

Abstract
Insulinomas are functional pancreatic neuroendocrine tumors that cause hypoglycemia and severe morbidity. The aim of our study was to identify gene mutations responsible for tumorigenesis of sporadic insulinoma. Whole exome sequencing analysis was performed on tumors and paired peripheral blood from three patients with insulinomas. After initial analysis, somatic mutations were obtained and a deleterious protein product was further predicted by various bioinformatic programs. Whole exome sequencing identified 55 rare somatic mutations among three insulinoma patients, including MEN1 gene nonsense mutations (c. 681C>G; p.Tyr227* in exon 4 of MEN1 and c. 346G>T; p.Glu116* in exon 2 of MEN1) in two different tumor samples. The mutations resulted in a significant truncation of the protein and a non‐functional gene product, which was involved in defective binding of menin to proteins implicated in genetic and epigenetic mechanisms. Our results extend the growing list of pathogenic MEN1 mutations in sporadic cases of insulinoma.
Keywords: insulinoma, MEN1, somatic mutation, whole exome sequencing
Abbreviations
- MEN‐1
multiple endocrine neoplasia type 1
- SNV
single nucleotide variant
Insulinoma is a rare and sporadically occurring neuroendocrine tumor that secretes an excess of insulin, resulting in symptoms of hypoglycemia in patients 1. Additionally, the catecholamines released from insulinoma produce several symptoms including sweating, nausea, weakness, anxiety and palpitation 2. Insulinoma is usually a benign neoplasm that is smaller than 2 cm in diameter, without signs of angiogenesis or metastases, and is easily curable by surgical resection 2. However, an understanding of the mechanism of the pathology of insulinoma is still needed. The small number of disease cases and lack of suitable animal models and cell strains have limited the study of the pathogenesis of insulinoma. The risk factors and related molecular mechanisms of insulinoma remain unclear.
Exome sequencing, which allows study of the complete protein‐coding regions in the genome, is valuable in searching for underlying genetic variation in disease. Moreover, extensive exome sequencing studies from human tumors have indicated that there are a large number of mutations in each tumor 3. Accumulating evidence suggests that genetic change is specific for insulinomas, such as high loss of heterozygosity rates on chromosome 22q and gain of 9q34 4, 5. Additionally, exome sequencing has revealed that there was a somatic mutation in the DNA‐binding zinc finger of the transcription factor Yin Yang 1 in insulinoma 6, 7, 8.
In order to further screen for potential genetic alterations in insulinoma, we selected the tumor tissue and matched blood of patients with insulinoma and performed exome sequencing and analysis of deleterious effects on the protein. In this study, we obtained a total of 55 gene mutations in insulinoma. Among them, mutations in MEN1 were most related to the pathology of insulinoma, which provides evidence for use in early disease screening and target treatment of insulinoma.
Materials and methods
Insulinoma patient samples
In our present study, three patients (INS1, INS2 and INS3) with primary insulinoma were enrolled from the Second Hospital of Hebei Medical University. The paired tumor and a peripheral blood sample for DNA extraction were collected from the patients after surgical removal. The insulinoma was diagnosed depending on current clinical guidelines and histopathological confirmation 9. The diagnosis was established using the following six tight criteria: (a) documented blood glucose levels ≤ 2.2 mmol·L−1 (≤ 40 mg·dL−1); (b) concomitant insulin levels ≥ 66 μU·L−1 (≥ 36 pmol·L−1; ≥ 3 μU·L−1); (c) C‐peptide levels ≥ 200 pmol·L−1; (d) proinsulin levels ≥ 5 pmol·L−1; (e) β‐hydroxybutyrate levels ≤ 2.7 mmol·L−1; (f) absence of sulfonylurea (metabolites) in the plasma and/or urine. Written consent forms were obtained from the enrolled participants, and the research protocol was approved by the ethics committee of the Second Hospital of Hebei Medical University (2017‐R086) and complied with the principles of the Declaration of Helsinki. Clinical information for the patients is shown in Table 1.
Table 1.
