

Abstract
The acquired resistance to bortezomib represents a major obstacle for multiple myeloma (MM) treatment. Studies revealed that the treatment with cyclic adenosine monophosphate (cAMP) may be a promising strategy for MM therapy. Therefore, the present study aimed to explore the mechanism of action of cAMP in MM cells. Our results showed that 8-CPT-cAMP and bortezomib synergistically induced growth inhibition and apoptosis in MM bortezomib-resistant cell lines and primary MM cells, in which protein kinase A (PKA) activation was involved. Furthermore, 8-CPT-cAMP induced the degradation of cyclinD1 and downregulation of myeloid cell leukemia-1 (Mcl-1). Moreover, 8-CPT-cAMP enhanced endoplasmic reticulum stress caused by bortezomib. A synergy between bortezomib and cAMP was also revealed in a murine MOPC315 xenograft model, which was evidenced by the significantly inhibited tumor growth and the improved multiple cancer-related parameters by the combination of the cAMP-elevating compound forskolin and bortezomib. Taken together, this study suggests that the treatment with cAMP may be a promising strategy for enhancing the therapeutic efficacy of bortezomib in MM treatment.
Keywords: Multiple myeloma, cAMP, bortezomib, apoptosis
Introduction
Multiple myeloma (MM) is one of the common hematologic malignancies, which is characterized by clonal B-cell-derived plasma cells accumulation in the bone marrow and accounts for approximately 1% of all neoplastic disorders [1,2]. Even though bortezomib therapy advanced the treatment for MM [3,4], off-target toxicities and drug-resistance development have been reported in MM treatment [5-7]. Therefore, further investigations are required to identify the drug resistance mechanisms and explore novel therapeutic strategies to overcome bortezomib resistance.
Recently, several studies have been focused on the development of a combination of bortezomib and inhibitors of signal transduction pathways to enhance antitumor efficacy and restore the sensitivity of MM cells to bortezomib [8,9]. Their results strongly supported the notion that combination therapy was an effective approach to overcome the pitfalls of the treatment with bortezomib only. Cyclic adenosine monophosphate (cAMP), one of the common second messengers, was found to be involved in hematopoietic cell proliferation, apoptosis, and differentiation [10]. Previous studies reported that cAMP signaling stimulation induced apoptosis in both steroid-sensitive and -resistant MM cells [11,12]. In addition, downregulation of myeloid cell leukemia-1 (Mcl-1) was involved in cAMP-induced apoptosis in MM [13]. Furthermore, it was suggested that intracellular cAMP elevation kills MM cells in vitro and inhibits MM development in vivo [14], and multiple mechanisms were involved in the process [15]. A recent examination revealed that the natural compound forskolin, a cAMP-elevating agent, synergized with dexamethasone to induce cell death in MM cells [16].
Therefore, we hypothesized that cAMP may sensitize MM cells to bortezomib, especially the bortezomib-resistant cells. In the present study, we found that cAMP induced cell apoptosis and overcame bortezomib resistance in MM both in vitro and in vivo.
Materials and methods
Cell culture and reagents
Human MM cell lines NCI-H929 (referred to as H929), U266, and the murine MM cell line MOPC-315, purchased from the American Type Culture Collection (ATCC, USA), were cultured in RPMI-1640, supplemented with 10% (v/v) fetal calf serum (FCS; Gibco, USA) and 1/100 penicillin-streptomycin (Gibco, USA). HEK-293T cell line was obtained from ATCC. Then, bone marrow stromal cells (BMSC) and HEK-293T were cultured in Dulbecco’s modified Eagle’s medium (DMEM, HyClone, Logan, UT, USA) supplemented with 10% (v/v) FCS and 1/100 penicillin-streptomycin. All cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
H929 bortezomib resistance (referred to as H929-R) cells and U266 bortezomib resistance (referred to as U266-R) cells were established by consecutive stimulation with a gradually increasing bortezomib dosage (See Supplementary material). In brief, H929 and U266 cells were gradually exposed to the increased dose of bortezomib (from an initial dose of 4 nM to a final dose of 50 nM within 12 months with a gradient of 4 nM/month). Further, the cells were maintained in 50 nM bortezomib for three months and cultured in a bortezomib-free medium for two weeks prior to further analysis.
