

Abstract
Poly(ethylene glycol) (PEG) can be functionalized and modified with various moieties allowing for a multitude of cross-linking chemistries. Here, we investigate how vinyl sulfone, acrylate, and maleimide functional end groups affect hydrogel formation, physical properties, viability of encapsulated cells, post-polymerization modification, and inflammatory response of the host. We have shown that PEG-VS hydrogels, in the presence of a co-monomer, N-vinyl-2-pyrrolidone (NVP), form more efficiently than PEG-Ac and PEG-Mal hydrogels, resulting in superior physical properties after 6 minutes of ultraviolet light exposure. PEG-VS hydrogels exhibited hydrolytic stability and non-fouling characteristics, as well as the ability to be modified with biological motifs, such as RGD, after polymerization. Additionally, unmodified PEG-VS hydrogels resulted in lesser inflammatory response, cellular infiltration, and macrophage recruitment after implantation for 28 days in mice. These findings show that altering the end group chemistry of PEG macromer impacts characteristics of the photo-polymerized network. We have developed a tunable non-degradable PEG system that is conducive for cell or tissue encapsulation and evokes a minimal inflammatory response, which could be utilized for future immunoisolation applications.
Keywords: poly(ethylene glycol), photo-polymerization, functional groups, RGD modification, inflammatory response
Graphical Abstract
1. Introduction
The use of synthetic hydrogels presents a unique opportunity for tissue engineering applications, because the polymer network can be tuned to mimic the microenvironment of the native tissue to achieve desired function [1–3]. Poly(ethylene glycol) (PEG) is one of the most studied and widely utilized biomaterial for synthetic hydrogels. Due to its high-water content and non-fouling properties, PEG based hydrogels are highly biocompatible and tunable [2,4]. Additionally, the viscoelastic properties of PEG hydrogels allow for expansion and growth of encapsulated tissue, which is vital for engineering a hydrogel based immunoisolating construct [5,6].
An immunoisolating construct or device creates a physical barrier around the implanted cells or tissues, precludes contact with immune cells from the host and prevents sensitization. For the immunoisolation of implanted allogeneic tissue, it is important for the network to be non-degradable yet porous enough to allow effective diffusion of nutrients and metabolites and, on the other hand, block transport of allo-antigens and cells out of the device and into the surrounding tissues of the recipient. An important design criteria when engineering an immunoisolating construct is to create an interface with the host tissues that elicits a minimal inflammatory response. Evoking a strong inflammatory response would result in a thick fibrotic capsule in addition to immune cell recruitment around the construct, thereby hindering function [7]. Due to its non-fouling properties, PEG coating and encapsulation has been shown to be a successful strategy to reduce acute inflammatory responses [8,9]. Additionally, PEG chain density, length, and conformation contribute to the ability to resist protein absorption [10–12].
Functionalization of the end hydroxyl groups of the otherwise inert PEG macromer with reactive end groups allows for a variety of crosslinking chemistries [13–17]. Multiple chemistries have been developed to functionalize PEG, such as vinyl sulfone, acrylate, amine, or maleimide end groups [18–22]. Presence of acrylate groups promotes the formation of a crosslinked network via photo-polymerization in the presence of a photo-initiator [13,23,24], while vinyl sulfone and maleimide end groups are used for Michael-type addition chemistry with cysteine containing enzyme sensitive peptides to create a proteolytically degradable network [16–18]. Additionally, PEG macromers can be modified with biological motifs such as integrin binding peptides to create a microenvironment conducive for cell attachment [25].
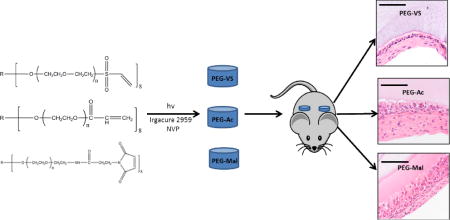
Crosslinking of hydrogels using photo-polymerization is expedient, results in hydrogels with superior physical properties for in situ encapsulation, and creates networks that are resistant to proteolytic degradation [23]. Furthermore, several groups have shown photo-polymerized systems are cytocompatible and useful for a multitude of engineering applications [26–34]. During the photo-polymerization process, light exposure creates free radicals in the presence of photo-initiators, which react as electron donors with the terminal double bonds and form crosslinked networks. Free radical mediated crosslinking is dependent on the reactivity of the free radical and the electrophilic nature of the available double bonds. Since this reaction is dependent on the presence of double bonds, we hypothesize that the chemistry of the functional end groups of a multi-arm PEG macromolecule will alter the susceptibility of double bonds to react with free radicals, therefore, affecting the integrity of the crosslinking network and physical properties of the hydrogel. To test this hypothesis, we prepared photo-polymerized vinyl sulfone (PEG-VS), acrylate (PEG-Ac), and maleimide (PEG-Mal) hydrogels (Figure 1) and assessed how changing chemistry of the functional end group of the multi-arm PEG backbone impacted the crosslinking efficiency, bulk properties, stability, cell compatibility, modification efficiency, and the degree of inflammatory response. Our end objective was to develop a tunable non-degradable PEG system that is conducive for cell or tissue encapsulation and evokes a minimal inflammatory response, which could be utilized for immunoisolation applications.
Figure 1.
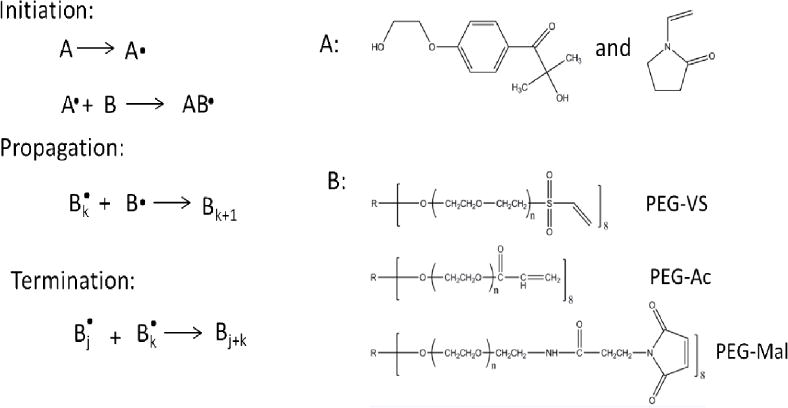
Chemical schematic of photo-polymerization of PEG-VS, PEG-Ac, and PEG-Mal in the presence of a photoinitiator.
2. Materials and Methods
2.1 Hydrogel Preparation
To form the hydrogel network, 2, 4, or 8-arm PEG-VS, PEG-Ac and PEG-Mal (JenKem Technology, Beijing, China) macromer powder was dissolved in sterile Dulbecco’s Phosphate Buffered Saline(DPBS) (pH 7.4, Gibco, USA) solution and 0.4 mg/100μL of photo-initiator Irgacure 2959 (Ciba, Basel, Switzerland, MW=224.3) to create a solid concentration of 5% w/v. Furthermore, a co-monomer, N-vinyl-2-pyrrolidone (NVP) (Sigma-Aldrich, USA), was added in some compositions at a final concentration of 0.1%, as NVP has been shown to enhance the gelation mechanism without impacting the cell compatibility [35]. To form the gels, the precursor solution (PEG, Irgacure 2959 and NVP (in some compositions)) was exposed to UV light at a constant intensity (1090 μW/cm2 at a distance of 4 cm) for a designated duration.
2.2 Gel Fractions of PEG- Hydrogels
5% w/v 2, 4, and 8-arm PEG-VS, PEG-Ac, and PEG-Mal hydrogels were tested with no NVP and no RGD (NVP(−), RGD(−)), with 0.1% v/v NVP and no RGD (NVP(+), RGD(−)), and with 0.1% v/v NVP and modified with 0.5 mM RGD (NVP(+), RGD(+)). PEG precursor solution of constructs containing 0.5 mM RGD was incubated with 0.5 mM RGD for 15 minutes in isotonic HEPES buffer prior to UV exposure. All precursor solutions were exposed to UV light (365 nm, 1090 μW/cm2 at a constant height of 4 cm) for 6 minutes and precursor solution containing NVP and no RGD were exposed for 6, 15, and 30 minutes. Gels were swelled overnight at room temperature to remove soluble monomers and lyophilized to remove water content. Dry mass was obtained and divided by theoretical solid content to quantify gel fraction (n=5 for all compositions).
