

Abstract
Exposure of blood to a variety of artificial surface induces contact activation, a process that contributes to the host innate response to foreign substances. On the foreign surface, the contact factors factor XII and plasma prekallikrein undergo reciprocal conversion to their fully active protease forms (factor XIIa and α-kallikrein, respectively) by a process supported by the cofactor high molecular weight kininogen. Contact activation can trigger blood coagulation by conversion of factor XI to the protease factor XIa. There is interest in developing therapeutic inhibitors to factor XIa and factor XIIa because these activated factors can contribute to thrombosis in certain situations. Drugs targeting these proteases may be particularly effective in thrombosis triggered by exposure of blood to the surfaces of implantable medical devices. Here we review clinical data supporting roles for factor XII and factor XI in thrombosis induced by medical devices, and pre-clinical data suggesting that therapeutic targeting of these proteins may limit surface-induced thrombosis.
Keywords: Contact activation, Factor XII, Factor XI, Plasma Prekallikrein, High Molecular Weight Kininogen
Introduction
Implantable medical devices that come into contact with flowing blood are widely used in clinical practice. Some, such as artificial heart valves, vascular grafts, ventricular assist devices (VADs), extracorporeal membrane oxygenation (ECMO) circuits, and central venous lines may be exposed to blood for months to years, while others including extracorporeal circuits used in hemodialysis and cardiopulmonary bypass (CPB) are exposed for shorter durations. Despite advances in development of biocompatible materials, most implantable devices induce blood clot formation that may lead to their failure or to thromboembolic sequelae.1–3 The device surfaces exposed to blood lack the endothelial cell layer that actively retards coagulation and platelet adhesion in normal blood vessels, and anticoagulation and/or anti-platelet therapy is required to prevent thrombus formation.
Plasma proteins rapidly adsorb onto most non-biologic materials, forming a layer several nanometers thick in which protein concentrations are several orders of magnitude higher than in circulating blood.3,4 Initially, abundant proteins such as albumin and fibrinogen deposit on the surface. Fibrinogen, with other adhesive proteins such as von Willebrand factor and fibronectin, anchor platelets and other blood cells to the surface. With time, the types of adherent proteins change, a process called the Vroman effect.5–8 Fibrinogen is replaced by surface recognition proteins involved in the innate immune response, including components of the complement system and the contact factors. The plasma contact factors are factor XII (FXII), plasma prekallikrein (PPK), and factor XI (FXI), the precursors of the proteases α-factor XIIa (FXIIa), α-kallikrein (P-Kal), and factor XIa (FXIa), respectively; and the non-enzyme glycoprotein high molecular weight kininogen (HK) (Figure 1).9–11 Assembly of the contact system on surfaces enhances several host-defense processes including kinin generation, complement activation and thrombin generation, promoting inflammation and coagulation (Figure 2).11–15
Figure 1. The Contact Factors.

Schematic diagrams showing domains in each contact factor. Sites of proteolysis during activation are indicated by arrows. The residues immediately N-terminal to each cleavage site are indicated. Trypsin-like protease domains are indicated in red. FXII is an 80 kDa polypeptide that is cleavage after Arg353 to form αFXIIa. Cleavage of αFXIIa after Arg334 separates the non-catalytic and catalytic domains, forming βFXIIa. The FXII non-catalytic domains are the fibronectin type 2 (F2), epidermal growth factor (EGF), fibronectin type 1 (F1), and kringle (K) domains, and a proline–rich region (PRR). PPK is a 93 kDa polypeptide that is cleaved after Arg371 to form α-kallikrein (P-Kal). FXI is a homodimer of 80 kDa polypeptides. Each subunit of the dimer is converted to FXIa by cleavage after Arg369. The non-catalytic portions of PPK and FXI contain four apple domains (A1 to A4). HK is comprised of 6 domains (D1–D4, D5H and D6H). The nanopeptide sequence for bradykinin (Arg363-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg371) is in D4. D5 facilitates binding to surfaces and D6 contains overlapping binding sites for FXI and PPK.
Figure 2. Contact Activation.

FXII binds to a surface (represented in gray) and undergoes autocatalytic conversion to FXIIa (reaction 1). FXIIa converts PPK to P-Kal (reaction 2) with HK serving as a co- factor. P-Kal propagates the process by activating additional FXII to FXIIa (reaction 3) and cleaves HK releasing bradykinin (reaction 4). FXIIa induces coagulation by converting FXI to FXIa (reaction 5) in an HK-dependent manner. P-Kal and a degradation product of FXII (β-FXIIa) can activate components of the complement cascade (black arrows).
There has been substantial interest over the past several years in developing therapeutic inhibitors of FXIIa, FXIa and P-Kal to treat or prevent thrombo-inflammatory processes.10,15–17 As these proteins are either not required (FXIIa and P-Kal), or serve a relatively minor role (FXIa), in the normal hemostatic response to blood vessel injury, it is anticipated that drugs targeting them would cause fewer bleeding complications than currently used anticoagulants. Given the presumed importance of contact factors to surface-induced coagulation, they may be ideal targets for prevention of thrombus formation on surfaces of medical devices. In this review, we discuss the role of contact activation in thrombosis associated with implantable devices and the pre-clinical data supporting FXII or FXI as targets for preventing device- induced thrombosis.
Contact Activation and Blood Coagulation
FXI and FXII were initially identified as factors missing in patients with defects in in vitro assays of surface-induced plasma coagulation (the predecessors of the activated partial thromboplastin time [aPTT] assay used in clinical practice today).18–20 In the cascade-waterfall model of coagulation clot formation is initiated by FXII conversion to FXIIa on a surface (Figure 3).18,21,22 FXIIa, activates FXI, setting off a series of proteolytic reactions leading to thrombin (factor IIa) generation, and clot formation. Subsequent work showed that PPK and HK contribute to the process. Surface-dependent activation of the contact factors (contact activation) is depicted in Figure 2.23,24 When blood is exposed to a surface, particularly one carrying a negative charge, FXII binds and undergoes autocatalytic conversion to FXIIa (Figure 1 and Figure 2, reaction 1).9–11 Recent data from our laboratory suggest that FXII in its single-chain “precursor” form expresses a low level of proteolytic activity that catalyzes initial conversion of FXII to FXIIa.25 Autocatalysis then accelerates as FXIIa accumulates. On the surface, FXIIa catalyzes PPK conversion to P-Kal (Figure 1 and Figure 2, reaction 1), which amplifies the process by converting additional FXII to FXIIa (Figure 2, reaction 3). The reciprocal surface-dependent contact activation of FXII and PPK is supported by HK, which facilitates PPK binding to the surface. In vivo, a variety of substances may serve as “surfaces” for inducing contact activation, including polyanions such as polymeric orthophosphate (polyphosphate)26,27 and nucleic acids28–31, collagen32, misfolded protein aggregates33, and the cell membranes/walls of microorganisms34–36.
Figure 3. Contact Activation-Initiated Coagulation.
