

Abstract
We aimed to evaluate the prevalence of familial hypercholesterolaemia (FH) in a subject with hypercholesterolaemia from two population-based cohorts in South Korea. A total of 283 subjects with total cholesterol levels of 290 mg/dL (7.5 mmol/L) or higher were selected from the Namwon and Dong-gu Studies. We used next generation sequencing (NGS) to detect mutations in low-density lipoprotein receptors (LDLR), apolipoprotein B (APOB) and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes. We have confirmed 17 different mutations of the LDLR, APOB and PCSK9 in 23 subjects (8.1%). Eleven LDLR variants and one APOB variant have been previously reported. One LDLR and two PCSK9 rare variants were identified in the variants database, but not in the FH mutation database. Two novel LDLR variants were found, p.Leu680Val, and p.Thr734Phe. No LDLR, APOB or PCSK9 deletions nor insertions were found. When the subjects were restricted to 110 subjects with a total cholesterol ≥310 mg/dL, only 10 variants were found in the 10 subjects (9.1%). These results suggest that given the low prevalence of FH mutations in subjects with high total cholesterol levels, NGS-based testing for a population-based approach to FH detection may not be cost-effective.
Keywords: Hyperlipoproteinemia Type II, Receptors, LDL, mutation, High-Throughput Nucleotide Sequencing
INTRODUCTION
Familial hypercholesterolaemia (FH, OMIM no.143890) is a common autosomal dominant disorder characterised by very high levels of low-density lipoprotein-cholesterol (LDL-C) and premature atherosclerosis and coronary heart disease.1
Most FH patients have mutations in the LDL receptor (LDLR) gene, while mutations in apolipoprotein B (ApoB) and proprotein convertase subtilisin/kexin type 9 (PCSK9) are rare. The LDLR gene is located on chromosome 19 with exon182 and encodes the LDLR protein, which normally removes LDL from the circulation, or ApoB, which is the part of the LDL that binds with the receptor. The prevalence of heterozygous FH is about 1/500–1/200 in most countries, homozygous FH is much rarer, occurring at a rate of 1 in a million.3 Mutation detection of these genes is one of most important for FH diagnosis along with total cholesterol levels (especially, LDL-C levels), tendon xanthomata, corneal arcus, and family history.4 More than 1,100 mutations in LDLR are described,5 and new mutations in LDLR and others are being updated (http://www.ucl.ac.uk/ldlr/Current/ and http://www.umd.be/LDLR/).
Based on extrapolations from these estimated 1/500–1/200 prevalences, it is estimated that there are 100,000–250,000 patients in South Korea. However, there is little data about the molecular basis of FH in Koreans which means that FH is vastly underdiagnosed and undertreated in South Korea. Diagnosis of FH may be overlooked. Many such cases remain undetected, and many patients with established FH do not receive appropriate treatment. Several recent reports have demonstrated that next generation sequencing (NGS) is very useful for the screening of FH mutation detection in large numbers of samples.6,7,8 Therefore, we examined the prevalence and distribution of FH in adults with high serum cholesterol from two population-based cohorts using targeted NGS-based testing.
MATERIALS AND METHODS
1. Subjects
In the Simon Broome Diagnostic criteria for FH, a cholesterol level of 290 mg/dL (7.5 mmol/L) was used as a cutoff to find the possible FH index patients. The European Atherosclerosis Society recommends that adults with total cholesterol ≥310 mg/dL (8 mmol/L) should be screened for FH. Therefore, a total of 283 patients with a total cholesterol level ≥290 mg/dL (7.5 mmol/liter) were selected from the Dong-gu Study (9,260 participants) and the Namwon Study (10,667 participants), which are ongoing prospective studies designed to investigate risk factors for cardiovascular disease, cognitive decline, and fracture in urban and rural populations, respectively. Details of the study subjects and measurements have been previously published.9 We did not use the clinical criteria of FH because self-reported family history of premature cardiovascular disease (CVD) may be highly inaccurate. The subjects with DNA were 9,234 in the Dong-Gu Study and 10,576 in the Namwon Study, of which total cholesterol of more than 290 mg/dL was 188 in the Dong-Gu Study and 95 in the Namwon study. This study was approved by the institutional review board of Chonnam National University Hospital (IRB No. I-2008-05-056), and informed consent was obtained from each subject.
