

Abstract
Diabetic kidney disease is the leading cause of kidney failure. However, studies of molecular mechanisms of early kidney damage are lacking. Here we examined for possible linkage between transcriptional regulation and quantitative structural damage in early diabetic kidney disease in Pima Indians with type 2 diabetes. Tissue obtained from protocol kidney biopsies underwent genome-wide compartment-specific gene expression profiling and quantitative morphometric analysis. The ultrastructural lesion most strongly associated with transcriptional regulation was cortical interstitial fractional volume (VvInt), an index of tubule-interstitial damage. Transcriptional co-expression network analysis identified 1843 transcripts that correlated significantly with VvInt. These transcripts were enriched for pathways associated with mitochondrial dysfunction, inflammation, migratory mechanisms, and tubular metabolic functions. Pathway network analysis identified IL-1β as a key upstream regulator of the inflammatory response and five transcription factors cooperating with p53 to regulate metabolic functions. VvInt-associated transcripts showed significant correlation with the urine albumin to creatinine ratio and measured glomerular filtration rate 10 years after biopsy, establishing a link between the early molecular events and long-term disease progression. Thus, molecular mechanisms active early in diabetic kidney disease were revealed by correlating intrarenal transcripts with quantitative morphometry and long-term outcomes. This provides a starting point for identification of urgently needed therapeutic targets and non-invasive biomarkers of early diabetic kidney disease.
Keywords: diabetic nephropathy, morphogenomics, longitudinal phenotype, transcriptomic profiling, pathway networks
INTRODUCTION
Diabetic kidney disease (DKD) is the leading cause of chronic kidney disease (CKD) and end-stage renal disease (ESRD) globally, contributing to spiraling healthcare costs and straining health care resources1–3. The current diagnostic approach to DKD using estimated glomerular filtration rate (eGFR) and urinary albumin:creatinine ratio (ACR) identifies individuals susceptible to progressive kidney disease and cardiovascular morbidity and mortality4–8. The trajectory of DKD, however, is often variable at the early stages9, and exposes the significant limitations of our current approach to identifying the at-risk population in early clinical disease10. Moreover, progression to ESRD in the absence of albuminuria11 and the frequent regression of microalbuminuria further aggravate the diagnostic and therapeutic challenges in early DKD12–15. The advent of high-throughput molecular profiling approaches provides a unique opportunity to advance our understanding of diabetic complications. The most commonly used methodology, genome-wide transcriptomic profiling, has been applied successfully in various kidney diseases in humans and model organisms, highlighting its potential for detecting and exploring relevant pathways in DKD16–19. A complementary approach to understanding pathophysiologic mechanisms leading to DKD is through quantitative morphometric studies of tissue from kidney biopsies. Combining these two approaches allows exploration of the molecular pathways associated with structural damage in DKD.
An overarching goal of our studies is to define the molecular correlates of early structural damage in DKD and their association with long-term outcomes. As tubulointerstitial changes are major determinants of progressive kidney damage20, the present study identified the transcriptional profiles associated with the degree of tubulointerstitial ultrastructural damage in protocol kidney biopsies from 49 Pima Indians with early DKD. The resulting morphogenomic signatures were then compared to transcriptional profiles active in established DKD, thereby linking early alterations to the long-term course of DKD.
RESULTS
An overview of the analytical strategy is shown in Figure 1. Baseline clinical, demographic, and morphometric characteristics at time of kidney biopsy are presented in Table 1. Mean measured glomerular filtration rate (iGFR; iothalamate) was 147 ml/min and median urine ACR was 35 mg/g. Of the 49 participants included in this study, 23 had normal ACR (<30 mg/g) at the time of biopsy, while 19 had microalbuminuria (30–299 mg/g) and 7 had macroalbuminuria (>300 mg/g). Cortical interstitial fractional volume (VvInt), expressed as a percentage of renal cortex, was 29.5±9.6% compared with 11.9±2.8% in non-diabetic living kidney donor biopsies (p <0.0001). Time course of iGFR and ACR over a median of 10 years of post-biopsy follow-up (median of 15.9 years of observation from enrollment) are provided in Supplementary Figure S1 (A and B).
Figure 1.
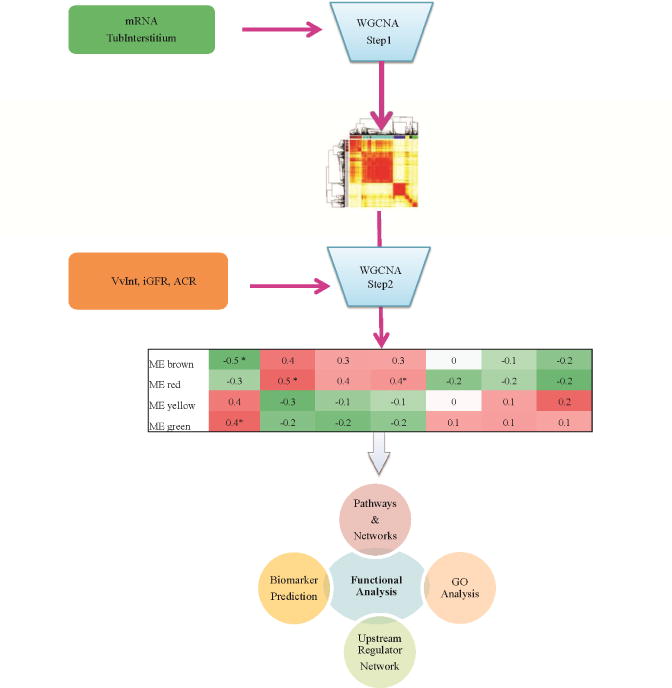
Schematic representation of the study design to identify VvInt associated co-expressed modules and long-term clinical outcome. In Step 1, modules or groups of genes with similar expression profiles are constructed from the transcriptome profile using a Weighted Gene Coexpression Analysis strategy. In Step 2, module eigengene (ME) is derived from each module and associated to clinical and structural parameters. The heatmap table illustrates the correlation coefficients of the module eigengene to the tested parameters. The significant modules (represented with asterisks) are then further investigated for functional enrichments.