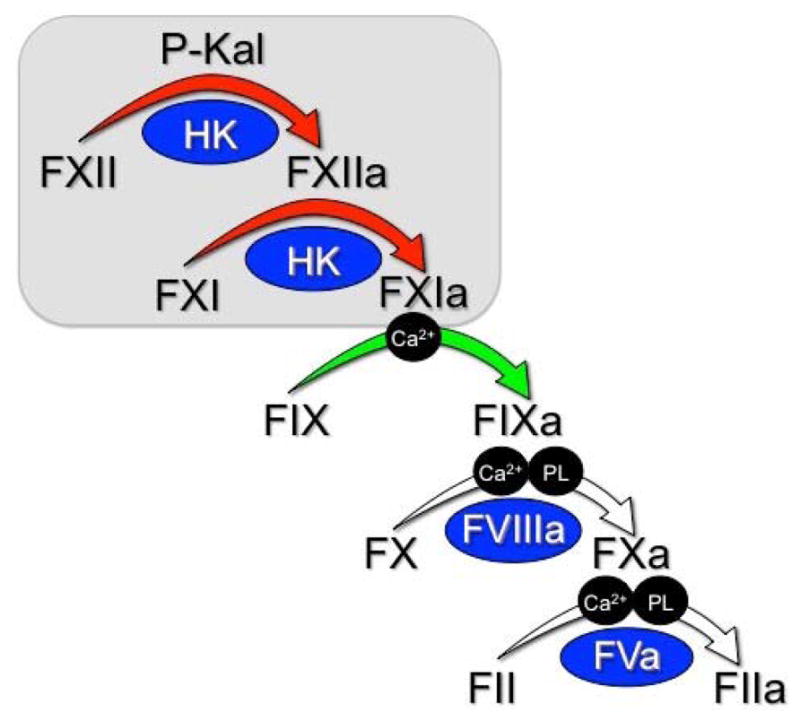
When plasma is exposed to a surface, the contact activation reactions shown in Figure 2 lead to accumulation of FXIa (red arrows). FXIa converts factor IX to factor IXaβ in a calcium dependent manner (green arrows). Factor IXaβ converts factor X to factor Xa in a calcium/phospholipid-dependent reaction requiring the cofactor factor VIIIa, and factor Xa in terms convers prothrombin (factor II) to thrombin (factor IIa) in the presence of the cofactor Va, phospholipid and calcium. Thrombin catalyzes multiple reactions, including fibrinogen conversion to fibrin and platelet activation through cleavage of protease activated receptors 1 and 4. The reactions depicted are required for normal coagulation in in vitro in aPTT assays.
FXIIa and P-Kal may contribute to several homeostatic and host-defense processes. P-Kal cleaves HK (Figure 2, reaction 4), releasing the proinflammatory peptide bradykinin (Figure 1).9–11 FXIIa may undergo proteolysis forming β-FXIIa, a protease that binds poorly to surfaces (Figure 1). P-Kal and β-FXIIa can activate components of the complement system (Figure 2, black arrows).11,13,37 FXIIa promotes thrombin generation and blood coagulation by converting FXI to FXIa (Figure 2, reaction 5).10,16 FXI is a homolog of PPK (Figure 1).38,39 In the aPTT assay, FXIIa activation of FXI, like FXIIa activation of PPK, involves HK (Figure 2). FXIa links contact activation to fibrin formation by efficiently converting the vitamin K-dependent zymogen factor IX to the protease factor IXaβ (Figure 3).10,40 While the aPTT is useful for identifying specific coagulation factor deficiencies in the clinic, it is clear that it is not an accurate model for hemostasis in vivo. Congenital absence of FXII, PPK or HK does not cause abnormal bleeding.18 FXI deficiency, in contrast, may cause excessive trauma-induced bleeding,41 indicating it can be activated independently of FXIIa. This hypothesis is supported by observations that FXI is activated by thrombin (Figure 4).42–44 The bleeding diathesis associated with FXI deficiency is relatively mild when compared with deficiency of its substrate factor IX,18 pointing to an ancillary role for FXI in hemostasis. However, despite its minor contribution to hemostasis, mounting evidence suggests FXI, and contact activation, make substantive contributions to thrombosis.
Figure 4. Factor XI as an Interface Between Contact Activation and Thrombin Generation.

The panel on the left (pink background) depicts our current understanding of thrombin generation in vivo. The process is initiated at a wound site by the factor VIIa/tissue factor (TF) complex, which converts factor X to factor Xa. Factor Xa in turn initiates prothrombin conversion (factor II) to thrombin (factor IIa). Factor VIIa/TF also converts factor IX to factor IXaβ, which sustains factor X activation. In some situations, additional factor IX activation through FXIa is required for hemostasis. It is thought that FXI activation in these cases is through a reaction that does not involve FXII. In the diagram, thrombin is shown activating FXI (gray arrow with dashed borders). The gray box displays the contact activation reactions shown in Figure 2 (red arrows). There is evidence that FXIa, likes its homolog P-Kal can activate FXII (green arrow with dashed borders). This suggest that FXI can function as a bidirectional interface connecting thrombin generation and contact activation, and allowing each system to influence the other. This differs from the scheme in Figure 3, which depicts a unidirectional path from FXII to thrombin generation.
Contact Activation and Thrombosis in Animal Models
In most in vivo thrombosis models, vessel occlusion is a rapid event induced in a healthy animal by acute injury (mechanical, chemical, photon) to a normal blood vessel, infusion of prothrombotic material (tissue factor, polyphosphate) into a vessel, or placement of a device (vascular graft, venous catheter, wire, thread) within a vessel. While it is questionable whether such models accurately reflect processes involved in human venous thromboembolism (VTE), stroke, or myocardial infarction (MI), some may be relevant for surface-induced coagulation associated with implantable medical devices. The contact factors contribute to venous and arterial occlusion in animals.10,15,26,45 Studies with mice lacking components of the contact system are illustrative. FXI and FXII deficient mice are as resistant to injury-induced thrombosis as are factor IX-deficient mice (a model of the hemorrhagic disorder hemophilia B).26,46–49 Mice lacking PPK or HK are also resistant to thrombus formation, consistent with a role for contact activation in thrombosis.49–53 In addition, PPK-deficient mice have reduced levels of tissue factor and higher levels of prostacyclin in their blood vessels than wild type mice, perhaps due to reduced bradykinin generation.51 This suggests that chronic suppression of contact activation could produce therapeutic effects by thrombin-dependent and thrombin-independent mechanisms.
The possibility that thrombotic processes in mice differ from those in primates is an important issue. When collagen-coated vascular grafts are placed within temporary arteriovenous fistulas in baboons, platelets and fibrin deposit in the grafts.10,55 Inhibition of FXI or FXII reduces thrombus growth,45,54–56 but in contrast to thrombus formation in mice, FXI inhibition in baboons limits thrombus formation to a greater extent than does FXII inhibition.10,54–56 These results suggest that FXI activation by FXII- independent processes is more important for thrombosis in primates than mice, and they imply that FXI may be a better antithrombotic target in humans. This premise is in line with data for venous thromboembolism (VTE), stroke, and myocardial infarction (MI) in humans, as discussed below.