2. Mutation detection
Genomic DNA was extracted from peripheral blood using a QIAamp DNA Mini Kit (Qiagen Inc., Chatsworth, CA, USA) according to the manufacturer's protocol. Design of amplicons, sample preparation and sequences for PCRbased Access Array (Fluidigm) were processed as described.10 Briefly, a total of 48 amplicons were designed to cover all exons of LDLR; four exons of PCSK9, containing the most common gain-of-function pathogenic mutation; and one spot of APOB including p.Arg3527Gln, APOE and SLCO1B1 containing rs4149056, the myopathy-associated variant. Samples were amplified using the 48.48 PCR-based Access Array (Fluidigm, San Francisco, USA) and purified pooled samples were sequenced on the Miseq sequencer (Illumina, Hayward, USA) according to the manufacturer's protocol.
3. Data analysis
Data analysis was performed on PCR Amplicon workflow using Miseq Reporter Software version 2.4.60 (Illumina, Hayward, USA), which used Burrows-Wheeler Aligner (BWA). We aligned reads against the GRCh37/hg19 reference genomes of targeted regions in the sample sheet. Integrative Genomics Viewer (IGV) was used to visualize the quality and variance of Bam and VCF files.11
Variants with a minor allele frequency of >1% in Ensemble,12 synonymous and intronic variants located outside exon/intron boundaries were excluded from further analysis. To classify pathogenic mutations, all non-synonymous variants were compared to a FH locus-specific database (http://www.ucl.ac.uk/ldlr/Current/ and http://www.umd.be/LDLR/) and the Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php) and were predicted for amino acid variations using predictive algorithms, SIFT, PolyPhen-2 and Mutation t@ster web servers.13,14,15 All variants in this study were classified as pathogenic mutations and variants of uncertain significance (VUS) as described.7 All rare variants were confirmed by conventional Sanger sequencing and high-resolution melting analysis.
4. Other measurements
Weight and height was measured in light clothing without shoes. Body-mass index (BMI) was calculated by dividing body weight by the square of the height (kg/m2). Venous blood samples were collected after an overnight fast. The serum levels of total cholesterol, HDL-cholesterol, and triglycerides were also measured with an automated analyzer using enzyme methods. LDL cholesterol was calculated by the Friedewald formula (LDL cholesterol (mg/dL)=total cholesterol−HDL cholesterol−triglyceride×1/5) when triglycerides are less than 400 mg/dL, but not in 27 subjects with triglycerides ≥400 mg/dL.
RESULTS
The characteristics of enrolled subjects in this study are shown in Table 1. Subjects were included according to two criteria for FH screening: total cholesterol (290 mg/dL or 310 mg/dL). In 283 subjects with a total cholesterol ≥290 mg/dL, the mean value of total cholesterol in both cohort studies was 312 mg/dL. Subjects with FH gene mutations had a higher percentage of a history of stroke than subjects without FH gene mutations. However, there was no difference in the other factors between the two groups. In 110 subjects with a total cholesterol ≥310 mg/dL, there was no difference in the general characteristics between subjects with FH gene mutations and those without FH gene mutations.
TABLE 1. General characteristics of study subjects with high cholesterol levels according to familial hypercholesterolemia mutation status.