TABLE 1.
Demographic, clinical, and morphometric characteristics at the time of kidney biopsy in 49 Pima Indians with type 2 diabetes.
| M/F (% Males) | 15/34 (30.6) |
| Age (years) | 46 ± 9.8 |
| Diabetes duration (yrs) | 15.7 ± 6.8 |
| BMI (kg/m2) | 35.2 ± 8.2 |
| HbA1c (%) | 9.20 ± 2 |
| Systolic Blood Pressure (mm/Hg) | 125 ± 14 |
| Diastolic blood Pressure (mm/Hg) | 78± 8 |
| ACR (mg/g) | 35.46 [90.21] |
| iGFR (ml/min) | 147 ± 45 |
| Serum creatinine (mg/dl) | 0.7 ± 0.2 |
| Cortical interstitial fractional volume (%) | 29.5±9.6 |
| Follow-up length post-biopsy (years) | 10.1[2.0] |
Data are presented as mean (±SD) or median [IQR] or proportions (%). M/F: Males/Females, BMI: body mass index, HbA1c: glycated hemoglobin A1c, ACR: urinary albumin-to-creatinine ratio, iGFR: iothalamate glomerular filtration rate, VvInt: cortical interstitial fractional volume.
Gene expression signatures associated with VvInt
Protocol kidney biopsy specimens were used for Affymetrix-based gene expression profiling of the tubulointerstitial compartment. Weighted Gene Co-expression Network Analysis (WGCNA)21 was used to define co-expressed gene sets (modules) and their relationships with VvInt, iGFR, and ACR (Figure 1). The protocol first identifies modules or groups of genes that preserve their correlation structure within each module, and then merges these modules based on their module eigengene (ME) profile (Step1). This approach reduces the dimension of the dataset and helps to define relevant functional associations. Using this method, we identified 11 functional modules in the tubulointerstitial expression data set. The module sizes ranged from 129 to 2378 transcripts. The MEs were then tested for their association with clinical and morphometric traits measured at the time of biopsy, (Step2), minimizing multiple testing penalties (Figure 2). MEs significantly associated with the structural parameter VvInt (p ≤0.05) are highlighted in Figure 2. Some MEs that showed significant correlation with VvInt were also associated with iGFR and ACR at the time of biopsy, though to a lesser extent.
Figure 2.
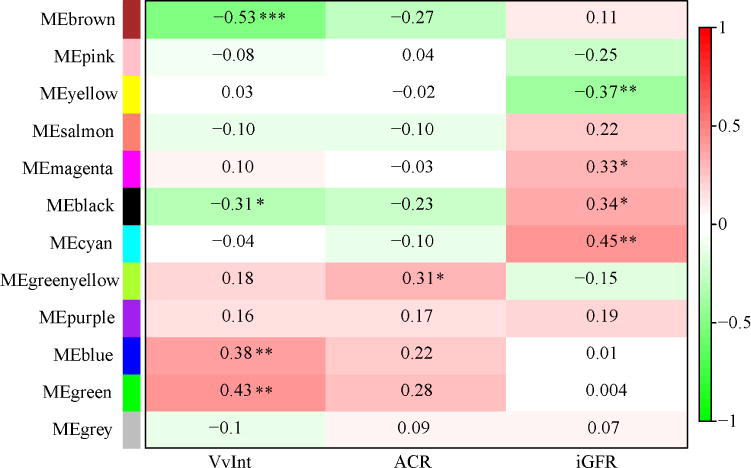
Association of module eigengene (ME) to VvInt and ACR/iGFR at the time of biopsy. 11 co-expressed modules were identified from tubulointerstitial gene expression profiles (n=49) in kidney tissue from Pima Indians. VvInt associated modules with a q-value ≤0.05 are considered significant (MEbrown, MEblack, MEblue and MEgreen). Green color indicates a negative correlation and red color indicates a positive correlation. The number inside each box is the correlation coefficient (Pearson correlation). Significance is denoted as *. The grey module collects the genes that do not share similar expression pattern.
Four modules in Figure 2 (Black, Blue, Brown and Green) showed the strongest eigenvector associations with VvInt (P value ≤ 0.05). These modules contained 484, 1664, 1617, and 1118 co-expressed genes respectively. Of the 1843 genes significantly correlated with VvInt (q-value ≤ 0.05) in these four modules, 913 co-expressed transcripts in the brown and black modules correlated negatively with VvInt and 930 transcripts found in the blue and green modules correlated positively with VvInt (Supplementary Table S1). Twenty-one genes from the black module that correlated negatively with VvInt showed positive correlation with iGFR at the time of biopsy; 123 genes from the green module that correlated positively with VvInt showed positive correlations with ACR (Supplementary Table S2).
Transcriptional interaction network and functional context of VvInt associated transcripts
VvInt associated transcripts from the 4 modules were then tested for known interactions using a co-citation network approach as implemented in Genomatix Pathway System (https://www.genomatix.de). Genes were scanned on PubMed indexed publications for association with each other on a sentence level through a functional attribute (e.g., “A induces B”). The 100 transcripts with the highest co-citation connectivity were displayed separately for positive and negative correlation with VvInt in co-citation networks (Figure 3 A and B). Major network subdomains included inflammatory signaling mediators (e.g., CCL2, and ICAM1), cell cycle control and proliferation mediators (e.g., TP53), and growth factor related signaling mechanisms (e.g., EGF and VEGF), suggesting activation of these transcriptional programs in the early stages of DKD. The VvInt associated gene sets were then analyzed for the gene ontology (GO) traits “cellular compartment” and “biological processes”. Transcripts correlated positively with VvInt were enriched with immune regulatory functions, cell activation, as well as cellular compartments including focal adhesion, extracellular matrix and space, and plasma membrane. Transcripts correlated negatively with VvInt were enriched with cellular pathways localized within specialized cellular organelles such as mitochondria and peroxisomes.