2.3 NMR Analysis to Characterize RGD Conjugation
We evaluated the effect of the end-group chemistries on the efficiency of Michael-type addition of RGD to 8-arm 5% PEG-VS, PEG-Ac, and PEG-Mal. 5% w/v PEG precursor solution was incubated with 5 mM RGD for 15 minutes in HEPES buffer. A 5% w/v 8-arm PEG solution contains 10 mM reactive arms; so theoretically, half of the arms should be reacted when 5 mM RGD is added. After incubation, 1 mL of the respective precursor solutions were transferred to a dialysis tubing (3.5-5 kDa MWCO, Spectrum Laboratories, California, USA). Dialysis against Milli-Q water was performed overnight to remove unreacted RGD molecules. The solutions were then lyophilized and the solid content was dissolved in 600 μL of deuterium oxide and transferred to a respective NMR tube. The NMR data was acquired on a 500 MHz Varian VNMR spectrometer equipped with two high band and one low band radio frequency channels. All spectra were acquired at 25 °C.
2.4 Fourier Transform Infrared Spectroscopy (FTIR) Analysis
Samples of pure chemicals and powdered dry hydrogels (xerogels) were submitted to FTIR analysis and the spectra were obtained using a Bomem MB 100 FTIR Spectrophotometer applying the Potassium Bromide (KBr) disc method. 1.5 mg of the powdered dry hydrogels were mixed and grinded (sample/KBr ratio was kept at 1/50 constant) and then compressed into a pellet at a pressure of 11 t for about a minute, using the Graseby Specac Model: 15.011. Spectra were obtained in the 4000–400 cm−1 wave number range at 25 °C and at 4 cm−1 spectral resolution.
2.5 Swelling Ratio
The swelling ratios of PEG hydrogels with varying compositions were determined. The hydrogels were formed as described above and submerged in DPBS for 24 hours. After 24 hours, excess DPBS was removed and the swollen mass of the hydrogels was obtained. These hydrogels were then air dried at room temperature and lyophilized to remove any moisture. Larger volumes (100 μL) of hydrogels were used to minimize error from weighing. The mass swelling ratio (Qm) was determined by dividing the mass of the swollen gels (Ms) by the mass of the dry gels (Md). Results shown are mean ± SD.
2.6 Rheology
The storage moduli of PEG hydrogels with varying compositions were investigated. After formation, the hydrogels were swollen in DPBS overnight. A TA HR-2 rheometer (TA Instruments) equipped with a 20 mm parallel plates and a Peltier stage was used for this study. To ensure the hydrogel was the same size as the geometry, 1 mm thick hydrogel slabs were made and a 20 mm diameter disc was punched out (n=3). A constant strain rate of 5% was maintained for all tested hydrogels and the storage modulus was obtained by performing an angular frequency sweep from 0.1 to 10 rad/s. Results shown are mean ± SD.
2.7 In Vitro Hydrolytic Degradation of PEG-VS, PEG-Ac and PEG-Mal Hydrogels
The sensitivity of PEG hydrogels with varying end groups to hydrolytic degradation was investigated by incubating the gels in DPBS (pH: 7.4) at 37 °C for a period of 60 days (n=5 for PEG-VS and PEG-Ac, n=4 for PEG-Mal). Fresh DPBS was added every other day. Degradation was measured by determining the swelling ratio which was calculated every fifth day relative to the average initial dry (polymer) mass. All swelling ratio measurements were normalized to the initial swelling ratio (Day 1) [36]. Results shown are mean ± SD.
2.8 Differential Scanning Calorimetry (DSC) Analysis
The DSC experiments were performed using a DSC 1 instrument (Mettler–Toledo, Switzerland). Dried hydrogel samples were crimped in standard 40 μl aluminum pans and tracings were performed between 0.0 and 350.0 °C, at a heating rate of 10 °C/min under constant nitrogen purge of 50 cm3/min.
2.9 Assessment of the Protective Effect of NVP during Photo-polymerization on Cell Viability
Mouse embryonic fibroblasts (MEFs) were encapsulated in 5% w/v 8-arm PEG-VS, PEG-Ac and PEG-Mal with, NVP(+), and without, NVP(−), 0.1% v/v NVP. Cells were encapsulated at a density of 250,000 cells/mL in a 10 μl gel. All cell containing precursor solutions were exposed to UV light for 6 minutes (n=3 for each condition). To assess cell viability, a Live/Dead assay (Thermofischer Scientific, Waltham, MA), based on membrane integrity was utilized. Briefly, 20 μl of 2 mM ethidium homodimer-1 and 5 μl of 4 mM calcein-AM were added to 10 mL of sterile PBS to make the stock solution. After 24 hours of culture, media was removed and 500 μl of Live/Dead stock solution was added to each sample and allowed to incubate for 30 minutes at 37 °C before imaging. Cell viability was quantified via Image J software.
2.10 Post-polymerization RGD Modification
We investigated whether photo-polymerized PEG hydrogels contained unreacted end group that allow post-polymerization modification with the RGD peptide. 100 μL precursor solution of 5% w/v 8-arm PEG-VS, PEG-Ac, and PEG-Mal with 0.1% v/v NVP was laid on the bottom of a 96 well plate and exposed to UV light for 6 minutes. Gels receiving post-polymerization RGD modification (RGD(+)) were incubated in a 5 mM RGD solution for 15 minutes. All gels were washed with sterile PBS and allowed to swell overnight. Mouse embryonic fibroblasts were then seeded onto the PEG layers at a density of 25,000 cells/mL and cultured for a period of 6 days to evaluate cell spreading. As previously described, cells were stained with a Live/Dead cytocompatibility kit to assess viability at day 6 of culture. Viability was quantified via Image J software.
2.11 Inflammatory Response to Implanted PEG-Hydrogels
To investigate the inflammatory response, we subcutaneously implanted PEG hydrogels with varying end-group chemistries with and without RGD modification. The Institutional Animal Care and Use Committee (IACUC) guidelines for survival surgery in rodents and Policy on Analgesic Use in Animals Undergoing Surgery were followed for all the procedures. Animal experiments for this work were performed in accordance with the protocol approved by the IACUC at the University of Michigan (PRO00005750). A small incision was made on the dorsal side of the anesthetized adult female B6CBAF1 mice and the PEG (-VS, -Ac and -Mal) hydrogels were implanted subcutaneously in the first group (n=3 mice/group). The second group of mice were implanted with PEG hydrogels modified with 0.5 mM RGD (n=3 mice/group). The skin was then closed using 5/0 absorbable sutures. The mice were placed in a clean warmed cage for recovery and monitored post-operatively for 10 days. The mice received Carprofen (RIMADYL, Zoetis, USA) (5 mg/kg body weight) for analgesia before the incision was made. The mice were euthanized after a period of 28 days following implantation and the PEG hydrogels were retrieved.
2.12 Histological Analysis
Retrieved PEG-VS, PEG-Ac and PEG-Mal hydrogels were fixed in Bouin’s fixative solution (Sigma-Aldrich) at 4 °C overnight and then transferred to 70% ethanol at 4°C until processing. After fixation, the samples were processed at the Histology Core in Microscopy & Image Analysis Laboratory at the University of Michigan. Samples were embedded in paraffin and serially sectioned at 5 μm thickness and were stained with hematoxylin and eosin. Stained sections were then examined for the presence of cells and host immune response under the light microscope.
2.13 Immunohistochemistry for Macrophages
To analyze the macrophage infiltration following implantation with PEG hydrogels, paraffin-sectioned slides were stained for mouse CD-68 and CD-163. First, sections were deparaffinized with Xylene and rehydrated. Slides were incubated in peroxide blocking reagent (BUF017B, BioRad, USA) for 30 min at room temperature to block any endogenous peroxidase activity. The slides were then incubated in antigen retrieval buffer, pH8.0 (BUF025A, BioRad) for 20 min at 96 °C and additional 20 min at room temperature for antigen retrieval. Following which the slides were incubated with normal goat serum (ab156046, abcam, USA) to block non-specific binding sites for 30 minutes at room temperature. The sections were incubated overnight at 4 °C with primary antibodies: rat anti-mouse CD-68 antibody (1:100 dilution, MCA1957; BioRad), rabbit anti-mouse CD-163 antibody (1:100 dilution, ab182422, abcam). The slides were subsequently incubated for 60 minutes at room temperature with secondary antibodies: goat anti-rat (1:100 dilution, STAR72, BioRad) for CD-68 and goat anti-rabbit (1:500 dilution, ab97051, abcam) for CD-163. Diaminobenzidine (ab94665, abcam) was used as a chromogen and hematoxylin as a counterstain. For negative-controls, paraffin sections were incubated without the primary antibody.
3. Statistical Methods
Statistical analysis was performed using R software. A Welch two-sample T-test was used to a evaluate differences in swelling ratio, storage modulus, and cell viability. The results were considered statistically significant when p<0.05.