Bortezomib (Sigma Aldrich, USA) and 8-CPT-cAMP (Sigma Aldrich, USA) were dissolved in PBS as a stock solution at concentrations of 10 mM and 10 mM, respectively. Forskolin was dissolved in dimethyl sulfoxide (DMSO) to obtain a stock solution at a concentration of 10 mM. H-89 (N-(2-[p-bromocinnamylamino] ethyl)-5-isoquinolinesulfonamide) was obtained from Calbiochem (San Diego, CA, USA) and dissolved in 50% ethanol with a stock solution at 10 mM.
Primary MM cells isolation
Mononuclear cells (MNCs) were isolated from the bone marrow of MM patients by Ficoll-Hypaque (Pharmacia, Piscataway, NJ, USA) density sedimentation. CD138+ cells were isolated from MNCs using EasyStep CD138+ magnetic nanoparticles (Stem Cell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions. Written informed consent was obtained from each patient. The study was conducted in accordance with the Declaration of Helsinki [17], and the study protocol was approved by the Clinical Investigational Reviewing Board of the Shanghai Jiao Tong University School of Medicine, China.
Cell apoptosis
The U266, H929, U266-R, and H929-R cells were exposed to bortezomib, 8-CPT-cAMP, or their combination. Cell apoptosis was measured by an Annexin-V apoptosis detection kit (BD Pharmingen, USA) following the manufacturer’s instructions. Data were collected and analyzed by FlowJo software (BD Biosciences, San Diego, CA, USA).
Synergism calculation
The synergism between bortezomib and 8-CPT-cAMP was determined by the combination index (CI) method using the CompuSyn software (ComboSyn Inc., Paramus, NJ, USA). Briefly, cell viability was determined by PI exclusion and expressed as the fractional inhibition by the individual drug or the drug combination. CI values greater than 1 indicated antagonism, a CI value of 1 indicated an additive effect, and CI values of less than 1 indicated synergy.
Western blot analysis
After treatment, cells were collected and lysed in RIPA lysis buffer (Beyotime, China). Protein concentration was determined by the Bradford assay. Equal amounts (10 µg/lane) of protein were separated by sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE, 10%-12%). Proteins were transferred to a PVDF membrane (Millipore, USA) and then blocked with 5% (w/v) non-fat milk at room temperature for 2 h. Afterwards, primary antibodies against PARP-1, caspase-3, PKA-cα, cyclinD1, Mcl-1, Bcl-2, Bax, CHOP, and ATF-4 with a dilution of 1:1,000 were incubated overnight at 4°C. Subsequently, horseradish peroxidase (HRP)-conjugated secondary antibodies were incubated at room temperature for 1 h. Protein bands were detected by electrochemiluminescence (ECL) (Amersham, USA) according to the manufacturer’s instructions. All antibodies (PARP-1, caspase-3, PKA-cα, cyclinD1, Mcl-1, Bcl-2, Bax, CHOP, and ATF-4) used in the present study were purchased from Santa Cruz Biotechnology, USA. Densitometry of proteins was performed by Quantity One software (Bio-Rad, Hercules, CA, USA).
Real-time PCR (RT-PCR)
Total RNA was extracted using the TRIzol (Invitrogen) method. Complementary DNA (cDNA) was transcribed using a reverse transcription kit (Promega, Japan) according to manufacturer’s instructions. The expression of CyclinD1, Mcl-1, CHOP, ATF-4, and GAPDH was detected by real time (RT-PCR) using the ABI PRISM 7900 system (Applied Biosystems, USA). The following RT-PCR cycling conditions were used: 95°C for 5 min; 40 cycles at 95°C for 5 s and at 60°C for 30 s; and then elongation was performed at 60°C for 10 min. The primers utilized are listed in Table 1. The relative mRNA expression of the target genes was analyzed by the 2-ΔΔCt method, and GAPDH served as an internal control.
Table 1.