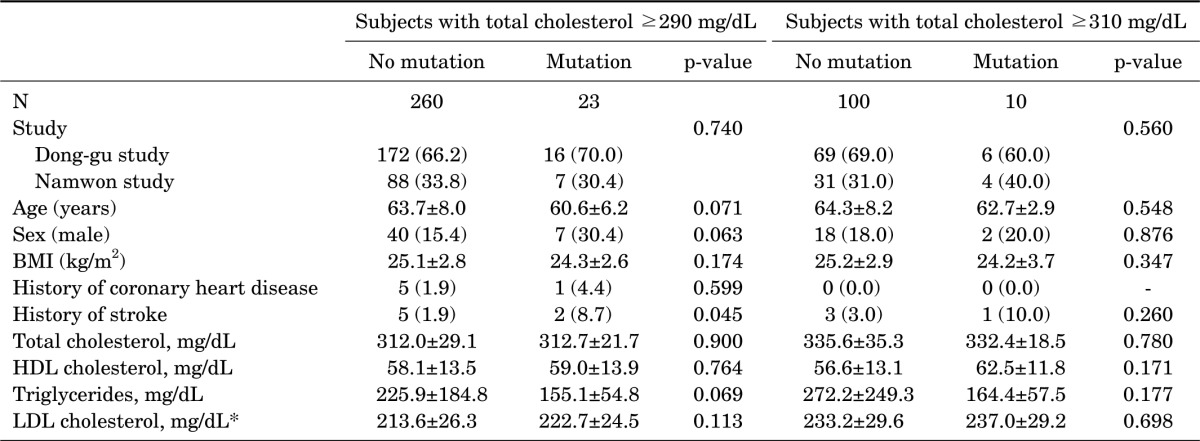
*LDL cholesterol was calculated by the Friedewald formula (LDL cholesterol (mg/dL)=total cholesterol−HDL cholesterol−triglyceride×1/5) when triglycerides are less than 400 mg/dL, but not in 27 subjects with triglycerides ≥400 mg/dL.
We have confirmed 17 different variants of the LDLR, PCSK9 and APOB in 23 subjects (8.1%) among the 283 subjects with a total cholesterol ≥290 mg/dL. Table 2 shows the distribution of the variants in LDLR, PCSK9 and APOB in the Dong-gu Study and the Namwon Study. Among these variants, eleven LDLR variants (p.Asp47Asn, p.Arg115His, p.Cys143Tyr, p.Asp221Asn, p.Glu228Lys, p. Arg257Trp, p.Cys352Phe, p.Gly516ser, p.Gly546Asp, p.His583Tyr and p.Asp589Asn) and one APOB variant (p. Arg3527Trp) in 18 (6.4%) subjects have been previously reported as disease-causing mutations in the FH locus-specific database and HGMD. Of the five variants of VUS, one rare variant (p.Arg132Gln) in LDLR and two rare variants (p.Glu85Lys and p.Arg499Cys) in PCSK9 were identified in the SNP database, not in FH database. Two novel LDLR variants (not reported in any variant database) were found, p.Leu680Val, and p.Thr734Phe. Among them, based on the prediction of three algorithms, SIFT, PolyPhen-2 and Mutation t@ster, p.Leu680Val, and p.Arg499Cys were considered as possibly pathogenic mutations. Further functional studies will be needed to identify the pathogenicity of these variants. No LDLR, APOB and PCSK9 deletions nor insertions were found. When the subjects were restricted to the 110 subjects with a total cholesterol of ≥310 mg/dL, we identified 10 different gene mutations in 10 subjects (9.1%) including 7 pathogenic FH-causing mutations in 7 subjects (6.4%) and three VUS in 3 subjects.
TABLE 2. The distribution of variants in LDLR, PCSK9 and APOB in the 283 subjects with a total cholesterol level ≥290 mg/dL from the Dong-gu Study and the Namwon Study.
