

Abstract
The glucagon-like peptide-1 receptor (GLP-1R) is a class B G protein-coupled receptor that is a major therapeutic target for the treatment of type 2 diabetes. Activation of this receptor promotes insulin secretion and blood glucose regulation. The GLP-1R can initiate signaling through several intracellular pathways upon activation by GLP-1. GLP-1R ligands that preferentially stimulate subsets among the natural signaling pathways (“biased agonists”) could be useful as tools for elucidating the consequences of specific pathways and might engender therapeutic agents with tailored effects. Using HEK-293 cells recombinantly expressing human GLP-1R, we have previously reported that backbone modification of GLP-1, via replacement of selected α-amino acid residues with β-amino acid residues, generates GLP-1 analogues with distinctive preferences for promoting G protein activation versus β-arrestin recruitment. Here, we have explored the influence of cell background across these two parameters and expanded our analysis to include affinity and other key signaling pathways (intracellular calcium mobilization and ERK phosphorylation) using recombinant human GLP-1R expressed in a CHO cell background, which has been used extensively to demonstrate biased agonism of GLP-1R ligands. The new data indicate that α/β-peptide analogues of GLP-1 exhibit a range of distinct bias profiles relative to GLP-1 and that broad assessment of signaling endpoints is required to reveal the spectrum of behavior of modified peptides. These results support the view that backbone modification via α→β amino acid replacement can enable rapid discovery of peptide hormone analogues that display substantial signal bias at a cognate GPCR.
Graphical abstract
1. Introduction
Type 2 diabetes mellitus is a chronic metabolic disorder characterized by insulin resistance, decreased insulin production, and the gradual failure of pancreatic β cells.1 These features result in consistently high glucose levels in patients,2 a condition that can lead to severe complications and premature death.1 Current diabetes treatments include insulin-sensitizing agents,3 exogenous insulin,4 and, more recently, agonists of the glucagon-like peptide 1 receptor (GLP-1R).5 This receptor has garnered interest because of its role not only in regulating blood glucose levels, but also in promoting other cellular and physiological outcomes that are impaired in diabetic patients; GLP-1R agonists increase satiety, decrease gastric emptying and enhance β cell health.6 The most potent native agonists of the GLP-1R are two closely related forms of the glucagon-like peptide-1, which are designated GLP-1(7–36)NH2 and GLP-1(7–37). Both are generated via processing of a longer precursor. These two peptides are referred to collectively as “GLP-1” below.
Binding of GLP-1 to the extracellular surface of the GLP-1R promotes recruitment of several G proteins, including Gαs, Gαq, Gαi and Gαo,7–9 as well as β-arrestin 1 and β-arrestin 2, to the cytoplasmic surface of the receptor.9, 10 While Gs stimulation is principally linked to activation of adenylate cyclase and cAMP formation, the canonical driver of GLP-1-stimulated insulin secretion,6 Gs, Gq and Gi/o proteins can each lead to mobilization of intracellular calcium and/or ERK1/2 phosphorylation, in a ligand- and cell-type-specific manner.7–9 β-arrestins can modulate cell proliferation and apoptosis, at least in part through activation of MAPKs such as ERK1/2,11, 12 while also playing a role in β-cell-mediated insulin secretion.11
The pleiotropy of signaling initiated by GPCRs allows for the potential of individual ligands of a specific receptor to generate distinct profiles of response, a phenomenon termed biased agonism.13–15 At a receptor level, bias is engendered by unique interactions between ligands and the receptor that, in turn, can stabilize distinct ensembles of conformations that promote differential engagement with effector proteins (e.g., a G protein or a β-arrestin).16, 17 Biased agonists have received substantial attention for their potential as tools for elucidating GPCR signaling mechanisms, and as therapeutic candidates that might exert focused physiological effects by minimizing activation of pathways other than those that offer therapeutic benefit.15, 18, 19
Both peptidic and non-peptidic ligands of the GLP-1R can exhibit biased agonism.20–24 For example, oxyntomodulin, a natural ligand for the GLP-1R, and the clinically approved agonist exendin-4 exhibit bias in canonical signaling pathways and for arrestin recruitment, relative to GLP-1 in recombinant expression systems9, 25 and in insulinoma cells that natively express the GLP-1R.9 Moreover, an N-terminally modified form of exendin-4, termed exendin P5, that exhibited bias away from arrestin recruitment (i.e., G protein-biased relative to exendin-4), had a distinct profile of response in rodent models of type 2 diabetes, providing evidence that biased agonists of the GLP-1R may provide novel opportunities for therapeutic intervention.23
Recently, we have begun to explore a non-traditional approach to generate new GLP-1 analogues, involving replacement of selected α-amino acid residues with β-amino acid residues (Figure 1A).24, 26 This backbone-modification strategy has produced “α/β-peptides” that function as β-arrestin-biased GLP-1R agonists in the context of HEK-293 cells recombinantly expressing the human GLP-1R.24 Backbone modification has received relatively little attention as an approach to the design of peptide hormone analogues, but holds significant promise for generation of novel peptides.22, 24, 26–33
Figure 1.
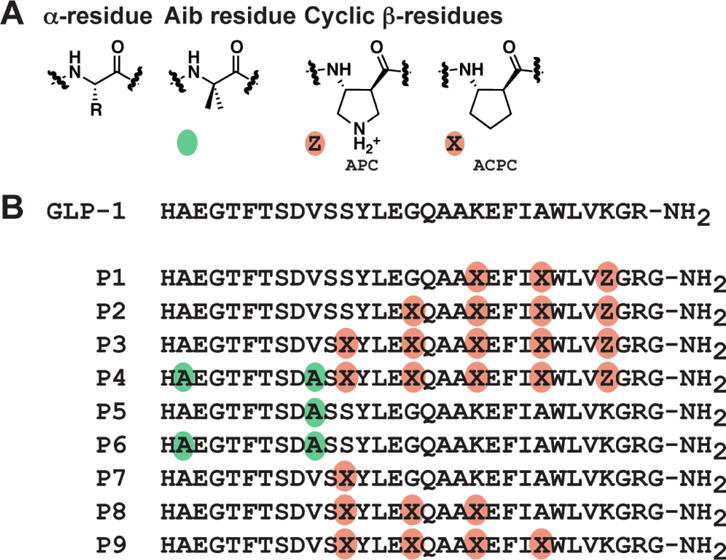
A. Amino acids used in this study. Colored circles indicate non-natural substitutions: green circles represent the non-proteinogenic α-residue Aib, and orange circles represent ring-constrained β-residues (X = ACPC, Z = APC). B. GLP-1(7–36)NH2 and α/β-peptide analogues 1 - 9 (based on GLP-1(7–37)NH2). Each peptide has a free N-terminus and a primary amide at the C-terminus.
Properly designed α/β-peptides can adopt a conformation that closely mimics the α-helix.34 α-Helix-mimetic α/β-peptides can bind tightly to specific target proteins.35 One advantage of α/β-peptides over their α-peptide counterparts is resistance to degradation by proteases;36, 37 proteolysis can limit the in vivo efficacy of α-peptides. The C-terminal portion of GLP-1 forms an α-helix in the GLP-1R-bound state,38 and this structural insight encouraged us to examine α/β-peptide analogues of GLP-1.26 Among previously described GLP-1 analogues P1 - P9 (Figure 1), the most highly substituted is α/β-peptide P4, which contains five α→β substitutions in the C-terminal region and two Aib (α-aminoisobutyric acid) substitutions in the N-terminal region. The Aib replacements protect the N-terminus from degradation by dipeptidyl peptidase 4 (DPP4) and neprilysin. Peptides P1 - P3 and P5 - P9 each contain a subset of the seven substitutions in P4. The α→β substitutions in P4 occur in a repeating αααβ pattern (β residue at every fourth position), with the specific β residue sites based on positions at which GLP-1 tolerates side chain incorporation into a lactam bridge.39, 40 Previously, we found that P1 - P9 could activate the GLP-1R to stimulate cAMP production24, 26 and to recruit β-arrestins, and a subset of these α/β-peptides displayed biased signaling relative to GLP-1.24
The ability to detect bias and indeed the observed direction of bias are dependent upon the breadth of endpoints studied and the cellular system used to explore this behavior. While the proximal driver for biased agonism may be at the level of receptor conformation, the expression of this bias (the observed bias) is critically dependent upon the expression, quantity and localization of effector and regulatory proteins within each cellular context.