Figure 3.

Transcriptional networks generated from the top 100 highly connected VvInt correlated transcripts. The connections or edges between the transcripts are established by a function word (inhibition, activation, regulation) derived from the known literature via Natural Language Processing. A) Positively-correlated VvInt network and B) Negatively-correlated VvInt network.
Pathway analysis was performed to define the functional context of the VvInt-associated transcripts. Significant enrichment with migratory, inflammatory and cell-cell/cell-matrix interaction pathways was found in the transcripts that correlated positively with VvInt. In contrast, transcripts that correlated negatively with VvInt showed significant enrichment for PXR/RXR Activation, FXR/RXR Activation pathways and metabolic pathways, turnover of amino acids, sugars, and lipids.
Pathway network associated with VvInt
The complex interaction structure among disease-associated pathways was visualized by constructing a VvInt-associated pathway network from the 53 significantly enriched pathways using the 1843 VvInt-related transcripts as the analysis input (Figure 4). In this representation, highly interconnected pathways (i.e. multiple shared genes) were aggregated into subclusters/domains. A clear bowtie structure of the VvInt-associated pathways emerged. Enzymatic and metabolic pathways co-aggregated, implying that their expression changes are associated with greater tubulointerstitial damage in early DKD. Additionally, amino acid and lipid metabolism pathways intersected with detoxification pathways. In the second subdomain, signaling mechanisms including extracellular matrix and growth factor signaling such as “Inhibition of Angiogenesis by TSP1” co-aggregated, also implicating the interaction of these pathways in early disease manifestation. In the VvInt pathway network, a number of pathways were densely interconnected throughout the entire network (Figure 4). This trend was also reflected in the hierarchy of genes shared across multiple pathways. Most of the genes were private or shared between only 2 or 3 pathways. A small subgroup, however, showed high connectivity among the VvInt-associated pathways, including PRKCQ, NFKB1, MAPK8, ALDH2, and RAC1. The capacity of these genes to connect multiple processes and signaling mechanisms suggests a more central role in coordinating phenotypic features and clinical traits of potential interest.
Figure 4.
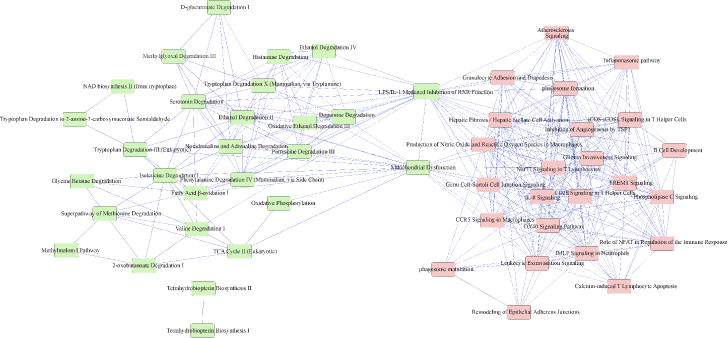
Ingenuity Pathway network showing significant pathways (P-value ≤0.05) enriched by VvInt correlated transcripts. Pathways are connected by 1 or more shared genes. The network displays two principal domains driven by negatively correlated (left) and positively correlated (right) transcripts. The nodes (pathways) are connected by (edges) genes shared between them
Identification of upstream regulators
The presence of a complex interconnected network suggests the possibility of a potential causal upstream mechanism activated in this dense network that affects the observed expression patterns in these downstream functions. We extracted 229 genes that were shared between more than one pathway (Supplementary Table S3). Using a causal network inference approach22 IL1β was identified as the master regulator of inflammation in our data set, affecting the expression of more than 50% of these shared gene sets via intermediate regulators. A separate set of five transcriptional regulators (PHF1, SOX2, NFAT5, TRIM29, HEY1), linked with the common intermediate transcriptional regulator TP53, can modulate downstream targets associated with differentiated tubule function and oxidative stress.
VvInt-correlated transcripts associated with long-term clinical disease course
To further elucidate the relationships between early structural damage, associated gene expression, and disease progression, VvInt expression modules were tested for their association with iGFR and ACR over time. The median [IQR] observation period for study participants was 15.9 [2.5] years, with a median follow up of 10.1 [2.0] years after kidney biopsy (Figure 5). From the 1843 VvInt-related transcripts within the four identified modules described above, 787 and 466 genes correlated with at least three iGFR and ACR measurements, respectively, over a median 10 year follow up after biopsy (P-value ≤ 0.05). We also evaluated the association of the VvInt modules with DKD progression defined by the slopes of iGFR and ACR. 33% of the VvInt associated genes in the black and brown module were associated significantly with iGFR slope (Supplementary Table S1). None of the four modules showed significant association with ACRslope.
Figure 5.
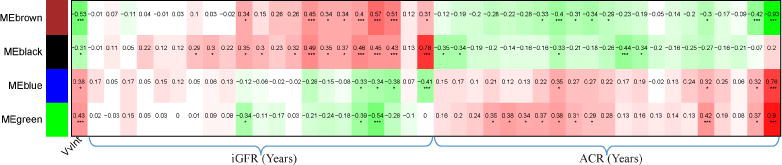
Association of four VvInt correlated modules and long-term clinical outcomes (ACR/iGFR). r ≥0.3 and P-value ≤0.05 are considered significant. Green color indicates a negative and red color indicates a positive correlation of modules to outcomes. The number inside each box is the correlation coefficient (Pearson correlation). Significance is denoted as *
To test the ability of this approach to identify non-invasive surrogates of VvInt linked to long-term clinical outcomes, a pilot study measured urinary EGF (uEGF) as a candidate non-invasive biomarker of renal function in the subset of patients with urine samples available from time of biopsy (n=46 out of 49 samples). Intra-renal EGF mRNA correlated positively (r=0.43, p-value <0.0001) with the urinary protein levels suggesting that intra-renal level of EGF is captured by the urinary protein. uEGF levels were strongly correlated with baseline kidney function, iGFR (r =0.47, p-value <0.0001) and with iGFR slope (r= 0.25, p-value =0.01), indicating its potential utility as a non-invasive biomarker capturing the disease progression.