Primers for RT-PCR
| Forward (5’-3’) | Reverse (5’-3’) | |
|---|---|---|
| CyclinD1 | GCGTACCCTGACACCAATCTC | CTCCTCTTCGCACTTCTGCTC |
| Mcl-1 | GGGCAGGATTGTGACTCTCATT | GATGCAGCTTTCTTGGTTTATGG |
| CHOP | AGGTCCTGTCCTCAGATGAAAT | CAGGGTCAAGAGTAGTGAAGGTTT |
| ATF-4 | CCTTCGACCAGTCGGGTTTG | CTGTCCCGGAAAAGGCATCC |
| GAPDH | CATCAAGAAGGTGGTGAAGC | ACCACCCTGTTGCTGTAG |
RNA interference and transfection
Complementary oligonucleotides pairs targeting PKA were designed (Top: 5’-GACAAACAGAAGGTGGTGATTCAAGAGATCACCACCTTCTGTTTGTCTTTTTTACGCGT-3’, Down: 5’-ACGCGTAAAAAAGACAAACAGAAGGTGGTGATCTCTTGAATCACCACCTTCTGTTTGTC-3’). Non-target control (NC) shRNA was synthesized by Transgene (Jiangsu, China) and was then annealed and ligated to the PSIREN-RetroQ Vector (Clontech, USA). shRNA-carrying retroviruses were produced in 293T cells and were further used to infect H929 cells. Stably transfected cells were selected by treatment with puromycin (2.0 μg/mL, Calbiochem, USA) for 96 h.
Co-culture
Bone marrow stromal cells (BMSCs) were isolated from bone marrow aspirates of MM patients as previously described [18]. For the cell co-culture experiment, 5 × 105 BMSCs cells were seeded in a 10-cm Petri dish. Next, 24 h after BMSCs seeding, 5 × 105 CD138+ cells were added into the dish, followed by 24-h incubation and collection of the cells for further use.
Cell morphology
After treatment, approximately 1 × 104 cells were cast on slides by using Thermo Shandon Cytospin (Thermo Fisher Scientific, USA). Cell morphology was analyzed by May-Grunwald-Giemsa staining and observed under a light microscope (Nikon, Japan).
Xenograft mouse model
Female BALB/c nu/nu mice aged 4-6 weeks, obtained from Silaike (Shanghai, China), were kept under specific-pathogen-free (SPF) conditions and used according to the Shanghai Medical Experimental Animal Care guidelines.
A number of 1 × 106 MOPC-315 cells were injected subcutaneously with matrix gel into the right hips. When tumor was palpable, the mice were divided into a control, bortezomib, forskolin, and bortezomib + forskolin groups (n = 4/group). Forskolin (5 mg/kg/d, intraperitoneal injection, i.p.), bortezomib (0.8 mg/kg, twice weekly by intravenous injection, i.v.), bortezomib plus forskolin (forskolin was injected four hours after bortezomib), or vehicle (8% DMSO and 92% saline) were administered. Body weight was monitored daily. Tumor sizes were measured with calipers, and the tumor volumes were calculated by the formula: tumor volume = 0.5 × a × b2, where “a” is the length and “b” is the width. Animal study protocols were approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University School of Medicine.
Histology
The expression of PCNA, cyclinD1, and Mcl-1 in MOPC-315 nude mice xenografts were analyzed using immunohistochemistry staining. Sections were deparaffinized, heat-treated with citrate buffer (pH 6.0) for 15 min. Next, endogenous peroxidase was blocked with 1% hydrogen peroxide for 30 min, and the non-specific-binding sites were blocked with 2% (w/v) goat serum for 30 min. The sections were then incubated with the primary antibodies against PCNA, TUNEL, cyclinD1, and Mcl-1 1:100 overnight at 4°C. Further, the biotinylated secondary antibody was incubated for 1 h. Avidin-biotin-peroxidase complex (Beyotime, China) was then added, and the color was developed by 3-3’-diaminobenzidine. Counterstaining was performed with hematoxylin and eosin (HE). All steps were performed at room temperature, and the sections were observed under a confocal microscope (Leica, Japan). Five random fields were selected.
Statistical analysis
The paired-samples t-test was applied to check the significance in the cell-line experiments using the PASW Statistic 18 software (IBM, Armonk, New York, USA) for Windows. The data were expressed as mean ± SEM. The Wilcoxon signed rank test was used in the in vivo experiments, to determine the significance of the between-group differences. A two-way P-value less than 0.05 was considered statistically significant.
