

Abstract
Biliary tract cancer (BTC) represents a heterogeneous disease with dismal outcome. Herein, we examined genotype and angiogenesis features in BTC. We applied genotyping (Sanger, qPCR, 101-gene panel NGS), mRNA relative quantification methods, and β-catenin immunohistochemistry in 84 FFPE BTC (55 gallbladder [GBC], 14 intrahepatic [ICC], 15 extrahepatic [ECC] carcinomas). We identified 541 mutations in 68 (81%) tumors. Top mutated genes were CTNNB1 (36%); PTEN (33%); TP53 (31%); PIK3R1 (29%); PIK3CA (13%); BRCA2 and KRAS (12%); BRCA1 (11%). Six GBCs were hypermutated [hm] displaying a distinct mutational pattern. Mutations in TP53 and PI3K, Wnt and RAS components were prevalent among non-hypermutated tumors. All hmGBCs carried mutations in BRCA2 and other homologous recombination repair (HRR) genes, in PD1, but not in CTNNB1 and KRAS. None of the pathogenic BRCA2 p.D2723G and BRCA1 p.Q563* and c.5266dupC was present at frequencies expected for germline mutations. We observed copy gains (>6 copies) in EGFR (9% of informative tumors), PRKAR1A (7%), PIK3CA (6%), ERBB2 (5%) and MET (4%). TP53 mutations were prevalent in GBC (P<0.001) and PRKAR1A copy gains in ICC (P=0.007). PTEN was frequently co-mutated with CTNNB1 (P<0.001). Unrelated to CTNNB1 mutations, nuclear β-catenin was detected in 45% of tumors, among them in 5/6 hmGBC. We observed strong mRNA expression correlation of the two neuropilins (NRP1 and NRP2) with each other (Spearman’s rho 0.59) and with the endothelin receptor (NRP2 rho 0.66; NRP2 rho 0.51), and between VEGFA and its receptors (FLT1 rho 0.49; KDR rho 0.45). All PIK3CA mutated tumors expressed endothelin 1 mRNA (P=0.010). Most tumors expressing nuclear β-catenin were negative for VEGFC (P=0.009) and FLT4 (P=0.002) mRNA expression. In conclusion, we confirmed the presence of known genomic aberrations in BTC and different genotypes between BTC subsets. Novel findings are the coexistence of PI3K and WNT pathway gene alterations in BTC, their association with angiogenesis, and the hypermutated GBCs with HRR gene mutations, all of which may be considered for new treatment options in this difficult to treat disease.
Keywords: Biliary tract cancer, gallbladder, TP53, RAS/PI3K, WNT pathway, homologous recombination repair, BRCA1, BRCA2
Introduction
Biliary tract cancer (BTC) comprises heterogeneous carcinomas portraying similarities but also differences in their molecular profiles, pathogenesis and disease behavior. These tumors, mostly adenocarcinomas, develop in the background of chronic inflammation as a result of malignant transformation of biliary epithelia and are anatomically classified as intrahepatic (ICC) or extrahepatic cholangiocarcinoma (ECC), and gallbladder carcinoma (GBC) [1,2]. The incidence of the disease, although relatively low and varying geographically, is rather increasing globally. Prognosis is dismal, with less than 18% of patients surviving after five years of diagnosis. GBC is the most common BTC, with the worst 5-year survival rate, below 10% [1-5].
Unravelling the genetic and epigenetic aberrations and their signaling pathways is key to identifying therapies for battling the challenging nature of the disease. Various genetic/molecular targets have been identified, involved in proliferation (RAS/MAPK), survival/apoptosis (PI3K/AKT), angiogenesis (VEGF), cell fate/proliferation (WNT/β-catenin) and tumor inflammation-associated (IL6/JAK/STAT3) pathways and recently, via whole-exome and next generation sequencing (NGS) studies, in SWI/SNF chromatin-remodeling complexes, including ARID1A, PBRM1 and BAP1, cell cycle regulation, mainly CDKN2A and CDKN2B, as well as DNA repair and FGFR pathways [1-4,6-14]. Hot-spot activating mutations in KRAS, BRAF and PIK3CA have been repeatedly reported in BTC, with variable frequencies between subsets of patients and studies. TP53 mutations are also well described in biliary tract cancers [1-4,6-13,15-20]. IDH1 and IDH2 mutations have been found in ICC, CTNNB1 mostly in GBC and ECC (4-9%), while PTEN mutations have been reported in GBC (4-7%), ECC (1-7%) and ICC (1-11%) [4,6,10-12,17,20-24].
BTCs harbor further alterations, including overexpression and/or amplification of EGFR, ERBB2 and MET, mediating abnormal activation of their downstream pathways. Overexpression and/or amplification of ERBB2 has been shown in GBC (14-16%) and ECC (5-20%), whereas of EGFR in all subtypes [5,10,25,26]. For EGFR, an association between overexpression and poor prognosis has been described in ICC. MET amplification has been shown recently in ICC [1-6,11,20]. Overexpression of PKA regulatory subunit 1 alpha (PRKAR1A) has also been shown in ICC, whereas gene fusions of the two catalytic subunits of PKA, PRKACA or PRKACB have been found in ECC [6,10,27]. Regarding angiogenesis, studies reveal increased expression levels of VEGFA in BTC and of endothelin receptors, EDNRA and EDNRB, in cholangiocarcinoma cell lines and biopsy samples. VEGFA expression has been associated with disease recurrence, metastasis and poor prognosis in BTC [1-4,6,28]. Herein, we interrogated mutations and copy number aberrations (CNAs) of genes shown to be altered in BTC or are members of cancer-related pathways in 84 archival tumor tissue samples from Greek BTC patients, mostly GBC. The mRNA profile of angiogenesis targets was also evaluated, together with protein expression of β-catenin via IHC. Genotyping and expression profiling results were correlated with each other and available demographic and histopathological features.
Methods
Patients, tissue blocks and samples
FFPE blocks from 84 patients were processed for genotyping, mRNA profiling and IHC. Patient demographics and histopathological parameters were collected from medical records and blocks were retrieved from the Hellenic Cooperative Oncology Group (HeCOG) tissue bank depository. Histological review and molecular analyses were implemented in the Laboratory of Molecular Oncology (AUTH-HeFCR). The research project was approved by the Board of Directors of “Agii Anargiri” General Oncology Hospital of Kifissia (02/04-1-2017).