Relationship of VvInt-associated transcripts in early DKD with differential expression in advanced DKD
To determine how gene expression patterns observed in the Pima early DKD biopsies compared with those of more advanced DKD, differential gene expression patterns of VvInt-associated transcripts from the Pima protocol biopsies were compared with tubulo-interstitial expression patterns in advanced clinically indicated biopsies (n=17) from a European (ERCB) DKD cohort. 1302 of the 1843 (71%) VvInt-related transcripts were significantly differentially expressed (q-value ≤ 0.05) in advanced DKD when compared with the living kidney donors (N=31). Strikingly, 1287 of the 1302 differentially-expressed transcripts in advanced DKD showed concordant changes with disease. Figure 6 highlights the preserved regulatory nodes from the VvInt-transcriptional network in advanced DKD.
Figure 6.
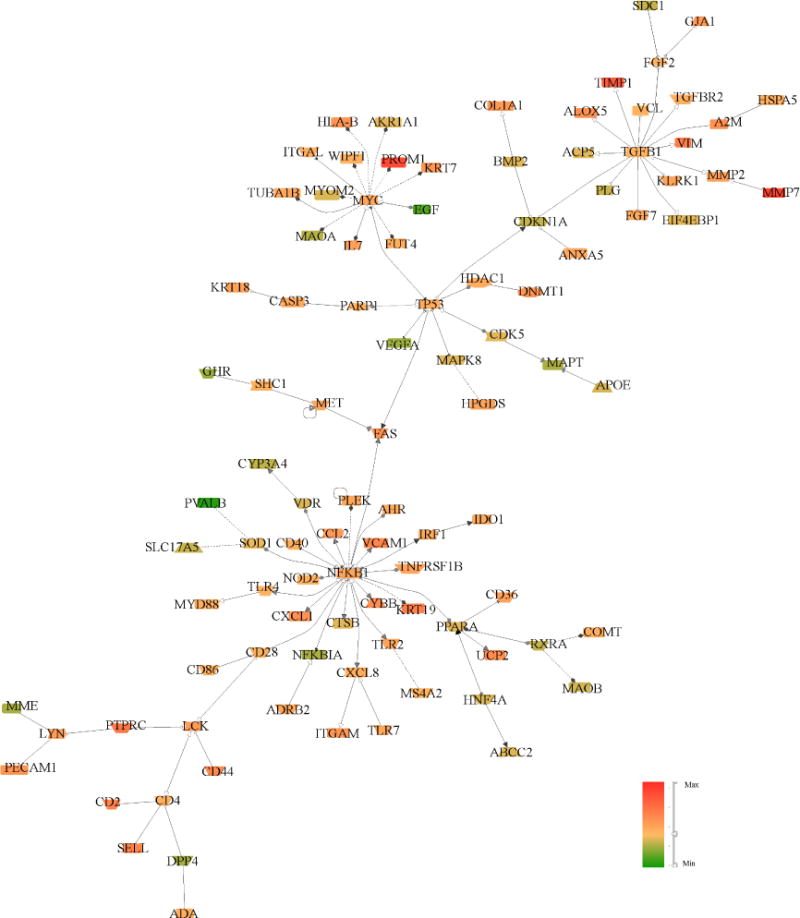
Transcriptional network generated from differentially expressed transcripts comparing ERCB DKD with healthy controls. The network represents the top 100 highly connected transcripts. The main nodes seen in the morphogenomic study in early DKD in Pima Indians are maintained and directionality of expression changes is conserved in 98.85% of nodes. The node color represents the direction of expression in DKD. Green = Decreased, Red = Increased levels compared to controls.
DISCUSSION
In this study, we developed a strategy combining genome-wide intra-renal gene expression profiling, quantitative morphometric analysis, and clinical outcome data to explore pathways underlying early “morphogenomic” changes in DKD. The protocol biopsies obtained from a relatively homogenous Pima Indian population permitted detection of signaling and metabolic interactions activated in early DKD. These interactions were then linked with both structural lesions in the cortical interstitium and long-term disease outcomes. Transcripts positively correlated with VvInt showed enrichment for inflammatory mechanisms, while those negatively correlated with VvInt showed enrichment for metabolic processes.
Our study links structure and function by combining molecular and morphometric data in early DKD with a goal of identifying potential targets for therapeutic intervention before irreversible loss of kidney function has occurred. The resulting gene expression sets were further assessed for disease relevance by testing the VvInt-linked transcripts for their associations with long-term functional outcomes (iGFR/ACR trajectories) over the next decade of follow-up post biopsy. The extended follow-up of the Pima protocol biopsy cohort made a direct replication of the findings in a study with parallel design difficult. However, a significant proportion of the implicated transcripts were also differentially expressed in a European cohort with more advanced DKD. The concordant regulation of a significant segment of the VvInt associated transcripts in a different ethnic and environmental background supports the relevance of the identified molecular mechanism beyond the Pima Indian cohort. This is particularly relevant as Pima Indians develop diabetes at an earlier age range from the typical Type 2 DM Caucasian patient with DKD and lack many of the co-morbidities seen in other populations. In addition, the Pima study cohort had a significant exposure to RAS blockade. These findings suggest that molecular-morphometric approaches can capture relevant regulatory events at an early stage of disease when intervention may be more effective than at later stages of disease progression.