Results
8-CPT-cAMP synergizes with bortezomib to induce MM cell apoptosis
A previous study indicated that 8-CPT-cAMP, a cell membrane-permeable cAMP analog, induced cell apoptosis and elevation of intracellular cAMP in MM cells, exhibiting attractive anti-cancer efficacy in MM animal models [14]. U266 and H929 cells treated with a combination of bortezomib and 8-CPT-cAMP showed an increasing proportion of condensed cytoplasms, ruptured nucleuses, and marginal chromatins as well as apoptotic bodies, which indicated the rising of cell death ratio. However, no prominent changes were observed in the presence of each agent alone (Figure 1A). This phenomenon was confirmed by Annexin V/PI staining, which suggests that 8-CPT-cAMP enhances the antitumor efficacy of bortezomib (Figure 1B). Moreover, the elevation of cleaved PARP and cleaved caspase-3 further reaffirmed these results (Figure 1C).
Figure 1.

The combination of 8-CPT-cAMP and bortezomib synergistically induced apoptosis in bortezomib-sensitive and -resistant multiple myeloma cells. A. Giemsa staining of U266, H929, U266-R, and H929-R cells following the treatment with 8-CPT-cAMP and bortezomib. Magnification: 400 ×; B. Cell apoptosis ratio of U266 and H929 cells exposed to 8-CPT-cAMP, bortezomib, or their combination for 48 h and 72 h, respectively. Cell apoptosis was evaluated by Annexin V/PI staining; C. Expression of PARP, caspase-3, and cleaved caspase-3 in U266 and H929 cells exposed to 8-CPT-cAMP, bortezomib, or their combination for 48 h; D. Cell apoptosis ratio of U266-R and H929-R cells exposed to 8-CPT-cAMP, bortezomib, or their combination for 48 h and 72 h, respectively; cell apoptosis was evaluated by Annexin V/PI staining; E. Expression of PARP, caspase-3, and cleaved caspase-3 in U266-R and H929-R cells exposed to 8-CPT-cAMP, bortezomib, or their combination for 48 h. U266-R, U266 bortezomib-resistant cells; H929-R, H929 bortezomib-resistant cells. The experiments were performed in triplicate. **P < 0.01, ***P < 0.001.
Next, we aimed to evaluate the effects of 8-CPT-cAMP in bortezomib-resistant MM cells. As expected, 8-CPT-cAMP synergized with bortezomib to induce marked morphological changes in U266-R and H929-R cells (Figure 1A). 8-CPT-cAMP alone did not significantly induce cell apoptosis in U266-R and H929-R cells. However, it dramatically enhanced the cell apoptotic effects of bortezomib (Figure 1D) and this was supported by the upregulation of cleaved PARP and cleaved caspase-3 (Figure 1E). To further verify that 8-CPT-cAMP was synergic with bortezomib in inducing MM cell apoptosis, bone marrow stromal cells (BMSCs) were isolated from three bortezomib-resistant MM patients. As depicted in Figure 2A, 2B, it is noteworthy that the combined treatment with 8-CPT-cAMP and bortezomib significantly promoted the apoptosis levels in BMSCs, which was confirmed by similar results, observed in CD138+ co-cultured BMSCs. The additive or synergistic cytotoxicity effect of the combination of bortezomib and 8-CPT-cAMP was further analyzed using the Chou-Talalay method. In the four tested MM cell lines (U266, H929, U266-R, and H929-R), the CI values between 8-CPT-cAMP and bortezomib treatments were less than 1 (Figure 3A, 3B), which suggested that the combination of these two drugs had synergistic effects in inducing MM cell apoptosis. Therefore, these results indicate that 8-CPT-cAMP synergizes with bortezomib in inducing MM cell apoptosis.
Figure 2.
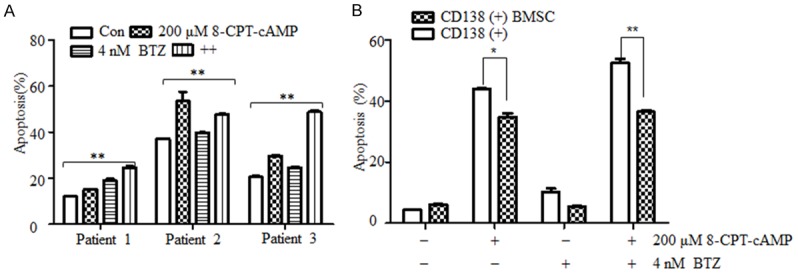
The combination of 8-CPT-cAMP and bortezomib synergistically induced apoptosis in primary CD138+ MM cells. A. Primary CD138+ cells isolated from MM patients were exposed to 8-CPT-cAMP, bortezomib, or their combinations for 24 h; cell apoptosis was evaluated by Annexin V/PI staining; B. CD138+ MM cell co-cultured with bone marrow stromal cells (BMSCs) for 24 h and treated with 8-CPT-cAMP, bortezomib, or their combination for 24 h; cell apoptosis was evaluated by Annexin V/PI staining. *P < 0.05, **P < 0.01. The experiments were performed in triplicate.