The studies described below provide a new and deeper understanding of the signaling properties of P1 - P9 at the GLP-1R by analyzing these peptides in a different cellular context (recombinantly expressed human GLP-1R in CHO cells, in contrast to the HEK293 cells used in previous studies), measuring peptide affinities, and broadening the range of signaling endpoints to include ERK1/2 phosphorylation and intracellular calcium mobilization. These latter endpoints are both relevant to the physiological signaling of the GLP-1R and have been characterized in this cell background in response to other biased agonists of the GLP-1R. Thus, the new data allow direct comparison of biased profiles of the α/β-peptides with bias profiles of previously studied peptides.
2. Materials and Methods
2.1 Materials
Dulbecco’s modified Eagle’s medium (DMEM), hygromycin-B, and Fluo-4 acetoxymethyl ester were purchased from Invitrogen (Carlsbad, CA, USA). Fetal bovine serum (FBS) was purchased from Thermo Fisher Scientific (Melbourne, Victoria, Australia). AlphaScreen™ reagents, and LANCE HTRF cAMP kit were purchased from PerkinElmer Life Sciences (Waltham, MA, USA). SureFire™ ERK1/2 reagents were generously supplied by TGR Biosciences (Adelaide, South Australia, Australia). GLP-1 was purchased from Mimotopes (Victoria, Australia).
All other reagents were purchased from Sigma (St. Louis, MO, USA) or BDH Merck (Melbourne, Vic, Australia) and were of an analytical grade.
2.2 Peptide Synthesis
Peptides were synthesized in house as previously described.24
2.3 Transfections and Cell Culture
Wildtype GLP-1R was isogenically integrated into FlpIn-Chinese hamster ovary (FlpInCHO) cells (Invitrogen), and selection of receptor-expressing cells was accomplished by treatment with 600 μg/mL hygromycin-B as previously reported.41 Transfected and parental FlpInCHO cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 5 % (v/v) FBS, 600 μg/mL hygromycin-B and incubated in a humidified environment at 37 °C in 5 % CO2. FlpInCHO cells stably expressing the GLP-1R were used at passages 18–32. FlpInCHO cell lines stably expressing GLP-1 receptor-Rluc8 and β-arrestin-1-Venus were used at passages 16–35. FlpInCHO cell lines stably expressing GLP-1 receptor-Rluc8 and β-arrestin-2-Venus were used at passages 15–33.
2.4 Radioligand Binding Assays
GLP-1R FlpInCHO were seeded at a density of 3 × 104 cells/well into 96-well culture plates and incubated overnight at 37°C in 5% CO2, and radioligand binding carried out as previously described using 125I-GLP-1 as the radioligand.42 Briefly, binding assays were performed on whole cells incubated overnight at 4°C with 0.05 nM 125I-GLP-1 tracer and increasing concentrations of unlabeled peptide. Cells were washed, solubilized in 0.1 M NaOH and radioactivity determined by γ-counting. For analysis, data were normalized to the specific binding for each individual experiment.
2.5 cAMP Accumulation
GLP-1R expressing FlpInCHO cells were seeded at a density of 3 × 104 cells/well into 96-well culture plates and incubated overnight at 37 °C in 5% CO2. Growth media was replaced with stimulation buffer (phenol-free DMEM containing 0.1 % (w/v) BSA, 5 mM HEPES and 0.5 mM 3-isobutyl-1-methylxanthine) and incubated for 30 min at 37 °C in 5 % CO2. Cells were stimulated with increasing concentrations of peptide ligand and incubated for 30 min at 37 °C in 5 % CO2. The reaction was terminated by rapid removal of the ligand-containing buffer and addition of 50 μL of ice-cold 100 % ethanol. After ethanol evaporation, 75 μL of lysis buffer (0.1 % (w/v) BSA, 0.3 % (v/v) Tween 20, and 5 mM HEPES, pH 7.4) was added, and 5 μL of lysate was transferred to a 384-well OptiPlate (PerkinElmer Life and Analytical Sciences).
The amount of cAMP present in each sample was determined using the Lance cAMP kit (PerkinElmer) with modifications to the manufacturer’s instructions. Briefly, 5 μL of the antibody solution (100-fold dilution of Alexa Fluor 647-anti cAMP antibody solution in detection buffer) was transferred into each well containing lysates/cAMP standard in reduced lighting conditions and incubated for 30 min at room temp. 10 μl of detection mix (1:1:124 of solution 1 (2.5 % v/v Eu-W8044 labeled streptavidin (Eu-SA)), solution 2 (8.75 % v/v Biotin- cAMP) and detection buffer respectively) was added to each well in reduced lighting conditions, and the plate was incubated at room temperature for 1 h.
HTRF (Homogeneous Time Resolved Fluorescence) for each sample was detected using an EnVision™ plate reader with excitation at 320 nm and emission at 615 nm. cAMP was determined for all samples via conversion to concentration of cAMP using a cAMP standard curve that was detected in parallel. Data were normalized to the maximal response elicited by GLP-1. 100 μM Forskolin was used as a positive control.
2.5 ERK1/2 Phosphorylation
GLP-1R expressing FlpInCHO cells were seeded at a density of 3 × 104 cells/well in DMEM with 5 % FBS into 96-well culture plates. The following day, the media was aspirated and the cells were washed twice with 100 μL PBS. 90 μL of serum free DMEM was then added and the cells were incubated overnight at 37 °C, 5 % CO2. Ligand-mediated pERK1/2 was determined using the AlphaScreen™ ERK1/2 SureFire™ protocol as previously described.43 Prior to generation of concentration response curves, initial pERK1/2 timecourse experiments were performed over 1 h using high concentrations of peptide ligand (1 μM) to determine the time at which pERK1/2 was maximal after stimulation by agonists. Concentration response curves were then generated at this peak time point for each ligand. The kinetics of pERK1/2 response were similar for all ligands, peaking at 6 min. Data were normalized to the maximal response elicited by GLP-1. 10% FBS, determined at 6 min was used as a positive control.
2.7 Intracellular Calcium Mobilization
GLP-1R expressing FlpInCHO cells stably were seeded in clear 96-well plates, at a density of 3 × 104 cells/well, in growth media and allowed to adhere overnight. On the day of assay, cells were washed twice with 100 μL modified Hanks buffered saline solution (HBSS containing; 150 mM NaCl, 2.6 mM KCl, 1.18 mM MgCl2, 10 mM D-glucose, 10 mM HEPES, 2.2 mM CaCl2, 2 mM probenecid, 0.5% (w/v) bovine serum albumin) and, in light diminished conditions, incubated for 1 h at 37°C with the cell permeant Ca2+ fluorophore, Fluo-4AM (final concentration of 10 μM). After incubation, the assay plates were transferred to a Molecular Devices FlexStation (Molecular Devices, Palo Alto, CA, USA), and robotic addition of ligands was performed. Fluorescence was determined immediately after peptide addition, with an excitation wavelength set to 485 nm and an emission wavelength set to 525 nm, and readings were taken every 1.36 sec for 120 sec. Peak magnitude was calculated using five-point smoothing, followed by correction against basal fluorescence. The peak value was used to create concentration-response curves. Data were normalized to the maximal response elicited by GLP-1. 100 μM ATP was used as a positive control.