H&E sections were evaluated by a pathologist (S.L. or M.B.) for confirmation of diagnosis, anatomic location, histologic type, grade and tumor cell content (TCC%). Tumor dense areas were selected for construction of tissue microarrays (TMA) containing two cores (1 mm in diameter) per tumor sample. Almost half the tumors (48%) had TCC≥50% but a few samples with 15% TCC were also processed. RNA and DNA were extracted from TMA sections (5 × 8 μm) with the VERSANT Sample 1.0 Reagent Kit (Siemens Healthcare Diagnostics, Tarrytown, NY), enabling simultaneous total RNA and DNA isolation [29]. For cDNA synthesis, random hexamers and SuperScript III Reverse Transcriptase were used, according to standard procedures (Invitrogen, cat. no. 48190011 and 18080044).
Mutation analysis
Mutation analysis was performed with NGS and for selected targets with Sanger sequencing or qPCR with Taqman-MGB genotyping assays.
Allelic discrimination with Taqman-MGB assays
Targeted mutation investigation of KRAS mutations p.G12S, p.G12R, p.G12C, p.G12D, p.G12A, p.G12V, p.G13D (exon 1, CCDS 8702.1), BRAF p.V600E (exon 15, CCDS 5863.1), PIK3CA p.E545K (exon 9, CCDS 43171.1), AKT1 p.E17K (exon 2, CCDS9994.1) and MEK1 p.Q56P and p.K57N (exon 2, CCDS10216.1) was performed with qPCR and duplex allelic discrimination Taqman-MGB (minor-groove-binders) assays in an ABI PRISM 7900HT Detection System (Applied Biosystems, Foster City, USA), as previously described [29,30].
Sanger sequencing
Screening of tumor FFPE DNA for PIK3R1, PIK3CA, CTNNB1, PTEN and IDH1 mutations was accomplished with dd-sequencing of PCR products, amplified with M13-coupled nested primers for CTNNB1 exons 1-5 (CCDS 2694.1), PTEN exons 1-9 (CCDS 31238.1), PIK3R1 exons 8-13 (CCDS 3993.1), PIK3CA exon 20 (CCDS 43171.1) and KRAS exon 2 (CCDS 8702.1) or with primers for IDH1 exon 2 (CCDS 2381.1). Sense and antisense sequencing was performed in a 10 ul reaction with the Big Dye Terminator kit v.1.1 [Applied Biosystems, Foster City, USA]. Following capillary electrophoresis in an ABI3130XL genetic analyzer, sequences were visualized and initially called with the Sequencing Analysis software v5.2 and further analyzed with the SeqScape software v2.5. Primer sequences and chromosome coordinates, as well as annealing temperatures are shown in Table S1.
NGS genotyping
For NGS analysis, a custom panel of 388 amplicons covering a total region of ~42.6 Kb was employed, targeting mutation relevant regions of 101 genes implicated in cancer-associated pathways (Table S2). Genotyping was accomplished in an Ion Proton Sequencer. Tumor DNA concentration was measured with the Qubit fluorometer (Life Technologies, Paisley, UK); eligibility criteria for processing samples for NGS were 2 ng/ul DNA amplifiable at Ct 32 for two different qPCR assays. For data retrieval, base calling was performed on the Torrent Server with Torrent Suite v5.0.2. Variants were called, annotated (Ion Reporter 5.2), and accepted for analysis upon read quality filtering at P<0.0001; position coverage >100; variant coverage >40 if position coverage between 100-200; position and variant coverage for + or - strand >50 and >20, respectively. Indels involving GC-stretches and non-annotated variants were also excluded from analysis, whereas for selected variants alignments were also visually validated with the Integrative Genomics Viewer (Broad Institute). Regarding NGS metrics of samples, median mean depth was 2718 (mean: 3279; min-max: 85-11885); Median uniformity 91 (mean: 90; Min-max: 54-95); Median number of variants 34 (mean: 51; Min-max: 17-471). Eligible variants were called mutations if these were amino acid changing, and if no reported minor allele frequency (MAF) or with MAF (dbSNP) or EMAF (5000 Exomes) <0.1% when annotated SNPs.
Gene copy number evaluation by qPCR
Gene copy number (CN) was evaluated by qPCR on the 7900HT with premade CNA assays (Life Technologies/Applied Biosystems, Paisley, UK). For all targets except DUSP6, CN was assessed with 2 different assays, targeting, if possible, regions close to the 5’ and 3’ end, in order to achieve better representation across the entire gene length. Assay IDs and relevant information are shown in Table S3. The method involved duplex PCR reactions with the TaqMan Copy Number Assay, containing gene-specific primers and a FAM dye-labeled MGB probe and the TaqMan Copy Number Reference Assay, containing primers for the reference single-copy gene and a VIC dye-labeled TAMRA probe. Reactions were run in duplicates for each target assay under default conditions. Two peripheral blood DNA samples from non-cancer patients were included in each run as calibrator samples, along with no-template controls (NTC). Results were obtained automatically with the CopyCaller Software v2.0 as predicted CN, in comparison to averaged calibrator values after setting the evaluation threshold for RNase P at CT=32. Samples were considered eligible for analysis if RNase P CT<32 and CN range between duplicates <2. For statistical purposes, once the predicted CN for each of the two assays per gene was retrieved from CopyCaller, total gene CN status was assessed as no-gain for quadruplicate average CN≤2.5 and as gain for average CN>2.5. A 4-scale classification was also used for a more detailed evaluation of CN changes, as follows: no gain (≤2.5 CN); marginal (3-4 CN); low gain (4.5-6 CN); high gain (≥6.5 CN). The 2.5 cut-off was favored for CN gain assessment in order to exclude DNA replication [29].
mRNA expression analysis
mRNA expression analysis of all targets was performed with premade exon-spanning Taqman-MGB assays (Applied Biosystems/Life Technologies), shown in Table S4 alongside a Taqman-MGB expression assay targeting β-glucuronidase (GUSB) (Assay ID: Hs99999908_m1; RefSeq: NM_000181.1; Probe exon Location: 11-12) to allow for relative quantification assessment. The commercially available TaqMan Control Total RNA (cat. no 4307281, Applied Biosystems) was applied as a positive control for inter-run evaluation of PCR assay efficiency, together with no-template controls. Samples were run in duplicates in the 7900HT system under default conditions. To obtain linear Relative Quantification (RQ) values, relative expression was assessed as (40-dCT), as previously [29]. Samples were considered eligible if GUSB CT<36 and deltaRQ for each duplicate pair (intra-run variation) of <1. The difference between inter-run RQ values for the reference RNA sample was <1 for all assays but NRP2, corresponding to 1.5, respectively.