Contact Activation and Thrombosis in Humans
There is a considerable body of work linking FXI to thrombosis in humans. The strongest association is between FXI and VTE. Risk of VTE is reduced in individuals with FXI-deficiency compared to the general population,57,58 and within the general population higher plasma FXI levels are associated with increased risk of VTE.59,60 Results of a phase 2 trial showing that reducing plasma FXI is more effective than standard of care (low molecular weight heparin) for preventing VTE in patients undergoing knee replacement surgery supports the epidemiologic data.61 A similar relationship between plasma FXI levels and thrombosis has been reported for ischemic stroke, with FXI deficiency reducing stroke risk,58,62 and higher FXI levels increase risk.63–65 The data supporting an associated between FXI and MI are less convincing. The incidence of MI in a small group of FXI-deficient patients was what would be expected in the population at-large.66 However, more recent data from a large group practice indicate FXI deficiency does lower MI risk.59 Data for non-FXI deficient individuals is mixed,64,65,67–69 but several studies point to an association between higher FXI levels and MI.67–69 Taken as a whole, results from epidemiology studies support a role for FXI in VTE and stroke in humans, with the association with VTE being particularly strong, and a possible role in MI.
The argument that FXII contributes to VTE, stroke or MI in humans, on the other hand, is relatively weak. The largest study of FXII-deficient subjects failed to reveal an association with VTE,70,71 and no differences in DVT incidence was noted across the range of FXII levels in the general population in the Leiden Thrombophilia71 and Longitudinal Investigation of Thromboembolism Etiology60 studies. FXII deficiency does not appear to provide protection from stroke,70 and there was no correlation between FXII level and stroke in the second Northwick Park Heart Study,72 Risk of Arterial Thrombosis In relation to Oral contraceptives (RATIO) study,65 or Atherosclerosis Risk in Communities (ARIC) study.64 Plasma FXII levels were not associated with MI in the RATIO65 and ARIC studies.64 Curiously, data from the Study of Myocardial Infarction Leiden cohort actually showed an inverse relationship between FXII levels and cardiovascular disease,67 a relationship also noted by Endler et al in a study from Austria.73
It seems reasonable to conclude that FXII contributes less to VTE and stroke, and perhaps MI, than does FXI. This in turn suggests that FXI activation via FXII-independent processes is more important than contact activation in these disorders, consistent with the impression that tissue factor-initiated thrombin generation is a major driver of thrombosis in humans.74,75 Further support for the conclusion that FXII makes relatively small contributions to VTE, stroke and MI comes from observations of patient lacking C1-Inhibitor (C1-INH), the major regulator of FXIIa and P-Kal. Congenital C1-INH deficiency causes hereditary angioedema (HAE), a disorder associated with activation of the contact system and bouts of soft tissue swelling due to excessive bradykinin generation.76,77 VTE, stroke or MI are not prominent features of HAE.76–78 The HAE phenotype indicates that reciprocal activation of FXII and PPK can occur in some, and perhaps most, situations without significant activation of FXI by FXIIa.
Evidence for Contact Activation Induced by Medical Devices
Components of implantable devices including vascular catheters,3,79,80 dialysis membranes,81,82 and artificial heart valves3,83 induce blood coagulation in vitro by a FXII-dependent mechanism. Yau et al. showed that the clotting time of normal plasma is shortened 3-fold by exposure to segments of coronary artery catheters.79 The same shortening occurs in plasma lacking factor VII (the protease responsible for tissue factor-initiated thrombin generation), but not in plasma lacking FXII or FXI. The FXIIa inhibitor corn trypsin inhibitor (CTI) blocks catheter-induced coagulation, consistent with the catheters inducing contact activation-initiated thrombin generation.80
Implantation of a variety of medical devices is associated with thrombosis in humans,1–3 but identifying evidence for contact activation as a causative factor in vivo is challenging. For many devices heparin is used to prevent thrombosis, and this may inhibit contact reactions, making them difficult to detect. Furthermore, FXIIa, FXIa and P-Kal concentrations in plasma may not reflect events on device surfaces.84,85 Finally, picomolar concentrations of proteases such as FXIa promote thrombin generation in vitro,86 indicating pathophysiologic FXIa levels in vivo may be below the detection thresholds for current assays. Markers of thrombin generation such as thrombin-antithrombin (TAT) complex may detect induction of coagulation, but do not answer the question of cause-and-effect. Three approaches have been used to assess contact activation in patient samples: (1) detecting reduction in plasma levels of unactivated contact factors, (2) measuring increases in free FXIIa and P-Kal activity with chromogenic substrates or ELISA, and (3) measuring increases in FXIIa, P-Kal or FXIa in complex with C1-INH or antithrombin.
Contact activation has been implicated in induction of blood coagulation in CPB, ECMO and renal dialysis, procedures that (1) involve exposure of blood to high surface area artificial membranes and (2) usually incorporate heparin as an anticoagulant. The association between CPB and blood coagulation is well recognized,87 but data on a role for contact activation in this procedure are mixed. Sontag et al. noted elevated FXIIa and reduced PPK in children undergoing CPB,88 and Wendel et al. reported decreased FXII and increased FXIIa and P-Kal activity during bypass.89 Watchfogel et al. detected increased P-Kal- C1-INH complexes,90 and Gallimore et al. measured reduced plasma HK levels during bypass,91 consistent with induction of contact activation. However, Boisclair et al. and Koster et al. did not find evidence for contact activation in patients undergoing CPB,92,93 and concluded tissue factor was largely responsible for thrombin generation in this setting. The discrepant data could reflect differences in assay sensitivities, or differences in oxygenator surfaces or protocols used in different places at different times. It is also important to keep in mind that the high heparin concentrations used in CPB may blunt contact activation. Observations that patients with FXII deficiency still have evidence of increased thrombin generation during CPB have been used to argue against a role for contact activation in CPB-induced coagulation.94 While the observation is consistent with tissue factor-induced thrombin production, this process must occur in patients undergoing surgery to prevent exsanguination. It does not necessarily follow that tissue factor-dependent thrombin generation is responsible for thrombosis in CPB.
There is limited data for ECMO, a procedure that uses lower heparin concentrations than in CPB. Using ex vivo ECMO circuits, Wendel et al. observed higher FXIIa and P-Kal levels in blood passing through non-heparin coated circuits than heparin-coated circuits,95 consistent with surface-induced contact activation by the ECMO surface and its inhibition by heparin. In ex vivo studies of renal dialysis circuits, Frank et al. observed thrombin generation (detected by measuring TAT complexes) when normal blood is exposed to different types of dialysis membranes.82 The most thrombogenic membrane (AN69XT Nephral 200) increased TAT in a FXII-dependent manner, with FXIIa-C1-INH complexes accumulating in normal blood exposed to the membrane.
VADs are becoming a common option for patients with advanced heart failure, either as a bridge to cardiac transplantation or as destination therapy. With these devices, blood is exposed to relatively small metal surfaces rather than large surface area membranes, and antithrombotic regimens are based primarily on warfarin and anti-platelet agents, which are unlikely to affect contact activation. Studies in animals and humans show reductions in plasma levels of FXII, FXI, PPK and HK after VAD placement,96–100 with increased levels of the activated forms of these proteins.101 FXII autoactivation and reciprocal FXII-PPK activation occurs in the protein layer adherent to the titanium-alloy surface of the devices.102,103 It is not clear if the surface promotes true contact activation through induction of conformational changes to surface-bound proteins, as in aPTT assays, or simply facilitates enzymatic reactions by concentrating proteins on the surface.