2.8 β-Arrestin Recruitment Assays
FlpInCHO cell lines stably expressing GLP-1 receptor-Rluc8 and either β-arrestin1- or β-arrestin2-Venus were generated using gateway technology. These cell lines were characterized and described previously.44 Cells were seeded in 96-well white culture plates at a density of 4 × 104 cells/well and cultured for 24 h. Cells were rinsed once with HBSS to remove traces of phenol red and incubated in fresh HBSS for a further 15 min. The Rluc substrate coelenterazine-h was added to reach a final concentration of 5 μM. After a 10 min incubation, the corresponding agonist was added, and bioluminescence resonance energy transfer (BRET) readings were collected using a LumiSTAR Omega instrument that allows sequential integration of signals detected in the 465–505 and 515–555 nm windows using filters with the appropriate band pass. The BRET signal was calculated by subtracting the ratio of 515–555 nm emission over 465–505 nm emission for a vehicle treated cell sample from the same ratio for the ligand treated cell sample. In this calculation, the vehicle treated cell sample represents background and results are expressed as ligand-induced BRET. This eliminates the requirement for measuring a donor only control sample. Initial time course experiments were performed over 20 min to determine the time at which β-arrestin1 and β-arrestin 2 recruitment was maximal for each ligand. Subsequent concentration response data were collected at this peak time. Data were normalized to the maximal response elicited by GLP-1.
2.9 Data Analysis
All data were analyzed using Prism 6 (GraphPad Software Inc., San Diego, CA, USA). For all analyses the data are unweighted, and each y value (mean of replicates for each individual experiment) is considered an individual point. To calculate IC50, EC50 and Emax values, concentration response signaling data were analysed as previously described41 using a three-parameter logistic equation.
Signaling bias was also quantified as previously described by analysis of concentration-response curves with nonlinear regression using an operational model of agonism modified to directly estimate the ratio of τ/KA.24, 25, 41
| Eq 1 |
where Em represents the maximal stimulation of the system, KA is the agonist-receptor dissociation constant, in molar concentration, [A] is the molar concentration of ligand and τ is the operational measure of efficacy in the system, which incorporates signaling efficacy and receptor density. All estimated τ/KA ratios included propagation of error for both τ and KA. Changes in τ/KA ratios with respect to GLP-1 for each novel peptide was used to quantitate bias between signalling pathways. Accordingly, bias factors included propagation of error from τ/KA ratios of each pathway.
2.10 Statistics
Changes in peptide affinity, potency, efficacy or bias of each peptide in comparison to the GLP-1 control were statistically analysed with one-way analysis of variance and Dunnett’s post test, and significance was accepted at p < 0.05.
3. Results
3.1 GLP-1R agonist affinities
We assessed the affinities of GLP-1 and peptides P1 - P9 for the GLP-1R expressed in FlpIn CHO cells via competition with a radiolabeled antagonist, 125I-exendin(9–39) (Table 1, Figure 2A). The resulting IC50 values represent an averaged response arising from multiple receptor conformations that are present because individual GLP-1R molecules are presumably engaged by different intracellular partners; in this context the predominant signaling effector complex will have the most impact.
Table 1.
Affinity and activity data for GLP-1 and α- and α/β-peptides P1 – P9 in GLP-1R binding, cAMP accumulation, Ca2+ mobilization, ERK1/2 phosphorylation, β-Arrestin-1 recruitment, and β-Arrestin-2 recruitment.
| Affinity | cAMP | β-Arrestin-1 | β-Arrestin-2 | iCa2+ | pERK1/2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| pIC50 (M) | pEC50 (M) | Emax | pEC50 (M) | Emax | pEC50 (M) | Emax | pEC50 (M) | Emax | pEC50 (M) | Emax | |
| GLP-1 | 8.1 ± 0.1 | 10.0 ± 0.1 | 100 | 7.4 ± 0.1 | 100 | 7.6 ± 0.1 | 100 | 7.5 ± 0.1 | 100 | 8.9 ± 0.1 | 100 |
|
| |||||||||||
| P1 | 7.0* ± 0.1 | 8.8* ± 0.1 | 130* ± 6 | 6.9 ± 0.2 | 85 ± 7 | 6.9 ± 0.1 | 108 ± 5 | 7.2 ± 0.3 | 89 ± 8 | 8.3 ± 0.2 | 112 ± 8 |
| P2 | 7.0* ± 0.1 | 8.2* ± 0.1 | 97 ± 6 | 6.0* ± 0.7 | 30* ± 20 | 5.9* ± 0.6 | 40* ± 20 | 6.4* ± 0.2 | 80 ± 10 | 7.9* ± 0.2 | 107 ± 9 |
| P3 | 5.9* ± 0.1 | 8.3* ± 0.2 | 89 ± 6 | 6.8 ± 0.3 | 50* ± 7 | 6.4* ± 0.3 | 61* ± 9 | 5.3* ± 0.3 | 100 ± 30 | 8.1 ± 0.2 | 92 ± 8 |
| P4 | 6.0* ± 0.1 | 7.6* ± 0.2 | 97 ± 7 | N.D. | N.D. | N.D. | N.D. | 5.2* ± 0.5 | 90 ± 40 | 8.3 ± 0.2 | 81 ± 8 |
| P5 | 7.6* ± 0.1 | 10.0 ± 0.1 | 102 ± 5 | 8.0 ± 0.1 | 87 ± 4 | 7.1 ± 0.1 | 102 ± 4 | 7.3 ± 0.2 | 82 ± 6 | 9.2 ± 0.3 | 89 ± 6 |
| P6 | 7.6* ± 0.1 | 9.9± 0.1 | 89 ± 5 | 7.9 ± 0.2 | 72 ± 4 | 7.4 ± 0.1 | 82 ± 5 | 7.4 ± 0.2 | 78 ± 5 | 8.9± 0.2 | 80 ± 7 |
| P7 | 6.9* ± 0.1 | 9.6 ± 0.1 | 89 ± 5 | 7.4 ± 0.4 | 36* ± 6 | 7.0 ± 0.2 | 45* ± 4 | 7.0 ± 0.2 | 60 ± 5 | 8.9 ± 0.2 | 89 ± 7 |
| P8 | 6.7* ± 0.1 | 8.5* ± 0.2 | 83 ± 5 | 7.3 ± 0.6 | 27* ± 9 | N.D. | N.D. | 5.7* ± 0.4 | 59* ± 7 | 8.2 ± 0.3 | 80 ± 8 |
| P9 | 7.0* ± 0.1 | 7.7* ± 0.1 | 107 ± 5 | 6.9 ± 0.5 | 23* ± 6 | N.D. | N.D. | N.D. | N.D. | 8.0* ± 0.3 | 73 ± 9 |
All experiments were performed in FlpIn CHO cells stably expressing the human GLP-1R. Data are normalized to the maximum response elicited by GLP-1 in each assay, and analyzed using a three-parameter logistic equation. pEC50 values are the logarithm of the concentration of agonist that produces half the maximal response. Emax represents the maximal response normalized to that of GLP-1. Values are the mean ± S.E.M. of three to four individual experiments, conducted in duplicate. Maximum response values for incomplete curves are the predicted maximum derived from curve fitting.
statistically significant difference from GLP-1 using one way analysis of variance followed by Dunnett’s test (P<0.05).
Figure 2.
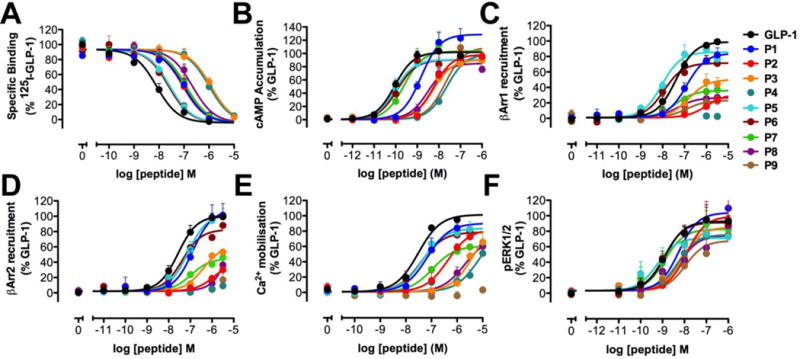
Binding and signaling profiles of GLP-1 and α- and α/β-peptides P1 – P9 in FlpInCHO cells stably expressing the human GLP-1R. Concentration-response curves for (A) GLP-1R binding, (B) cAMP accumulation, (C) Ca2+ mobilization, (D) ERK1/2 phosphorylation, (E) β-Arrestin-1 recruitment, and (F) β-Arrestin-2 recruitment. Data are normalized to the maximum response elicited by GLP-1 in each assay, and analyzed using a three-parameter logistic equation. Values are the mean + S.E.M. of three to four individual experiments, conducted in duplicate.