Immunohistochemistry
Immunohistochemical staining for nuclear, cytoplasmic and membrane β-catenin was performed according to standard protocols on 2.5 μm thick TMA sections with the Polymer Refine Detection system (DS9800; Leica Microsystems, Germany/Menarini Diagnostics, Athens, Greece). The sections were stained with antibodies against β-catenin, clone 17C2 (Novocastra, Newcastle, UK) at dilution 1:350 overnight. DAB (3,3-diaminobenzidine) was used as a chromogen and hematoxylin as counterstain. Each core was assessed for the % of cells with strong (intensity 3), moderate (intensity 2) and weak (intensity 1) staining. For nuclear, cytoplasmic or membrane β-catenin, staining was considered positive if ≥1% tumor cells were stained at any intensity, as proposed by two previous studies, given that no validated scoring system exists for interpreting IHC data for β-catenin [7].
Statistical analysis
Categorical data are presented as counts and respective percentages, whereas continuous data are presented as means, medians and ranges. For categorical variables, associations were examined with the Fisher’s exact test. A P<0.05 was considered statistically significant. For statistical analysis, gallbladder carcinomas were either grouped together with bile duct and ampulla of Vater carcinomas or examined separately. Cut-offs for continuous mRNA RQ values of each gene target were selected where available, according to natural breaks in their distributional profiles in order to categorize tumor samples as either expressing or not expressing for each mRNA target. Correlations for continuous mRNA RQ and average CN values in paired gene comparisons were assessed using the non-parametric Spearman’s correlation coefficient method and P<0.05 was considered significant. Statistical analyses were performed with SAS software (SAS for Windows, version 9.3, SAS Institute Inc., Cary, NC). Histograms and color correlation maps were created with SPSS (version 20.0 for Windows, SPSS Inc.) or JMP software (version 10.0.0, SAS Institute Inc., Cary, NC).
Results
Patient demographic and histopathological characteristics are shown in Table 1. Information on lymph node status and tumor size was only available for 27 (32%) and 47 (56%) patients; hence, these parameters were excluded from analysis.
Table 1.
Basic demographic and clinicopathological data of 84 patients with BTC
| N | % | |
|---|---|---|
| Age (years; median cut-off)& | ||
| ≥68 | 42 | 50.0 |
| <68 | 37 | 44.1 |
| Missing data | 5 | 5.9 |
| Sex | ||
| Men | 38 | 45.2 |
| Women | 46 | 54.8 |
| Surgery type | ||
| Biopsy only | 14 | 16.7 |
| Cholecystectomy (CE) | 55 | 65.5 |
| Partial hepatectomy | 3 | 3.6 |
| Partial hepatectomy & CE | 4 | 4.8 |
| Pancreato-duodenectomy | 7 | 8.3 |
| Missing data | 1 | 1.2 |
| Tumor size (cm) | ||
| ≤2 | 17 | 20.2 |
| 2-5 | 24 | 28.6 |
| >5 | 6 | 7.1 |
| Missing data | 37 | 44.0 |
| Lymph node status | ||
| Negative | 14 | 16.7 |
| Positive | 13 | 15.5 |
| Missing data | 57 | 67.9 |
| Site of primary disease | ||
| Intrahepatic cholangiocarcinoma (ICC) | 14* | 16.7 |
| Extrahepatic cholangiocarcinoma (ECC) | 15*,^ | 17.9 |
| Gallbladder carcinoma (GBC) | 55 | 65.4 |
| Histologic differentiation | ||
| High | 8 | 9.5 |
| Moderate | 31 | 36.9 |
| Poor | 34 | 40.5 |
| Missing data | 11 | 13.1 |
| Adenocarcinoma, histologic specifications | ||
| In situ carcinoma (ISC) | 2 | 2.4 |
| Adenocarcinoma NOS | 65* | 77.4 |
| Mucinous | 2 | 2.4 |
| Papillary | 3 | 3.6 |
| Tubular | 4 | 4.8 |
| Mixed histology# | 8 | 9.5 |
range: 48-91 yrs;
differential diagnosis (DDx) between primary BTC and metastatic adenocarcinoma; there were 5 such cases, 4 DDx vs. ICC and 1 DDx vs. ECC;
1 Vater ampullary carcinoma; NOS: not otherwise specified, including the 5 DDx tumors.
including 2 infiltrative adenocarcinomas with ISC areas, 4 tubular & papillary, and 2 tubular & cribriform adenocarcinomas.
Genotyping and associations between genotypic markers and with patient characteristics
The REMARK diagram in Figure 1 displays the study outline and informative results per target and per method. Sanger sequencing/qPCR yielded informative results in 63 tumors; of these 47 harbored 114 mutations in 5 genes. NGS was informative in 81 tumors; of these, 51 carried 436 coding mutations (14% of all eligible variants) in 77 out of 101 panel genes. The detailed list of mutations detected with Sanger/qPCR and NGS is shown in Tables S5 and S6, respectively. Collectively, genotyping with all methods yielded 541 mutations in 80 genes and in 68 tumors (81%); no mutations were found in 16 tumors. Most mutations were missense (93%), 6% were nonsense and 1% were frameshift indels.
Figure 1.
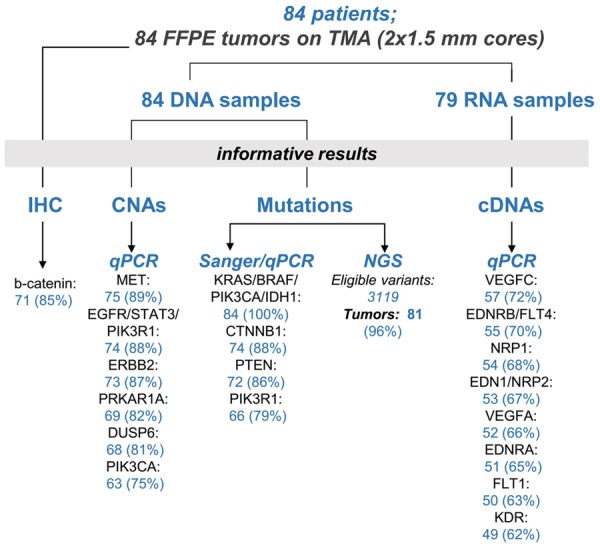
REMARK diagram. Study outline showing informative results per molecular target and per method of detection for FFPE tumor DNA and RNA samples, and for β-catenin immunohistochemistry (IHC). TMA: tissue microarray; CNA: copy number alterations.