Clinical information for the patients with insulinoma. INS1, INS2 and INS3 represent the three patients with insulinoma
| Sample | Gender | Age at diagnosis (years) | Grade | Metastatic disease | Tumor size (cm) | Ki67 |
|---|---|---|---|---|---|---|
| INS1 | Male | 64 | G2 | No | 0.6 | 2% |
| INS2 | Female | 75 | G1 | No | 0.8 | 2% |
| INS3 | Female | 57 | G2 | No | 1.0 | 2% |
Exome sequencing and data analysis
Genomic DNA was isolated from blood and tissue and was controlled for quality by measuring its concentration using a Nanodrop 2000 (Illumina, San Diego, CA, USA) and measuring fragmentation by agarose gel electrophoresis. Qualified Genomic DNA was prepared for exome sequencing with an Agilent SureSelect Human All Exon 50 Mb Exon Kit (Agilent Technologies, Santa Clara, CA, USA). The genomic DNA of each sample was fragmented and captured for exome sequencing with the Illumina HiSeq 2500 Sequencer platform (Illumina). For each sample, sequencing reads with 125‐bp paired‐end and Q 30 > 92% were generated.
After filtering the low quality and contaminating reads, sequence reads were mapped to the human genome sequence (hg19) using the Burrows–Wheeler alignment tool (http://bio-bwa.sourceforge.net/), which generated the sequence alignment/map file. The PCR duplicate reads were further removed using the picard software program.
Single nucleotide variant detection and annotation
To obtain the important candidate genes, the mutect software 10 was used to detect single nucleotide variants (SNVs). Variants were filtered for minimum genotype quality of 50 and minimum coverage depths of 10. Then, the software annovar (http://www.openbioinformatics.org/annovar/) was applied to annotate the qualified variants. Finally, the variants were obtained and the deleteriousness of variants was subsequently predicted by various bioinformatics programs (e.g. sift, polyphen2, lrt, mutationtaster, mutationassessor, fathmm, radialsvm, lr).
Results
General characteristics of the patients
We studied three patients with insulinoma, two female and one male. None had a family history of insulinoma. Moreover, we also excluded multiple endocrine neoplasia type 1 (MEN‐1 syndrome) from non‐tumor tissue. The WHO grading classification of pancreatic neuroendocrine tumors updated in 2010 includes neuroendocrine tumor G1, neuroendocrine tumor G2, neuroendocrine carcinoma G3 and mixed adenoneuroendocrine carcinoma 11. Depending on the classification system 12, two patients (INS1 and INS3) were classified as Grade II and one was classified as Grade I in this study. All presented with signs and symptoms of hypoglycemia. The hypoglycemia was corrected in all cases after surgical removal. General pathological and demographic characteristics for the three patients are shown in Table 1.
Genetic analysis
Genomic DNA from insulinomas and matched blood samples was subjected to whole exome sequencing. After mapping of the human genome sequence (hg19), a total of > 86% of the exome region was covered (Table 2). Exome sequencing analysis overall identified 40 210, 40 272 and 41 910 SNVs for tumor tissue, and 41 106, 40 050 and 41 451 SNVs for the matched blood samples. We identified 55 rare somatic mutations among the three patients, of which 39 were non‐synonymous, four were nonsense and 12 were synonymous. An overview of detected somatic mutations after exome sequencing is provided in Table 3. MEN1 gene nonsense mutations occurred in two different tumor samples. A c. 681C>G; p.Tyr227* mutation was found in exon 4 of the MEN1 gene in INS1, and c. 346G>T; p.Glu116* mutation was found in exon 2 of the MEN1 gene in INS2. The mutations were not present in the corresponding leukocyte DNA. Additionally, these mutations were predicted to be damaging by sift or lrt.
Table 2.