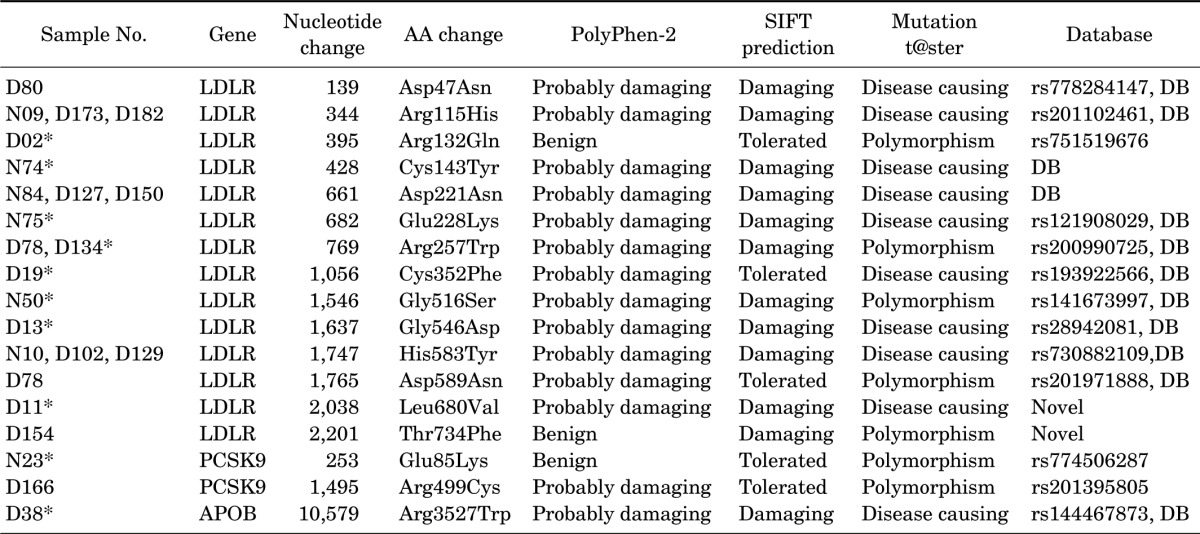
*Subject with a total cholesterol level ≥310 mg/dL.
DISCUSSION
Recent studies have shown that NGS-based assays are useful and cost effective in genetic diagnosis of FH in primary care.6,7,8 In this study, we used PCR-based 48.48 Access Array microfluidic technology (Fluidigm) to identify FH variants (LDLR, PCSK9 and APOB) in high serum cholesterol groups from two population-based cohorts. In 283 subjects with a cholesterol level of 290 mg/dL or higher, we identified 17 different gene mutations in 23 subjects (8.1%) including 12 pathogenic FH-causing mutations in 18 subjects (6.4%) and six VUS in 5 subjects.
Stroke is less common in FH than in premature CHD.16 However, in our study, there was no difference in the percentage of ischemic heart disease with or without gene mutation, but the percentage of stroke history was higher in the group with gene mutation. In particular, one case with pathogenic FH-causing mutations had for both p.Arg257Trp and p.Asp589Asn. Elevated LDL cholesterol (LDL-C) lead to coronary heart disease and stroke.17,18,19 There were few studies of associations with LDLR, PCSK9 and ApoB mutations and stroke.20,21 These results are not consistent because of the small sample size and only several targeted mutations, not whole exons. Further studies with larger cases and mutations of whole exons will be needed.
The pathogenic FH mutation rate in this study was 6.4%, which was slightly lower than 7.6% in the Latvian population8 and higher than 2.1% in the Generation Scotland7. These inconsistent results between our results and those of previous studies in different populations may due to the different ranges of cholesterol levels for cut-off, and different NGS methods6 and clinical information. Since large insertions/ deletions could not be detected by the Access Array based NGS used in this study, the mutation rate in our study could be higher, although the rate of large insertions/ deletions is very row. MLPA analysis is needed for large indel detection.
Of 12 pathogenic FH-causing mutations, 9 mutations have previously been reported in Asia including China, Japan and Korea22,23,24,25,26,27,28,29,30 (UK and French database). Mutations, p.Arg115His and p.Glu228Lys in all three countries and p.Arg257Trp and p.Asp589Tyr in China and Korea have been found. In addition, mutations p.Asp47Asn in only Japan, p.Cys143Tyr and p.His583Tyr in only China and p.Asp221Asn and APOB p. Arg 3527Trp Asn in only Korea have been reported, respectively. The Arg115His, Asp221Asn and His583Tyr mutations in our study were found in 3 cases, respectively, these mutations might occur frequently in Korean.
