F10 Murine Melanoma Cell Culture and Membrane Derivation
B16–F10 mouse melanoma cells (CRL-6475; American Type Culture Collection) were cultured at 37 °C with 5% CO2 in T175 tissue culture flasks (Becton Dickinson) with Dulbecco’s Modified Eagle Medium (DMEM; Mediatech) supplemented with 10% bovine growth serum (Hyclone) and 1% penicillin-streptomycin (Gibco). At 80–90% confluency, ~16–18 million cells per flask were collected in phosphate buffered saline (PBS; Mediatech) by scraping, pelleted at 700 ×g for 7 min in a Sorvall Legend Micro21R centrifuge, then resuspended in a 50:50 solution of cryopreservation medium (Hyclone) and complete DMEM. Cell aliquots were stored at −20 °C before use. To derive membrane, cells were first washed in a starting buffer containing 30 mM Tris-HCl pH 7.0 (Quality Biological) with 0.0759 M sucrose (Sigma Aldrich) and 0.225 M D-mannitol (Sigma Aldrich), then mechanically disrupted in the presence of phosphatase inhibitor and protease inhibitor cocktails (Sigma Aldrich) using a Kinematica Polytron PT 10/35 probe homogenizer at 70% power for 15 passes. Membrane was separated from the resulting homogenate by differential centrifugation using a Beckman Coulter Optima L-90K Ultracentrifuge. Homogenate was pelleted at 10,000 ×g for 25 min, and the supernatant was then pelleted at 150,000 ×g for 35 min. The resulting pellet of cell membrane was washed in 0.2 mM ethylenediaminetetraacetic acid (EDTA; USB Corporation) in DNase free/RNase free water (Invitrogen) and stored in the same solution at −20 °C until use. Total membrane protein content was quantified by a BCA protein assay kit (Pierce).
Cancer Cell Membrane-Coated Nanoparticle Preparation and Characterization
Polymeric cores were prepared using 0.18 dL g−1 carboxyl-terminated 50:50 poly(lactic-co-glycolic) acid (PLGA; LACTEL Absorbable Polymers) using a double emulsion process. PLGA was dissolved in dichloromethane (DCM) at a concentration of 50 mg mL−1. 500 µL of polymer was added to 100 µL of 200 mM Tris-HCl pH 8 and sonicated using a Fisher Scientific 150E Sonic Dismembrator at 70% power pulsed (2 seconds on/1 second off) for 1 min. An outer aqueous phase consisting of 5 mL of 10 mM Tris-HCl pH 8 was added to the polymer solution and sonicated at the same setting for 2 min. The emulsion was then added to 10 mL of 10 mM Tris-HCl pH 8 and magnetically stirred at 700 ×g for 2.5 h. After stirring, the particles were pelleted at 21,100 ×g for 8 min, and washed twice in 10 mM Tris-HCl pH 8. Adjuvant-loaded polymeric cores (CpG-NPs) were made by including CpG oligodeoxynucleotide 1826 (CpG), synthesized using the sequence 5’-TCCATGACGTTCCTGACGTT-3 with all phosphorothioate bonds (Integrated DNA Technologies), at 500 µM to the inner phase of the double emulsion during nanoparticle synthesis. To optimize the loading, CpG-NPs were made with CpG inputs of 250, 500, 1000, and 2000 pmol per 1 mg of PLGA. Each formulation was lyophilized overnight, then resuspended in 1 mL of acetone. PLGA was precipitated and pelleted with the addition of 1 mL water followed by centrifugation at 21,100 ×g for 20 min. CpG concentration of the supernatants were measured using a Quant-iT Oligreen ssDNA quantification kit (Invitrogen) according to manufacturer’s instructions. Further studies employed an initial input of 1000 pmol CpG per 1 mg of PLGA.