Figure 3.
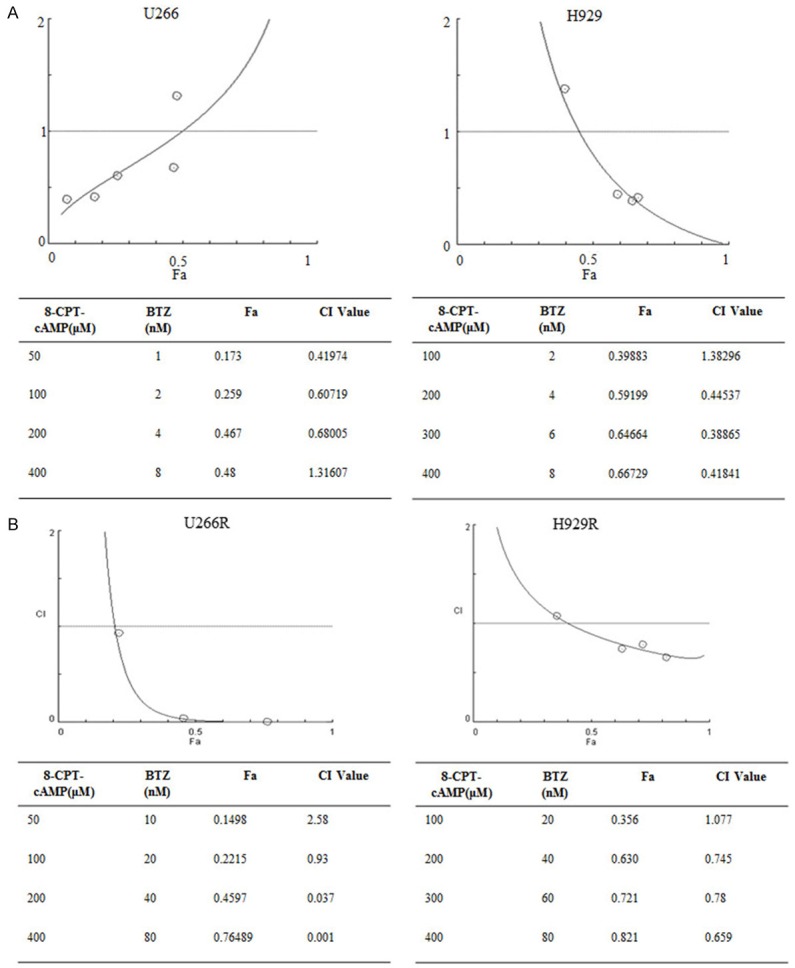
8-CPT-cAMP and bortezomib have synergetic effects. CI analysis of the combination of 8-CPT-cAMP and bortezomib in (A) U266 and H929 cells and in (B) U266-R and H929-R cells. U266-R, U266 bortezomib-resistant cells; H929-R, H929 bortezomib-resistant cells. The experiments were performed in triplicate.
PKA activation was involved in bortezomib and cAMP-induced cell growth inhibition and apoptosis
As known, protein kinase A (PKA) is the main downstream effector protein in triggering biological responses. Therefore, the activators of PKA (6-Bnz-cAMP) and exchange protein directly activated by cAMP (Epac, 8-pCPT-2’-O-Me-cAMP) were further used to examine the potential role of PKA in the cell growth inhibition and apoptosis induced by cAMP and bortezomib. As can be seen in Figure 4B, 6-Bnz-cAMP inhibited the proliferation of both H929 and H929-R cells and enhanced bortezomib-induced cell apoptosis as indicated by Annexin V/PI staining, the upregulation of cleaved caspase-3, and cleaved PARP (Figure 4C, 4D), as well as by the morphological changes observed (data not shown). Conversely, 8-pCPT-2’-O-Me-cAMP, a specific Epac activator, did not synergize with bortezomib to induce cell apoptosis (Figure 4A). These observations suggest that the activation of PKA was involved in the synergistic effect of the combination of cAMP and bortezomib in inducing apoptosis of MM cells. To confirm the validity of this hypothesis, the expression of PKA in H929 cells was silenced (Figure 4E). As expected, PKA knockdown significantly decreased the 8-CPT-cAMP- and bortezomib-induced cell apoptosis (Figure 4F). Furthermore, the addition of H89 (a specific PKA inhibitor) blocked the apoptosis-inducing effect triggered by bortezomib and 8-CPT-cAMP (Figure 4G). Taken together, these results suggested that PKA was involved in 8-CPT-cAMP- and bortezomib-induced apoptosis.