Summary of sequencing data. C1, C2 and C3: tissues of three patients with insulinoma; N1, N2 and N3: blood of three patients with insulinoma
| Sample | Reads | Read length (bp) | > Q 30 (%) | Exome size (bp) | Exome coverage |
|---|---|---|---|---|---|
| C1 | 104 472 186 | 125 | 94.82 | 74 856 280 | 87.36% |
| C2 | 107 751 402 | 125 | 92.34 | 74 856 280 | 87.79% |
| C3 | 112 709 600 | 125 | 94.69 | 74 856 280 | 88.19% |
| N1 | 110 592 066 | 125 | 94.5 | 74 856 280 | 87.90% |
| N2 | 98 905 302 | 125 | 94.49 | 74 856 280 | 86.08% |
| N3 | 109 660 740 | 125 | 94.58 | 74 856 280 | 88.44% |
Table 3.
Overview of the gene mutations found by exome sequencing in the three patients with insulinoma
| Sample | Chr | Start | Ref | Alt | Gene | Mutation type | AA change |
|---|---|---|---|---|---|---|---|
| INS1 | 1 | 1.1 × 108 | G | A | PSRC1 | Nonsynonymous | p.Ala227Val |
| INS1 | 2 | 1 926 848 | A | G | MYT1L | Synonymous | p.Asn231Asn |
| INS1 | 2 | 2.38 × 108 | T | C | COL6A3 | Nonsynonymous | p.Glu1565Gly |
| INS1 | 3 | 1.47 × 108 | C | T | ZIC4 | Nonsynonymous | p.Arg246His |
| INS1 | 3 | 1.89 × 108 | C | T | TPRG1 | Synonymous | p.Leu120Leu |
| INS1 | 5 | 1.46 × 108 | G | T | PPP2R2B | Nonsynonymous | p.Pro235Thr |
| INS1 | 6 | 32 548 632 | T | A | HLA‐DRB1 | Nonsynonymous | p.Arg218Ser |
| INS1 | 7 | 1 542 657 | G | A | INTS1 | Nonsynonymous | p.Arg77Cys |
| INS1 | 7 | 4 249 780 | T | A | SDK1 | Nonsense | p.Leu329* |
| INS1 | 7 | 1.48 × 108 | A | G | CNTNAP2 | Nonsynonymous | p.Lys1156Glu |
| INS1 | 7 | 1.57 × 108 | G | T | PTPRN2 | Nonsynonymous | p.Ser827Tyr |
| INS1 | 9 | 99 157 190 | A | G | ZNF367 | Synonymous | p.Cys202Cys |
| INS1 | 10 | 99 153 502 | C | A | RRP12 | Nonsynonymous | p.Ala157 Ser |
| INS1 | 10 | 1.3 × 108 | C | G | MKI67 | Nonsynonymous | p.Gly477Ala |
| INS1 | 11 | 64 575 141 | G | C | MEN1 | Nonsense | p.Tyr227* |
| INS1 | 11 | 1.02 × 108 | A | G | CEP126 | Nonsynonymous | p.Ile674Val |
| INS1 | 13 | 95 887 080 | G | A | ABCC4 | Synonymous | p.Ala105Ala |
| INS1 | 14 | 74 968 287 | G | T | LTBP2 | Nonsynonymous | p.Pro1726Gln |
| INS1 | 16 | 28 943 787 | G | C | CD19 | Nonsynonymous | p.Gly70Ala |
| INS1 | 16 | 31 405 651 | C | A | ITGAD | Nonsynonymous | p.Phe42Leu |
| INS1 | 17 | 21 318 727 | A | T | KCNJ12 | Nonsynonymous | p.Met25Leu |
| INS1 | 17 | 57 761 285 | A | G | CLTC | Nonsynonymous | p.His1462Arg |
| INS1 | 19 | 10 799 330 | G | A | ILF3 | Nonsynonymous | p.Gly843Ser |
| INS2 | 1 | 22 332 008 | T | C | CELA3A | Synonymous | p.Asp66Asp |
| INS2 | 1 | 40 229 393 | G | A | BMP8B | Synonymous | p.Leu313Leu |
| INS2 | 6 | 32 007 839 | G | T | CYP21A2 | Nonsynonymous | p.Ala236Ser |