Incorporation of one (P7), three (P1, P8) or four (P2, P9) β amino acid residues, regardless of position, led to a ~10-fold reduction in affinity, relative to GLP-1, for the GLP-1R. Addition of a fifth β residue (P3, P4) further reduced affinity by 10-fold relative to other α/β-peptides in this set. α/β-Peptide P4 contains two Aib substitutions, which seem to have little impact on affinity for the GLP-1R, because P3 and P4 are indistinguishable. This conclusion is supported by the observation that α-peptides P5 and P6, which contain one or both of the Aib substitutions in P4, display only slightly reduced affinity for the GLP-1R relative to GLP-1.
3.2 Evaluation of cAMP production stimulated by P1 - P9
We measured cAMP accumulation in response to P1 - P9 in FlpIn CHO cells stably expressing the human GLP-1R (Table 1, Figure 2B). We observed a modest decline in potency arising from α→β replacement (P1→P2, P3), and a further decline upon Aib replacements (P3→P4). Peptides P5 - P7 were similar in potency to GLP-1, while P8 and P9 displayed substantially reduced potency relative to GLP-1. We previously assessed the activities of P1 - P9 in HEK293 cells transiently transfected with the human GLP-1R, using a kinetic GloSensor assay.24, 45 The cAMP potencies for P1 - P9 in the current study using an AlphaScreen assay are similar to those measured in HEK cells using the GloSensor assay.24 The similarity between these two assays, involving different cell types, provides confidence that the trends are robust.
3.3 Evaluation of β-arrestin recruitment stimulated by P1 - P9
We assessed β-arrestin-1 and β-arrestin-2 recruitment to the GLP-1R for P1 - P9 using β-arrestin-1 and β-arrestin-2 BRET assays in FlpInCHO cells stably expressing GLP-1R-Rluc8 and either β-arrestin-1 or β-arrestin-2-Venus (Table 1, Figure 2C,D). Among α/β-peptides P1 - P4, we observed substantial declines in the recruitment of β-arrestins-1 and -2 upon introduction of β residues, relative to GLP-1, with little or no β-arrestin recruitment by the maximally modified P4. α-Peptides P5 and P6 were similar to GLP-1 in terms of recruiting both β-arrestins-1 and -2 to the GLP-1R. α/β-Peptides P7, P8 and P9 exhibited substantial depressions in the maximum level of β-arrestin-1 and -2 recruited by the GLP-1R relative to GLP-1. The trends in β-arrestin recruitment are generally similar between the current set of assays and those reported previously,24 and the inter-assay differences in peptide behavior are likely due to differences in cellular background (the original BRET assays involved transfected HEK293FT cells, while the new BRET assay were conducted in transfected FlpIn CHO cells) and differential expression of regulatory proteins between the two experiments. The previous BRET assays were conducted with cells that had been co-transfected with GRK5, which enhances the affinity of β-arrestins for GLP-1R by promoting receptor phosphorylation.10, 46 In contrast, GRK5 was not employed in the current BRET assays; thus, coupling between the receptor and each β-arrestin should be weakened in the new assays relative to the original assays. A second difference is specific to the β-arrestin-2 recruitment assay. The original BRET assay employed a mutated β-arrestin-2 plasmid (R393E, R395E), which is expected to enhance the BRET signal by preventing clathrin binding and subsequent receptor internalization.10, 46 In contrast, native β-arrestin-2 was used for the current assay.
3.4 Intracellular calcium mobilization stimulated by P1 - P9
To further explore how the different pathways activated by GLP-1 are affected by α→β replacements, we measured the abilities of P1 - P9 to promote intracellular calcium mobilization, which reports on Gαq and Gαs activation,9, 47–49 in FlpIn CHO cells stably expressing the human GLP-1R (Table 1, Figure 2E). Overall, α→β replacements led to a reduction in calcium mobilization. Incorporation of three β-amino acid residues (P1) into the C-terminal region of GLP-1 had the smallest impact on activity, with the decrease in activity becoming more pronounced for analogues containing additional α→β replacements extending toward the N-terminus of GLP-1 (P2 - P4). Incorporation of a single β residue at position 18 (7) led to a slight decrease in activity; further α→β substitutions in the central region of GLP-1, to generate P9, essentially abolished calcium mobilization. Neither of the two Aib replacements (P5 and P6) had a substantial effect on calcium mobilization in terms of potency or maximum response relative to GLP-1.
3.5 Stimulation of ERK1/2 phosphorylation stimulated by P1 - P9
As a complement to characterizing the activity of analogues 1–9 in activating various pathways directly mediated by interaction between the GLP-1R and intracellular effector proteins (i.e. G proteins Gαs and Gαq, β-arrestin-1 or β-arrestin-2), we assessed the activity of P1 - P9 in promoting ERK1/2 phosphorylation (Table 1, Figure 2F). GLP-1-mediated ERK1/2 phosphorylation is dependent on both G protein and β-arrestin activity,9, 11, 12, 49 which led us to explore how α→β replacement would affect signaling in this composite pathway, particularly for the β-arrestin-biased α/β-peptides P3, P8 and P9. Our data indicate that ERK1/2 phosphorylation was less strongly affected by α→β replacements than was cAMP production, β-arrestin recruitment or calcium mobilization.
3.6 Stimulus bias induced by P1 - P9
To determine whether peptides among P1 - P9 display signaling bias relative to GLP-1 in the expanded set of signaling pathways characterized, and to compare any bias between cAMP accumulation and β-arrestin recruitment in the CHO cell background to the β-arrestin bias we observed for α/β-peptides P3, P8 and P9 in the HEK293 cell, we analyzed the efficacy of each analogue in assays for cAMP accumulation, calcium mobilization, ERK1/2 phosphorylation, β-arrestin-1 recruitment or β-arrestin-2 recruitment using the operational model of agonism.50, 51 Transduction coefficients (log(τ/KA)) for each analogue were extracted from concentration-response curves and compared with transduction coefficients for GLP-1 in each effector pathway. These comparisons allowed us to calculate a bias factor (Δlog(τ/KA)) for each peptide in terms of calcium mobilization, ERK1/2 phosphorylation, β-arrestin-1 recruitment or β-arrestin-2 recruitment relative to cAMP accumulation (Figure 3, Table 2). We also determined bias factors for each peptide in terms of β-arrestin-1 recruitment or β-arrestin-2 recruitment relative to either calcium mobilization or ERK1/2 phosphorylation (Figure 3, Table 2).
Figure 3.