4. Results
4.1 The end group chemistry in PEG hydrogels impacts gelation efficiency and physical properties
We hypothesized that altering the functional end group attached to a multi-arm PEG backbone would alter the gelation efficiency and physical properties of the resultant photo-polymerized hydrogel due to a change in the reaction environment and reactivity of the respective group. To determine the effect of functional end groups on efficiency of gelation, we exposed the respective precursor solutions to 6, 15, and 30 minutes of UV light and measured gel fraction. When comparing across the three functional groups, PEG-VS hydrogels formed most efficiently, as demonstrated by the largest gel fraction at all exposure times and compositions (Figure 2A, Table 1). After 6 minutes exposure to UV light (1090 μW/cm2), 8-arm PEG-VS, PEG-Ac, and PEG-Mal (NVP(+), RGD(−)) hydrogels formed with 99%, 89%, and 63% efficiency, respectively. All tested multi-arm PEG-VS hydrogels that included NVP formed completely by 6 minutes of exposure, while PEG-Ac and PEG-Mal hydrogels did not form completely up to 30 minutes of UV light exposure (Figure 2). All multi-armed PEG-VS hydrogels incorporated 99% of solid content by 6 minutes, while PEG-Ac and PEG-Mal hydrogels incorporated up to 89% and 70% solid content by 30 minutes, respectively (Figure 2).
Figure 2.
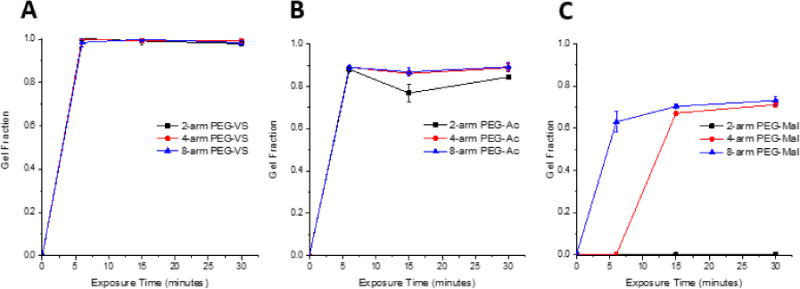
Gel fraction of 2-, 4-, and -8-arm 5% w/v (A) PEG-VS, (B) PEG-Ac, and (C) PEG-Mal hydrogels with 0.1% v/v NVP exposed to UV light (1090 μW/cm2) for 6, 15, and 30 minutes (n=5 for all compositions).
Table 1.
Gel fraction of -2, -4, and -8-arm 5% w/v PEG-VS, PEG-Ac, and PEG-Mal hydrogels with .1% v/v NVP and without RGD modification (n=15 for 8-arm, n=5 for 2- and 4-arm compositions), without NVP and without RGD modification (n=5 for all compositions), and with .1% v/v NVP and with .5mM RGD (n=5 for all compositions). All gels were exposed to UV light for 6 minutes. “–” indicates the hydrogels did not form.
| NVP(−), RGD(−) | NVP(+), RGD(−) | NVP(+), RGD(+) | |
|---|---|---|---|
| 8-arm PEG-VS | 0.99±0.02 | 0.99±0.02 | 0.73±0.05 |
| 8-arm PEG-Ac | 0.88±0.01 | 0.89±0.02 | 0.71±0.01 |
| 8-arm PEG-Mal | – | 0.63±0.05 | 0.52±0.02 |
| 4-arm PEG-VS | 1.0±0.01 | 1.0±0.0 | – |
| 4-arm PEG-Ac | 0.88±0.01 | 0.89±0.01 | 0.64±0.02 |
| 4-arm PEG-Mal | – | – | – |
| 2-arm PEG-VS | – | 1.0±0.01 | – |
| 2-arm PEG-Ac | 0.81±0.01 | 0.88±0.01 | 0.64±0.01 |
| 2-arm PEG-Mal | – | – | – |
Formation of PEG-VS photo-crosslinked hydrogels is the result of the reaction between the electron deficient activated vinyl group and free radicals, similar to the nucleophile-catalyzed thiol Michael-type addition reaction of PEG hydrogels. The stronger reactivity of the vinyl sulfone group is due to the vinyl moiety being more electron deficient than that of the acrylate or maleimide[24], which was confirmed by the downfield shifting of the VS protons in the PEG-VS 1H NMR spectrum (Supplemental Figure 1A). The sulfone group of the PEG-VS is a stronger electron withdrawing group compared to the neighboring carbonyl group of acrylate and amide, attracting more electrons away from the neighboring double bond, causing electron deficiency. Thus, the double bond in vinyl sulfone is more susceptible to reacting with free radicals and the rate of propagation is increased. During propagation, the electron withdrawing force of the sulfone moiety further destabilizes the secondary free radical resulting in an increase in reactivity [37]. Interestingly, PEG-Mal did not crosslink as quickly or efficiently as PEG-VS or PEG-Ac, across all compositions. One of the proposed explanations could be that the reaction of free radicals with the double bond resulted in bond dimerization, thereby, stabilizing the resultant free radical via resonance [38]. To further investigate the impact the functional end groups had on the network, swelling ratio (Figure 3) and storage modulus (Figure 4A) of PEG-VS, PEG-Ac, and PEG-Mal hydrogels were investigated. Altering the functional end group in a multi-arm PEG macromer impacted the crosslinking network as shown by the significant difference between swelling ratio (Figure 3B) and storage moduli (Figure 4A) of 5% w/v 8-arm PEG-VS, PEG-Ac, and PEG-Mal (NVP(+), RGD(−)) hydrogels. Photo-crosslinked PEG-Mal exhibited the highest swelling ratio (Qm=22) which corroborated that the PEG-Mal network was less dense compared to that of PEG-VS (Qm=19) and PEG-Ac (Qm=15) (Figure 3B). PEG-VS and PEG-Ac hydrogels exhibited a storage modulus of 4500-5000 Pa, while PEG-Mal hydrogels had a storage modulus of 2400 Pa, indicating a less mechanically strong network (Figure 4A).
Figure 3.
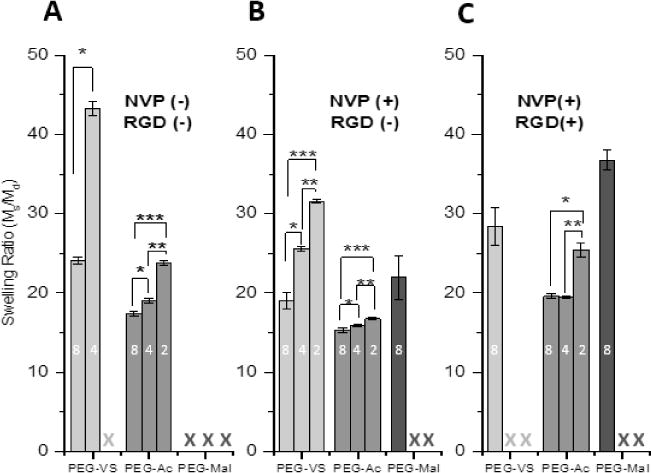
Swelling ratio of (A) 5% w/v 2-, 4-, and -8-arm 5% w/v PEG-VS, PEG-Ac, and PEG-Mal hydrogels without NVP and without RGD modification (n=5 for all compositions), (B) with NVP and without RGD modification (n=15 for 8-arm, n=5 for 2- and 4-arm), and (C) with NVP and with .5mM RGD (n=5 for all compositions) after 6 minutes exposure to UV light (1090 μW/cm2). Numbers (2,4, or 8) within columns indicate arms around PEG macromer. “X” indicates the hydrogel composition did not form. Differing letters (a, b, c) and indicate statistical significance (p<0.05). Significance was determined by a Welch two-sample t-test.
Figure 4.
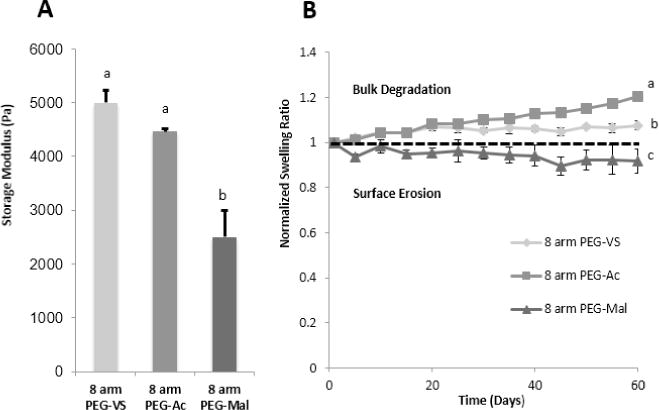
(A) Storage modulus of 5% 8-arm w/v PEG-VS (n=3), PEG-Ac (n=3), and PEG-Mal (n=3) with NVP and without RGD modification after 6 minutes exposure to UV light and (B) Degradation over 60 days in PBS of 8-arm 5% w/v PEG-VS (n=5), PEG-Ac (n=5), and PEG-Mal (n=4) hydrogels with NVP and without RGD modification after 6 minutes exposure to UV light. “*” indicate statistical significance (p<0.05). Significance was determined by a Welch two-sample t-test.