To our knowledge, this is the first study of the mutation detection for FH based on the general population of Asia. However, the data reported here has several limitations. First, in the Dutch Lipid Clinic Network criteria for FH,31 the LDL cholesterol level was used as a diagnostic criterion along with the clinical criteria, but because we could not directly measure LDL cholesterol, we used total cholesterol as the criterion for FH selection. Second, our study did not show the clinical characteristics including the tendon xanthomata of patients with familial hypercholesterolemia. Third, we found VUS in five (1.4%) of the 283 subjects with a cholesterol levels of 290 mg/dL or higher. The interpretation of VUCS remains a challenge and the ambiguous nature of a VUS may limit its clinical utility with NGS.32 Further segregation and functional studies will be needed to clarify the pathogenicity of these variants.
In conclusion, we have confirmed 17 different mutations of the LDLR, APOB and PCSK9 in 23 subjects (8.1%) in 283 subjects with a total cholesterol of 290 mg/dL or higher from two large population-based cohorts. These results suggest that given the low prevalence of FH mutations in subjects with high total cholesterol levels, NGS-based testing for a population-based approach to FH detection may not be cost-effective.
ACKNOWLEDGEMENTS
This study was financially supported by Chonnam National University (Grant number: 2013-2622).
Footnotes
CONFLICT OF INTEREST STATEMENT: None declared.
References
- 1.Marks D, Thorogood M, Neil HA, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis. 2003;168:1–14. doi: 10.1016/s0021-9150(02)00330-1. [DOI] [PubMed] [Google Scholar]
- 2.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 3.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–1803. doi: 10.1172/JCI18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scriver CR, Beaudet AL, Sly WS, Valle D. The metabolic and molecular bases of inherited disease. 8th ed. New York: Mc-Graw-Hill; 2001. [Google Scholar]
- 5.Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet. 2008;72:485–498. doi: 10.1111/j.1469-1809.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- 6.Vandrovcova J, Thomas ER, Atanur SS, Norsworthy PJ, Neuwirth C, Tan Y, et al. The use of next-generation sequencing in clinical diagnosis of familial hypercholesterolemia. Genet Med. 2013;15:948–957. doi: 10.1038/gim.2013.55. [DOI] [PubMed] [Google Scholar]
- 7.Norsworthy PJ, Vandrovcova J, Thomas ER, Campbell A, Kerr SM, Biggs J, et al. Targeted genetic testing for familial hypercholesterolaemia using next generation sequencing: a population-based study. BMC Med Genet. 2014;15:70. doi: 10.1186/1471-2350-15-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radovica-Spalvina I, Latkovskis G, Silamikelis I, Fridmanis D, Elbere I, Ventins K, et al. Next-generation-sequencing-based identification of familial hypercholesterolemia-related mutations in subjects with increased LDL-C levels in a latvian population. BMC Med Genet. 2015;16:86. doi: 10.1186/s12881-015-0230-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kweon SS, Shin MH, Jeong SK, Nam HS, Lee YH, Park KS, et al. Cohort profile: the Namwon study and the Dong-gu study. Int J Epidemiol. 2014;43:558–567. doi: 10.1093/ije/dys244. [DOI] [PubMed] [Google Scholar]
- 10.Wu WF, Sun LY, Pan XD, Yang SW, Wang LY. Use of targeted exome sequencing in genetic diagnosis of Chinese familial hypercholesterolemia. PLoS One. 2014;9:e94697. doi: 10.1371/journal.pone.0094697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visual ization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, et al. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–D716. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 15.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. Mutation-Taster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 16.Huxley RR, Hawkins MH, Humphries SE, Karpe F, Neil HA Simon Broome Familial Hyperlipidaemia Register Group and Scientific Steering Committee. Risk of fatal stroke in patients with treated familial hypercholesterolemia: a prospective registry study. Stroke. 2003;34:22–25. doi: 10.1161/01.str.0000047123.14312.3e. [DOI] [PubMed] [Google Scholar]