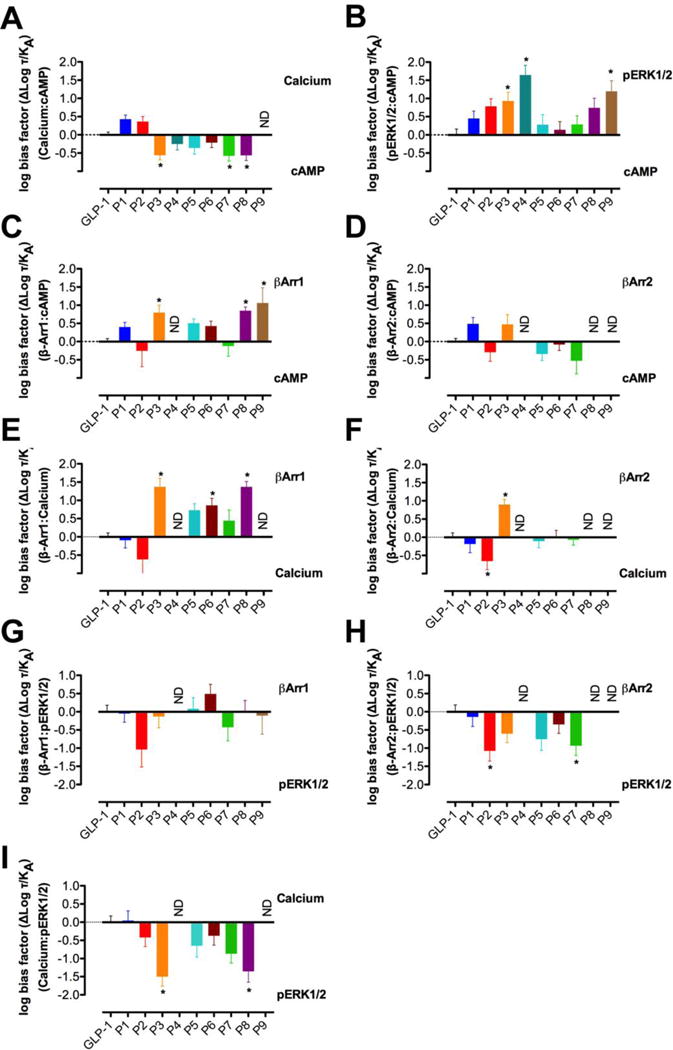
Bias factors for α- and α/β-peptides P1 – P9 relative to GLP-1 in Ca2+ mobilization relative to cAMP accumulation (A), ERK1/2 phosphorylation relative to cAMP accumulation (B), β-Arrestin-1 recruitment relative to cAMP accumulation (C), β-Arrestin-2 recruitment relative to cAMP accumulation (D), β-Arrestin-1 recruitment relative to Ca2+ mobilization (E), β-Arrestin-2 recruitment relative to Ca2+ mobilization (F), β-Arrestin-1 recruitment relative to ERK1/2 phosphorylation (G), and β-Arrestin-2 recruitment relative to ERK1/2 phosphorylation (H). Changes in log (τ/KA) were calculated to provide a measure of the degree of stimulus bias exhibited between different signaling pathways relative to that of the reference agonist GLP-1. Values are the mean ± SEM of three to four individual experiments, conducted in duplicate. * statistically significant difference from GLP-1 using one-way analysis of variance followed by Dunnett’s test.
Table 2.
Stimulus bias exhibited by α- and α/β-peptides P1 – P9 relative to the reference agonist GLP-1.
| Δlog(τ/KA)
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Ca2+ vs cAMP | pERK1/2 vs cAMP | βArr1 vs cAMP | βArr2 vs cAMP | βArr1 vs Ca2+ | βArr2 vs Ca2+ | βArr1 vs pERK1/2 | βArr2 vs pERK1/2 | |
| GLP-1 | 0.0 ± 0.1 | 0.0 ± 0.2 | 0.0 ± 0.1 | 0.0 ± 0.1 | 0.0 ± 0.1 | 0.0 ± 0.1 | 0.0 ± 0.2 | 0.0 ± 0.2 |
| P1 | 0.4 ± 0.1 | 0.5 ± 0.2 | 0.4 ± 0.1 | 0.5 ± 0.2 | −0.1 ± 0.2 | −0.2 ± 0.2 | 0.0 ± 0.2 | −0.1 ± 0.3 |
| P2 | 0.4 ± 0.1 | 0.8 ± 0.2 | −0.3 ± 0.4 | −0.3 ± 0.3 | −0.7 ± 0.5 | −0.7* ± 0.2 | −1.0 ± 0.5 | −1.0* ± 0.3 |
| P3 | −0.6* ± 0.1 | 0.9* ± 0.2 | 0.8* ± 0.2 | 0.5 ± 0.3 | 1.4* ± 0.2 | 0.9* ± 0.1 | −0.1 ± 0.3 | −0.6 ± 0.2 |
| P4 | −0.2 ± 0.2 | 1.6* ± 0.3 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| P5 | −0.4 ± 0.2 | 0.3 ± 0.3 | 0.5 ± 0.1 | −0.3 ± 0.2 | 0.7 ± 0.2 | −0.1 ± 0.2 | 0.1 ± 0.3 | −0.8 ± 0.3 |
| P6 | −0.1 ± 0.2 | 0.1 ± 0.2 | 0.4 ± 0.1 | −0.1 ± 0.2 | 0.9* ± 0.2 | 0.0 ± 0.2 | 0.5 ± 0.3 | −0.3 ± 0.2 |
| P7 | −0.6* ± 0.1 | 0.3 ± 0.2 | −0.1 ± 0.3 | −0.5 ± 0.4 | 0.4 ± 0.3 | −0.1 ± 0.1 | −0.4 ± 0.4 | −0.9* ± 0.3 |
| P8 | −0.6* ± 0.1 | 0.7 ± 0.3 | 0.8* ± 0.1 | N.D. | 1.4* ± 0.1 | N.D. | 0.0 ± 0.3 | N.D. |
| P9 | N.D. | 1.2* ± 0.3 | 1.1* ± 0.4 | N.D. | N.D. | N.D. | −0.1 ± 0.5 | N.D. |
Stimulus bias exhibited by P1 – P9 relative to GLP-1in Ca2+ mobilization relative to cAMP accumulation, ERK1/2 phosphorylation relative to cAMP accumulation, β-Arrestin-1 recruitment relative to cAMP accumulation, β-Arrestin-2 recruitment relative to cAMP accumulation, β-Arrestin-1 recruitment relative to Ca2+ mobilization, β-Arrestin-2 recruitment relative to Ca2+ mobilization, β-Arrestin-1 recruitment relative to ERK1/2 phosphorylation, and β-Arrestin-2 recruitment relative to ERK1/2 phosphorylation. Changes in log (τ/KA) were calculated to provide a measure of the degree of stimulus bias exhibited between different signaling pathways relative to that of the reference agonist GLP-1. Values are the mean ± SEM of three to four individual experiments, conducted in duplicate.
statistically significant difference from GLP-1 using one-way analysis of variance followed by Dunnett’s test.
The bias factors summarized in Figure 3 and Table 2 reveal that α/β-peptides P3, P4, P7, P8 and P9 manifest significant bias in at least one pathway. For example, peptides P3, P7 and P8 are weakly biased toward cAMP accumulation relative to calcium mobilization (Figure 3A). Peptides P3, P4 and P9 are biased toward ERK1/2 phosphorylation relative to cAMP accumulation (Figure 3B). Peptides P3, P8 and P9 are biased toward β-arrestin-1 recruitment relative to cAMP production (Figure 3A). None among P1 - P9 displayed bias toward or away from β-arrestin-2 recruitment relative to cAMP accumulation (Figure 3D); however, bias factors could not be calculated for P4, P8 and P9 due to weak β-arrestin-2 responses to these peptides (Figure 2, Table 1). For this reason, it was impossible to robustly compare β-arrestin-2 recruitment with other signaling pathways.
4. Discussion and Conclusions
The characterization of P1 - P9 in the current study was performed in CHO cells, while our initial studies with these analogues were performed in HEK293 cells; thus, these two studies collectively allow one to assess the impact of cellular background on the manifestation of biased agonism.24 Moreover, because the current studies evaluate bias for P1 - P9 in terms of a more diverse set of signaling and regulatory endpoints relative to the previous study, the data reported here allow a more complete understanding of the activity profiles of these GLP-1 analogues, and these data can be used to compare the bias profiles of P1 - P9 to the profiles of known agonists of the GLP-1R.