In addition to the functional end group, the number of reactive ends in the multi-arm PEG macromer affected the properties of the photo-crosslinked hydrogels. Decreasing the number of arms around a PEG macromer from 8 to 4 to 2 arms decreased the efficiency of the photo-initiated reaction, similar to what we observed when the hydrogels were formed using Michael-type crosslinking chemistry [39]. The most dramatic effect of decreasing number of arms was observed in PEG-Mal hydrogels with NVP, because 2 and 4-arm PEG-Mal did not form after 6 minutes of UV light exposure (Figure 2C, Table 1, Figure 3B); this finding further confirmed that maleimide end functional group does in fact stabilize the double bond and sterically hinder free radical attack more than vinyl sulfone or acrylate groups. To further investigate why the 8-arm PEG-Mal network was not fully formed after 6 minutes of exposure and whether the irradiation time was the limiting factor, we increased the UV exposure time to 30 minutes. The storage moduli of 8-arm PEG-Mal hydrogel after 30 minutes of exposure reached on average 9470 Pa (n=3). The volume of the crosslinked hydrogel remained unchanged and we concluded that the 3.5-fold increase in the storage modulus compared to shorter irradiation times was due to improved network formation and not due to the evaporation of solvent. These results indicated incomplete network formation after 6 minutes of UV exposure, as corroborated by the gel fraction (Figure 2C). When considering 8-arm and 4-arm PEG-VS and acrylate gels, with the increase in functionality (number of arms) of the macromer, the fraction of arms that must become crosslinked in order to produce a gel decreases, allowing macromer structure to set the fractional conversion of end groups that must be achieved to reach the gel point [40].
To validate network formation, FTIR analysis was performed (Supplemental Figure 2). The observed peak at 1420 cm−1 represents C‒H of C=C, which is related to formation of free radicals after decomposition of the photo-initiator (Supplemental Figure 2A–C) [41,42]. All FTIR spectra showed a broad absorption band around 3430 cm−1 due to the −OH stretching frequencies [42]. Peaks around 2920 and 2864 cm−1 are related to asymmetric and symmetric behavior (stretching) of methyl group (namely =C–H vibrations) (Supplemental Figure 2A–C) [43], as complemented by the NMR spectra (Supplemental Figure 1). The spectrum of 8-arm PEG-Ac (NVP(+)) gels suggests network formation due to the formation of C‒C bonds between PEG polymer backbone chains in the hydrogel (Supplemental Figure 2B) that affects the mobility of the PEG backbone [44], as observed by the spectrum peak broadening. This observation was supported by the finding that PEG-Ac hydrogels have a swelling ratio of approximately 15 independent of the number of arms around the macromer (Figure 3B). A peak around 1720 cm−1 in the spectra of hydrogels indicates double bond conversion (Supplemental Figure 2B, C) [45]. The FTIR spectra of 8-arm PEG-Ac and 8-arm PEG-Mal hydrogel had a stronger peak compared to the spectrum of 8-arm PEG-VS gel (Supplemental Figure 2A), suggesting that a greater portion of double bonds stay intact, which presents an option of post crosslinking modification of the unreacted double bonds.
4.2 Inclusion of NVP alters hydrogel physical properties and has a protective effect on cell viability during encapsulation
It has been demonstrated NVP accelerates gelation due to the increase in vinyl group concentration and diffusion of free radicals [35]. To investigate this effect on gelation and network formation after 6 minutes of UV light exposure, gel fraction and swelling ratio with and without NVP was acquired (Table 1, Figure 3A, B). The presence of NVP in the precursor solution is essential for formation of 2-arm PEG-VS gels in 6 minutes, but had a less pronounced effect on gelation of 4- and 8-arm PEG-VS networks (Table 1). Inclusion of NVP impacted the physical properties of the network as indicated by the significant decrease in swelling ratio (Figure 3A, B). NVP demonstrated no impact on PEG-Ac network formation and a minimal impact on the physical properties. For all PEG-Mal networks, the inclusion of NVP is essential for gelation (Table 1, Figure 3A). Furthermore, steric hindrance of the free radicals on the macromer ends would explain the improved gelation in the presence of NVP [46]. In radical polymerization, the reactivity of a free radical depends upon the electrons’ delocalization, polarity and volume nature of the side groups linked to the radical carbon [47]. The effect was more pronounced for vinyl sulfone, meaning that the radicals at the end of the chain in PEG-VS are more sterically hindered than those of acrylates. We hypothesize the electron deficiency of the vinyl moiety and destabilization of the radical in PEG-VS is able to overcome steric hindrance to efficiently form hydrogels via free radical reaction (Figure 3A, Table 1). In this reaction, NVP acts as a facilitator and creates more free radicals that are accessible by larger sterically hindered VS and Mal groups. We demonstrated that the degree to which NVP facilitates the reaction was dependent on the chemistry and structure around the vinyl groups in a multi-arm PEG macromer.
Additionally, for any cell/tissue encapsulation, the ability for the cells to survive the encapsulation process is vital. To investigate whether cells or tissue can survive the encapsulation process, we encapsulated mouse embryonic fibroblasts (MEFs) in PEG-VS, PEG-Ac, and PEG-Mal hydrogels with and without NVP and assessed survival after one day in culture. Similar to Hao et al. [35], we observed that after the addition of NVP to the precursor solution the encapsulated cells exhibited greater viability (Figure 5A). Cells encapsulated in PEG-VS, PEG-Ac, and PEG-Mal hydrogels with NVP exhibited a viability of 87%, 89%, and 68%,respectively, which decreased significantly to 60% and 40%, when NVP was not included in PEG-VS and PEG-Ac hydrogels(Figure 5A(F)). PEG-Mal hydrogels did not form when NVP was not included. It was hypothesized that the protective effect of NVP was due to improved formation kinetics of the network, decreasing the time and absolute exposure of the cells to free radicals. Overall, we showed that NVP can be added in the precursor solution to increase the kinetics of the reaction, the physical properties of the network, and does not negatively impact cell viability upon encapsulation. When comparing cellular response to encapsulation between the end functional groups of PEG macromers, cells encapsulated in PEG-VS and PEG-Ac exhibited a significantly higher viability (87% and 89%, respectively) than cells encapsulated in PEG-Mal hydrogels (68% viability) (Figure 5A(F)). We hypothesize that this is due to the kinetics of the reaction and cells being exposed to more absolute free radicals during the PEG-Mal encapsulation, as well as toxicity from residual maleimide groups.
Figure 5.
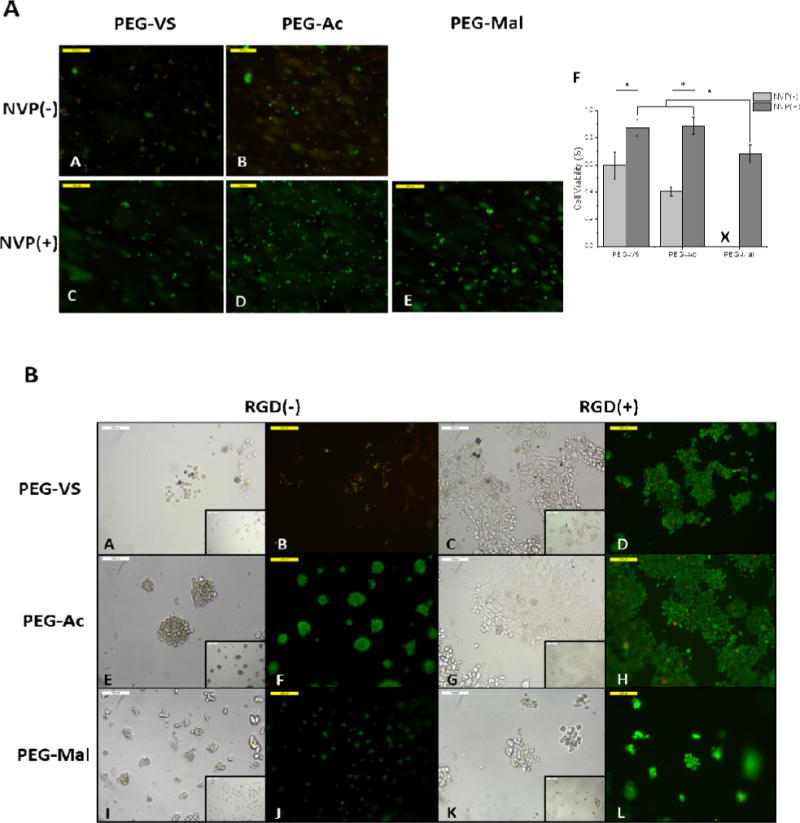
(5A)Fluorescent images MEFs encapsulated in 5% 8-arm PEG-VS and PEG-Ac without NVP (A,B), respectively and 5% 8-arm PEG-VS, PEG-Ac, and PEG-Mal with .1% v/v NVP (C,D,E), respectively. Cells were stained with calcein AM and ethidium homodimer-1 for 30 minutes. Magnification 10×. (F)Quantification of cell viability in PEG-VS, PEG-Ac, and PEG-Mal hydrogels (n=3). “*” indicates statistical significance (p<0.05). “X” indicates the hydrogel did not form. (5B) Brightfield and fluorescent images of MEFS seeded on 5% 8-arm PEG-VS, PEG-Ac, and PEG-Mal hydrogels without RGD modification (A,B,E,F,I,J) and 5% 8-arm PEG-VS, PEG-Ac, and PEG-Mal hydrogels incubated in a 5mM RGD solution after polymerization (C,D,G,H,K,L). Magnification 20×, inset magnification 10×.