With respect to method concordance, we obtained 100% consistent results for the absence of BRAF p.V600E in all tumors and for the presence of PIK3CA p.E542K and p.E545K. PIK3CA p.H1047R was retrieved only with NGS. NGS detected 3 out of 6 KRAS codon 12 or 13 mutations that were observed with qPCR and none of 2 mutations found with Sanger in codons 59 and 63. Four out of 5 discordant findings concerned DNA samples with TCC>60%, suggesting distinct subclones in the tumor; in one sample with TCC<20%, a low frequency mutation would be below the default sensitivity of and would have been missed by the NGS method. Classic pathogenic or likely pathogenic TP53 (p.R175H), PIK3CA (p.E542K, p.E545K) and KRAS (p.G12A) mutations (ClinVar), were present in ≥2 tumors. Three BRCA2 mutations (p.D2723G, p.A1233T, A627V), the first one reported as pathogenic when inherited, were also recurrent but were present at low frequencies, not anticipated for germline mutations. Further recurrent mutations registered in COSMIC and in dbSNP with MAFs<10^-4 were observed in TP53, PTEN, KMT2D, BRCA2, KIT and ABL1 (Tables S5, S6).
Figure 2A shows the distribution of genomic variants across tumors. The number of SNPs did not vary among the NGS informative tumors, but the number of mutations did, ranging from 0 to 160 per tumor. More than half (56%) of mutated tumors harbored 1 or, occasionally, ≥2 mutations in 1 or 2 genes; 17 (25%) in 3 or 4 genes; 13 (19%) in ≥5 of the interrogated genes. Among the latter, 6 GBCs possessed mostly multiple mutations in >10 genes, accountable for 64% of all mutations detected with NGS; hence, we considered these 6 GBC as hypermutated (hm). Hypermutation or absence of mutations could not be considered as artifacts, based on the constant SNP number, the good quality metrics of the samples and the perfect Integrative Genome Viewer (IGV) reads. Although the number of hm tumors was very small for performing reliable statistics, we observed a distinct pattern of mutations between hm and non-hm tumors. All hmGBC carried mutations in multiple homologous recombination repair (HHR) genes, particularly BRCA2 (6/6), BRCA1 (5/6), BAP1 (5/6), PALB2 (3/6), which were infrequent or absent in non-hm tumors. Further, all hmGBC demonstrated mutations in the PD1 gene (PDCD1), while only 1 non-hm tumor demonstrated the same feature. By contrast, none of the hmGBC carried mutations in CTNNB1 and KRAS, which were exclusively present in non-hm tumors.
Figure 2.

Distribution of mutations and copy number alterations (CNAs) across tumors. *Black asterisks represent non informative tumors for mutations, nuclear β-catenin presence and CNAs. A. For mutations, tumors are clustered together according to site of primary disease and denoted with distinct colors; yellow corresponds to a single ampullary carcinoma grouped together with bile duct carcinomas as extrahepatic. Numbers 0-≥5 and the related-5 color scale represent the number of mutations per gene per tumor. Genes mutated in >1 tumors are shown. Hypermutated GBCs are clustered together within a grey rectangular. Upper bar charts show the number of detected mutations vs. SNPs for each tumor, and the difference between number of mutations vs. number of mutated genes, as indicated. Tumors with a difference >5, harboring multiple mutations in >10 genes were considered hypermutated (arrow). Blue asterisk: truncated Y-axes. For tumors without SNP bars, mutations were detected with Sanger sequencing only. Black cells below the bar charts denote tumors positive for nuclear β-catenin protein with IHC. Diagram on the left shows prevalence of mutated genes and diagram on the right prevalence of clonal mutations (VAF>25%) in hypermutated GBCs and all other mutated tumors, as indicated; red asterisks: genes not interrogated with NGS. CTNNB1 (36%), PTEN (33%), TP53 (31%) and PIK3R1 (29%) were the most frequently mutated genes, followed by PIK3CA (13%), BRCA2 and KRAS (12%), and BRCA1 (11%) while all other mutated genes were present in <10% of tumors. Mutations in TP53 and members of PI3K, Wnt and RAS pathways were prevalent in all mutated tumors, excluding the hypermutated GBCs. Mutations in DNA repair, chromatin modification, immune response genes together with TP53 were predominant in hypermutated GBCs, while KRAS and CTNNB1 mutations were absent. B. For CNAs, CN status per gene is described by a 4-color scale, as indicated. Corresponding CN prevalence is shown in the diagram on the left. EGFR was the most frequently amplified gene, with 35% of all tumors showing gain, either high or low, followed by PIK3CA (27%), PRKAR1A (21%), MET (17%), PIK3R1 (8%) and ERBB2 (6%).
Most NGS mutations (89%) were present at VAFs<25%, a feature most prominent in hmGBC (Table S6). Top genes with clonal mutations (VAFs>25%) were TP53 and PIK3CA, found only in GBC (Figure 2A). All TP53 (p.R175H, p.R273H, p.H179Y) and most PIK3CA (p.E545K, p.E542K and p.H1047R) clonal mutations were known pathogenic or likely pathogenic, according to ClinVar. In non-hm tumors, we found 19 clonal mutations (VAFs>25%) in 9 genes and in 15 tumors (33% of non-hm cases). In 4 hmGBCs we detected 28 clonal mutations, corresponding to 20 genes (Figure 2A). Of note, a POLE clonal mutation (p.Pro486Ser; not registered, outside the exonuclease domain) was found at 30% VAF in a hmGBC with 160 mutations in 56 genes; the germline pathogenic BRCA1 p.Q1777fs (VAF 26%) and p.Q563* (VAF 40%) were present in another but at frequencies not expected for germline mutations, while 1 MSH3 mutation and 4 mutations in HRR genes (2 PALB2, 1 BRCA1 and 1 BRCA2) were also identified at VAFs>25% in a single hmGBC (Figure 2A; Table S6).
Regarding CNAs, 38 tumors (45%) were informative for all examined targets (Figure 1). The distribution of CN changes across tumors is shown in Figure 2B. For all genes, most aberrations were marginal gains. EGFR was by far the most frequently amplified gene, while CN gains of PRKAR1A, PIK3CA, ERBB2 and MET were also observed (Figure 2B). Associations and p values of genetic alterations are shown in Table 2. Most PTEN mutations coincided with CTNNB1 mutations. TP53 mutations were more prevalent in GBC, whereas PRKAR1A CN gains in ICC. All KIT mutations were found in patients >68 yrs old and a female preponderance for CTNNB1 mutations, although both these findings may be biased by the overall limited sample size.
Table 2.