In summary, while some data from human patients point to a role for contact activation as a contributor to implantable device-induced thrombo-inflammation, others do not. In our opinion, a major limiting factor in these studies is the use of anticoagulants such as heparin that inhibit the very processes under study. Animal models are probably better suited for addressing this issue, as studies can include non-treatment control arms.
Pre-clinical Studies Targeting FXII and FXI for Thromboprophylaxis with Medical-Device
Several compelling studies have been published in recent years that convincingly implicate the contact system in implantable device-induced thrombosis in animal models. These studies have provided proof-of-concept for testing inhibitors of FXII and FXI in humans. Larsson et al. studied the effects of therapeutic FXIIa inhibition in rabbits connected to pediatric ECMO circuits.104,105 In the absence of an anticoagulant, the circuit rapidly occluded with heavy deposition of fibrin on the surface of the oxygenator elements. Treating the animals with unfractionated heparin, as expected, prevented circuit occlusion. Animals on heparin demonstrated significant bleeding with injury to the skin or cuticle, consistent with the known effects of this drug on factor Xa and thrombin. Treatment with a monoclonal IgG (3F7) that specifically recognizes the active site of FXIIa prevented fibrin deposition and circuit occlusion, with an effect at least as good as that of heparin. As importantly, 3F7 did not prolong bleeding times in either injury model, despite prolonging the aPTT, consistent with the clinical impression that FXII-deficiency does not compromise hemostasis.18
Yau et al. studied the effects of factor VII, FXI, FXII or HK reduction on thrombus formation induced by polyurethane catheters inserted into the jugular veins of rabbits. The protein levels were reduced by ~90% using factor-specific antisense oligonucleotides that inhibit production of the proteins in the liver.106 FXII or FXI reduction prolonged the time to catheter-induced thrombus formation more than two-fold, while factor VII or HK reduction had little effect. At first glance, the negative result with HK reduction suggests that a process distinct from classic contact activation may contribute to thrombosis in this model. However, the normal plasma level of HK is >600 nM and even 10% residual protein may provide enough cofactor activity to support contact activation.
Recently, David et al. described monoclonal IgGs that specifically block the catalytic activity of FXIa, and tested their ability to prevent jugular vein thrombosis induced by placement of a thrombogenic thread within the lumen of the blood vessel.107 The antibody DEF prevented vessel occlusion with an efficacy comparable to that of the factor Xa active site inhibitor rivaroxaban. At a concentration 10-fold higher than that required to prevent vessel occlusion, DEF did not increase the cuticle bleeding in rabbits, in contrast to therapeutic doses of rivaroxaban, which caused a significant increase in bleeding.
Conclusions and Future Directions
A variety of compounds (antisense oligonucleotides, antibodies, small molecular active site inhibitors) targeting FXI or FXIa are entering early clinical trials.108 Therapies targeting FXII and FXIIa are also under development.108 Based on pre-clinical studies, these agents will probably be tested first as primary or secondary thrombosis prophylaxis, rather than treatment of acute thrombotic events. While the phase 2 trial demonstrating that decreasing FXI level prevents venous thrombus formation in patients undergoing knee arthroplasty is an encouraging start,62 our understanding of the roles of FXIa and FXIIa in human thrombosis and the types of thrombotic events that may respond to inhibitors of these proteases is incomplete. Among their possible roles, FXIIa and FXIa appear to function as part of the innate host response to infection, assembling and activating on surfaces of microorganisms.34–36 Given this, it is not surprising that the system would assemble on foreign materials used in implantable medical devices. Prevention of artificial surface-induced thrombosis with FXIIa and FXIa inhibitors seems to be an obvious path to pursue.
A major advantage of FXIa or FXIIa inhibition over currently available anticoagulants is likely to be in the area of safety. These proteins serve, at most, supportive roles in hemostasis and, hypothetically, complete inhibition of either should be well tolerated under most circumstances. This could be of great value in certain groups of patients with implantable devices who are prone to excessive bleeding. For example, many patients on ECMO are infants who bleed with heparin therapy even as they are at risk for thromboembolic events. Patients who have undergone VAD implantation develop an acquired von Willebrand syndrome due to mechanical and shear-induced degradation of von Willebrand factor.109–110 This defect, superimposed on warfarin and anti-platelet agents used to prevent device-induced thrombosis, leaves these patients prone to severe bleeding, particularly from the gastrointestinal tract. Targeted inhibition of FXIa or FXIIa may reduce thrombotic risk in a manner that would leave hemostasis largely intact. These agents could also be combined with standard drugs to reduce the required intensity of the standard therapy.
It remains to be established if FXII or FXI is the better target for preventing implantable device- induced thrombosis. Both have potential advantages and disadvantages. For FXII, the fact that it is not required for hemostasis and that it triggers surface-induced coagulation seems to make it an ideal target. Furthermore, FXII inhibition may limit PPK activation, blunting inflammation. Population-based studies raise the possibility that FXII may serve a less important role in thrombosis in humans than does FXI. However, while this may be the case for common disorders such as MI, stroke, and VTE, it does imply that FXII does not participate substantively in thrombosis on non-biologic surfaces. Inhibition of FXI, hypothetically, would interfere with contact activation-induced thrombin generation and the thrombin- initiated feedback loop that sustains factor IX activation (Figure 4), producing a more potent antithrombotic effect than would inhibition of FXII. Interestingly, recent data also point to a role for FXI in inflammation.111–113 While the cascade/waterfall hypothesis (Figure 3) describes FXIa as a product of contact activation, we observed that FXIa stimulates contact activation in vitro and in vivo by enhancing FXII activation.112 We postulate that FXI is a bidirectional interface connecting thrombin generation and contact activation, as shown in Figure 4, and its inhibition could prevent cross-talk between the two systems.10 A concern with a FXI inhibitor is bleeding. Indeed, some patients with FXI deficiency do experience excessive trauma or surgery induced bleeding.18 However, bleeding is rarely spontaneous in FXI-deficient patients and tends to involve specific tissues.18 The observation that low FXI levels did not compromise hemostasis in knee replacement surgery61 suggests FXI inhibition should cause fewer hemorrhagic problems compared to heparin or warfarin. As agents targeting the contact factors become available, we will be able to test individual and combination drug therapies to determine if they will have utility in preventing thrombosis in patients with implantable devices.
Acknowledgments
The authors acknowledge support from awards HL81326 and HL8837 (from the National Heart, Lung and Blood Institute.
Footnotes
CONFLICTS OF INTEREST
B.T. has no conflicts to report. D.G. is a consultant for several pharmaceutical companies with an interest in developing therapeutics directed against factor XI, factor XII and PPK.