4.3 The functional end group of a PEG macromer impacts the efficiency of RGD modification
We determined the efficiency of RGD conjugation to PEG-VS and PEG-Ac by analyzing NMR spectra of the macromers before and after the conjugation reaction. By reacting 5% 8-arm PEG monomers, which have 10 mM reaction arms with 5 mM RGD we expected to see a 50% depletion of vinyls. Using NMR and assigning a vinyl peak an integration value of 1.0, the ratio between the protons in the double bond and in the PEG backbone can be calculated and compared between unmodified and RGD modified PEG macromers (Supplemental Figure 1). For PEG-VS, there was a 53.5% reduction in vinyl protons when comparing modified to unmodified, indicating 100% binding efficiency. For 8-arm PEG-Ac, there was a 24.5% reduction of vinyls from unmodified to modified. The double bonds in PEG-Mal undergone hydrolysis during dialysis and the ratio between the modified and unmodified macromers could not be determined (Supplemental Figure 1). Based on these results, we concluded that RGD binds with 100% efficiency to vinyl sulfone moieties because of the vinyls’ electron deficiency and susceptibility to thiol-mediated reactions.
RGD modification of multi-armed PEG impacted gelation and the physical properties of the resultant hydrogel. When 0.5mM RGD was added to PEG-VS, PEG-Ac, and PEG-Mal hydrogels, solid content incorporation decreased after 6 minutes of gelation (Table 1) and swelling ratio increased compared to unmodified hydrogels (Figure 3B,C), indicating a less dense network. The most drastic impact was seen in PEG-VS hydrogels as 2- and 4-arm PEG-VS hydrogels modified with .5mM RGD did not form after 6 minutes of gelation (Table 1).
4.4 Inclusion of NVP alters has a protective effect on cell viability during encapsulation
Additionally, for any cell/tissue encapsulation, the ability for the cells to survive the encapsulation process is vital. To investigate whether cells or tissue can survive the encapsulation process, we encapsulated mouse embryonic fibroblasts (MEFs) in PEG-VS, PEG-Ac, and PEG-Mal hydrogels with and without NVP and assessed survival after one day in culture. Similar to Hao et al. [35], we observed that after the addition of NVP to the precursor solution the encapsulated cells exhibited greater viability (Figure 5A). Cells encapsulated in PEG-VS, PEG-Ac, and PEG-Mal hydrogels with NVP exhibited a viability of 87%, 89%, and 68%,respectively, which decreased significantly to 60% and 40%, when NVP was not included in PEG-VS and PEG-Ac hydrogels(Figure 5A(F)). PEG-Mal hydrogels did not form when NVP was not included. It was hypothesized that the protective effect of NVP was due to improved formation kinetics of the network, decreasing the time and absolute exposure of the cells to free radicals. Overall, we showed that NVP can be added in the precursor solution to increase the kinetics of the reaction, the physical properties of the network, and does not negatively impact cell viability upon encapsulation. When comparing cellular response to encapsulation between the end functional groups of PEG macromers, cells encapsulated in PEG-VS and PEG-Ac exhibited a significantly higher viability (87% and 89%, respectively) than cells encapsulated in PEG-Mal hydrogels (68% viability) (Figure 5A(F)). We hypothesize that this is due to the kinetics of the reaction and cells being exposed to more absolute free radicals during the PEG-Mal encapsulation.
4.5 Available double bonds allow for post-polymerization RGD modification of PEG hydrogel networks
To investigate whether post-polymerization modification of the crosslinked hydrogels with RGD was possible we incubated the gels with 5mM RGD solution for 15 min. After a thorough washing step, we seeded mouse embryonic fibroblasts (MEFs) on top of the gels. We compared cell adhesion to 5% w/v 8-arm PEG-VS, PEG-Ac, and PEG-Mal hydrogels with RGD modification (RGD(+)) and without RGD (RGD(−)) added after polymerization (Figure 5B). Cells seeded on the hydrogels treated with RGD post-polymerization attached to the gel surface and demonstrated a spindle-shaped morphology. Without the RGD modification, cells seeded on the gels formed clusters and did not spread. Post-polymerization modification with RGD also improved the viability of the seeded cells for PEG-VS hydrogels, which was 49% without and 95% with RGD modification. Larger clusters and 100% viability was observed when cells were seeded on unmodified PEG-Ac and PEG-Mal hydrogels (Figure 5B). This can be explained by the fact that vinyl sulfone groups are more hydrophilic than acrylate and maleimide groups and hardly promote any protein absorption, increasing the non-fouling properties [8]. The possibility of modifying a hydrogel with bioactive peptides after polymerization enables the creation of a bio-responsive surface without drastically impacting the physical properties of the network.
4.6 Hydrolytic stability of photo-crosslinked PEG hydrogels for immunoisolation application
In immunoisolation applications, proteolytic or hydrolytic degradation of the hydrogel leads to exposure of the recipient immune system to the donor tissue and may result in sensitization of the host and rejection of the implanted tissue causing premature graft failure. For this reason, an immunoisolating hydrogel system must be designed to be non-degradable and stable in vivo. Photo-polymerization of functionalized PEG forms crosslinks between the carbon atoms, through the double bonds at the end-groups of the otherwise biologically inert macromer. Similar to previous findings demonstrating significant hydrolytic degradation of PEG-Ac hydrogel [36], we found that in physiological conditions, PEG-VS hydrogels were more stable than PEG-Ac hydrogels (Figure 4B). PEG-VS hydrogel swelling ratio changed the smallest percentage over a span of 60 days in DPBS. The presence of carbonyls in PEG-Ac and PEG-Mal hydrogels make them more susceptible to hydrolytic attack of hydroxyl groups. In addition to being hydrolytically stable, differential scanning calorimetry (DSC) demonstrated PEG-VS samples show good thermal behavior within the application temperature (Supplemental Figure 3). Within the application temperature, no significant changes are seen, since the two curves, before and after gelation, are similar. The Tm of the polymer has been shifted from 51.3 °C to 54.6 °C, as a result of UV polymerization. As a consequence of network formation, and hence more complex, less flexible structure, the decrease in endothermic peak is evident (Supplemental Figure 3).
4.7 Inflammatory response of the surrounding host tissues to the implanted PEG hydrogels
Attenuated inflammatory response at the interface between the immunoisolating device and the host tissues contributes to extended immunoisolation and survival of the graft. We investigated the local interactions at the interface between the hydrogel implant and host tissues. We implanted subcutaneously for 28 days PEG-VS, PEG-Ac, and PEG-Mal hydrogels, and performed macro- and microscopic characterization. Interestingly, hydrogels at the retrieval showed blood vessel development around the hydrogels, yet the hydrogels were intact and were easily removable. PEG-Mal hydrogels with and without RGD induced a strong response from the host, compared to other groups showing dense collagen deposition with stratified cells. In addition to the presence of multinucleated giant cells, PEG-Mal based hydrogels with and without RGD showed clear infiltration of cells into the hydrogel (Figure 6Q, R, U and V) with presence of macrophages M1 and M2, as was confirmed with CD-68 and CD-163 antibody staining (Figure 6S, T, W and X), which will hinder the immunoisolating properties of the hydrogel (Figure 6Q, R, U and V). Similarly, PEG-Ac hydrogels became encapsulated in a pseudostratified cell lining with giant cells, with some infiltration into the hydrogel (Figure 6I, J, M and N). Neovascularization was observed in the surrounding dense collagen with increased macrophage infiltration in PEG-Ac gels without RGD compared to those with RGD (Figure 6I, J, K, L, M, N, O, P). PEG-VS hydrogels showed the least inflammatory response with an attenuated cell lining and a thinner but dense collagen deposition with only occasional giant cells. No cellular infiltration in PEG-VS hydrogels occurred after 28 days (Figure 6A, B) compared to PEG-VS-RGD hydrogels (Figure 6E, F). This observation corroborated with what was seen in Figure 5B, as unmodified PEG-VS hydrogels elicited minimal cellular attachment and spreading compared to PEG-Ac and PEG-Mal hydrogels (Figure 5B), indicating less protein absorption. Interestingly, PEG-VS hydrogels with and without RGD showed stronger presence of macrophage M2 (Figure 6D, H) and lesser presence of M1 (Figure 6C, G) compared to PEG-Ac and PEG-Mal hydrogels, possibly indicating a healing response.