B16–F10 cancer cell membrane-coated CpG-NPs (CpG-CCNPs) were made by pelleting the CpG-NP cores and resuspending them in solution containing B16–F10 cell membrane. The mixture was sonicated in a 1.5 mL disposable sizing cuvette (Brandtech) using a Fisher Scientific FS30D bath sonicator at a frequency of 42 kHz and a power of 100 W for 2 min. The nanoparticles were washed twice in 10 mM Tris-HCl pH 8, and resuspended to a concentration of 25 mg polymer per 1 mL of solution in 5 mM Tris-HCl pH 7.5 and 0.2 mM EDTA in DNase free/RNase free water for in vitro studies or in 10% sucrose with the same buffer concentrations for in vivo studies. If not used immediately, particles were stored at −20 °C. In the study, CpG-CCNPs were fabricated with 100 µg of membrane protein per 1 mg of PLGA. Size and surface zeta potential of CCNPs were determined through dynamic light scattering (DLS) measurements using a Malvern ZEN 3600 Zetasizer. To test the stability of CCNPs in the 10% sucrose solution, particles were stored at 4 °C for two weeks with size measured by DLS every other day. The morphology of CCNPs was examined by transmission electron microscopy using a Zeiss Libra 120 PLUS EF-TEM. Samples were resuspended in 10 mM Tris-HCl pH 8, deposited onto a glow discharged carbon-coated 400 square mesh copper grid (Electron Microscopy Sciences), and negatively stained with 1 wt% uranyl acetate (Electron Microscopy Sciences).
Membrane Antigen Retention
Identification of characteristic B16–F10 tumor antigens was completed via western blotting. B16–F10 whole cells were collected from culture by scraping, lysed using 0.2% Triton X-100 (Sigma Aldrich) in water, and sonicated. B16–F10 lysed cells, B16–F10 membrane, and CpG CCNPs were analyzed for protein content using a BCA assay, then each diluted to 0.2 mg mL−1 in water. Each sample was then mixed with NuPAGE 4× lithium dodecyl sulfate (LDS) sample loading buffer (Novex) and heated for 10 min at 70 °C. 25 µL of each sample was loaded into 12-well Bolt 4–12% Bis-Tris gels (Invitrogen) and run at 165 V for 45 min in MOPS running buffer (Novex). Proteins were transferred to 0.45 µm nitrocellulose membrane (Pierce) in Bolt transfer buffer (Novex) at 10 V for 60 min. After blocking with 5% milk (Genesee Scientific) in PBS with 0.05% Tween 20 (National Scientific), blots were immunostained with mouse anti-mouse gp100 (EP4863(2); Abcam), rabbit anti-mouse TRP2 (E-10; Santa Cruz Biotechnology), or mouse anti-mouse MART1 (A103; Santa Cruz Biotechnology). The appropriate horseradish peroxidase-conjugated secondary (Biolegend) was used for secondary staining. Membranes were developed with ECL western blotting substrate (Pierce) in an ImageWorks Mini-Medical/90 Developer.
In Vitro Uptake and Activity
All animal studies were designed and proceeded in compliance to the University of California, San Diego Institutional Animal Care and Use Committee. Female C57BL/6NHsd mice were obtained at 6–10 weeks old from Envigo Harlan. CO2 asphyxiation followed by cervical dislocation was used for euthanizing mice. Bone marrow-derived dendritic cell (BMDC) culture was adapted from a previously published protocol.[25] Healthy mice were euthanized using carbon dioxide asphyxiation followed by cervical dislocation. Both femurs were dissected, cleaned in 70% ethanol, and cut on both ends. Bone marrow was then flushed out of the bone with a 1 mL sterile syringe using warm BMDC basal media consisting of 500 mL Isocove’s Modification of DMEM with 2 mM L-Glutamine and 25 mM HEPES (Mediatech) supplemented with 50 mL USDA certified fetal bovine serum (Omega Scientific), 500 µL 55 mM β-mercaptoethanol (Gibco), 5 mL 200 mM L-Glutamine (Gibco), and 5 mL penicillin-streptomycin. Cells were then pelleted at 700 ×g for 5 min, resuspended in BMDC growth media, consisting of the basal media further supplemented with 10 ng mL−1 granulocyte/macrophage-colony stimulating factor (GM-CSF; Biolegend), to a concentration of 1 × 106 cells mL−1, and plated into petri plates at 2 × 106 cells per plate. On the third day of culture, 10 mL of BMDC growth media was added to each plate.