The bias factors for P1 - P9 in terms of β-arrestin recruitment relative to cAMP production, shown in Figure 4 and Table 2, are reasonably consistent with those of our previous study,24 particularly for β-arrestin-1. Small differences in bias factor are evident for analogues that do not display strong bias, but analogues that display strong bias, such as P8 and P9, are biased toward β-arrestin-1 recruitment over cAMP (either accumulation or production) in both sets of experiments. Analogue P3 is significantly biased in the CHO cell background, and trends towards bias in the HEK cells, although the latter effect is not statistically significant. The consistency in bias factors for β-arrestin-1 recruitment relative to cAMP indicates that, for β-arrestin-1 recruitment, differences in cell background between the two sets of experiments do not significantly impact the identification of an analogue as either biased or not biased, even if changes in cellular background do slightly alter the relative efficacy for individual analogues between experiments. The β-arrestin-1 bias factors calculated for P3, P8 and P9 in CHO cells are consistently larger than those calculated for these peptides in HEK293 cells, suggesting more efficient coupling in the CHO cells, despite the overexpression of GRK5 in the HEK293 cells.24 Thus, the current studies imply that GRKs other than GRK5 are the predominant sources of receptor phosphorylation in the CHO cell background.
Figure 4.
Comparison of the bias factors for α- and α/β-peptides P1 – P9 relative to GLP-1 for β-Arrestin-1 recruitment versus cAMP accumulation between FlpInCHO cells (A) and HEK293 cells24 (B) and for β-Arrestin-2 recruitment versus cAMP accumulation between FlpInCHO cells (C) and HEK293 cells24 (D). Changes in log (τ/KA) were calculated to provide a measure of the degree of stimulus bias exhibited between different signaling pathways relative to that of the reference agonist GLP-1. * statistically significant difference from GLP-1 using one way analysis of variance followed by Dunnett’s test (P<0.05).
In our previous study, P3, P8 and P9 exhibited bias toward β-arrestin-2 recruitment over cAMP production,24 but in the new study activity was too low in the β-arrestin-2 recruitment assays to allow bias factor calculation for these α/β-peptides. The previous system was engineered to enhance β-arrestin-2 coupling through a combination of overexpression of GRK5 and mutation (R393E, R395E) of the arrestin that enhance the BRET signal by preventing clathrin binding and subsequent receptor internalization.10, 46 In the current assay, we examined recruitment of native β-arrestin-2, which was poorly recruited by lower-efficacy peptides. The lack of quantitative signal for these peptides makes interpretation of potential changes to signaling bias between the two cell types problematic. Overall, the pattern of bias changes in the enhanced β-arrestin-2 assay in HEK293 cells, along with the β-arrestin-1 profiles in both CHO and HEK cells, indicate fundamental differences in the properties of the P3, P8 and P9 α/β-peptides relative to the GLP-1 itself. Comparing the bias profiles of P1 - P9 between CHO and HEK293 cells highlights that the utility of using recombinant systems lies in probing bias and in fingerprinting the activity profiles of different agonists, but not in making specific claims about the relevance of observed bias to physiological effects manifested in native cells and whole organisms.
Expanding the diversity of pathways characterized in the current study, relative to the previous report, reveals more extensive bias within P1 - P9 beyond bias toward β-arrestin recruitment over cAMP (Table 2, Figure 3). Among P3, P4, P7, P8 and P9, each α/β-peptide manifests significant bias in at least one pathway. P3, P7 and P8 are all weakly biased toward cAMP accumulation relative to calcium mobilization, though these analogues are only weakly active in both pathways. P3, P4 and P9 are biased toward ERK1/2 phosphorylation over cAMP accumulation; weak but statistically insignificant trends of this type are observed for other peptides, including P2 and P8. Overall, backbone modification has only limited impact on ERK1/2 phosphorylation, leading to bias towards this pathway over those for which substantial changes in response are observed. The pathway that most closely parallels the trend in bias for ERK1/2 phosphorylation is β-arrestin-1 recruitment, toward which P3, P8 and P9 are biased over cAMP accumulation. The ERK1/2 phosphorylation signal in CHO cells is a composite of β-arrestin- and G protein-dependent signaling,9, 11, 12 and the correlation between ERK1/2 phosphorylation bias and β-arrestin-1 bias suggests that the β-arrestin pathway is predominant for P3, P8 and P9 for causing ERK1/2 phosphorylation. However, the proposed β-arrestin pathway dominance may not pertain to all α/β-peptides. Among the ERK1/2 phosphorylation-biased compounds, P4 is the most strongly biased toward ERK1/2 phosphorylation over cAMP production, but P4 caused no measurable signal in the β-arrestin-recruitment assays, despite the robust pERK1/2 response. Understanding the relative bias of P4 for ERK1/2 phosphorylation versus β-arrestin recruitment will require more sensitive assays of β-arrestin recruitment.
Some among P1 - P9 display selective bias toward or away from either β-arrestin-1 recruitment or β-arrestin-2 recruitment when compared with various other pathways. P6, for example, is biased toward β-arrestin-1 recruitment over calcium mobilization, but not does not favor β-arrestin-2 recruitment over calcium mobilization. Moreover, P7 favors ERK1/2 phosphorylation over β-arrestin-2 recruitment, while not favoring ERK1/2 phosphorylation over β-arrestin-1 recruitment or vice versa. Cases in which GLP-1 analogues selectively favor or disfavor either β-arrestin-1 or β-arrestin-2 recruitment suggest the intriguing possibility that these analogues could serve as starting points for more strongly biased GLP-1 agonists that could be used to parse the roles of β-arrestin-1 and β-arrestin-2 activity at the GLP-1R. However, this possibility would need to be carefully assessed in physiological target cells, because the efficiency of recruitment of each β-arrestin is likely to be influenced by the complement of GRKs that are expressed.
One interesting outlier in the comparison of ERK1/2 phosphorylation and β-arrestin-1 and -2 recruitment for our α/β-peptides is P2, which is significantly biased toward ERK1/2 phosphorylation over β-arrestin-2 recruitment (in addition, P2 trends towards bias of ERK1/2 phosphorylation over β-arrestin-1 recruitment). P2 is also biased towards calcium mobilization over β-arrestin-2 recruitment (with a trend in this direction for β-arrestin-1), suggesting that the P2 α/β-peptide may have a novel G protein bias profile.
Several well-studied peptides, including exendin-4 and oxyntomodulin, have been identified as biased agonists of the GLP-1R.9, 25 Both exendin-4 and oxyntomodulin bias the GLP-1R toward β-arrestin-1 recruitment and β-arrestin-2 recruitment over cAMP accumulation in experiments performed in FlpIn CHO cells.9, 25 Oxyntomodulin also biases the GLP-1R toward ERK1/2 phosphorylation over cAMP accumulation, but exendin-4 does not exhibit bias toward ERK1/2 phosphorylation.9, 25 The observation that exendin-4 and oxyntomodulin are both biased toward β-arrestin recruitment but only oxyntomodulin is also biased toward ERK1/2 phosphorylation may be explained by different degrees of contribution from β-arrestin-1 and β-arrestin-2 activity in mediating downstream ERK1/2 phosphorylation.9 The distinct bias profiles for these two peptides indicate different modes of activation of the GLP-1R in response to either oxyntomodulin or exendin-4.
Because the bias factors calculated for P1 - P9 in this work are derived from experiments performed in the same FlpIn CHO cells that were used in the experiments to determine bias for exendin-4 and oxyntomodulin, the bias for P1 - P9 can be compared to that observed for exendin-4 and oxyntomodulin without concerns that either differences in cellular background or in assay format are the cause of distinct bias profiles between discrete agonists. Figure 5 provides a graphical summary of bias effects that allows ready comparison of P1 - P9 (Figure 5A, B) or exendin-49 and oxyntomodulin9 (Figure 5C) with GLP-1 in terms of all five of the GLP-1R signaling outcomes we monitored. Each “web of bias” is constructed to convey bias relative to the cAMP production pathway. P4 is illustrated in both Figure 5A, which highlights ERK1/2 phosphorylation-biased ligands, and 5B as a reference for the α/β peptides.
Figure 5.
Webs of bias for α- and α/β-peptides P1 – P9 (A, B) and known biased agonists exendin-4 and oxyntomodulin9 (C) relative to GLP-1 in FlpInCHO cells stably expressing the human GLP-1R. Circles represent data that are significantly biased. Triangles represent data where no value could be defined. The τ/KA ratio extracted from standard concentration-response data is used to calculate bias factors (ΔΔ(τ/KA) through normalization of the transduction coefficient (τ/KA) to a reference ligand (GLP-1) and reference pathway (cAMP accumulation).