| INS2 | 8 | 52 733 231 | G | A | PCMTD1 | Nonsense | p.Arg176* |
| INS2 | 8 | 88 298 821 | T | A | CNBD1 | Nonsynonymous | p.Tyr322Asn |
| INS2 | 8 | 1.25 × 108 | G | A | TMEM65 | Nonsynonymous | p.Ala179Val |
| INS2 | 8 | 1.25 × 108 | T | C | TMEM65 | Nonsynonymous | p.Tyr176Cys |
| INS2 | 9 | 1.13 × 108 | C | A | SVEP1 | Nonsynonymous | p.Arg902Leu |
| INS2 | 11 | 64 577 236 | C | A | MEN1 | Nonsense | p.Glu116* |
| INS2 | 11 | 89 018 006 | C | A | TYR | Nonsynonymous | p. Pro417His |
| INS2 | 12 | 6 787 522 | G | A | ZNF384 | Nonsynonymous | p.Pro153Ser |
| INS2 | 12 | 9 243 947 | A | G | A2M | Synonymous | p.Ser773Ser |
| INS2 | 12 | 1.25 × 108 | A | G | UBC | Synonymous | p.Asp647Asp |
| INS2 | 17 | 7 671 513 | G | A | DNAH2 | Nonsynonymous | p.Arg1290His |
| INS2 | 17 | 7 834 438 | C | G | TRAPPC1 | Nonsynonymous | p. Ser67Thr |
| INS2 | 17 | 34 797 666 | G | A | TBC1D3B | Synonymous | p.Ala490Ala |
| INS2 | 19 | 41 355 849 | A | G | CYP2A6 | Synonymous | p. Leu73Leu |
| INS2 | X | 37 027 691 | T | C | FAM47C | Nonsynonymous | p.Phe403Ser |
| INS3 | 1 | 12 854 188 | T | C | PRAMEF1 | Nonsynonymous | p.Cys138Arg |
| INS3 | 2 | 1.28 × 108 | G | A | MYO7B | Nonsynonymous | p.Gly163Asp |
| INS3 | 4 | 1.52 × 108 | C | A | RPS3A | Nonsynonymous | p.Asn147Lys |
| INS3 | 4 | 1.52 × 108 | A | C | RPS3A | Nonsynonymous | p.Gln157Pro |
| INS3 | 5 | 1.4 × 108 | T | C | PCDHA5 | Nonsynonymous | p.Val596Ala |
| INS3 | 6 | 99 850 428 | T | C | PNISR | Nonsynonymous | p.Lys439Glu |
| INS3 | 6 | 1.38 × 108 | C | T | OLIG3 | Nonsynonymous | p.Ala175Thr |
| INS3 | 8 | 33 449 689 | C | A | DUSP26 | Nonsynonymous | p.Ala160Ser |
| INS3 | 10 | 21 903 830 | T | G | MLLT10 | Nonsynonymous | p.Cys194Gly |
| INS3 | 11 | 1 093 437 | G | C | MUC2 | Synonymous | p.Thr1752Thr |
| INS3 | 11 | 1.26 × 108 | C | T | PUS3 | Nonsynonymous | p.Glu143Lys |
| INS3 | 16 | 7 629 904 | C | T | RBFOX1 | Synonymous | p.Asp152Asp |
| INS3 | 17 | 79 667 512 | G | A | HGS | Nonsynonymous | p.Ser633Asn |
| INS3 | 19 | 19 030 142 | G | A | COPE | Nonsynonymous | p.Pro6Ser |
Mutation of MEN1 gene
The p.Tyr227* mutation (SWLYLKGSYMRCDRKMEV) and p.Glu116* mutation (VSSRELVKKVSDVIWNSL) both resulted in a significant truncation of menin, which may destroy the functional domain and affect its function. Amino acids of p.Tyr227* and p.Glu116* mutations are highly conserved across multiple species (Fig. 1A,B). Moreover, two nonsense mutations locate in the functional domain of MEN1, indicating that it may affect the binding of lysine methyltransferase 2A (Fig. 1C). In the current study, the nonsense mutations happened early in the sequence of MEN1, which may obviously result in a non‐functional gene product (Fig. 1D).