Figure 6.
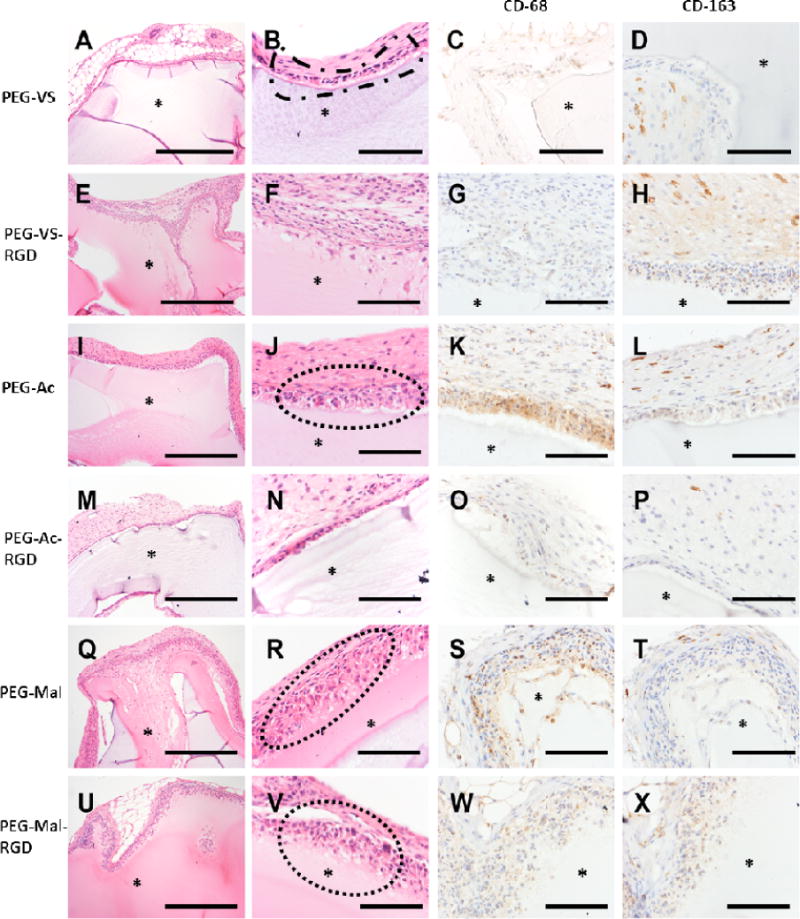
Histological images of 8-arm PEG types after implantation for 28 days. (A, B) PEG-VS, (E, F) PEG-VS-RGD, (I, J) PEG-Ac, (M, N) PEG-Ac-RGD, (Q, R) PEG-Mal and (U, V) PEG-Mal-RGD. Host response to implanted PEG types as observed: (B) PEG-VS implant with retraction artifact and loosely adherent capsule composed of attenuated epithelioid cells (dashed outline). (J) PEG-Ac implant with multilayered cellular capsule overlying granulation tissue with prominent angiogenesis and frequent multinucleate giant cells (dashed outline). (R, V) PEG-Mal implant with infiltration into and partial resorption of the hydrogel matrix (dotted outline). (C, G) Presence of macrophage M1 (CD-68 staining) were observed in (G, K) PEG-Ac and (S, W) PEG-Mal hydrogels. Whereas macrophage M2 (CD-163 staining) observed in weak to strong presence between PEG hydrogel types (P, T, X, D, H and L). (*) PEG hydrogel. Magnification 10× (A, E, I, M, Q, U) scale bar, 500 μm. Magnification 40× (B, C, D, F, G, H, J, K, L, N, O, P, R, S, T, V, W, X), scale bar, 50 μm.
5. Discussion
The design of an immunoisolating hydrogel system has to address cytocompatibility upon encapsulation, the physical properties of the network that match the physiological host environment, resistance to proteolytic and hydrolytic degradation, and minimal inflammatory response. In a previous study, we investigated the ability of TheraCyte and alginate to support and protect ovarian tissue in a syngeneic and allogeneic model [48]. We found that alginate was not able to withstand the volumetric expansion of ovarian tissue, compromising its immunoisolating capability. Immunoisolating non-degradable PEG hydrogels have been used for encapsulating pancreatic cells [49–52] using crosslinking chemistries with non-degradable peptides [52–54], scramble peptides [55–57], and photo-polymerization [23,35,58]. However, non-degradable and scramble peptides can require longer gelation times compared to photo-polymerization[54], and gelation upon monomer-peptide introduction may limit advanced patterning opportunities. During photo-crosslinking, expedient polymerization only upon light exposure presents the opportunity for advanced patterning and spatiotemporal control. To our knowledge, non-degradable PEG-VS hydrogels have not been developed using solely photo-polymerization. Photo-polymerized PEG-VS based hydrogels, described here, meet the design criteria for an immunoisolating device and are advantageous towards microencapsulation due to the high degree of tunability, biocompatibility, and non-fouling surface [2,4]. We hypothesized that utilizing PEG-VS as the backbone polymer would create a hydrolytically stable hydrogel system in a physiological environment without impacting tissue viability upon encapsulation. In this study, we investigated how changing the end functional group around a PEG macromer impacts gelation, physical properties, and compatibility, with the objective of designing a hydrogel system that could be used towards immunoisolating applications.
We first compared how vinyl sulfone, acrylate, and maleimide impacted gelation efficiency and physical properties of the resultant PEG hydrogels. PEG-VS hydrogels form most efficiently across the functional groups given the same exposure time, because double bonds in the vinyl sulfone moiety are more electron deficient [24], leading to faster free radical reaction and propagation. The physical properties of PEG-VS hydrogels were also superior to that of PEG-Ac and PEG-Mal given the same crosslinking time, which would be vital for tissue encapsulation and tuning the mesh size for immunoisolation applications. A shorter crosslinking time to complete the encapsulation minimizes the impact on cell viability. Additionally, including NVP, a co-monomer, at a low concentration demonstrated a protective effect on cell viability upon encapsulation. We hypothesized that in the presence of NVP the cells were exposed to a decreased absolute number of free radicals for a shorter time due to the improved gelation kinetics. Overall, we have demonstrated that addition of NVP improves gelation, physical properties, and cell cytocompatibility.
Unmodified PEG hydrogels are inert to protein absorption and cell interactions. We showed the efficiency of RGD binding was impacted by the functional end group that was present during the thiol-mediated reaction. In this reaction the thiol present in the side chains of RGD reacts with vinyl groups present in the functional group on the PEG arms. PEG-VS incorporated RGD most efficiently due to the electron deficiency of the double bond and availability to react with thiol groups. We have been able to demonstrate that RGD can be incorporated into PEG-VS and PEG-Ac hydrogel networks after photo-polymerization due to the presence of unreacted double bonds. Post-crosslinking modification could be important for tissue engineering applications as one can create a hydrogel system with the desired physical properties and then modify the network with bioactive motifs post-polymerization.
Resistance to hydrolytic degradation is an important design criterion for an immunoisolating device. Photo-polymerization was used as the crosslinking chemistry to minimize the extent of proteolytic degradation. However, hydrolytic degradation of ester and maleimide bonds may lead to premature graft rejection. PEG-Ac and PEG-Mal hydrogels are susceptible to hydrolytic degradation due to the presence of carbonyls and/or ring opening, while PEG-VS hydrogels are more stable in physiological conditions. We confirmed that PEG-VS was the most hydrolytically stable over a span of 60 days. Additionally, biocompatibility of an implanted device with the surrounding tissues of the host and a minimal inflammatory response contribute to the graft longevity. Macrophages play a key role in inflammatory response after implantation of biomaterials [59]. Upon implantation, PEG-VS hydrogels had the least amount of cellular infiltration compared to PEG-Ac and PEG-Mal hydrogels. RGD incorporation in all PEG hydrogel types further facilitated cell infiltration. Interestingly, PEG-Ac-RGD hydrogels attracted less macrophage reaction when compared to the non-modified PEG-Ac, which corroborates with Blakney et al. who used PEG-DA hydrogels [60]. Overall, we demonstrated that PEG-VS hydrogels can form efficiently via photo-polymerization, are hydrolytically stable, maintain cell viability upon encapsulation, and elicit a lesser inflammatory response compared to PEG-Ac and PEG-Mal hydrogels.