Significant associations of mutations, copy number aberrations and mRNAexpression of angiogenesis related genes
| Genomic change | No genomic change | p value | |
|
| |||
| Site | TP53 mut | TP53 wt | |
| ECC | 0 | 14 (100%) | <0.001 |
| GBC | 23 (43%) | 31 (57%) | |
| ICC | 2 (15%) | 11 (85%) | |
| Site | PRKAR1A copy gain | PRKAR1A no gain | |
| ECC & GBC | 26 (45%) | 32 (55%) | 0.007 |
| ICC | 10 (91%) | 1 (9%) | |
| Sex | CTNNB1 mut | CTNNB1 wt | |
| Men | 20 (49%) | 21 (51%) | 0.017 |
| Women | 7 (21%) | 26 (79%) | |
| PTEN | CTNNB1 mut | CTNNB1 wt | |
| Mutated (mut) | 17 (63%) | 6 (14%) | <0.001 |
| Wild-type (wt) | 10 (37%) | 38 (86%) | |
| Age | KIT mut | KIT wt | |
| ≥68 yrs | 6 (15%) | 35 (85%) | 0.028 |
| <68 yrs | 0 | 35 (100%) | |
|
| |||
| mRNA RQ positive | mRNA RQ negative | ||
|
| |||
| Nuclear β-catenin | EDNRB | ||
| Positive | 15 (40%) | 9 (75%) | 0.047 |
| Negative | 23 (60%) | 3 (25%) | |
| Nuclear β-catenin | FLT4 | ||
| Positive | 6 (25%) | 18 (69%) | 0.002 |
| Negative | 18 (75%) | 8 (31%) | |
| Nuclear β-catenin | VEGFC | ||
| Positive | 4 (20%) | 19 (59%) | 0.009 |
| Negative | 16 (80%) | 13 (41%) | |
| CTNNB1 | EDNRB | ||
| Mutated | 10 (25%) | 7 (70%) | 0.021 |
| Wild-type | 30 (75%) | 3 (30%) | |
| PIK3R1 | EDNRA | ||
| Mutated | 5 (17%) | 5 (6%) | 0.032 |
| Wild-type | 25 (83%) | 4 (44%) | |
| PTEN | EDNRA | ||
| Mutated | 7 (20%) | 7 (70%) | 0.005 |
| Wild-type | 28 (80%) | 3 (30%) | |
| PIK3CA | EDN1 | ||
| Mutated | 9 (27%) | 0 | 0.010 |
| Wild-type | 24 (73%) | 20 (100%) | |
| ERBB2 | FLT4 | ||
| Copy gain | 10 (36%) | 2 (9%) | 0.045 |
| No copy gain | 18 (64%) | 20 (91%) | |
ICC, ECC: intra- and extrahepatic cholangiocarcinoma, respectivley; GBC: gallbladder carcinoma; RQ: relative quantification value.
β-catenin protein expression
Immunohistochemical staining for β-catenin was informative in 71 tumors. Membranous or cytoplasmic β-catenin staining was seen in 69 tumors (97%), while nuclear β-catenin in 32 tumors (45%) and in 5 out of 6 hmGBC (Figure 2A). No association was observed between nuclear b-catenin and CTNNB1 mutations.
mRNA expression profiling and associations
In total, 79 RNA samples were available for mRNA expression analysis and 34 (43%) yielded informative results for all targets (Figure 1). The distribution of tumor RQ values per target is displayed in Figure 3. Most tumors expressed relatively high mRNA levels of VEGFA, its receptors FLT1 and KDR and co-receptors NRP1 and NRP2, as well as of endothelin EDN1 and its receptors EDNRA and EDNRB (Figure 3). We observed strong mRNA expression correlations between NRP2 and EDNRA, but also between the two neuropilins (NRP1 and NRP2), NRP1 and EDNRA, as well as between VEGFA and its receptors (Figure 4).
Figure 3.

Distribution of mRNA relative quantification (RQ) values (40-dCT) for each target. Red lines correspond to designated cut-offs; Mean for EDN1, FLT4, VEGFC; natural cut-off for EDNRB, FLT1, KDR, NRP1, NRP2, and VEGFA; 25th percentile for EDNRA. Percentages left or right to the red line define expressing or not expressing tumors according to cutoff. Mean (M); Median (Mdn); ± SD values are shown.
Figure 4.
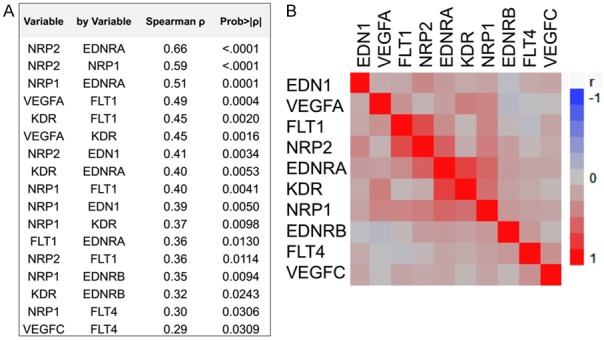
Correlations of mRNA targets with each other. Relative quantification values were compared as continuous variables. A. Spearman’s correlations and statistically significant p values are shown. B. Correlation map (clustered).
All PIK3CA mutated tumors expressed EDN1 mRNA, while the majority of EDNRA expressing tumors lacked PTEN or PIK3R1 mutations. Most tumors expressing nuclear β-catenin were negative for VEGFC and FLT4 mRNA expression but positive for EDNRB, while most EDNRB expressing tumors lacked CTNNB1 mutations. Finally, the majority of tumors with ERBB2 CN gain expressed FLT4 (Table 2). Spearman’s coefficient values depicting significant associations between angiogenesis targets and clustered correlation maps are shown in Figure 4. We did not observe any associations between clinicopathological parameters and mRNA expression of the examined genes.
Discussion
Mutations in TP53 and in genes of the PI3K, Wnt and RAS pathways were prevalent in our BTC cohort. The frequency and predominance of TP53 mutations in the present GBC is in line with the proposed role of TP53 aberrations early in GBC pathogenesis [4,10,12,31]. The herein higher than previously frequency of CTNNB1, PTEN and PIK3R1 mutations could be partly due to the screening method, since we performed Sanger sequencing on nested PCR products. A high incidence of CTNNB1 mutations is reported in adenomas of the gallbladder but these mutations are not considered as major contributors towards gallbladder carcinogenesis [12,23,32]. Here, the detection of nuclear β-catenin protein in almost half the tumors suggests canonical Wnt pathway activation, complying with earlier work [7]. Although CTNNB1 mutations were present in half the tumors with nuclear β-catenin expression, these two parameters were not associated, implying that the Wnt pathway may be activated through different factors in BTC. In support to this option, experimental studies show that Wnt pathway activation in cholangiocarcinoma is sustained by Wnt ligand-secreting inflammatory macrophages [6,13,33]. The co-existence of CTNNB1 and PTEN mutations is novel in BTC; this pattern is seen in ovarian and endometrial endometrioid carcinomas implying Wnt and PI3K pathway cross-talk [34]. In agreement with the involvement of inactivating PTEN mutations in GBC and ICC development, almost half of PTEN mutations in our BTC were predicted pathogenic [10-12,20,21,23,24,32]. The incidence of PIK3CA mutations abides by previous studies [1,2,11,31]. Despite the high incidence of PIK3R1 mutations recorded here, few studies have detected inactivating PIK3R1 mutations in BTC [10,24]. Nevertheless, the herein reported high incidence of clonal and known pathogenic mutations in the PI3K pathway support the rationale for testing BKM120, MK2206 and everolimus (PI3K, AKT and mTOR inhibitors, respectively) in phase I/II studies for their efficacy as targeted therapies for refractory and advanced disease [2,4,6,11,31].