Comparing the bias profiles for oxyntomodulin and exendin-4 to those for P3, P4, P8 and P9, which each display significant bias toward either β-arrestin-1 or ERK1/2 phosphorylation or both over cAMP,9, 24, 25 we can categorize each GLP-1 analogue as being either “oxyntomodulin-like” or “exendin-4-like” in terms of its bias profile. (This categorization is imperfect, because both oxyntomodulin and exendin-4 are also biased toward β-arrestin-2 over cAMP,9, 25 while no β-arrestin-2 bias factors could be calculated for any among P3, P4, P8 or P9.) P3 and P9 are both biased toward β-arrestin-1 and ERK1/2 phosphorylation over cAMP, making them “oxyntomodulin-like” biased agonists of the GLP-1R. P8 is biased toward β-arrestin-1 over cAMP but not toward ERK1/2 phosphorylation over cAMP, and is therefore an “exendin-4-like” biased agonist of the GLP-1R. The differences in bias profiles for P8 compared to P3 and P9 indicate that these sets of analogues differ in how they activate the GLP-1R. P4 is biased toward ERK1/2 phosphorylation over cAMP production, but no bias factor could be determined for P4 in terms of β-arrestin-1 over cAMP; thus, the bias profile of P4 is unique because it differs from the profile of either exendin-4 or oxyntomodulin.
We have previously shown that modifying the backbone of GLP-1 via incorporation of β-amino acid residues can generate agonists that engender significant bias toward β-arrestin-1 and/or β-arrestin-2 recruitment over cAMP production relative to GLP-1 itself.24 Here, we expand the characterization of these biased peptides to include receptor-affinity measurements and additional signaling endpoint measurements. Our new data show that several among the α- and α/β-peptides we characterized are biased toward additional signaling outcomes beyond β-arrestin recruitment, thereby highlighting the importance of monitoring a diverse set of signaling and regulatory endpoints when characterizing novel agonists to identify biased agonists. These new results strengthen the conclusion that α→β residue replacement can alter receptor signaling relative to the parent α-peptide. Thus, α→β residue replacement may prove to be a general method by which receptor selectivity can be engineered into a peptide agonist that activates its cognate receptor to initiate different signaling pathways. The α/β-peptides characterized in this work may have utility as tools to probe the roles of β-arrestin recruitment and ERK1/2 phosphorylation in GLP-1R signaling. Moreover, these α/β-peptides could provide a platform to develop pathway-selective therapeutic agents targeting the GLP-1R.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (NIGMS) (GM056414, to S.H.G.) and the National Health and Medical Research Council of Australia (NHMRC) (project grants [1061044] and [1065410], and NHMRC program grant [1055134] to P.M.S. and D.W.); P.M.S. is a NHMRC Principal Research Fellow. D.W. is a NHMRC Career Development Fellow. M.V.H. was supported in part by a Chemical Biology Interface Training Grant from NIGMS (T32 GM008505). Support for this research was provided by the University of Wisconsin–Madison, Office of the Vice Chancellor for Research and Graduate Education with funding from the Wisconsin Alumni Research Foundation.
Footnotes
Conflict of interest statement
The authors declare the following competing financial interest(s): S.H.G. is an inventor on a patent application covering GLP-1 analogues described here; S.H.G. is a cofounder of Longevity Biotech, Inc., which is pursuing biomedical applications of α/β-peptides.
References
- 1.Zaccardi F, Webb DR, Yates T, Davies MJ. Pathophysiology of type 1 and type 2 diabetes mellitus: a 90-year perspective. Postgrad Med J. 2016;92:63–69. doi: 10.1136/postgradmedj-2015-133281. [DOI] [PubMed] [Google Scholar]
- 2.DeFronzo RA. Pathogensis of Type 2 (non-insulin dependent) diabetes mellitus: a balanced overview. Diabetologia. 1992;35:389–397. doi: 10.1007/BF00401208. [DOI] [PubMed] [Google Scholar]
- 3.Hundal RS, Inzucchi SE. Metformin: new understandings, new uses. Drugs. 2003;63:1879–1894. doi: 10.2165/00003495-200363180-00001. [DOI] [PubMed] [Google Scholar]
- 4.Swinnen SG, Hoekstra JB, DeVries JH. Insulin therapy for type 2 diabetes. Diabetes Care. 2009;32(Suppl. 2):S253–259. doi: 10.2337/dc09-S318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holst JJ, Knop FK, Vilsboll T, Krarup T, Madsbad S. Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care. 2011;34(Suppl 2):S251–257. doi: 10.2337/dc11-s227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baggio LL, Drucker DJ. (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 7.Montrose-Rafizadeh C, Avdonin P, Garant MJ, Rodgers BD, Kole S, Yang H, et al. Pancreatic Glucagon-Like Peptide-1 Receptor Couples to Multiple G Proteins and Activates Mitogen-Activated Protein Kinase Pathways in Chinese Hamster Ovary Cells. Endocrinology. 1999;140:132–1140. doi: 10.1210/endo.140.3.6550. [DOI] [PubMed] [Google Scholar]
- 8.Hallbrink M, Homqvist T, Olsson M, Ostenson CG, Efendic S, Langel U. Different domains in the third intracellular loop of the GLP-1 receptor are responsible for Gas Gai/Gao activation. Bichim Biophys Acta. 2001;1546:79–86. doi: 10.1016/s0167-4838(00)00270-3. [DOI] [PubMed] [Google Scholar]
- 9.Wootten D, Reynolds CA, Smith KJ, Mobarec JC, Koole C, Savage EE, et al. The Extracellular Surface of the GLP-1 Receptor Is a Molecular Trigger for Biased Agonism. Cell. 2016;165:1632–1643. doi: 10.1016/j.cell.2016.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jorgensen R, Kubale V, Vrecl M, Schwartz TW, Elling CE. Oxyntomodulin differentially affects glucagon-like peptide-1 receptor beta-arrestin recruitment and signaling through Galpha(s) J Pharmaocl Exp Ther. 2007;322:148–154. doi: 10.1124/jpet.107.120006. [DOI] [PubMed] [Google Scholar]
- 11.Sonoda N, Imamura T, Yoshizaki T, Babendure JL, Lu JC, Olefsky JM. Beta-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic beta cells. Proc Natl Acad Sci USA. 2008;105:6614–6619. doi: 10.1073/pnas.0710402105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quoyer J, Longuet C, Broca C, Linck N, Costes S, Varin E, et al. GLP-1 mediates antiapoptotic effect by phosphorylating Bad through a beta-arrestin 1-mediated ERK1/2 activation in pancreatic beta-cells. J Biol Chem. 2010;285:1989–2002. doi: 10.1074/jbc.M109.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharmacol Toxicol. 2002;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 16.Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Kenakin T. Ligand-selective receptor conformations revisited: the promise and the problem. Trends Pharmacol Sci. 2003;24:346–354. doi: 10.1016/S0165-6147(03)00167-6. [DOI] [PubMed] [Google Scholar]
- 18.Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol Med. 2011;17:126–139. doi: 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Appleton KM, Luttrell LM. Emergent biological properties of arrestin pathway-selective biased agonism. J Recept Signal Transduct Res. 2013;33:153–161. doi: 10.3109/10799893.2013.769004. [DOI] [PubMed] [Google Scholar]
- 20.Koole C, Wootten D, Simms J, Valant C, Sridhar R, Woodman OL, et al. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol Pharmacol. 2010;78:456–465. doi: 10.1124/mol.110.065664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wootten D, Savage EE, Willard FS, Bueno AB, Sloop KW, Christopoulos A, et al. Differential activation and modulation of the glucagon-like peptide-1 receptor by small molecule ligands. Mol Pharmacol. 2013;83:822–834. doi: 10.1124/mol.112.084525. [DOI] [PubMed] [Google Scholar]