Figure 1.
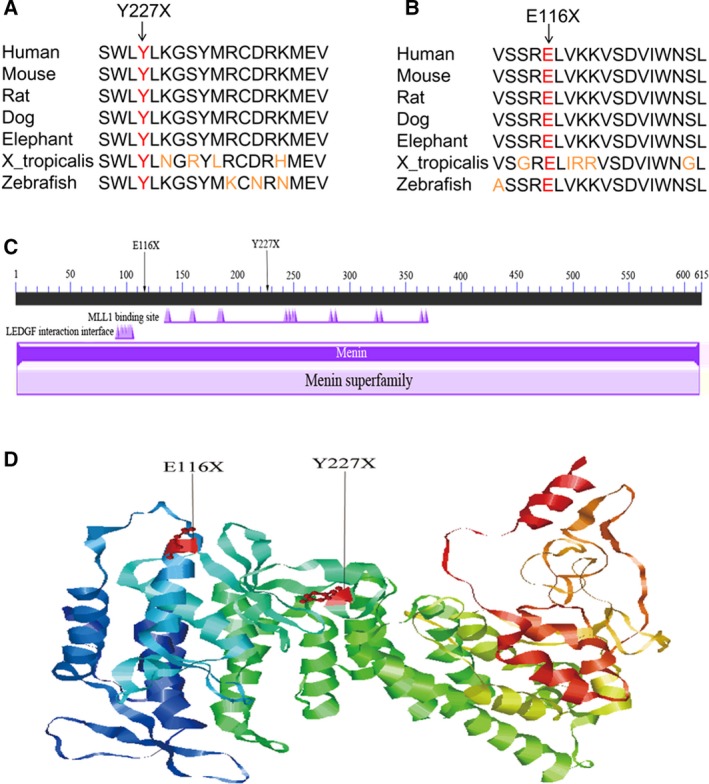
Nonsense mutation in MEN1. (A) Conservation of p.Tyr227* in the MEN1 protein. (B) Conservation of p.Glu116* in the MEN1 protein. (C) Locations of p.Tyr227* and p.Glu116* mutations in the conserved domain in the MEN1 protein. (D) Location of p.Tyr227* and p.Glu116* mutations on the crystal structure of the MEN1 protein.
Discussion
Insulinoma is a common neuroendocrine tumor with an incidence of four in every 1 million persons annually 13. Moreover, most of the insulinomas arise sporadically. Although rare, it has the potential to produce profound metabolic derangements that require early recognition and treatment. In this study, exome sequencing was performed on three sporadic insulinoma cases to delineate genetic contributors to this rare endocrine tumor. Although the overall frequency of somatic mutations was low and predicted to be damaging, there were two nonsense variants that occurred in MEN1 in two of the three patients, namely a c. 681C>G; p.Tyr227* mutation in exon 4 of MEN1 and a c. 346G>T; p.Glu116* mutation in exon 2 of MEN1. Our result showed that the mutations in MEN1 may play an important role in the development of insulinoma. However, further functional research is needed to validate their roles.
MEN1 consists of 10 exons 9 and encodes a protein with 615 amino acids 14; it is considered a putative tumor suppressor gene associated with neuroendocrine tumors 15. Menin, the encoded protein of MEN1, is a typical GTPase stimulated by nm23. It is mostly found in the nucleus and can regulate gene expression in a positive or negative way, and it has been demonstrated to interact with transcription activators, transcription repressors, cell signaling proteins and various other proteins. In addition, it plays major roles in DNA repair, cell cycle regulation and chromatin remodeling. Generally, menin is considered as a transcriptional regulator and interacts with a number of nuclear and cytosolic proteins, which indicates that it may participate in various biological pathways of tumor formation 16, 17, 18. Additionally, a number of sporadic endocrine tumors, including parathyroid adenomas, pancreatic insulinomas and pituitary prolactinomas, have somatic mutations of MEN1 alleles, suggesting that MEN1 may play a role in non‐hereditary endocrine tumors 15, 19, 20. Our results showed that MEN1 mutations were found in two of the three insulinoma patients, which provided further evidence that MEN1 might be an important factor in the pathological process of insulinoma.