A novelty in this study is the described subset of hmGBC, with prevalent HRR gene mutations that were present in all these tumors, and occasionally with multiple clonal mutations in genes from different DNA repair pathways (POLE, MMR). Inactivating mutations in HRR genes have been previously described in hypermutated BTC and in rare cases with familial cholangiocarcinoma [10,11]. Germline DNA was not available from our patients; therefore, we cannot proof the inherited or acquired nature of HRR mutations. However, since their frequencies were in general lower than expected for germline mutations in tissues, we rather speculate that our hmGBC mostly carried somatic mutations in HRR genes. Since DNA repair deficient tumors display sensitivity to PD1 blockade with immune checkpoint inhibitors (pembrolizumab), immunotherapy or treatment with synthetic lethality drugs (e.g., PARP inhibitors) might prove beneficiary for BTC patients with the respective mutational profile [6,11,35]. Of note, all hmGBC carried mutations in the PD1 and half of them in the PD-L1 gene. PD-L1 mutations were clonal, perhaps justifying the reported increased expression of PD-L1 in hypermutated BTC [10]. However, PD1 mutations presented at low frequencies and might have derived from the tumor microenvironment and not necessarily from the cancer cells. At present, the effect of such mutations on the PD1/PD-L1 axis and especially on the PD1 protein is unknown. This finding merits further investigation, since pembrolizumab for example, which is already trialed in BTC, particularly targets the PD1 protein.
Mutations in epigenetic regulators have been reported in BTC, in concert with our findings [2,9,10,12,24]. Limited knowledge exists on the prognostic significance of genetic aberrations in chromatin remodeling genes, except for BAP1, associated with aggressive disease, suggesting that histone deacetylase inhibitors might hold promise as therapeutic targets [11]. The demonstrated incidence of KRAS mutations agrees with earlier work. In addition, although FGFR2 is considered as a therapeutic target in BTC, particularly in ICC [6,10], we only observed 1 FGFR2 mutated ICC probably due to the limited number of such tumors in the present series. The latter may also explain the absence of IDH1 and BRAF V600E mutations which are mostly reported in ICC [2,4,18,22].
We detected gene copy gains more frequently in EGFR and less so in PRKAR1A, PIK3CA, PIK3R1, MET and ERBB2. Consistent with this finding, EGFR protein expression has been described in BTC and associated with amplification [1,6,11,26]. EGFR inhibitors have been employed in BTC treatment, mostly phase II and 1 phase III study, with only modest success, mainly due to downstream KRAS mutations or other markers associated with response to EGFR therapy [2,6]. ERBB2 may also be overexpressed and amplified in GBC and ECC [1,2,6,25,26]. Treatment with trastuzumab has shown beneficiary results in GBC patients with ERBB2 amplification or overexpression [5]. MET amplification is seen in ICC, while MET protein overexpression in GBC, the latter associated with decreased overall survival [1,20]. Contrary to the PIK3CA copy gains observed here, amplification has not been described in BTC [36]. In comparison, the present novel finding on PRKAR1A copy gains in ICC may explain the previously reported PRKAR1A mRNA and protein overexpression in these tumors [27]. PRKAR1A knockdown reduces growth and induces apoptosis in cholangiocarcinoma cell lines, with a concomitant decrease in PI3K/Akt, JAK/STAT and Wnt signaling, suggesting pathway crosstalk and a role for PRKAR1A as a target for therapy [27].
In agreement with previous work, we observed increased mRNA expression of VEGFA and other angiogenesis factors, and positive correlations between mRNA expression of VEGFA and its receptors [1,3,6,37]. Inhibitors against VEGFA (bevacizumab), or VEGFA receptors, including sorafenib, sunitinib and vandetanib, have been tested for their effectiveness in BTC treatment in various phase I/II trials; only the former, depending on regimen, yielded a 7 or 8.1 months benefit in median PFS [1-4,6]. About half the tumors in our cohort showed low to moderate VEGFC and FLT4 mRNA expression, while we also report on a novel inverse association between nuclear β-catenin expression and lymphangiogenesis, which merits functional proof. A recent study demonstrated VEGFC protein expression in cholangiocarcinoma, shown to be higher in those with lymph node metastasis [38]. Unfortunately, missing information on lymph node status here prevented examination of associations between VEGFC mRNA expression and lymph node metastasis.
Our observations on high mRNA expression of neuropilins NRP1 and NRP2, complies with their described expression in gastrointestinal adenocarcinomas, gallbladder and ampulla of Vater carcinomas and their roles in tumor progression, metastasis, angiogenesis and lymphangiogenesis, suggesting they are targets for therapy [39,40]. SEMA3B, a tumor suppressor and inhibitory ligand of NRP2, is frequently hypermethylated in cholangiocarcinoma [4,39]. Our findings of moderate mRNA expression of EDN1 and its receptors EDNRA and EDNRB agrees with previous findings displaying EDNRA and EDNRB protein expression in cholangiocarcinoma tissue at higher levels than normal tissue. EDNRA and EDNRB have emerged as druggable targets in cancer treatment; they are overexpressed in various cancers and their activation mediates PI3K/AKT, β-catenin and Ras/Raf/MEK cascades, promoting tumor proliferation, angiogenesis and metastasis. In epithelial ovarian carcinoma, the EDN1/EDNRA axis facilitates nuclear β-catenin translocation and subsequent transcriptional activation, dependent on β-arrestin/EGFR cross-talk and also induces VEGFA expression via HIF-1α [41]. Clearly, this retrospective descriptive study has certain limitations, including small sample size and missing data on tumor size and nodal status. Nevertheless, the herein observed interplay between PI3K pathway or Wnt pathway aberrations and the mRNA expression of angiogenesis factors is a novelty that necessitates validation within a larger patient population.
In conclusion, we retrospectively validated in our cohort of BTC patients genetic aberrations and mRNA profiles of genes proposed to function as oncogenic drivers, are members of pathways activated in BTC or are tested as drug targets [8-11,24]. We also confirmed the presence of distinct genotypes among GBC, ICC and ECC. Novel findings include the presence of PIK3CA and PRKAR1A copy gains, the prevalence of the latter in ICC, the co-existence of PI3K and WNT pathway gene mutations, their association with angiogenesis, and the hypermutated GBC subtype with HRR gene mutations. The latter may be considered for new treatment options in this challenging to treat disease.