To make CpG-CCNPs with fluorescently labeled polymeric cores, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD, ex/em = 644/663 nm; Biotium) was added to the PLGA solution at 0.1 wt% of the polymer during nanoparticle synthesis. For the nanoparticle uptake study, BMDCs were collected on day 5 using 1 mM EDTA in PBS. Cells were washed once in PBS, resuspended in BMDC basal media, and plated into a 24-well suspension plates. DiD-labeled CpG-CCNPs were added at a final concentration of 1.4 mg mL−1. At each timepoint (0, 15 min, 30 min, 1 h, 2 h, 6 h, 12 h, 24 h), media was removed, and the cells were detached with trypsin-EDTA (Gibco). Cells were collected, washed once in trypsin-EDTA, washed twice in PBS, and resuspended in 200 µL of 10% phosphate buffered formalin (Fisher). The adjuvant uptake study was conducted similarly, instead employing CpG-CCNPs synthesized with CpG containing a 5' 6-FAM modification (Integrated DNA Technologies). Free dye-labeled CpG was used at an equivalent concentration for comparison. For all experiments, after each time point was collected and processed, 1 drop of NucBlue Live ReadyProbe Reagent UV stain (Molecular Probes) was added and data was collected using a Becton Dickinson FacsCanto-II flow cytometer. All data was analyzed using Flowjo software.
The activity of delivered CpG was examined using a BMDC cytokine release assay. BMDCs were plated on day 6 into 96-well plates at a concentration of 8 × 104 cells mL−1 in BMDC growth media. Dilutions of CpG-CCNP or free CpG were added to the cells. After 2 h of incubation, the cells were washed three times with fresh BMDC growth media and cultured for another two days. Supernatant was then collected and measured for the presence of pro-inflammatory cytokines using mouse IL-6 and IL-12p40 ELISA kits (Biolegend) according to manufacturer’s instructions.
Antigen and adjuvant colocalization was visualized by imaging BMDCs incubated with dual-labeled CpG-CCNPs. B16–F10 membrane was labeled using CF647 succinimidyl ester dye (Biotium) and used to coat CpG-CCNPs fabricated with FAM-modified CpG. BMDCs were seeded into 8-well chamber slides at 7.5 × 104 cells mL−1 and incubated with the nanoparticles for 15 min at 0.7 mg mL−1. Cells were then washed three times with PBS, fixed with 10% formalin for 30 min, then washed again three times with PBS and mounted onto coverslips using VECTASHIELD mounting media with DAPI (Vector Laboratories). Samples were imaged on a Deltavision RT Deconvolution Microscope at 60× magnification.
In Vivo Cellular Localization and Dendritic Cell Activation
To assess in vivo localization, DiD-labeled CpG-CCNPs were injected subcutaneously into each hock of female C57BL/6NHsd mice. After 24 h, the popliteal lymph nodes were collected into 500 µL of dissociation buffer consisting of 1 mg mL−1 collagenase D from Clostridium histolyticum (Roche) and 1 mg mL−1 DNase I grade II, from bovine pancreas (Roche) in Dulbecco’s PBS with calcium and magnesium (Gibco). Lymph nodes were dissociated manually by pipetting and then were stained with FITC-labeled antibodies for dendritic cells (anti-mouse CD11c, N418; Biolegend), macrophages (anti-mouse F4/80, BM8; Biolegend), T cells (anti-mouse CD3, 17A2; Biolegend), B cells (anti-mouse CD19, 6D5; Biolegend), and granulocytes (anti-mouse Ly-6G/Ly-6C, RB6-8C5; Biolegend) for 30 min. Appropriate dye-labeled antibody isotypes (Biolegend) were used for gating purposes with cells from an untreated lymph node. After washing, dead cells were labeled with propidium iodide (Biolegend). Data was collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed using FlowJo software.