It is noted that insulinomas can occur sporadically or in combination with MEN‐1 syndrome. Moreover, the MEN1 gene is the first gene that has been identified as a candidate gene in the tumorigenesis of insulinoma. MEN‐1 syndrome represents an autosomal dominant disorder related to mutations in the MEN1 gene mapped to chromosome 11q13 21, 22. Generally, simple and local tumor enucleation of MEN‐1 syndrome‐associated insulinomas is not likely to be curative. Although genetic testing for MEN1 fails to detect mutation rate of 10–25%, it plays a vital role in identifying patients with hereditary insulinomas 23, 24. Therefore, genetic testing for MEN‐1 syndrome is beneficial to clinical diagnosis. Okamoto et al. 25 suggested that a novel six‐nucleotide insertion in exon 4 of the MEN1 gene might contribute to the familial insulinoma. A prior study indicated that MEN1 gene mutations were lacking in 27 sporadic insulinomas 26. By evaluating a large family with malignant insulinoma and hyperparathyroidism, Hasani‐Ranjbar et al. 27 found a novel MEN1 gene frameshift germline mutation, which was associated with malignant insulinoma. Moreover, a recent study found several novel pathogenic MEN1 mutations in sporadic cases of insulinoma 28. Herein, we found mutations in MEN1 on chromosome 11 in patients with insulinoma, which had a damaging role in the function of the encoded protein. Our finding was consistent with other studies indicating the role of MEN1 mutation in human sporadic insulinomas 26, 29, 30, 31, 32, which provided a crucial clue in the treatment of insulinoma. Interestingly, Waldmann et al. 33 found the p.E116X mutation in exon 2 of the MEN1 gene in 21 patients with MEN‐1 syndrome and adrenal lesions. In addition, Turner et al. 34 identified the p.Y227X mutation in exon 4 of the MEN1 gene in multiple endocrine neoplasia type 1. This further suggested that MEN1 was significantly associated with insulinoma.
Conclusions
In the current study, exome sequencing for three sporadic insulinomas identified two somatic nonsense mutations in MEN1 (c. 681C>G; p.Tyr227* and c. 346G>T; p.Glu116*), which might induce a non‐functional gene product and contribute to the oncogenesis of sporadic insulinoma. However, there were some limitations in this study. The samples selected were small and larger samples are further needed to validate their roles in insulinoma. Further studies are needed to precisely explore the role of genetic mutations of MEN1 in clinical manifestations of these patients. In addition, validation of MEN1 mutations (such as by Sanger sequencing) and study of the underlying biological function of the MEN1 mutations is needed in a further study.
Author contributions
JD, QS and MW analyzed and interpreted the data. CQ was the major contributor in writing the manuscript. CY designed the project. All authors read and approved the final manuscript.