Acknowledgements
This study was supported by an internal Hellenic Cooperative Oncology Group (HeCOG) translational research grant (HE TRANS_37/17). The authors wish to thank Mrs. Sophia Chrisafi for tumor data management and Mrs. Emily Daskalaki and Mrs. Elpida Charalambous for technical assistance with molecular methods. Special thanks to Dr. Eleftherios Panteris for his contribution in the presentation of results.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Marks EI, Yee NS. Molecular genetics and targeted therapeutics in biliary tract carcinoma. World J Gastroenterol. 2016;22:1335–1347. doi: 10.3748/wjg.v22.i4.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bridgewater JA, Goodman KA, Kalyan A, Mulcahy MF. Biliary tract cancer: epidemiology, radiotherapy, and molecular profiling. Am Soc Clin Oncol Educ Book. 2016;35:e194–203. doi: 10.1200/EDBK_160831. [DOI] [PubMed] [Google Scholar]
- 3.Merla A, Liu KG, Rajdev L. Targeted therapy in biliary tract cancers. Curr Treat Options Oncol. 2015;16:48. doi: 10.1007/s11864-015-0366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oyasiji T, Zhang J, Kuvshinoff B, Iyer R, Hochwald SN. Molecular targets in biliary carcinogenesis and implications for therapy. Oncologist. 2015;20:742–751. doi: 10.1634/theoncologist.2014-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Javle M, Churi C, Kang HC, Shroff R, Janku F, Surapaneni R, Zuo M, Barrera C, Alshamsi H, Krishnan S, Mishra L, Wolff RA, Kaseb AO, Thomas MB, Siegel AB. HER2/neu-directed therapy for biliary tract cancer. J Hematol Oncol. 2015;8:58. doi: 10.1186/s13045-015-0155-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chong DQ, Zhu AX. The landscape of targeted therapies for cholangiocarcinoma: current status and emerging targets. Oncotarget. 2016;7:46750–46767. doi: 10.18632/oncotarget.8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen W, Liang J, Huang L, Cai J, Lei Y, Lai J, Liang L, Zhang K. Characterizing the activation of the Wnt signaling pathway in hilar cholangiocarcinoma using a tissue microarray approach. Eur J Histochem. 2016;60:2536. doi: 10.4081/ejh.2016.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie D, Ren Z, Fan J, Gao Q. Genetic profiling of intrahepatic cholangiocarcinoma and its clinical implication in targeted therapy. Am J Cancer Res. 2016;6:577–586. [PMC free article] [PubMed] [Google Scholar]
- 9.Churi CR, Shroff R, Wang Y, Rashid A, Kang HC, Weatherly J, Zuo M, Zinner R, Hong D, Meric-Bernstam F, Janku F, Crane CH, Mishra L, Vauthey JN, Wolff RA, Mills G, Javle M. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One. 2014;9:e115383. doi: 10.1371/journal.pone.0115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, Hama N, Hosoda F, Urushidate T, Ohashi S, Hiraoka N, Ojima H, Shimada K, Okusaka T, Kosuge T, Miyagawa S, Shibata T. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47:1003–1010. doi: 10.1038/ng.3375. [DOI] [PubMed] [Google Scholar]
- 11.Jain A, Javle M. Molecular profiling of biliary tract cancer: a target rich disease. J Gastrointest Oncol. 2016;7:797–803. doi: 10.21037/jgo.2016.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simbolo M, Fassan M, Ruzzenente A, Mafficini A, Wood LD, Corbo V, Melisi D, Malleo G, Vicentini C, Malpeli G, Antonello D, Sperandio N, Capelli P, Tomezzoli A, Iacono C, Lawlor RT, Bassi C, Hruban RH, Guglielmi A, Tortora G, de Braud F, Scarpa A. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget. 2014;5:2839–2852. doi: 10.18632/oncotarget.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boulter L, Guest RV, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ, Ridgway RA, Samuel K, Van Rooijen N, Barry ST, Wigmore SJ, Sansom OJ, Forbes SJ. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest. 2015;125:1269–1285. doi: 10.1172/JCI76452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P, Lind GE, Folseraas T, Forbes SJ, Fouassier L, Geier A, Calvisi DF, Mertens JC, Trauner M, Benedetti A, Maroni L, Vaquero J, Macias RI, Raggi C, Perugorria MJ, Gaudio E, Boberg KM, Marin JJ, Alvaro D. Expert consensus document: cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European network for the study of cholangiocarcinoma (ENS-CCA) Nat Rev Gastroenterol Hepatol. 2016;13:261–280. doi: 10.1038/nrgastro.2016.51. [DOI] [PubMed] [Google Scholar]
- 15.Javle M, Rashid A, Churi C, Kar S, Zuo M, Eterovic AK, Nogueras-Gonzalez GM, Janku F, Shroff RT, Aloia TA, Vauthey JN, Curley S, Mills G, Roa I. Molecular characterization of gallbladder cancer using somatic mutation profiling. Hum Pathol. 2014;45:701–708. doi: 10.1016/j.humpath.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voss JS, Holtegaard LM, Kerr SE, Fritcher EG, Roberts LR, Gores GJ, Zhang J, Highsmith WE, Halling KC, Kipp BR. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol. 2013;44:1216–1222. doi: 10.1016/j.humpath.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Zhu AX, Borger DR, Kim Y, Cosgrove D, Ejaz A, Alexandrescu S, Groeschl RT, Deshpande V, Lindberg JM, Ferrone C, Sempoux C, Yau T, Poon R, Popescu I, Bauer TW, Gamblin TC, Gigot JF, Anders RA, Pawlik TM. Genomic profiling of intrahepatic cholangiocarcinoma: refining prognosis and identifying therapeutic targets. Ann Surg Oncol. 2014;21:3827–3834. doi: 10.1245/s10434-014-3828-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goeppert B, Frauenschuh L, Renner M, Roessler S, Stenzinger A, Klauschen F, Warth A, Vogel MN, Mehrabi A, Hafezi M, Boehmer K, von Deimling A, Schirmacher P, Weichert W, Capper D. BRAF V600E-specific immunohistochemistry reveals low mutation rates in biliary tract cancer and restriction to intrahepatic cholangiocarcinoma. Mod Pathol. 2014;27:1028–1034. doi: 10.1038/modpathol.2013.206. [DOI] [PubMed] [Google Scholar]