References
- 1.Nielsen VG, Kirklin JK, Holman WL, et al. Mechanical circulatory device thrombosis: a new paradigm linking hypercoagulation and hypofibrinolysis. ASAIO J Am Soc Artif Intern Organs 1992. 2008;54:351–358. doi: 10.1097/MAT.0b013e31817f3e03. [DOI] [PubMed] [Google Scholar]
- 2.Ekdahl KN, Lambris JD, Elwing H, et al. Innate immunity activation on biomaterial surfaces: a mechanistic model and coping strategies. Adv Drug Deliv Rev. 2011;63:1042–1050. doi: 10.1016/j.addr.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaffer IH, Fredenburgh JC, Hirsh J, Weitz JI. Medical device-induced thrombosis: what causes it and how can we prevent it? J Thromb Haemost JTH. 2015;13(Suppl 1):S72–81. doi: 10.1111/jth.12961. [DOI] [PubMed] [Google Scholar]
- 4.Wilson CJ, Clegg RE, Leavesley DI, Pearcy MJ. Mediation of biomaterial-cell interactions by adsorbed proteins: a review. Tissue Eng. 2005;11:1–18. doi: 10.1089/ten.2005.11.1. [DOI] [PubMed] [Google Scholar]
- 5.Turbill P, Beugeling T, Poot AA. Proteins involved in the Vroman effect during exposure of human blood plasma to glass and polyethylene. Biomaterials. 1996;17:1279–1287. [PubMed] [Google Scholar]
- 6.Leonard EF, Vroman L. Is the Vroman effect of importance in the interaction of blood with artificial materials? J Biomater Sci Polym Ed. 1991;3:95–107. doi: 10.1163/156856292x00105. [DOI] [PubMed] [Google Scholar]
- 7.Scott CF. Mechanism of the participation of the contact system in the Vroman effect. Review and summary. J Biomater Sci Polym Ed. 1991;2:173–181. doi: 10.1080/09205063.1991.9756658. [DOI] [PubMed] [Google Scholar]
- 8.Hirsh SL, McKenzie DR, Nosworthy NJ, Denman JA, Sezerman OU, Bilek MMM. The Vroman effect: competitive protein exchange with dynamic multilayer protein aggregates. Colloids Surf B Biointerfaces. 2013;103:395–404. doi: 10.1016/j.colsurfb.2012.10.039. [DOI] [PubMed] [Google Scholar]
- 9.Vogler EA, Siedlecki CA. Contact activation of blood-plasma coagulation. Biomaterials. 2009;30(10):1857–1869. doi: 10.1016/j.biomaterials.2008.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gailani D, Gruber A. Factor XI as a therapeutic target. Arterioscler Thromb Vasc Biol. 2016;36:1316–1322. doi: 10.1161/ATVBAHA.116.306925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14:28–39. doi: 10.1111/jth.13194. [DOI] [PubMed] [Google Scholar]
- 12.Jukema BN, de Maat S, Maas C. Processing of Factor XII during Inflammatory Reactions. Front Med. 2016;3:52. doi: 10.3389/fmed.2016.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghebrehiwet B, Kaplan AP, Joseph K, Peerschke EIB. The complement and contact activation systems: partnership in pathogenesis beyond angioedema. Immunol Rev. 2016;274:281–289. doi: 10.1111/imr.12469. [DOI] [PubMed] [Google Scholar]
- 14.Lin L, Wu M, Zhao J. The initiation and effects of plasma contact activation: an overview. Int J Hematol. 2017;105:235–243. doi: 10.1007/s12185-016-2132-x. [DOI] [PubMed] [Google Scholar]
- 15.Nickel KF, Long AT, Fuchs TA, Butler LM, Renné T. Factor XII as a Therapeutic Target in Thromboembolic and Inflammatory Diseases. Arterioscler Thromb Vasc Biol. 2017;37:13–20. doi: 10.1161/ATVBAHA.116.308595. [DOI] [PubMed] [Google Scholar]
- 16.Schmaier AH. Antithrombotic potential of the contact activation pathway. Curr Opin Hematol. 2016;23:445–452. doi: 10.1097/MOH.0000000000000271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weitz JI, Fredenburgh JC. Factors XI and XII as Targets for New Anticoagulants. Front Med. 2017;4:19. doi: 10.3389/fmed.2017.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gailani D, Neff AT. Hematology: Basic Principles and Practice. 6. Philadelphia, PA: 2013. Rare Coagulation Factor Deficiencies; pp. 1971–1986. [Google Scholar]
- 19.Ratnoff OD, Colopy JE. A familial hemorrhagic trait associated with a deficiency of a clot- promoting fraction of plasma. J Clin Invest. 1955;34:602–613. doi: 10.1172/JCI103109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenthal RL, Dreskin OH, Rosenthal N. New hemophilia-like disease caused by deficiency of a third plasma thromboplastin factor. Proc Soc Exp Biol Med Soc Exp Biol Med N Y N. 1953;82:171– 174. doi: 10.3181/00379727-82-20057. [DOI] [PubMed] [Google Scholar]
- 21.Macfarlane RG. AN ENZYME CASCADE IN THE BLOOD CLOTTING MECHANISM, AND ITS FUNCTION AS A BIOCHEMICAL AMPLIFIER. Nature. 1964;202:498–499. doi: 10.1038/202498a0. [DOI] [PubMed] [Google Scholar]
- 22.Davie EW, Ratnoff OD. WATERFALL SEQUENCE FOR INTRINSIC BLOOD CLOTTING. Science. 1964;145:1310–1312. doi: 10.1126/science.145.3638.1310. [DOI] [PubMed] [Google Scholar]
- 23.Hathaway WE, Belhasen LP, Hathaway HS. Evidence for a new plasma thromboplastin factor. I. Case report, coagulation studies and physicochemical properties. Blood. 1965;26:521–532. [PubMed] [Google Scholar]
- 24.Colman RW, Bagdasarian A, Talamo RC, et al. Williams trait. Human kininogen deficiency with diminished levels of plasminogen proactivator and prekallikrein associated with abnormalities of the Hageman factor-dependent pathways. J Clin Invest. 1975;56:1650–1662. doi: 10.1172/JCI108247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov I, Matafonov A, Sun M-F, et al. Proteolytic properties of single-chain factor XII: a mechanism for triggering contact activation. Blood. 2017;129:1527–1537. doi: 10.1182/blood-2016-10-744110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Müller F, Mutch NJ, Schenk WA, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143–1156. doi: 10.1016/j.cell.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrissey JH, Smith SA. Polyphosphate as modulator of hemostasis, thrombosis, and inflammation. J Thromb Haemost JTH. 2015;13(Suppl 1):S92–97. doi: 10.1111/jth.12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kannemeier C, Shibamiya A, Nakazawa F, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A. 2007;104:6388–6393. doi: 10.1073/pnas.0608647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gould TJ, Vu TT, Swystun LL, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol. 2014;34:1977–1984. doi: 10.1161/ATVBAHA.114.304114. [DOI] [PubMed] [Google Scholar]
- 31.Ivanov I, Shakhawat R, Sun M-F, et al. Nucleic acids as cofactors for factor XI and prekallikrein activation: Different roles for high-molecular-weight kininogen. Thromb Haemost. 2017;117:671– 681. doi: 10.1160/TH16-09-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Meijden PEJ, Munnix ICA, Auger JM, et al. Dual role of collagen in factor XII-dependent thrombus formation. Blood. 2009;114:881–890. doi: 10.1182/blood-2008-07-171066. [DOI] [PubMed] [Google Scholar]