- 17.Youngblom E, Pariani M, Knowles JW. Familial hypercholesterolemia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, editors. GeneReviews®. Seattle: University of Washington; 2014. [Google Scholar]
- 18.Raal FJ, Pilcher GJ, Panz VR, van Deventer HE, Brice BC, Blom DJ, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124:2202–2207. doi: 10.1161/CIRCULATIONAHA.111.042523. [DOI] [PubMed] [Google Scholar]
- 19.Elis A, Zhou R, Stein EA. Effect of lipid-lowering treatment on natural history of heterozygous familial hypercholesterolemia in past three decades. Am J Cardiol. 2011;108:223–226. doi: 10.1016/j.amjcard.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 20.Tybjaerg-Hansen A, Steffensen R, Meinertz H, Schnohr P, Nordestgaard BG. Association of mutations in the apolipoprotein B gene with hypercholesterolemia and the risk of ischemic heart disease. N Engl J Med. 1998;338:1577–1584. doi: 10.1056/NEJM199805283382203. [DOI] [PubMed] [Google Scholar]
- 21.Frikke-Schmidt R, Arlien-Søborg P, Thorsen S, Jensen HK, Vorstrup S. LDL receptor mutations and ApoB mutations are not risk factors for ischemic cerebrovascular disease of the young, but lipids and lipoproteins are. Eur J Neurol. 1999;6:691–696. doi: 10.1046/j.1468-1331.1999.660691.x. [DOI] [PubMed] [Google Scholar]
- 22.Chiou KR, Charng MJ. Common mutations of familial hypercholesterolemia patients in Taiwan: characteristics and implications of migrations from southeast China. Gene. 2012;498:100–106. doi: 10.1016/j.gene.2012.01.092. [DOI] [PubMed] [Google Scholar]
- 23.Gouni-Berthold I, Berthold HK. Familial hypercholesterolemia: etiology, diagnosis and new treatment options. Curr Pharm Des. 2014;20:6220–6229. doi: 10.2174/1381612820666140620125213. [DOI] [PubMed] [Google Scholar]
- 24.Han SM, Hwang B, Park TG, Kim DI, Rhee MY, Lee BK, et al. Genetic testing of Korean familial hypercholesterolemia using whole-exome sequencing. PLoS One. 2015;10:e0126706. doi: 10.1371/journal.pone.0126706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinchcliffe M, Le H, Fimmel A, Molloy L, Freeman L, Sullivan D, et al. Diagnostic validation of a familial hypercholesterolaemia cohort provides a model for using targeted next generation DNA sequencing in the clinical setting. Pathology. 2014;46:60–68. doi: 10.1097/PAT.0000000000000026. [DOI] [PubMed] [Google Scholar]
- 26.Jiang L, Sun LY, Dai YF, Yang SW, Zhang F, Wang LY. The distribution and characteristics of LDL receptor mutations in China: A systematic review. Sci Rep. 2015;5:17272. doi: 10.1038/srep17272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri MA, Inoue T, et al. Genotypic and phenotypic features in homozygous familial hypercholesterolemia caused by proprotein convertase subtilisin/kexin type 9 (PCSK9) gain-of-function mutation. Atherosclerosis. 2014;236:54–61. doi: 10.1016/j.atherosclerosis.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Miyake Y, Yamamura T, Sakai N, Miyata T, Kokubo Y, Yamamoto A. Update of Japanese common LDLR gene mutations and their phenotypes: Mild type mutation L547V might predominate in the Japanese population. Atherosclerosis. 2009;203:153–160. doi: 10.1016/j.atherosclerosis.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Yao RE, Wang J, Geng J, Zheng Z, Yu T, Yu Y, et al. Identification of LDLR mutations in two Chinese pedigrees with familial hypercholesterolemia. J Pediatr Endocrinol Metab. 2012;25:769–773. doi: 10.1515/jpem-2012-0024. [DOI] [PubMed] [Google Scholar]
- 30.Yu W, Nohara A, Higashikata T, Lu H, Inazu A, Mabuchi H. Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis. 2002;165:335–342. doi: 10.1016/s0021-9150(02)00249-6. [DOI] [PubMed] [Google Scholar]
- 31.Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490a. doi: 10.1093/eurheartj/eht273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med. 2015;17:444–451. doi: 10.1038/gim.2014.122. [DOI] [PubMed] [Google Scholar]