- 19.Zhao S, Cao Y, Liu SB, Wang XA, Bao RF, Shu YJ, Hu YP, Zhang YJ, Jiang L, Zhang F, Liang HB, Li HF, Ma Q, Xu Y, Wang Z, Zhang YC, Chen L, Zhou J, Liu YB. The E545K mutation of PIK3CA promotes gallbladder carcinoma progression through enhanced binding to EGFR. J Exp Clin Cancer Res. 2016;35:97. doi: 10.1186/s13046-016-0370-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ross JS, Wang K, Gay L, Al-Rohil R, Rand JV, Jones DM, Lee HJ, Sheehan CE, Otto GA, Palmer G, Yelensky R, Lipson D, Morosini D, Hawryluk M, Catenacci DV, Miller VA, Churi C, Ali S, Stephens PJ. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist. 2014;19:235–242. doi: 10.1634/theoncologist.2013-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ali A, Mishra PK, Sharma S, Arora A, Saluja SS. Effects of PTEN gene alteration in patients with gallbladder cancer. Cancer Genet. 2015;208:587–594. doi: 10.1016/j.cancergen.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 22.Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, Kwak EL, Clark JW, Ryan DP, Deshpande V, Dias-Santagata D, Ellisen LW, Zhu AX, Iafrate AJ. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17:72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumari N, Corless CL, Warrick A, Beadling C, Nelson D, Neff T, Krishnani N, Kapoor VK. Mutation profiling in gallbladder cancer in Indian population. Indian J Pathol Microbiol. 2014;57:9–12. doi: 10.4103/0377-4929.130849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou S, Li J, Zhou H, Frech C, Jiang X, Chu JS, Zhao X, Li Y, Li Q, Wang H, Hu J, Kong G, Wu M, Ding C, Chen N, Hu H. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun. 2014;5:5696. doi: 10.1038/ncomms6696. [DOI] [PubMed] [Google Scholar]
- 25.Roa I, de Toro G, Schalper K, de Aretxabala X, Churi C, Javle M. Overexpression of the HER2/neu gene: a new therapeutic possibility for patients with advanced gallbladder cancer. Gastrointest Cancer Res. 2014;7:42–48. [PMC free article] [PubMed] [Google Scholar]
- 26.Yang X, Wang W, Wang C, Wang L, Yang M, Qi M, Su H, Sun X, Liu Z, Zhang J, Qin X, Han B. Characterization of EGFR family gene aberrations in cholangiocarcinoma. Oncol Rep. 2014;32:700–708. doi: 10.3892/or.2014.3261. [DOI] [PubMed] [Google Scholar]
- 27.Loilome W, Juntana S, Namwat N, Bhudhisawasdi V, Puapairoj A, Sripa B, Miwa M, Saya H, Riggins GJ, Yongvanit P. PRKAR1A is overexpressed and represents a possible therapeutic target in human cholangiocarcinoma. Int J Cancer. 2011;129:34–44. doi: 10.1002/ijc.25646. [DOI] [PubMed] [Google Scholar]
- 28.Fava G, Demorrow S, Gaudio E, Franchitto A, Onori P, Carpino G, Glaser S, Francis H, Coufal M, Marucci L, Alvaro D, Marzioni M, Horst T, Mancinelli R, Benedetti A, Alpini G. Endothelin inhibits cholangiocarcinoma growth by a decrease in the vascular endothelial growth factor expression. Liver Int. 2009;29:1031–1042. doi: 10.1111/j.1478-3231.2009.01997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gogas H, Kotoula V, Alexopoulou Z, Christodoulou C, Kostopoulos I, Bobos M, Raptou G, Charalambous E, Tsolaki E, Xanthakis I, Pentheroudakis G, Koutras A, Bafaloukos D, Papakostas P, Aravantinos G, Psyrri A, Petraki K, Kalogeras KT, Pectasides D, Fountzilas G. MYC copy gain, chromosomal instability and PI3K activation as potential markers of unfavourable outcome in trastuzumab-treated patients with metastatic breast cancer. J Transl Med. 2016;14:136. doi: 10.1186/s12967-016-0883-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kotoula V, Charalambous E, Biesmans B, Malousi A, Vrettou E, Fountzilas G, Karkavelas G. Targeted KRAS mutation assessment on patient tumor histologic material in real time diagnostics. PLoS One. 2009;4:e7746. doi: 10.1371/journal.pone.0007746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bizama C, Garcia P, Espinoza JA, Weber H, Leal P, Nervi B, Roa JC. Targeting specific molecular pathways holds promise for advanced gallbladder cancer therapy. Cancer Treat Rev. 2015;41:222–234. doi: 10.1016/j.ctrv.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Yoo KH, Kim NK, Kwon WI, Lee C, Kim SY, Jang J, Ahn J, Kang M, Jang H, Kim ST, Ahn S, Jang KT, Park YS, Park WY, Lee J, Heo JS, Park JO. Genomic alterations in biliary tract cancer using targeted sequencing. Transl Oncol. 2016;9:173–178. doi: 10.1016/j.tranon.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–1473. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McConechy MK, Ding J, Senz J, Yang W, Melnyk N, Tone AA, Prentice LM, Wiegand KC, McAlpine JN, Shah SP, Lee CH, Goodfellow PJ, Gilks CB, Huntsman DG. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod Pathol. 2014;27:128–134. doi: 10.1038/modpathol.2013.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldstein D, Lemech C, Valle J. New molecular and immunotherapeutic approaches in biliary cancer. ESMO Open. 2017;2:e000152. doi: 10.1136/esmoopen-2016-000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang SD, McCrudden CM, Meng C, Lin Y, Kwok HF. The significance of combining VEGFA, FLT1, and KDR expressions in colon cancer patient prognosis and predicting response to bevacizumab. Onco Targets Ther. 2015;8:835–843. doi: 10.2147/OTT.S80518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao R, Chang Y, Liu Z, Liu Y, Guo S, Yu J, Wang J. Effect of vascular endothelial growth factor-C expression on lymph node metastasis in human cholangiocarcinoma. Oncol Lett. 2015;10:1011–1015. doi: 10.3892/ol.2015.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Migliozzi MT, Mucka P, Bielenberg DR. Lymphangiogenesis and metastasis--a closer look at the neuropilin/semaphorin3 axis. Microvasc Res. 2014;96:68–76. doi: 10.1016/j.mvr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hansel DE, Wilentz RE, Yeo CJ, Schulick RD, Montgomery E, Maitra A. Expression of neuropilin-1 in high-grade dysplasia, invasive cancer, and metastases of the human gastrointestinal tract. Am J Surg Pathol. 2004;28:347–356. doi: 10.1097/00000478-200403000-00007. [DOI] [PubMed] [Google Scholar]
- 41.Rosano L, Bagnato A. β-arrestin1 at the cross-road of endothelin-1 signaling in cancer. J Exp Clin Cancer Res. 2016;35:121. doi: 10.1186/s13046-016-0401-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.