- 33.Maas C, Govers-Riemslag JWP, Bouma B, et al. Misfolded proteins activate factor XII in humans, leading to kallikrein formation without initiating coagulation. J Clin Invest. 2008;118:3208–3218. doi: 10.1172/JCI35424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frick I-M, Akesson P, Herwald H, et al. The contact system--a novel branch of innate immunity generating antibacterial peptides. EMBO J. 2006;25:5569–5578. doi: 10.1038/sj.emboj.7601422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frick I-M, Björck L, Herwald H. The dual role of the contact system in bacterial infectious disease. Thromb Haemost. 2007;98:497–502. [PubMed] [Google Scholar]
- 36.Oehmcke S, Herwald H. Contact system activation in severe infectious diseases. J Mol Med Berl Ger. 2010;88:121–126. doi: 10.1007/s00109-009-0564-y. [DOI] [PubMed] [Google Scholar]
- 37.Foley JH, Conway EM. Cross Talk Pathways Between Coagulation and Inflammation. Circ Res. 2016;118:1392–1408. doi: 10.1161/CIRCRESAHA.116.306853. [DOI] [PubMed] [Google Scholar]
- 38.Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35–40. doi: 10.1101/sqb.2009.74.001. [DOI] [PubMed] [Google Scholar]
- 39.Ponczek MB, Gailani D, Doolittle RF. Evolution of the contact phase of vertebrate blood coagulation. J Thromb Haemost JTH. 2008;6:1876–1883. doi: 10.1111/j.1538-7836.2008.03143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gailani D, Geng Y, Verhamme I, et al. The mechanism underlying activation of factor IX by factor XIa. Thromb Res. 2014;133(Suppl 1):S48–51. doi: 10.1016/j.thromres.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol. 2016;9:629–637. doi: 10.1080/17474086.2016.1191944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naito K, Fujikawa K. Activation of human blood coagulation factor XI independent of factor XII. Factor XI is activated by thrombin and factor XIa in the presence of negatively charged surfaces. J Biol Chem. 1991;266:7353–7358. [PubMed] [Google Scholar]
- 43.Gailani D, Broze GJ. Factor XI activation in a revised model of blood coagulation. Science. 1991;253:909–912. doi: 10.1126/science.1652157. [DOI] [PubMed] [Google Scholar]
- 44.Matafonov A, Sarilla S, Sun M, et al. Activation of factor XI by products of prothrombin activation. Blood. 2011;118:437–445. doi: 10.1182/blood-2010-10-312983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Montfoort ML, Meijers JCM. Recent insights into the role of the contact pathway in thrombo- inflammatory disorders. Hematol Am Soc Hematol Educ Program. 2014;2014:60–65. doi: 10.1182/asheducation-2014.1.60. [DOI] [PubMed] [Google Scholar]
- 46.Wang X, Cheng Q, Xu L, et al. Effects of factor IX or factor XI deficiency on ferric chloride- induced carotid artery occlusion in mice. J Thromb Haemost JTH. 2005;3:695–702. doi: 10.1111/j.1538-7836.2005.01236.x. [DOI] [PubMed] [Google Scholar]
- 47.Renné T, Pozgajová M, Grüner S, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202:271–281. doi: 10.1084/jem.20050664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng Q, Tucker EI, Pine MS, et al. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood. 2010;116:3981–3989. doi: 10.1182/blood-2010-02-270918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Revenko AS, Gao D, Crosby JR, et al. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood. 2011;118:5302–5311. doi: 10.1182/blood-2011-05-355248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bird JE, Smith PL, Wang X, et al. Effects of plasma kallikrein deficiency on haemostasis and thrombosis in mice: murine ortholog of the Fletcher trait. Thromb Haemost. 2012;107:1141–1150. doi: 10.1160/th-11-10-0682. [DOI] [PubMed] [Google Scholar]
- 51.Stavrou EX, Fang C, Merkulova A, et al. Reduced thrombosis in Klkb1−/− mice is mediated by increased Mas receptor, prostacyclin, Sirt1, and KLF4 and decreased tissue factor. Blood. 2015;125:710–719. doi: 10.1182/blood-2014-01-550285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kokoye Y, Ivanov I, Cheng Q, et al. A comparison of the effects of factor XII deficiency and prekallikrein deficiency on thrombus formation. Thromb Res. 2016;140:118–124. doi: 10.1016/j.thromres.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Merkulov S, Zhang W-M, Komar AA, et al. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood. 2008;111:1274–1281. doi: 10.1182/blood-2007-06-092338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gruber A, Hanson SR. Factor XI-dependence of surface- and tissue factor-initiated thrombus propagation in primates. Blood. 2003;102:953–955. doi: 10.1182/blood-2003-01-0324. [DOI] [PubMed] [Google Scholar]
- 55.Tucker EI, Marzec UM, White TC, et al. Prevention of vascular graft occlusion and thrombus- associated thrombin generation by inhibition of factor XI. Blood. 2009;113:936–944. doi: 10.1182/blood-2008-06-163675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matafonov A, Leung PY, Gailani AE, et al. Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood. 2014;123:1739–1746. doi: 10.1182/blood-2013-04-499111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011;105:269– 273. doi: 10.1160/TH10-05-0307. [DOI] [PubMed] [Google Scholar]
- 58.Preis M, Hirsch J, Kotler A, et al. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017;129:1210–1215. doi: 10.1182/blood-2016-09-742262. [DOI] [PubMed] [Google Scholar]
- 59.Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342:696–701. doi: 10.1056/NEJM200003093421004. [DOI] [PubMed] [Google Scholar]
- 60.Cushman M, O’Meara ES, Folsom AR, Heckbert SR. Coagulation factors IX through XIII and the risk of future venous thrombosis: the Longitudinal Investigation of Thromboembolism Etiology. Blood. 2009;114:2878–2883. doi: 10.1182/blood-2009-05-219915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Büller HR, C, Bhanot S, et al. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015;372:232–240. doi: 10.1056/NEJMoa1405760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008;111:4113–4117. doi: 10.1182/blood-2007-10-120139. [DOI] [PubMed] [Google Scholar]
- 63.Yang DT, Flanders MM, Kim H, Rodgers GM. Elevated factor XI activity levels are associated with an increased odds ratio for cerebrovascular events. Am J Clin Pathol. 2006;126:411–415. doi: 10.1309/QC259F09UNMKVP0R. [DOI] [PubMed] [Google Scholar]