Dendritic cell activation following immunization with CpG-CCNPs, CpG-NPs, CCNPs, or additional controls was determined by testing dendritic cell maturation and lymph node cytokine secretion. To test vaccines with antigens and adjuvants delivered as separate components, additional controls of CCNP with free CpG and B16-F10 whole lysate with free CpG were also administered. The CCNPs with free CpG formulation was made by mixing the two components such that the final ratio was 25 mg of PLGA per 3.5 nmol of CpG. Whole cell lysate was prepared by three freeze-thaw cycles at −80 °C for 10 min followed by 10 min at 37 °C. The amount of protein used for the formulation was normalized by the amount of Na+K+-ATPase, a characteristic membrane protein, compared with CCNPs as determined by immunoblotting (see Supplementary Methods). To examine dendritic cell maturation in vivo, 50 µL of each formulation at 25 mg mL−1 of nanoparticle, or equivalent, was injected into the hock. After 24 h, the popliteal lymph nodes of all treated mice were collected into 500 µL dissociation buffer and manually dissociated. Cells were stained using FITC anti-mouse CD11c with either Alexa647-conjugated anti-mouse CD40 (HM40-3; Biolegend), CD80 (12-10A1; Biolegend), CD86 (GL-1; Biolegend), or MHC-II (M5/114.15.2; Biolegend). Appropriate dye-labeled antibody isotypes (Biolegend) were used for gating purposes with cells from an untreated lymph node. After 30 min of incubation at 4 °C, the cells were washed and stained with CellTrace™ Calcein Violet, AM (Molecular Probes) in PBS according to manufacturer’s instructions. Data was collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed using FlowJo software. To analyze cytokine production, lymph node-derived single cell suspensions were plated with 500 µL of BMDC growth media in 24-well tissue culture plates. After 48 h, supernatant was collected and analyzed for cytokine content using IL-6 and IL-12p40 ELISA kits according to the manufacturer's instructions.
Adoptive T Cell Proliferation and Native T Cell Generation
B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J (pmel-1) transgenic mice were obtained from the Jackson Laboratory at 4–6 weeks old. The spleen, popliteal lymph node, and inguinal lymph nodes of one pmel-1 mouse were collected for dissociation into single cell suspensions. The red blood cells in the spleen were removed using lysis buffer (Biolegend), and all remaining cells were pooled together. CD8+ T cells were separated out using CD8a (Ly-2) microbeads (Miltenyi Biotec) on Miltenyi Biotec MACS LS separation columns per manufacturer’s instructions. After separation, cells were washed in PBS and stained with carboxyfluorescein succinimidyl ester (CFSE; eBiosciences). Cells were then diluted to 2.5 × 106 cells mL−1 and 200 µL was transferred to naïve C57BL/6NHsd recipients. 2 h post-injection, each mouse was injected with 50 µL of various vaccine formulations in both hocks. 4 days after treatment, the spleens were collected and dissociated into single cell suspensions. Adoptively transferred T cells were stained for using APC-conjugated anti-mouse CD8a (53-6.7; Biolegend) and Pacific Blue-conjugated anti-mouse CD90.1 (OX-7; Biolegend). Data was collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed using FlowJo software. CFSE dilution was used to assess the degree of T cell proliferation.
To assess the native generation of antigen-specific T cells, C57BL/6NHsd mice were vaccinated subcutaneously with 50 µL of the different formulations in each hock on days 0, 2, and 4. On day 10, spleens were collected and processed into single cell suspensions using mechanical dissociation. After lysing the red blood cells, 5 × 106 splenocytes were plated into 6-well suspension plates and pulsed with either 1 µg mL−1 of mouse gp100 peptide with sequence EGSRNQDWL (Anaspec) or 1 µg mL−1 of TRP2 peptide with sequence SVYDFFVWL (Anaspec) in BMDC growth media. After 7 days, cells were collected, washed in PBS, and stained with APC-conjugated anti-mouse CD8a and either PE-labeled H-2Db gp100 tetramer (MBL International) or H-2Kb TRP2 tetramer (MBL International). Data was collected using a Becton Dickinson FACSCanto-II flow cytometer and analyzed using FlowJo software.