Combining differentially expressed genes into functional categories of coordinated regulation, referred to as pathway analysis, facilitates the discovery of individual system components in a given tissue. This approach has been applied in previous gene expression studies in human and murine DKD, which focused primarily on differentially expression between DKD from indication biopsies and normal kidneys16,17,19,23. These prior studies elucidated the role of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and Jak/STAT signaling which was established as a pathogenetic contributor to DKD progression16,17,23–25. Generating DKD pathway maps from gene expression studies provided part of the rationale for testing the JAK1 and JAK2 inhibitor, Baricitinib, as a potential therapy in advanced DKD (ClinicalTrials.gov number NCT01683409). Baricitinib significantly reduced the level of urine albumin excretion in a dose dependent manner compared with placebo after six months of treatment, with a sustained benefit noted four weeks after discontinuing Baricitinib26. In addition, reduction in blood and urinary markers of JAK-STAT activation predicted from the gene expression profiling studies preceded the albuminuria reduction, demonstrating the power of this approach to identify targets together with their engagement biomarkers.
A single process or pathway is rarely the sole determinant of disease progression. Through the simultaneous assessment and integration of information across multiple pathways, it is possible to describe relevant pathway interactions in complex disease processes. This approach facilitates the identification of regulatory bottlenecks that could be evaluated as potential drug targets or as markers of integrated disease activity. In our study, the individual structure-function pathways were mapped to each other using a matrix of shared genes among the pathways, establishing a pathway network of early DKD. This network illustrated in Figure 4 shows a bowtie-shaped structure tying together two distinct clusters of gene activity, linking tubular dedifferentiation and inflammation processes with two pathways, ‘mitochondrial dysfunction’ and ‘LPS/IL-1 mediated inhibition of RXR function’.
In the inflammation cluster, IL1β was identified as the dominant upstream regulator affecting multiple pathways. IL1β-dependent mechanisms are well characterized in DKD model systems of advanced tubular cell dysfunction27,28, where they link apoptosis and innate immune activation. IL1β-dependent transcripts are well established downstream signaling elements of the inflammasome, which is activated in progressive loss of tubule function29.
In the dedifferentiation cluster, a set of five transcription factors was identified as potential upstream regulators of the tubular differentiation pathways. The interplay between the 5 master regulators (PHF1, SOX2, NFAT5, TRIM29, and HEY1) and TP53 could affect downstream targets involved in oxidative phosphorylation, mitochondrial dysfunction, and production of nitric oxide and reactive oxygen species in macrophages. Indeed, proximal tubule cell-specific TP53 deletion resulted in decreased oxidative stress, reduced macrophage infiltration and tubule structural damage30. An additional speculative association involves NFAT5, a transcription factor which has both tonicity-dependent and independent functions31. NFAT5 might play an important role in linking diabetic hyperosmotic stress to disease progression by instigating the release of pro-inflammatory cytokines from tissue resident immune and epithelial cells.
Gene expression studies capture the transcriptional response of the tissue to the local and systemic effects of the disease processes currently active in DKD. ‘Causal inference’ network approaches can be used to infer the functional state of the transcriptional regulatory mechanism, linking gene expression changes with activation of upstream regulators22. Using these approaches, we were able to link kidney mRNA with urinary protein profiles in cohorts with established CKD, including DKD, and identify and cross-validate novel disease progression predictors32. Thus, by using this strategy, the transcriptional profile of early DKD reported in this study for the first time can be used as a framework to guide future experimental efforts. The preliminary data presented above linking tissue EGF mRNA with urinary EGF levels, cross-sectional iGFR and prospective loss of renal function support this strategy.
By anchoring our gene signatures on a single morphometric feature (VvInt-cortical interstitial fractional volume) in patients with early DKD and key outcome-related clinical variables (e.g. ACR, iGFR) over an extended observation period, we provided a functional context for the expression changes. The significant association of VvInt–linked genes with the future changes of ACR and iGFR may enable identification of urinary markers reflecting the intra–renal disease processes active in patients with disease activity undetectable by conventional clinical means.
In summary, we applied an integrated strategy that combines genome-wide intra-renal gene expression profiling, quantitative morphometric analysis, and clinical outcomes data, to explore potential novel pathways underlying early structural changes in DKD. VvInt-related transcripts showed significant enrichment for inflammatory mechanisms and for substrate metabolic processes in the renal tubule. The association of VvInt–linked genes with long-term clinical outcomes (ACR and iGFR) validated the relevance of the early processes to DKD progression. Critically, this approach may enhance our ability to identify urinary markers of intra–renal disease processes active in patients with minimal or clinically undetectedDKD.
MATERIALS AND METHODS
From 1965–2007, Pima Indians from the Gila River Indian Community participated in a longitudinal study of diabetes and its complications. 169 adults with type 2 diabetes from this population participated in a prospective randomized, double-blinded, placebo-controlled interventional clinical trial (ClinicalTrials.gov number, NCT00340678). The purpose of the trial was to evaluate the renoprotective efficacy of the angiotensin receptor blocker losartan in type 2 diabetes. The clinical trial included annual measurements of GFR by the urinary clearance of iothalamate9,33. iGFR was expressed in ml/min and was not adjusted for body surface area (BSA) to avoid underestimating GFR due to significant obesity, as reported elsewhere34–37. A kidney biopsy was performed at the end of the 6-year treatment period38. Upon completion of the clinical trial, participants were returned to the care of their primary physicians and annual research examinations that included measurement of iGFR were continued.
Of the 111 participants who underwent ultrasound guided protocol kidney biopsy at the end of the treatment period, 49 provided sufficient tissue for tubule-interstitial gene expression analysis used in this study. For morphometric assessments, unbiased random sampling methods were used to measure structural parameters. Measurements were performed by an investigator (EJW) who was masked to the clinical data. Tissue was processed and embedded in epoxy resin (Epon 812). Digital light and electron micrographs were used to make measurements using formal stereologic methods to account for two-dimensional sampling of three-dimensional objects39
Twelve predefined renal parameters were assessed including cortical interstitial fractional volume (VvInt). VvInt was measured using light microscopy by intercept counting and expressed as percentage of the total cortical area. Fifteen photomicrographs were selected randomly from the renal cortex using a 15-μm counting grid containing 225 cells. A total of 3375 intercepts were counted per subject38,40. Morphometric variables for each individual were calculated as the mean of all measurements for that individual.