References
- 1. Shin JJ, Gorden P and Libutti SK (2010) Insulinoma: pathophysiology, localization and management. Future Oncol 6, 229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fendrich V, Waldmann J, Bartsch DK and Langer P (2009) Surgical management of pancreatic endocrine tumors. Nat Rev Clin Oncol 6, 419–428. [DOI] [PubMed] [Google Scholar]
- 3. Kennedy SR, Loeb LA and Herr AJ (2012) Somatic mutations in aging, cancer and neurodegeneration. Mech Ageing Dev 133, 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wild A, Langer P, Ramaswamy A, Chaloupka B and Bartsch DK (2001) A novel insulinoma tumor suppressor gene locus on chromosome 22q with potential prognostic implications. J Clin Endocrinol Metab 86, 5782–5787. [DOI] [PubMed] [Google Scholar]
- 5. Speel EJ, Scheidweiler AF, Zhao J, Matter C, Saremaslani P, Roth J, Heitz PU and Komminoth P (2001) Genetic evidence for early divergence of small functioning and nonfunctioning endocrine pancreatic tumors: gain of 9Q34 is an early event in insulinomas. Cancer Res 61, 5186–5192. [PubMed] [Google Scholar]
- 6. Cromer MK, Choi M, Nelson‐Williams C, Fonseca AL, Kunstman JW, Korah RM, Overton JD, Mane S, Kenney B, Malchoff CD et al (2015) Neomorphic effects of recurrent somatic mutations in Yin Yang 1 in insulin‐producing adenomas. Proc Natl Acad Sci USA 112, 4062–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cao Y, Gao Z, Li L, Jiang X, Shan A, Cai J, Peng Y, Li Y, Jiang X, Huang X et al (2013) Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat Commun 4, 2810. [DOI] [PubMed] [Google Scholar]
- 8. Lichtenauer UD, Di Dalmazi G, Slater EP, Wieland T, Kuebart A, Schmittfull A, Schwarzmayr T, Diener S, Wiese D, Thasler WE et al (2015) Frequency and clinical correlates of somatic Ying Yang 1 mutations in sporadic insulinomas. J Clin Endocrinol Metab 100, E776–E782. [DOI] [PubMed] [Google Scholar]
- 9. de Herder WW, Niederle B, Scoazec JY, Pauwels S, Kloppel G, Falconi M, Kwekkeboom DJ, Oberg K, Eriksson B, Wiedenmann B et al (2006) Well‐differentiated pancreatic tumor/carcinoma: insulinoma. Neuroendocrinology 84, 183–188. [DOI] [PubMed] [Google Scholar]
- 10. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES and Getz G (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bosman F, Carneiro F, Hruban R and Theise N (2010) WHO Classification of Tumours of the Digestive System, 4th edn WHO, Geneva, Switzerland. [Google Scholar]
- 12. Sobin LH (1974) Histological Classification of Tumours. Springer, Berlin, Heidelberg, Germany. [Google Scholar]
- 13. Service FJ, McMahon MM, O'Brien PC and Ballard DJ (1991) Functioning insulinoma–incidence, recurrence, and long‐term survival of patients: a 60‐year study. Mayo Clin Proc 66, 711–719. [DOI] [PubMed] [Google Scholar]
- 14. Guru SC, Manickam P, Crabtree JS, Olufemi SE, Agarwal SK and Debelenko LV (1998) Identification and characterization of the multiple endocrine neoplasia type 1 (MEN1) gene. J Intern Med 243, 433–439. [DOI] [PubMed] [Google Scholar]
- 15. Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert‐Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA et al (1997) Positional cloning of the gene for multiple endocrine neoplasia‐type 1. Science 276, 404–407. [DOI] [PubMed] [Google Scholar]
- 16. Shen HC, He M, Powell A, Adem A, Lorang D, Heller C, Grover AC, Ylaya K, Hewitt SM, Marx SJ et al (2009) Recapitulation of pancreatic neuroendocrine tumors in human multiple endocrine neoplasia type I syndrome via Pdx1‐directed inactivation of Men1. Cancer Res 69, 1858–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Agarwal SK, Kennedy PA, Scacheri PC, Novotny EA, Hickman AB, Cerrato A, Rice TS, Moore JB, Rao S, Ji Y et al (2005) Menin molecular interactions: insights into normal functions and tumorigenesis. Horm Metab Res 37, 369–374. [DOI] [PubMed] [Google Scholar]