- 22.Broichhagen J, Podewin T, Meyer-Berg H, von Ohlen Y, Johnston NR, Jones BJ, et al. Optical Control of Insulin Secretion Using an Incretin Switch. Angew Chem Intl Ed Engl. 2015;54:15565–15569. doi: 10.1002/anie.201506384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Sturchler E, Zhu J, Nieto A, Cistrone PA, Xie J, et al. Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects. Nat Commun. 2015;6:8918. doi: 10.1038/ncomms9918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hager MV, Johnson LM, Wootten D, Sexton PM, Gellman SH. beta-Arrestin-Biased Agonists of the GLP-1 Receptor from beta-Amino Acid Residue Incorporation into GLP-1 Analogues. J Am Chem Soc. 2016;138:14970–14979. doi: 10.1021/jacs.6b08323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wootten D, Simms J, Miller LJ, Christopoulos A, Sexton PM. Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc Natl Acad Sci USA. 2013;110:5211–5216. doi: 10.1073/pnas.1221585110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson LM, Barrick S, Hager MV, McFedries A, Homan EA, Rabaglia ME, et al. A potent alpha/beta-peptide analogue of GLP-1 with prolonged action in vivo. J Am Chem Soc. 2014;136:12848–12851. doi: 10.1021/ja507168t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denton EV, Craig CJ, Pongratz RL, Appelbaum JS, Doerner AE, Narayanan A, et al. A beta‐Peptide Agonist of the GLP‐1 Receptor, a Class B GPCR. Org Lett. 2013;15:5318–5321. doi: 10.1021/ol402568j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai X, Niu Y, Zhu J, Yang AQ, Wu YF, Ye XS. A new GLP-1 analogue with prolonged glucose-lowering activity in vivo via backbone-based modification at the N-terminus. Bioorg Med Chem. 2016;24:1163–1170. doi: 10.1016/j.bmc.2016.01.036. [DOI] [PubMed] [Google Scholar]
- 29.Peggion E, Mammi S, Schievano E, Silvestri L, Schiebler L, Bisello A, et al. Structure-Function Studies of Analogues of Parathyroid Hormone (PTH)-1–34 Containing beta-Amino Acid Residues in Positions 11–13. Biochemistry. 2002;41:8162–8175. doi: 10.1021/bi0200155. [DOI] [PubMed] [Google Scholar]
- 30.Schievano E, Mammi S, Carretta E, Fiori N, Corich M, Bisello A, et al. Conformational and Biological Characterization of Human Parathyroid Hormone hPTH(1–34) Analogues Containing beta-Amino Acid Residues in Positions 17–19. Biopolymers. 2003;70:534–547. doi: 10.1002/bip.10508. [DOI] [PubMed] [Google Scholar]
- 31.Cheloha RW, Maeda A, Dean T, Gardella TJ, Gellman SH. Backbone modification of a polypeptide drug alters duration of action in vivo. Nat Biotechnol. 2014;32:653–655. doi: 10.1038/nbt.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olson KE, Kosloski-Bilek LM, Anderson KM, Diggs BJ, Clark BE, Gledhill JM, Jr, et al. Selective VIP Receptor Agonists Facilitate Immune Transformation for Dopaminergic Neuroprotection in MPTP-Intoxicated Mice. J Neurosci. 2015;35:16463–16478. doi: 10.1523/JNEUROSCI.2131-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheloha RW, Watanabe T, Dean T, Gellman SH, Gardella TJ. Backbone Modification of a Parathyroid Hormone Receptor-1 Antagonist/Inverse Agonist, ACS Chem. Biol. 2016;11:2752–2762. doi: 10.1021/acschembio.6b00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, et al. Structural and biological mimicry of protein surface recognition by alpha/beta-peptide foldamers. Proc Natl Acad Sci USA. 2009;106:14751–14756. doi: 10.1073/pnas.0902663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Checco JW, Gellman SH. Targeting recognition surfaces on natural proteins with peptidic foldamers. Curr Opin Struct Biol. 2016;39:96–105. doi: 10.1016/j.sbi.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horne WS, Boersma MD, Windsor MA, Gellman SH. Sequence-based design of alpha/beta-peptide foldamers that mimic BH3 domains. Angew Chem Intl Ed Engl. 2008;47:2853–2856. doi: 10.1002/anie.200705315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steer DS, Lew RA, Perlmutter P, Smith AI, Aguilar M-I. Beta-amino acids: versatile peptidomimetics. Curr Med Chem. 2002;9:811–822. doi: 10.2174/0929867024606759. [DOI] [PubMed] [Google Scholar]
- 38.Underwood CR, Garibay P, Knudsen LB, Hastrup S, Peters GH, Rudolph R, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J Biol Chem. 2010;285:723–730. doi: 10.1074/jbc.M109.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miranda LP, Winters KA, Gegg CV, Patel A, Aral J, Long J, et al. Design and Synthesis of Conformationally Constrained Glucagon-Like Peptide-1 Derivatives with Increased Plasma Stability and Prolonged in Vivo Activity. J Med Chem. 2008;51:2758–2765. doi: 10.1021/jm701522b. [DOI] [PubMed] [Google Scholar]
- 40.Murage EN, Schroeder JC, Beinborn M, Ahn JM. Search for alpha-helical propensity in the receptor-bound conformation of glucagon-like peptide-1. Bioorg Med Chem. 2008;16:10106–10112. doi: 10.1016/j.bmc.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 41.Koole C, Wootten D, Simms J, Miller LJ, Christopoulos A, Sexton PM. Second extracellular loop of human glucagon-like peptide-1 receptor (GLP-1R) has a critical role in GLP-1 peptide binding and receptor activation. J Biol Chem. 2012;287:3642–3658. doi: 10.1074/jbc.M111.309328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koole C, Wootten D, Simms J, Valant C, Miller LJ, Christopoulos A, et al. Polymorphism and ligand dependent changes in human glucagon-like peptide-1 receptor (GLP-1R) function: allosteric rescue of loss of function mutation. Mol Pharmacol. 2011;80:486–497. doi: 10.1124/mol.111.072884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.May LT, Avlani VA, Langmead CJ, Herdon HJ, Wood MD, Sexton PM, et al. Structure-function studies of allosteric agonism at M2 muscarinic acetylcholine receptors. Mol Pharmacol. 2007;72:463–476. doi: 10.1124/mol.107.037630. [DOI] [PubMed] [Google Scholar]
- 44.Savage EE, Wootten D, Christopoulos A, Sexton PM, Furness SG. A simple method to generate stable cell lines for the analysis of transient protein-protein interactions. BioTechniques. 2013;54:217–221. doi: 10.2144/000114013. [DOI] [PubMed] [Google Scholar]
- 45.Binkowski BF, Butler BL, Stecha PF, Eggers CT, Otto P, Zimmerman K, et al. A luminescent biosensor with increased dynamic range for intracellular cAMP. ACS Chem Biol. 2011;6:1193–1197. doi: 10.1021/cb200248h. [DOI] [PubMed] [Google Scholar]
- 46.Jorgensen R, Martini L, Schwartz TW, Elling CE. Characterization of glucagon-like peptide-1 receptor beta-arrestin 2 interaction: a high-affinity receptor phenotype. Mol Endocrinol. 2005;19:812–823. doi: 10.1210/me.2004-0312. [DOI] [PubMed] [Google Scholar]
- 47.Wheeler MB, Lu M, Dillon JS, Leng XH, Chen C, Boyd AE. Functional expression of the rat glucagon-like peptide-1 receptor, and evidence for coupling to both adenylyl cyclase and phospholipase-C. Endocrinology. 1993;133:57–62. doi: 10.1210/endo.133.1.8391428. [DOI] [PubMed] [Google Scholar]
- 48.Holz GG. Epac: a new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes. 2004;53:5–13. doi: 10.2337/diabetes.53.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson A, Kanamarlapudi V. Agonist-induced internalisation of the glucagon-like peptide-1 receptor is mediated by the Galphaq pathway. Biochemical pharmacology. 2015;93:72–84. doi: 10.1016/j.bcp.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 50.Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- 51.Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS chemical neuroscience. 2012;3:193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]