6. Conclusion
This study has demonstrated that reactive functional end groups of the multi-arm PEG macromers impact free radical mediated network formation. The PEG-VS network formed more efficiently via photo-polymerization, impacting bulk properties, was most stable in physiological conditions, and elicited an attenuated inflammatory response compared to PEG-Ac and PEG-Mal hydrogels. Further, NVP can be added to the precursor solution to expedite the cross-linking process without impacting cellular response upon encapsulation. One of the potential applications of these hydrogels would be their use for immunoisolation methods. A shorter crosslinking time would improve the biocompatibility of a hydrogel when used with live cells or in proximity to live tissues and should have the least effect on the viability of cell or tissue encapsulated within the photo-polymerized hydrogel. These attributes are crucial for efficient encapsulation of tissue or cells, including those of allogeneic origin, for long-term immunoisolating applications.
Supplementary Material
Statement of Significance.
The objective of this study was to develop a tunable non-degradable PEG system that is conducive for encapsulation and evokes a minimal inflammatory response, which could be utilized for immunoisolation applications. This study has demonstrated that reactive functional groups of the PEG macromers impact free radical mediated network formation. Here, we show PEG-VS hydrogels meet the design criteria for an immunoisolating device as PEG-VS hydrogels form efficiently via photo-polymerization, impacting bulk properties, was stable in physiological conditions, and elicited a minimal inflammatory response. Further, NVP can be added to the precursor solution to expedite the cross-linking process without impacting cellular response upon encapsulation. These findings present an additional approach/chemistry to encapsulate cells or tissue for immunoisolation applications.
Acknowledgments
This work was supported by The Hartwell Foundation (14-PAF00981), the National Institute of Biomedical Imaging and Bioengineering to AS and MC (R01 EB022033), NIAD K23AI108951 to EAF, the Ronald and Eileen Weiser Center for Europe and Eurasia support to NM, and the National Science Foundation Graduate Research Fellowship Program (DGE 1256260) to JRD. The authors would like to thank the University of Michigan NMR Facility for assisting in the acquisition of the NMR spectra, as well as Dr. Bojan Calija from the University of Belgrade for his help in DSC spectra acquisition.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
James R. Day, University of Michigan, Department of Biomedical Engineering, Ann Arbor 48109, Michigan, USA.
Anu David, University of Michigan, Department of Biomedical Engineering, Ann Arbor 48109, Michigan, USA.
Jiwon Kim, University of Michigan, Department of Biomedical Engineering, Ann Arbor 48109, Michigan, USA.
Evan A. Farkash, University of Michigan Medical School, Department of Pathology, Ann Arbor 48109, Michigan, USA.
Marilia Cascalho, Associate Professor of Surgery, Associate Professor of Microbiology and Immunology, Transplantation Biology, Medical Science Research Building I, 1150 West Medical Center Drive, A520B, Ann Arbor 48109 Michigan USA.
Nikola Milašinović, Department of Forensics, Academy of Criminalistic and Police Studies, Cara Dušana 196, 11080 Belgrade, Serbia.
Ariella Shikanov, University of Michigan, Department of Biomedical Engineering, Department of Macromolecular Sciences and Engineering, 1101 Beal Avenue, Ann Arbor 48109, Michigan, USA.
References
- 1.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 2.Tibbitt MW, Anseth AS. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol Bioeng. 2009;103:655–63. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee KY, Mooney DJ. Hydrogels for tissue engineering. Chem Rev. 2001;101:1869–79. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 4.Bryant SJ, Bender RJ, Durand KL, Anseth KS. Encapsulating chondrocytes in degrading PEG hydrogels with high modulus: engineering gel structural changes to facilitate cartilaginous tissue production. Biotechnol Bioeng. 2004;86:747–55. doi: 10.1002/bit.20160. [DOI] [PubMed] [Google Scholar]
- 5.Moutos FT, Estes BT, Guilak F. Multifunctional hybrid three-dimensionally woven scaffolds for cartilage tissue engineering. Macromol Biosci. 2010;10:1355–64. doi: 10.1002/mabi.201000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts JJ, Earnshaw A, Ferguson VL, Bryant SJ. Comparative study of the viscoelastic mechanical behavior of agarose and poly(ethylene glycol) hydrogels. J Biomed Mater Res B Appl Biomater. 2011;99:158–69. doi: 10.1002/jbm.b.31883. [DOI] [PubMed] [Google Scholar]
- 7.Anderson JM, Rodriguez A, Chang DT. Foreign body reaction to biomaterials. Semin Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bridges AW, Garcia AJ. Anti-inflammatory polymeric coatings for implantable biomaterials and devices. J Diabetes Sci Technol. 2008;2:984–94. doi: 10.1177/193229680800200628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kingshott P, Griesser HJ. Surfaces that resist bioadhesion. Curr Opin Solid State Mater Sci. 1999;4:403–412. [Google Scholar]
- 10.Morra M. On the molecular basis of fouling resistance. J Biomater Sci Polym Ed. 2000;11:547–69. doi: 10.1163/156856200743869. [DOI] [PubMed] [Google Scholar]
- 11.Szleifer I. Protein adsorption on tethered polymer layers: effect of polymer chain architecture and composition. Physica A. 1997;244:370–388. [Google Scholar]
- 12.Szleifer I. Polymers and proteins: interactions at interfaces. Curr Opin Solid State Mat Sci. 1997;2:337–344. [Google Scholar]
- 13.Elisseeff J, Anseth K, Sims D, McIntosh W, Randolph M, Langer R. Transdermal photopolymerization for minimally invasive implantation. Proc Natl Acad Sci USA. 1999;96:3104–7. doi: 10.1073/pnas.96.6.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buxton AN, Zhu J, Marchant R, West JL, Yoo JU, Johnstone B. Design and characterization of poly(ethylene glycol) photopolymerizable semi-interpenetrating networks for chondrogenesis of human mesenchymal stem cells. Tissue Eng. 2007;13:2549–60. doi: 10.1089/ten.2007.0075. [DOI] [PubMed] [Google Scholar]
- 15.Hubbell JA. Synthetic biodegradable polymers for tissue engineering and drug delivery. Curr Opin Solid State Mater Sci. 1998;3:246–251. [Google Scholar]
- 16.Metters A, Hubbell JA. Network formation and degradation behavior of hydrogels formed by Michael-type addition reactions. Biomacromolecules. 2005;6:290–301. doi: 10.1021/bm049607o. [DOI] [PubMed] [Google Scholar]
- 17.Park Y, Lutolf MP, Hubbell JA, Hunziker EB, Wong M. Bovine primary chondrocyte culture in synthetic matrix metalloproteinase-sensitive poly(ethylene glycol)-based hydrogels as a scaffold for cartilage repair. Tissue Eng. 2004;10:515–22. doi: 10.1089/107632704323061870. [DOI] [PubMed] [Google Scholar]
- 18.Lutolf MP, Hubbell JA. Synthesis and physicochemical characterization of end-linked poly(ethylene glycol)-co-peptide hydrogels formed by Michael-type addition. Biomacromolecules. 2003;4:713–22. doi: 10.1021/bm025744e. [DOI] [PubMed] [Google Scholar]
- 19.Morpurgo M, Veronese FM, Kachensky D, Harris JM. Preparation and Characterization of Poly(ethylene glycol) Vinyl Sulfone. Bioconjugate Chem. 1996;7:363–368. doi: 10.1021/bc9600224. [DOI] [PubMed] [Google Scholar]
- 20.Milton JM, Zhao X. Degradable heterobifunctional poly(ethylene glycol) acrylates and gels and conjugates derived therefrom. US6362276 B1 USA. 2002 Mar 26; [Google Scholar]
- 21.Harris JM, Sedaghat-Herati MR. Preparation and use of polyethylene glycol propionaldehyde. US 5252714 A USA. 1993 Oct 12; [Google Scholar]
- 22.Nhu K, Hyun C, Lee J, Pak Y. Preparation method of peg-maleimide derivatives. US 6828401 B2 USA. 2004 Dec 7; [Google Scholar]
- 23.Shih H, Lin CC. Cross-linking and degradation of step-growth hydrogels formed by thiol ene photoclick chemistry. Biomacromolecules. 2012;13:2003–12. doi: 10.1021/bm300752j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bowman CN, Nair DP, Podgorski M, Chatani S. Thiol-containing dual cure polymers and methods using same. US9340636 B2 USA. 2016 May 17; [Google Scholar]