In Vivo Immunity and Therapeutic Efficacy
To study the protection conferred by vaccination, C57BL/6NHsd mice were vaccinated with 50 µL of the different formulations at 25 mg mL−1 of PLGA, or equivalent, on days 0, 7, and 14. On day 20, the right flank of each mouse was shaved and, on day 21, mice were challenged with 2 × 105 B16–F10 cells subcutaneously on the right flank. Tumors were measured every other day and the experimental endpoint was defined as either death or tumor size greater than 200 mm2.
To study the antitumor therapeutic effect, C57BL/6NHsd mice were first challenged on the right flank with 5 × 104 B16–F10 cells on day 0. On days 1, 2, 4, and 7, mice were vaccinated subcutaneously in the same flank with 200 µL of the nanoparticulate formulations. The subcutaneous route was chosen in this case to accommodate the larger dosage that was employed. The checkpoint blockade cocktail, consisting of 100 µg anti-CTLA4 (9H10; BioXCell) and 200 µg anti-PD1 (RMP1-14; BioXCell) was administered intraperitoneally on the same days. Tumors were measured every other day and the experimental endpoint was defined as either death or tumor size greater than 200 mm2.
Supplementary Material
Acknowledgments
This work is supported by the National Institutes of Health under Award Number R01CA200574 and the National Science Foundation Grant DMR-1505699.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. Nat. Rev. Clin. Oncol. 2016;13:394. doi: 10.1038/nrclinonc.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang Y. J. Clin. Invest. 2015;125:3335. doi: 10.1172/JCI83871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mellman I, Coukos G, Dranoff G. Nature. 2011;480:480. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabi Z, Man S. Adv. Drug Deliv. Rev. 2006;58:902. doi: 10.1016/j.addr.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Finn OJ. Nat. Rev. Immunol. 2003;3:630. doi: 10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- 6.Buonaguro L, Petrizzo A, Tornesello ML, Buonaguro FM. Clin. Vaccine Immunol. 2011;18:23. doi: 10.1128/CVI.00286-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim R, Emi M, Tanabe K. Immunology. 2007;121:1. doi: 10.1111/j.1365-2567.2007.02587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mittal D, Gubin MM, Schreiber RD, Smyth MJ. Curr. Opin. Immunol. 2014;27:16. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parmiani G, Castelli C, Dalerba P, Mortarini R, Rivoltini L, Marincola FM, Anichini A. J. Natl. Cancer Inst. 2002;94:805. doi: 10.1093/jnci/94.11.805. [DOI] [PubMed] [Google Scholar]
- 10.Slingluff CL., Jr Cancer J. 2011;17:343. doi: 10.1097/PPO.0b013e318233e5b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottesman MM. Annu. Rev. Med. 2002;53:615. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 12.Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, Paucar A, Yang H, Ohashi M, Zhu S, Wykosky J, Reed R, Nelson SF, Cloughesy TF, James CD, Rao PN, Kornblum HI, Heath JR, Cavenee WK, Furnari FB, Mischel PS. Science. 2014;343:72. doi: 10.1126/science.1241328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keenan BP, Jaffee EM. Semin. Oncol. 2012;39:276. doi: 10.1053/j.seminoncol.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Gruijl TD, van den Eertwegh AJM, Pinedo HM, Scheper RJ. Cancer Immunol. Immunother. 2008;57:1569. doi: 10.1007/s00262-008-0536-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lokhov PG, Balashova EE. J. Cancer. 2010;1:230. doi: 10.7150/jca.1.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palucka K, Banchereau J. Nat. Rev. Cancer. 2012;12:265. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vandenberk L, Belmans J, Van Woensel M, Riva M, Van Gool SW. Front. Immunol. 2015;6:663. doi: 10.3389/fimmu.2015.00663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabinovich GA, Gabrilovich D, Sotomayor EM. Annu. Rev. Immunol. 2007;25:267. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou W. Nat. Rev. Cancer. 2005;5:263. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 20.