Demographics, anthropomorphic measurements, and kidney function data were assessed at enrollment and on an annual basis thereafter. Urine albumin excretion was expressed as the ACR in mg/g creatinine. This study was approved by the Institutional Review Board of the National Institute of Diabetes and Digestive and Kidney Diseases. Each participant signed an informed consent document.
Microarray target preparation, quality assessment and data preprocessing
Kidney biopsy tissue procurement and gene expression profiling was performed as described previously17,41 on Affymetrix GeneChip Array Human Genome series U133A and Plus 2.0 (Affymetrix, Inc., Santa Clara, CA). Affymetrix image files were obtained, processed, normalized and batch corrected as described previously42. Owing to ethical considerations, privacy protection, and to avoid identifying individual study participants in this vulnerable population, the Institutional Review Board of the National Institute of Diabetes and Digestive and Kidney Diseases has stipulated that individual-level gene expression and genotype data from this study cannot be made publicly available.
Construction of Gene Co-expression modules
WGCNA implemented in R to construct gene co-expression modules21. This method employs a systematic sequential approach starting with pairwise Pearson correlations among all the genes in the expression matrix. Modules (i.e. clusters) of highly interconnected genes are constructed from a Topological Overlap Matrix (TOM) and subsequent hierarchical clustering. The pairs that share higher topological overlap are combined into 11 co-expression modules and one module containing all remaining genes. The next step constructs a hypothetical module eigengene ME, a summarized vector for each module to represent the gene expression profiles within each module. Finally, we correlated the MEs to clinical traits and focused further analysis on those modules with MEs significantly correlated with the trait (P-value ≤ 0.05). Transcripts contained in these significant modules with significant trait associations are used for downstream functional analysis.
In our compartment-specific analysis, the tubulointerstitial ultrastructural lesion most strongly associated with transcriptional regulation was cortical interstitial fractional volume (VvInt), an index of interstitial fibrosis and the focus of this study. Ultrastructural glomerular alterations are considered to be early hallmark changes of DKD, but interstitial fibrosis and associated tubular damage are also well-established parameters linked with long-term disease progression, especially in type 2 patients43–45.
Pathway analysis and upstream regulator analysis approach
Ingenuity Pathway System software tool (Ingenuity Systems, Redwood City, CA) was used to define functional concepts and pathways enriched in the retrieved gene sets, Causal Network Analysis (CNA) implemented in Ingenuity Pathway Analysis was used to derive the upstream regulators affecting the observed expression changes in the shared genes. The shared genes were extracted from the pathway network, where at least two pathways were involved. The CNA method screens for possible upstream regulators that explain the expression changes in the downstream targets through more than one intermediate regulator. Transcriptional networks integrating literature-derived knowledge were established using GePS from the Genomatix software suite (https://www.genomatix.de).
Differential Analysis
We used the Significance Analysis of Microarrays (SAM) implemented in MeV (mev.tm4.org) suite to identify differentially expressed gene sets with an FDR of ≤0.05 as the significance threshold.
Supplementary Material
Supplementary Figure S1: Time course plot of (A) iGFR (ml/min, log2 transformed) and (B) ACR (mg/g, log2 transformed) over time in 49 Pima Indians.
Supplementary Table S1: 1843 tubulointerstitial transcripts significantly correlated with cortical interstitial fractional volume from four modules in Pima Indians. r, Pearson product-moment correlation coefficient; sorted first on module color and then by P-value ≤ 0.05. Asterisk (*) in the Gene Symbol column indicates the gene is also significantly correlated with GFRslope.
Supplementary Table S2: Subset of the VvInt correlated transcripts associated with iGFR (21 genes) and ACR (123 genes) at time of biopsy in Pima Indians. r, Pearson product-moment correlation coefficient; P-value ≤ 0.05.
Supplementary Table S3: The list of 229 genes extracted from the cortical interstitial fractional volume enriched pathway network and their assignment to the enriched pathways. These genes are shared between more than one pathway.
{kind=link}
Acknowledgments
This work was performed in partial fulfillment of the requirements for the doctoral work of V.N. from the Medical Faculty of Ludwig-Maximilians-University Munich, Germany. This work was supported by the German Research Foundation (KO 4266/1-1) and by an American Heart Association Postdoctoral Fellowship (13POST14400000) to C.V.K., by an American Diabetes Association Clinical Science Award (1-08-CR-42) to R.G.N, by the National Institute of Diabetes and Digestive and Kidney Diseases to M.K. (DK083912, DK082841, DK020572, DK092926), by Boehringer Ingelheim and by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases. This research project received further bioinformatics support from the Applied Systems Biology Core of the University of Michigan George M. O`Brien Kidney Research Core Center (P30-DK081943).
The authors are indebted to the members of the Gila River Indian Community for participating in this investigation, and facilitating this study. We gratefully acknowledge the nurses, doctors and support staff of the Diabetes Epidemiology and Clinical Research Section involved in collecting and processing the data. We further acknowledge the assistance of Michael Mauer, MD, and his team in the Department of Pediatric Nephrology at the University of Minnesota, Minneapolis, MN, and the Beckman Center at Stanford University for processing and imaging the kidney tissue. We also acknowledge funding from the Else-Kroener Fresenius Foundation and all participating centers of the European Renal cDNA Bank – Kroener-Fresenius Biopsy Bank (ERCB–KFB). We acknowledge Shahaan Smith for the urinary EGF measurements using the ELISA technology
M.S. and C.M.B.K. have been employees of Boehringer-Ingelheim at time of conduct of this study. Boehringer-Ingelheim provided an unrestricted research grant to MK for part of the study. E.J.W and R.G.N are employees of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
The remaining authors declare no competing financial interest relevant to this research article.