- 18. Nikfarjam M, Warshaw AL, Axelrod L, Deshpande V, Thayer SP, Ferrone CR and Fernandez‐del Castillo C (2008) Improved contemporary surgical management of insulinomas: a 25‐year experience at the Massachusetts General Hospital. Ann Surg 247, 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ludwig L, Schleithoff L, Kessler H, Wagner PK, Boehm BO and Karges W (1999) Loss of wild‐type MEN1 gene expression in multiple endocrine neoplasia type 1‐associated parathyroid adenoma. Endocr J 46, 539–544. [DOI] [PubMed] [Google Scholar]
- 20. Libutti SK, Crabtree JS, Lorang D, Burns AL, Mazzanti C, Hewitt SM, O'Connor S, Ward JM, Emmert‐Buck MR, Remaley A et al (2003) Parathyroid gland‐specific deletion of the mouse Men1 gene results in parathyroid neoplasia and hypercalcemic hyperparathyroidism. Cancer Res 63, 8022–8028. [PubMed] [Google Scholar]
- 21. Wermer P (1963) Endocrine adenomatosis and peptic ulcer in a large kindred. Inherited multiple tumors and mosaic pleiotropism in man. Am J Med 35, 205–212. [DOI] [PubMed] [Google Scholar]
- 22. Larsson C, Skogseid B, Oberg K, Nakamura Y and Nordenskjold M (1988) Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 332, 85–87. [DOI] [PubMed] [Google Scholar]
- 23. Callender GG, Rich TA and Perrier ND (2008) Multiple endocrine neoplasia syndromes. Surg Clin North Am 88, 863–895. [DOI] [PubMed] [Google Scholar]
- 24. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck‐Peccoz P, Bordi C, Conte‐Devolx B, Falchetti A, Gheri RG, Libroia A et al (2001) Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86, 5658–5671. [DOI] [PubMed] [Google Scholar]
- 25. Okamoto H, Tamada A, Hai N, Doi M, Uchimura I, Hirata Y and Kosugi S (2002) A novel six‐nucleotide insertion in exon 4 of the MEN1 gene, 878insCTGCAG, in three patients with familial insulinoma and primary hyperparathyroidism. Jpn J Clin Oncol 32, 368–370. [DOI] [PubMed] [Google Scholar]
- 26. Cupisti K, Hoppner W, Dotzenrath C, Simon D, Berndt I, Roher HD and Goretzki PE (2000) Lack of MEN1 gene mutations in 27 sporadic insulinomas. Eur J Clin Invest 30, 325–329. [DOI] [PubMed] [Google Scholar]
- 27. Hasani‐Ranjbar S, Amoli MM, Ebrahim‐Habibi A, Gozashti MH, Khalili N, Sayyahpour FA, Hafeziyeh J, Soltani A and Larijani B (2011) A new frameshift MEN1 gene mutation associated with familial malignant insulinomas. Fam Cancer 10, 343–348. [DOI] [PubMed] [Google Scholar]
- 28. Jyotsna VP, Malik E, Birla S and Sharma A (2015) Novel MEN 1 gene findings in rare sporadic insulinoma–a case control study. BMC Endocr Disord 15, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gortz B, Roth J, Krahenmann A, de Krijger RR, Muletta‐Feurer S, Rutimann K, Saremaslani P, Speel EJ, Heitz PU and Komminoth P (1999) Mutations and allelic deletions of the MEN1 gene are associated with a subset of sporadic endocrine pancreatic and neuroendocrine tumors and not restricted to foregut neoplasms. Am J Pathol 154, 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhuang Z, Vortmeyer AO, Pack S, Huang S, Pham TA, Wang C, Park WS, Agarwal SK, Debelenko LV, Kester M et al (1997) Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res 57, 4682–4686. [PubMed] [Google Scholar]
- 31. Moore PS, Beghelli S, Zamboni G and Scarpa A (2003) Genetic abnormalities in pancreatic cancer. Mol Cancer 2, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gumbs AA, Moore PS, Falconi M, Bassi C, Beghelli S, Modlin I and Scarpa A (2002) Review of the clinical, histological, and molecular aspects of pancreatic endocrine neoplasms. J Surg Oncol 81, 45–53; discussion 54. [DOI] [PubMed] [Google Scholar]
- 33. Waldmann J, Bartsch DK, Kann PH, Fendrich V, Rothmund M and Langer P (2007) Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg 392, 437–443. [DOI] [PubMed] [Google Scholar]
- 34. Turner JJ, Leotlela PD, Pannett AA, Forbes SA, Bassett JH, Harding B, Christie PT, Bowen‐Jones D, Ellard S, Hattersley A et al (2002) Frequent occurrence of an intron 4 mutation in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 87, 2688–2693. [DOI] [PubMed] [Google Scholar]