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER, Linette GP. Science. 2015;348:803. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castle JC, Kreiter S, Diekmann J, Löwer M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, Koslowski M, Kuhn AN, Britten CM, Huber C, Türeci O, Sahin U. Cancer Res. 2012;72:1081. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 22.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, Chan TA. N. Engl. J. Med. 2014;371:2189. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA. Science. 2015;348:124. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu C-MJ, Fang RH, Wang K-C, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L. Nature. 2015;526:118. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang RH, Hu C-MJ, Luk BT, Gao W, Copp JA, Tai Y, O’Connor DE, Zhang L. Nano Lett. 2014;14:2181. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Proc. Natl. Acad. Sci. U. S. A. 2011;108:10980. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schlößer HA, Theurich S, Shimabukuro-Vornhagen A, Holtick U, Stippel DL, von Bergwelt-Baildon M. Immunotherapy. 2014;6:973. doi: 10.2217/imt.14.58. [DOI] [PubMed] [Google Scholar]
- 28.Ghirelli C, Hagemann T. J. Clin. Invest. 2013;123:2355. doi: 10.1172/JCI69999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HMCS, Signori E, Honoki K, Georgakilas AG, Amin A, Helferich WG, Boosani CS, Guha G, Ciriolo MR, Chen S, Mohammed SI, Azmi AS, Keith WN, Bilsland A, Bhakta D, Halicka D, Fujii H, Aquilano K, Ashraf SS, Nowsheen S, Yang X, Choi BK, Kwon BS. Semin. Cancer Biol. 2015;35(Suppl):S185. doi: 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Novellino L, Castelli C, Parmiani G. Cancer Immunol. Immunother. 2005;54:187. doi: 10.1007/s00262-004-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. Expert Rev. Vaccines. 2011;10:499. doi: 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shang L, Nienhaus K, Nienhaus GU. J. Nanobiotechnology. 2014;12:5. doi: 10.1186/1477-3155-12-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oh N, Park J-H. Int. J. Nanomedicine. 2014;9(Suppl 1):51. doi: 10.2147/IJN.S26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaturvedi A, Pierce SK. Traffic. 2009;10:621. doi: 10.1111/j.1600-0854.2009.00899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vollmer J, Weeratna R, Payette P, Jurk M, Schetter C, Laucht M, Wader T, Tluk S, Liu M, Davis HL, Krieg AM. Eur. J. Immunol. 2004;34:251. doi: 10.1002/eji.200324032. [DOI] [PubMed] [Google Scholar]
- 36.Seliger B, Wollscheid U, Momburg F, Blankenstein T, Huber C. Cancer Res. 2001;61:1095. [PubMed] [Google Scholar]
- 37.Xu D, Gu P, Pan P-Y, Li Q, Sato AI, Chen S-H. Int. J. Cancer. 2004;109:499. doi: 10.1002/ijc.11696. [DOI] [PubMed] [Google Scholar]
- 38.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJM. Nat. Rev. Cancer. 2016;16:219. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 39.Curran MA, Montalvo W, Yagita H, Allison JP. Proc. Natl. Acad. Sci. U. S. A. 2010;107:4275. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH. J. Clin. Invest. 2015;125:3401. doi: 10.1172/JCI80009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Herati RS, Mansfield KD, Patsch D, Amaravadi RK, Schuchter LM, Ishwaran H, Mick R, Pryma DA, Xu X, Feldman MD, Gangadhar TC, Hahn SM, Wherry EJ, Vonderheide RH, Minn AJ. Nature. 2015;520:373. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morse MA, Lyerly HK. Vaccine. 2015;33:7377. doi: 10.1016/j.vaccine.2015.10.057. [DOI] [PubMed] [Google Scholar]
- 43.Kleponis J, Skelton R, Zheng L. Cancer Biol. Med. 2015;12:201. doi: 10.7497/j.issn.2095-3941.2015.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibney GT, Kudchadkar RR, DeConti RC, Thebeau MS, Czupryn MP, Tetteh L, Eysmans C, Richards A, Schell MJ, Fisher KJ, Horak CE, Inzunza HD, Yu B, Martinez AJ, Younos I, Weber JS. Clin. Cancer Res. 2015;21:712. doi: 10.1158/1078-0432.CCR-14-2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD. N. Engl. J. Med. 2015;373:23. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.