Author Contributions V.N, C.V.K., P.J.N., M.K., and R.G.N. designed and performed experiments, analyzed data and wrote the manuscript. B.Y., K.V.L, E.J.W. provided human biosamples, collected epidemiological study data and edited the manuscript. B.G processed the tissue and edited the manuscript. V.N and W.J designed and analyzed the urinary EGF data M.S., C.M.B.K., J.H. and J.L.H interpreted and edited the manuscript. M.K. and R.G.N. are the guarantors of the work as a whole including the study design, data access/analysis, and the final decision to submit and publish the research, as such, take full responsibility for the integrity of the data and the accuracy of the data analysis. All the authors read the manuscript, discussed the results, and approved the manuscript. Supplementary information is available at Kidney International’s website
References
- 1.Collins AJ, Foley RN, Herzog C, et al. US Renal Data System 2012 Annual Data Report. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2013;61(1 Suppl 1):A7–e1. doi: 10.1053/j.ajkd.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 2.Saran R, Li Y, Robinson B, et al. US Renal Data System 2014 Annual Data Report: Epidemiology of Kidney Disease in the United States. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2015;66(1 Suppl 1):Svii, S1–305. doi: 10.1053/j.ajkd.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhuo X, Zhang P, Barker L, et al. The lifetime cost of diabetes and its implications for diabetes prevention. Diabetes care. 2014;37(9):2557–2564. doi: 10.2337/dc13-2484. [DOI] [PubMed] [Google Scholar]
- 4.de Boer IH, Rue TC, Cleary PA, et al. Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: an analysis of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications cohort. Archives of internal medicine. 2011;171(5):412–420. doi: 10.1001/archinternmed.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Boer IH, Rue TC, Hall YN, et al. Temporal trends in the prevalence of diabetic kidney disease in the United States. Jama. 2011;305(24):2532–2539. doi: 10.1001/jama.2011.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fox CS, Matsushita K, Woodward M, et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without diabetes: a meta-analysis. Lancet (London, England) 2012;380(9854):1662–1673. doi: 10.1016/S0140-6736(12)61350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groop PH, Thomas MC, Moran JL, et al. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes. 2009;58(7):1651–1658. doi: 10.2337/db08-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orchard TJ, Secrest AM, Miller RG, Costacou T. In the absence of renal disease, 20 year mortality risk in type 1 diabetes is comparable to that of the general population: a report from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetologia. 2010;53(11):2312–2319. doi: 10.1007/s00125-010-1860-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lemley KV, Boothroyd DB, Blouch KL, et al. Modeling GFR trajectories in diabetic nephropathy. American journal of physiology Renal physiology. 2005;289(4):F863–870. doi: 10.1152/ajprenal.00068.2004. [DOI] [PubMed] [Google Scholar]
- 10.Levey AS, Cattran D, Friedman A, et al. Proteinuria as a surrogate outcome in CKD: report of a scientific workshop sponsored by the National Kidney Foundation and the US Food and Drug Administration. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2009;54(2):205–226. doi: 10.1053/j.ajkd.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 11.Kramer HJ, Nguyen QD, Curhan G, Hsu CY. Renal insufficiency in the absence of albuminuria and retinopathy among adults with type 2 diabetes mellitus. Jama. 2003;289(24):3273–3277. doi: 10.1001/jama.289.24.3273. [DOI] [PubMed] [Google Scholar]
- 12.Lemley KV, Abdullah I, Myers BD, et al. Evolution of incipient nephropathy in type 2 diabetes mellitus. Kidney international. 2000;58(3):1228–1237. doi: 10.1046/j.1523-1755.2000.00223.x. [DOI] [PubMed] [Google Scholar]
- 13.Pavkov ME, Knowler WC, Hanson RL, et al. Predictive power of sequential measures of albuminuria for progression to ESRD or death in Pima Indians with type 2 diabetes. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2008;51(5):759–766. doi: 10.1053/j.ajkd.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavkov ME, Mason CC, Bennett PH, et al. Change in the distribution of albuminuria according to estimated glomerular filtration rate in Pima Indians with type 2 diabetes. Diabetes care. 2009;32(10):1845–1850. doi: 10.2337/dc08-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perkins BA, Ficociello LH, Silva KH, et al. Regression of microalbuminuria in type 1 diabetes. The New England journal of medicine. 2003;348(23):2285–2293. doi: 10.1056/NEJMoa021835. [DOI] [PubMed] [Google Scholar]
- 16.Berthier CC, Zhang H, Schin M, et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes. 2009;58(2):469–477. doi: 10.2337/db08-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmid H, Boucherot A, Yasuda Y, et al. Modular activation of nuclear factor-kappaB transcriptional programs in human diabetic nephropathy. Diabetes. 2006;55(11):2993–3003. doi: 10.2337/db06-0477. [DOI] [PubMed] [Google Scholar]
- 18.Wiggins JE, Patel SR, Shedden KA, et al. NFkappaB promotes inflammation, coagulation, and fibrosis in the aging glomerulus. Journal of the American Society of Nephrology: JASN. 2010;21(4):587–597. doi: 10.1681/ASN.2009060663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woroniecka KI, Park AS, Mohtat D, et al. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60(9):2354–2369. doi: 10.2337/db10-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. American journal of kidney diseases: the official journal of the National Kidney Foundation. 1992;20(1):1–17. doi: 10.1016/s0272-6386(12)80312-x. [DOI] [PubMed] [Google Scholar]