- 64.Suri MFK, Yamagishi K, Aleksic N, Hannan PJ, Folsom AR. Novel hemostatic factor levels and risk of ischemic stroke: the Atherosclerosis Risk in Communities (ARIC) Study. Cerebrovasc Dis Basel Switz. 2010;29:497–502. doi: 10.1159/000297966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Siegerink B, Govers-Riemslag JWP, Rosendaal FR, Ten Cate H, Algra A. Intrinsic coagulation activation and the risk of arterial thrombosis in young women: results from the Risk of Arterial Thrombosis in relation to Oral contraceptives (RATIO) case-control study. Circulation. 2010;122:1854–1861. doi: 10.1161/CIRCULATIONAHA.110.943738. [DOI] [PubMed] [Google Scholar]
- 66.Salomon O, Steinberg DM, Dardik R, et al. Inherited factor XI deficiency confers no protection against acute myocardial infarction. J Thromb Haemost JTH. 2003;1:658–661. doi: 10.1046/j.1538-7836.2003.00195.x. [DOI] [PubMed] [Google Scholar]
- 67.Doggen CJM, Rosendaal FR, Meijers JCM. Levels of intrinsic coagulation factors and the risk of myocardial infarction among men: Opposite and synergistic effects of factors XI and XII. Blood. 2006;108:4045–4051. doi: 10.1182/blood-2005-12-023697. [DOI] [PubMed] [Google Scholar]
- 68.Butenas S, Undas A, Gissel MT, Szuldrzynski K, Zmudka K, Mann KG. Factor XIa and tissue factor activity in patients with coronary artery disease. Thromb Haemost. 2008;99:142–149. doi: 10.1160/TH07-08-0499. [DOI] [PubMed] [Google Scholar]
- 69.Berliner JI, Rybicki AC, Kaplan RC, Monrad ES, Freeman R, Billett HH. Elevated levels of Factor XI are associated with cardiovascular disease in women. Thromb Res. 2002;107:55–60. doi: 10.1016/s0049-3848(02)00190-1. [DOI] [PubMed] [Google Scholar]
- 70.Zeerleder S, Schloesser M, Redondo M, et al. Reevaluation of the incidence of thromboembolic complications in congenital factor XII deficiency--a study on 73 subjects from 14 Swiss families. Thromb Haemost. 1999;82:1240–1246. [PubMed] [Google Scholar]
- 71.Koster T, Rosendaal FR, Briët E, Vandenbroucke JP. John Hageman’s factor and deep-vein thrombosis: Leiden thrombophilia Study. Br J Haematol. 1994;87:422–424. doi: 10.1111/j.1365-2141.1994.tb04937.x. [DOI] [PubMed] [Google Scholar]
- 72.Govers-Riemslag JWP, Smid M, Cooper JA, et al. The plasma kallikrein-kinin system and risk of cardiovascular disease in men. J Thromb Haemost JTH. 2007;5:1896–1903. doi: 10.1111/j.1538-7836.2007.02687.x. [DOI] [PubMed] [Google Scholar]
- 73.Endler G, Marsik C, Jilma B, Schickbauer T, Quehenberger P, Mannhalter C. Evidence of a U- shaped association between factor XII activity and overall survival. J Thromb Haemost JTH. 2007;5:1143–1148. doi: 10.1111/j.1538-7836.2007.02530.x. [DOI] [PubMed] [Google Scholar]
- 74.Tatsumi K, Mackman N. Tissue Factor and Atherothrombosis. J Atheroscler Thromb. 2015;22:543–549. doi: 10.5551/jat.30940. [DOI] [PubMed] [Google Scholar]
- 75.Manly DA, Boles J, Mackman N. Role of tissue factor in venous thrombosis. Annu Rev Physiol. 2011;73:515–525. doi: 10.1146/annurev-physiol-042210-121137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zuraw BL, Christiansen SC. HAE Pathophysiology and Underlying Mechanisms. Clin Rev Allergy Immunol. 2016;51:216–229. doi: 10.1007/s12016-016-8561-8. [DOI] [PubMed] [Google Scholar]
- 77.Naudin C, Burillo E, Blankenberg S, Butler L, Renné T. Factor XII Contact Activation. Semin Thromb Hemost. 2017 Mar; doi: 10.1055/s-0036-1598003. [DOI] [PubMed] [Google Scholar]
- 78.Reshef A, Zanichelli A, Longhurst H, Relan A, Hack CE. Elevated D-dimers in attacks of hereditary angioedema are not associated with increased thrombotic risk. Allergy. 2015;70:506–513. doi: 10.1111/all.12587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yau JW, Stafford AR, Liao P, Fredenburgh JC, Roberts R, Weitz JI. Mechanism of catheter thrombosis: comparison of the antithrombotic activities of fondaparinux, enoxaparin, and heparin in vitro and in vivo. Blood. 2011;118:6667–6674. doi: 10.1182/blood-2011-07-364141. [DOI] [PubMed] [Google Scholar]
- 80.Yau JW, Stafford AR, Liao P, et al. Corn trypsin inhibitor coating attenuates the prothrombotic properties of catheters in vitro and in vivo. Acta Biomater. 2012;8:4092–4100. doi: 10.1016/j.actbio.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 81.Ekdahl KN, Soveri I, Hilborn J, Fellström B, Nilsson B. Cardiovascular disease in haemodialysis: role of the intravascular innate immune system. Nat Rev Nephrol. 2017 Feb; doi: 10.1038/nrneph.2017.17. [DOI] [PubMed] [Google Scholar]
- 82.Frank RD, Weber J, Dresbach H, Thelen H, Weiss C, Floege J. Role of contact system activation in hemodialyzer-induced thrombogenicity. Kidney Int. 2001;60:1972–1981. doi: 10.1046/j.1523-1755.2001.00009.x. [DOI] [PubMed] [Google Scholar]
- 83.Bernacca GM, Gulbransen MJ, Wilkinson R, Wheatley DJ. In vitro blood compatibility of surface- modified polyurethanes. Biomaterials. 1998;19:1151–1165. doi: 10.1016/s0142-9612(98)00016-7. [DOI] [PubMed] [Google Scholar]
- 84.Matata BM, Wark S, Sundaram S, et al. In vitro contact phase activation with haemodialysis membranes: role of pharmaceutical agents. Biomaterials. 1995;16:1305–1312. doi: 10.1016/0142-9612(95)91045-z. [DOI] [PubMed] [Google Scholar]
- 85.Matata BM, Courtney JM, Sundaram S, et al. Determination of contact phase activation by the measurement of the activity of supernatant and membrane surface-adsorbed factor XII (FXII): its relevance as a useful parameter for the in vitro assessment of haemodialysis membranes. J Biomed Mater Res. 1996;31:63–70. doi: 10.1002/(SICI)1097-4636(199605)31:1<63::AID-JBM8>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 86.Kravtsov DV, Matafonov A, Tucker EI, et al. Factor XI contributes to thrombin generation in the absence of factor XII. Blood. 2009;114:452–458. doi: 10.1182/blood-2009-02-203604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sniecinski RM, Chandler WL. Activation of the hemostatic system during cardiopulmonary bypass. Anesth Analg. 2011;113:1319–1333. doi: 10.1213/ANE.0b013e3182354b7e. [DOI] [PubMed] [Google Scholar]
- 88.Sonntag J, Dähnert I, Stiller B, Hetzer R, Lange PE. Complement and contact activation during cardiovascular operations in infants. Ann Thorac Surg. 1998;65:525–531. doi: 10.1016/s0003-4975(97)01340-4. [DOI] [PubMed] [Google Scholar]