- 25.Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309:30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- 26.Bryant SJ, Nuttelman CR, Anseth KS. Cytocompatibility of UV and visible light photoinitiating systems on cultured NIH/3T3 fibroblasts in vitro. J Biomater Sci Polym Ed. 2000;11:439–457. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 27.Elisseeff J, Anseth K, Sims D, McIntosh W, Randolph M, Yaremchuk M, Langer R. Transdermal photopolymerization of poly(ethylene oxide)-based injectable hydrogels for tissue-engineered cartilage. Plast Reconstr Surg. 1999;104:1014–1022. doi: 10.1097/00006534-199909040-00017. [DOI] [PubMed] [Google Scholar]
- 28.Silverman RP, Elisseeff J, Passaretti D, Huang W, Randolph MA, Yaremchuk MJ. Transdermal photopolymerized adhesive for seroma prevention. Plast Reconstr Surg. 1999;103:531–535. doi: 10.1097/00006534-199902000-00025. [DOI] [PubMed] [Google Scholar]
- 29.Elisseeff J, McIntosh W, Anseth K, Riley S, Ragan P, Langer R. Photoencapsulation of chondrocytes in poly(ethylene oxide)-based semi-interpenetrating networks. J Biomed Mater Res. 2000;51:164–171. doi: 10.1002/(sici)1097-4636(200008)51:2<164::aid-jbm4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 30.Burdick JA, Anseth KS. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 2002;23:4315–4323. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 31.Burdick JA, Anseth KS. Delivery of osteoinductive growth factors from degradable PEG hydrogels influences osteoblast differentiation and mineralization. J Control Release. 2002;83:53. doi: 10.1016/s0168-3659(02)00181-5. [DOI] [PubMed] [Google Scholar]
- 32.Bryant SJ, Nuttelman CR, Anseth KS. The effects of crosslinking density on cartilage formation in photocrosslinkable hydrogels. Biomed Sci Instrum. 1999;35:309–314. [PubMed] [Google Scholar]
- 33.Mann BK, Gobin AS, Tsai AT, Schmedlen RH, West JL. Smooth muscle cell growth in photopolymerized hydrogels with cell adhesive and proteolytically degradable domains: synthetic ECM analogs for tissue engineering. Biomaterials. 2001;22:3045–3051. doi: 10.1016/s0142-9612(01)00051-5. [DOI] [PubMed] [Google Scholar]
- 34.Williams CG, Malik AN, Kim TK, Manson PN, Elisseeff JH. Variable cytocompatibility of six cell lines with photoinitiators used for polymerizing hydrogels and cell encapsulation. Biomaterials. 2005;26:1211–8. doi: 10.1016/j.biomaterials.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 35.Hao Y, Shih H, Munoz Z, Kemp A, Lin CC. Visible light cured thiol-vinyl hydrogels with tunable degradation for 3D cell culture. Acta Biomater. 2014;10:104–14. doi: 10.1016/j.actbio.2013.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Browning MB, Cereceres SN, Luong PT, Cosgriff-Hernandez EM. Determination of the in vivo degradation mechanism of PEGDA hydrogels. J Biomed Mater Res A. 2014;102:4244–4251. doi: 10.1002/jbm.a.35096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nyugen MT, Kryachko ES, Vanquickenborne LG. The chemistry of phenols Part 2. Chichester, England: 2003. p. 140. Chapter 1. [Google Scholar]
- 38.Roberts MJ, Bently MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2002;54:459–76. doi: 10.1016/s0169-409x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 39.Kim J, Kong YP, Niedzielski SM, Singh RK, Putnam AJ, Shikanov A. Characterization of the crosslinking kinetics of multi-arm poly(ethylene glycol) hydrogels formed via Michael-type addition. Soft Matter. 2016:2076–85. doi: 10.1039/c5sm02668g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sperinde JJ, Griffith LG. Control and Prediction of Gelation Kinetics in Enzymatically Cross-Linked Poly(ethylene glycol) Hydrogels. Macromolecules. 2000;33:5476–5480. [Google Scholar]
- 41.Ma Z, Niu X, Xu Z, Guo J. Synthesis of novel macrophotoinitiator for the photopolymerization of acrylate. J Appl Polym Sci 2014 [Google Scholar]
- 42.Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, Garcia AJ. Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Adv Mater. 2012;24:64–70. doi: 10.1002/adma.201103574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milašinović N, Čalija B, Vidović B, Sakač MC, Vujić Z, Knežević-Jugović Z. Sustained release of α-lipoic acid from chitosan microbeads synthetized by inverse emulsion method. J Taiwan Inst Chem Eng. 2016;60:106–112. [Google Scholar]
- 44.Bae M, Divan R, Suthar KJ, Macini DC, Gemeinhart RA. Fabrication of Poly(ethylene glycol) Hydrogel Structures for Pharmaceutical Applications using Electron beam and Optical Lithography. J Vac Sci Technol B Microelectron Nanometer Struct Process Meas Phenom. 2010;28:C6P24–C6P29. doi: 10.1116/1.3517716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia H, Barros AS, Goncalves C, Gama FM, Gil AM. Characterization of dextrin hydrogels by FTIR spectroscopy and solid. Eur Polym J. 2008;44:2318–2329. [Google Scholar]
- 46.Gatica N, Fernandez N, Opazo A, Alegria S, Gargallo L, Radic D. Synthesis and characterization of functionalized vinyl copolymers. Electronegativity and comonomer reactivity in radical copolymerization. Polym Int. 2003;52:1280–1286. [Google Scholar]
- 47.Vijaykumar S, Prasannkumar S, Sherigara BS, Shelke NB, Aminabhavi TM, Reddy BSR. Copolymerization ofN-vinyl pyrrolidone with functionalized vinyl monomers: Synthesis, characterization and reactivity relationships. Macromol Research. 2009;17:1003–1009. [Google Scholar]
- 48.David A, Day JR, Cichon AL, Lefferts A, Cascalho M, Shikanov A. Restoring Ovarian Endocrine Function with Encapsulated Ovarian Allograft in Immune Competent Mice. Ann Biomed Eng. 2017;45:1685–1696. doi: 10.1007/s10439-016-1780-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cruise GM, Hegre OD, Scharp DS, Hubbell JA. A sensitivity study of the key parameters in the interfacial photopolymerization of poly(ethylene glycol) diacrylate upon porcine islets. Biotechnol Bioeng. 1998;57:655–65. doi: 10.1002/(sici)1097-0290(19980320)57:6<655::aid-bit3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 50.Cruise GM, Hegre OD, Lamberti FV, Hager SR, Hill R, Scharp DS, Hubbell JA. In vitro and in vivo performance of porcine islets encapsulated in interfacially photopolymerized poly (ethylene glycol) diacrylate membranes. Cell Tranplant. 1999;8:293–306. doi: 10.1177/096368979900800310. [DOI] [PubMed] [Google Scholar]
- 51.Weber LM, Cheung CY, Anseth KS. Multifunctional pancreatic islet encapsulation barriers achieved via multilayer PEG hydrogels. Cell Transplant. 2008;16:1049–57. [PubMed] [Google Scholar]
- 52.Headen DM, Aubry G, Lu H, Garcia AJ. Microfluidic-based generation of size-controlled, biofunctionalized synthetic polymer microgels for cell encapsulation. Adv Mater. 2014;26:3003–8. doi: 10.1002/adma.201304880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qiu Y, Lim JJ, Scott L, Jr, Adams RC, Bui HT, Temenoff JS. PEG-based hydrogels with tunable degradation characteristics to control delivery of marrow stromal cells for tendon overuse injuries. Acta Biomater. 2011;7:959–66. doi: 10.1016/j.actbio.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 54.Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, Garcia AJ. Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in-situ delivery. Adv Mater. 2012;24:64–2. doi: 10.1002/adma.201103574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shikanov A, Smith RM, Xu M, Woodruff TK, Shea LD. Hydrogel network design using multifunctional macromers to coordinate tissue maturation in ovarian follicle culture. Biomaterials. 2011;32:2524–2531. doi: 10.1016/j.biomaterials.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schechte I, Berger A. On the size of the active size in proteases. I. Papain. Biochem Bioph Res Co. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 57.Weiss MS, Penalver Bernabe B, Shikanov A, Bluver DA, Mui MD, Shin S, Broadbelt LJ, Shea LD. The impact of adhesion peptides within hydrogels on the phenotype and signaling of normal and cancerous mammary epithelial cells. Biomaterials. 2012;33:3548–3559. doi: 10.1016/j.biomaterials.2012.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nguyen KT, West JL. Photopolymerizable hydrogels for tissue engineering applications. Biomaterials. 2002;23:4307–14. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 59.Zaveri TD, Lewis JS, Dolgova NV, Clare-Salzler MJ, Keselowsky BG. Integrin-directed modulation of macrophage responses to biomaterials. Biomaterials. 2014;35:3504–15. doi: 10.1016/j.biomaterials.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blakney AK, Swartzlander MD, Bryant SJ. The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)-based hydrogels. J Biomed Mater Res A. 2012;100:1375–86. doi: 10.1002/jbm.a.34104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.