- 21.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics (Oxford, England) 2014;30(4):523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hodgin JB, Nair V, Zhang H, et al. Identification of cross-species shared transcriptional networks of diabetic nephropathy in human and mouse glomeruli. Diabetes. 2013;62(1):299–308. doi: 10.2337/db11-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nature reviews Nephrology. 2011;7(6):327–340. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- 25.Ortiz-Munoz G, Lopez-Parra V, Lopez-Franco O, et al. Suppressors of cytokine signaling abrogate diabetic nephropathy. Journal of the American Society of Nephrology: JASN. 2010;21(5):763–772. doi: 10.1681/ASN.2009060625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brosius FC, Tuttle KR, Kretzler M. JAK inhibition in the treatment of diabetic kidney disease. Diabetologia. 2016;59(8):1624–1627. doi: 10.1007/s00125-016-4021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorenz G, Darisipudi MN, Anders HJ. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2014;29(1):41–48. doi: 10.1093/ndt/gft332. [DOI] [PubMed] [Google Scholar]
- 28.Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nature reviews Nephrology. 2016;12(1):13–26. doi: 10.1038/nrneph.2015.175. [DOI] [PubMed] [Google Scholar]
- 29.Anders HJ. Of Inflammasomes and Alarmins: IL-1beta and IL-1alpha in Kidney Disease. Journal of the American Society of Nephrology: JASN. 2016;27(9):2564–2575. doi: 10.1681/ASN.2016020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ying Y, Kim J, Westphal SN, et al. Targeted deletion of p53 in the proximal tubule prevents ischemic renal injury. Journal of the American Society of Nephrology: JASN. 2014;25(12):2707–2716. doi: 10.1681/ASN.2013121270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neuhofer W. Role of NFAT5 in inflammatory disorders associated with osmotic stress. Current genomics. 2010;11(8):584–590. doi: 10.2174/138920210793360961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ju W, Nair V, Smith S, et al. Tissue transcriptome-driven identification of epidermal growth factor as a chronic kidney disease biomarker. Science translational medicine. 2015;7(316):316ra193. doi: 10.1126/scitranslmed.aac7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemley KV, Blouch K, Abdullah I, et al. Glomerular permselectivity at the onset of nephropathy in type 2 diabetes mellitus. Journal of the American Society of Nephrology: JASN. 2000;11(11):2095–2105. doi: 10.1681/ASN.V11112095. [DOI] [PubMed] [Google Scholar]
- 34.Chagnac A, Weinstein T, Korzets A, et al. Glomerular hemodynamics in severe obesity. American journal of physiology Renal physiology. 2000;278(5):F817–822. doi: 10.1152/ajprenal.2000.278.5.F817. [DOI] [PubMed] [Google Scholar]
- 35.Delanaye P, Radermecker RP, Rorive M, et al. Indexing glomerular filtration rate for body surface area in obese patients is misleading: concept and example. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2005;20(10):2024–2028. doi: 10.1093/ndt/gfh983. [DOI] [PubMed] [Google Scholar]
- 36.Levey AS, Kramer H. Obesity, glomerular hyperfiltration, and the surface area correction. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2010;56(2):255–258. doi: 10.1053/j.ajkd.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 37.Schmieder RE, Beil AH, Weihprecht H, Messerli FH. How should renal hemodynamic data be indexed in obesity? Journal of the American Society of Nephrology: JASN. 1995;5(9):1709–1713. doi: 10.1681/ASN.V591709. [DOI] [PubMed] [Google Scholar]
- 38.Weil EJ, Fufaa G, Jones LI, et al. Effect of losartan on prevention and progression of early diabetic nephropathy in American Indians with type 2 diabetes. Diabetes. 2013;62(9):3224–3231. doi: 10.2337/db12-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weibel ER. Practical Methods for Biological Morphometry. New York: Academic Press; 1979. [Google Scholar]
- 40.Weil EJ, Lemley VK, Mason CC, et al. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney International. 2012;8(9):1010–1017. doi: 10.1038/ki.2012.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cohen CD, Frach K, Schlondorff D, Kretzler M. Quantitative gene expression analysis in renal biopsies: a novel protocol for a high-throughput multicenter application. Kidney international. 2002;61(1):133–140. doi: 10.1046/j.1523-1755.2002.00113.x. [DOI] [PubMed] [Google Scholar]
- 42.Martini S, Nair V, Keller BJ, et al. Integrative biology identifies shared transcriptional networks in CKD. Journal of the American Society of Nephrology: JASN. 2014;25(11):2559–2572. doi: 10.1681/ASN.2013080906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bader R, Bader H, Grund KE, et al. Structure and function of the kidney in diabetic glomerulosclerosis. Correlations between morphological and functional parameters. Pathology, research and practice. 1980;167(2-4):204–216. doi: 10.1016/S0344-0338(80)80051-3. [DOI] [PubMed] [Google Scholar]
- 44.Bohle A, Wehrmann M, Bogenschutz O, et al. The pathogenesis of chronic renal failure in diabetic nephropathy. Investigation of 488 cases of diabetic glomerulosclerosis. Pathology, research and practice. 1991;187(2-3):251–259. doi: 10.1016/s0344-0338(11)80780-6. [DOI] [PubMed] [Google Scholar]
- 45.Bonventre JV. Can we target tubular damage to prevent renal function decline in diabetes? Seminars in nephrology. 2012;32(5):452–462. doi: 10.1016/j.semnephrol.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1: Time course plot of (A) iGFR (ml/min, log2 transformed) and (B) ACR (mg/g, log2 transformed) over time in 49 Pima Indians.
Supplementary Table S1: 1843 tubulointerstitial transcripts significantly correlated with cortical interstitial fractional volume from four modules in Pima Indians. r, Pearson product-moment correlation coefficient; sorted first on module color and then by P-value ≤ 0.05. Asterisk (*) in the Gene Symbol column indicates the gene is also significantly correlated with GFRslope.
Supplementary Table S2: Subset of the VvInt correlated transcripts associated with iGFR (21 genes) and ACR (123 genes) at time of biopsy in Pima Indians. r, Pearson product-moment correlation coefficient; P-value ≤ 0.05.
Supplementary Table S3: The list of 229 genes extracted from the cortical interstitial fractional volume enriched pathway network and their assignment to the enriched pathways. These genes are shared between more than one pathway.