- 89.Wendel HP, Jones DW, Gallimore MJ. FXII levels, FXIIa-like activities and kallikrein activities in normal subjects and patients undergoing cardiac surgery. Immunopharmacology. 1999;45:141–144. doi: 10.1016/s0162-3109(99)00067-3. [DOI] [PubMed] [Google Scholar]
- 90.Wachtfogel YT, Harpel PC, Edmunds LH, Colman RW. Formation of C1s-C1-inhibitor, kallikrein- C1-inhibitor, and plasmin-alpha 2-plasmin-inhibitor complexes during cardiopulmonary bypass. Blood. 1989;73:468–471. [PubMed] [Google Scholar]
- 91.Gallimore MJ, Jones DW, Winter M, Wendel HP. Changes in high molecular weight kininogen levels during and after cardiopulmonary bypass surgery measured using a chromogenic peptide substrate assay. Blood Coagul Fibrinolysis Int J Haemost Thromb. 2002;13:561–568. doi: 10.1097/00001721-200209000-00012. [DOI] [PubMed] [Google Scholar]
- 92.Boisclair MD, Lane DA, Philippou H, et al. Mechanisms of thrombin generation during surgery and cardiopulmonary bypass. Blood. 1993;82:3350–3357. [PubMed] [Google Scholar]
- 93.Koster A, Fischer T, Gruendel M, et al. Management of heparin resistance during cardiopulmonary bypass: the effect of five different anticoagulation strategies on hemostatic activation. J Cardiothorac Vasc Anesth. 2003;17:171–175. doi: 10.1053/jcan.2003.42. [DOI] [PubMed] [Google Scholar]
- 94.Burman JF, Chung HI, Lane DA, Philippou H, Adami A, Lincoln JC. Role of factor XII in thrombin generation and fibrinolysis during cardiopulmonary bypass. Lancet Lond Engl. 1994;344:1192–1193. doi: 10.1016/s0140-6736(94)90509-6. [DOI] [PubMed] [Google Scholar]
- 95.Wendel HP, Scheule AM, Eckstein FS, Ziemer G. Haemocompatibility of paediatric membrane oxygenators with heparin-coated surfaces. Perfusion. 1999;14:21–28. doi: 10.1177/026765919901400104. [DOI] [PubMed] [Google Scholar]
- 96.Taenaka Y, Matsuda T, Takano H, Umezu M, Akutsu T. Influences of ventricular assist device pumping on blood coagulation. ASAIO Trans. 1989;35:396–398. doi: 10.1097/00002480-198907000-00072. [DOI] [PubMed] [Google Scholar]
- 97.Takahama T, Kanai F, Hiraishi M, et al. Significance of various anticoagulation therapies during use of a left ventricular assist device. ASAIO Trans. 1989;35:426–429. doi: 10.1097/00002480-198907000-00082. [DOI] [PubMed] [Google Scholar]
- 98.Tanaka K, Wada K, Morimoto T, et al. Hemostatic alterations caused by ventricular assist devices for postcardiotomy heart failure. Artif Organs. 1991;15:59–65. doi: 10.1111/j.1525-1594.1991.tb00760.x. [DOI] [PubMed] [Google Scholar]
- 99.Tanaka K, Sato T, Kondo C, et al. Hematological problems during the use of cardiac assist devices: clinical experiences in Japan. Artif Organs. 1992;16:182–188. doi: 10.1111/j.1525-1594.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 100.Himmelreich G, Ullmann H, Riess H, et al. Pathophysiologic role of contact activation in bleeding followed by thromboembolic complications after implantation of a ventricular assist device. ASAIO J Am Soc Artif Intern Organs 1992. 1995;41:M790–794. doi: 10.1097/00002480-199507000-00122. [DOI] [PubMed] [Google Scholar]
- 101.Koster A, Loebe M, Hansen R, et al. Alterations in coagulation after implantation of a pulsatile Novacor LVAD and the axial flow MicroMed DeBakey LVAD. Ann Thorac Surg. 2000;70:533–537. doi: 10.1016/s0003-4975(00)01404-1. [DOI] [PubMed] [Google Scholar]
- 102.Guo Z, Bussard KM, Chatterjee K, Miller R, Vogler EA, Siedlecki CA. Mathematical modeling of material-induced blood plasma coagulation. Biomaterials. 2006;27:796–806. doi: 10.1016/j.biomaterials.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 103.Chatterjee K, Vogler EA, Siedlecki CA. Procoagulant activity of surface-immobilized Hageman factor. Biomaterials. 2006;27:5643–5650. doi: 10.1016/j.biomaterials.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 104.Larsson M, Rayzman V, Nolte MW, et al. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med. 2014;6:222ra17. doi: 10.1126/scitranslmed.3006804. [DOI] [PubMed] [Google Scholar]
- 105.Worm M, Köhler EC, Panda R, et al. The factor XIIa blocking antibody 3F7: a safe anticoagulant with anti-inflammatory activities. Ann Transl Med. 2015;3:247. doi: 10.3978/j.issn.2305-5839.2015.09.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yau JW, Liao P, Fredenburgh JC, et al. Selective depletion of factor XI or factor XII with antisense oligonucleotides attenuates catheter thrombosis in rabbits. Blood. 2014;123:2102–2107. doi: 10.1182/blood-2013-12-540872. [DOI] [PubMed] [Google Scholar]
- 107.David T, Kim YC, Ely LK, et al. Factor XIa–specific IgG and a reversal agent to probe factor XI function in thrombosis and hemostasis. Sci Transl Med. 2016;8:353ra112–353ra112. doi: 10.1126/scitranslmed.aaf4331. [DOI] [PubMed] [Google Scholar]
- 108.Gailani D, Bane CE, Gruber A. Factor XI and contact activation as targets for antithrombotic therapy. J Thromb Haemost. 2015;13:1383–1395. doi: 10.1111/jth.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eckman PM, John R. Bleeding and thrombosis in patients with continuous-flow ventricular assist devices. Circulation. 2012;125:3038–3047. doi: 10.1161/CIRCULATIONAHA.111.040246. [DOI] [PubMed] [Google Scholar]
- 110.Meyer AL, Malehsa D, Budde U, Bara C, Haverich A, Strueber M. Acquired von willebrand syndrome in patients with a centrifugal or axial continuous flow left ventricular assist device. JACC Heart Fail. 2014;2:141–145. doi: 10.1016/j.jchf.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 111.Tucker EI, Verbout NG, Leung PY, Hurst S, McCarty OJ, Gailani D, Gruber A. Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood. 2012;119:4762–4768. doi: 10.1182/blood-2011-10-386185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bane CE, Jr, Ivanov I, Matafonov A, Boyd KL, Cheng Q, Sherwood ER, Tucker EI, Smiley ST, McCarty OJ, Gruber A, Gailani D. Factor XI Deficiency Alters the Cytokine Response and Activation of Contact Proteases during Polymicrobial Sepsis in Mice. PLoS One. 2016;11:e0152968. doi: 10.1371/journal.pone.0152968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shnerb Ganor R, Harats D, Schiby G, Gailani D, Levkovitz H, Avivi C, Tamarin I, Shaish A, Salomon O. Factor XI Deficiency Protects Against Atherogenesis in Apolipoprotein E/Factor XI Double Knockout Mice. Arterioscler Thromb Vasc Biol. 2016;36:475–481. doi: 10.1161/ATVBAHA.115.306954. [DOI] [PMC free article] [PubMed] [Google Scholar]