Figure 4.
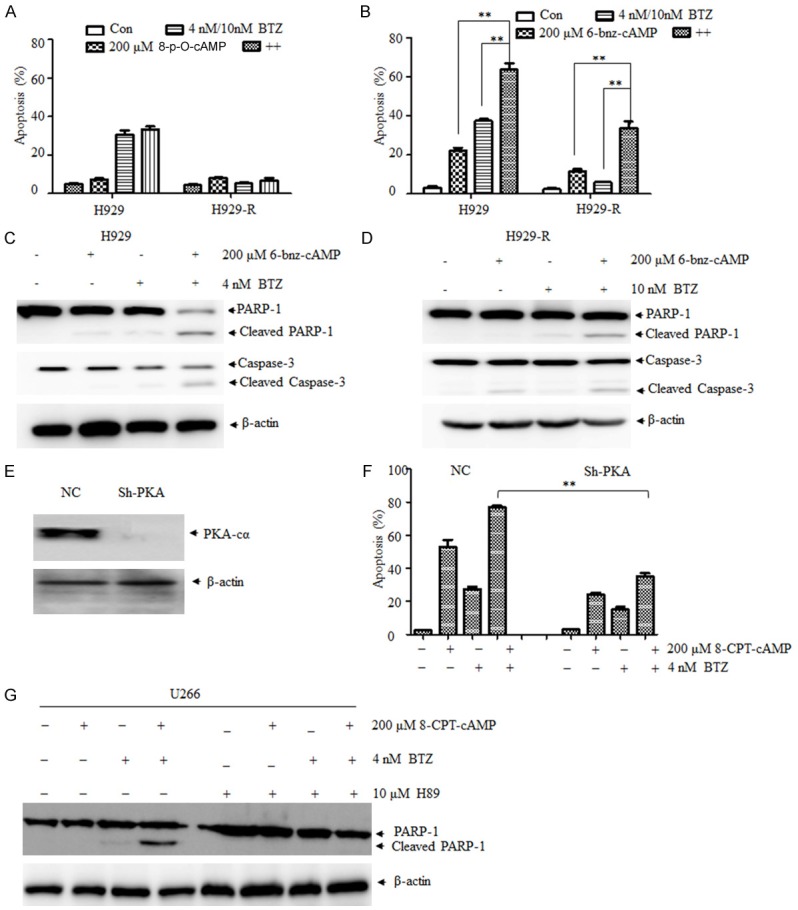
PKA is involved in the cell growth inhibition and apoptosis induced by bortezomib and cAMP. A. Cell apoptosis ratio of H929 and H929-R cells exposed to 8-p-O-cAMP (Epac activator), bortezomib, or their combination for 48 h, respectively. Cell apoptosis was evaluated by Annexin V/PI staining; B. Cell apoptosis ratio of H929 and H929-R cells exposed to 6-bnz-cAMP (PKA activator), bortezomib, or their combination for 48 h, respectively. Apoptosis was evaluated by Annexin V/PI staining; C. H929 cells were exposed to bortezomib, 6-bnz-cAMP, or their combination for 48 h, and the expression of PARP and caspase-3 was measured by Western blot; D. H929-R cells were exposed to bortezomib, 6-bnz-cAMP, or their combination for 48 h, and the expression of PARP and caspase-3 was measured by Western blot; E. Knockdown of PKA in H929 cells by stably transfected with non-specific shRNA pairs (normal control, NC) or shRNA pairs against PKA-Cα; F. PKA-silenced H929 cells were exposed to 8-CPT-cAMP, bortezomib, or their combination for 48 h. Apoptosis was evaluated by Annexin V/PI staining; G. The expression of PARP and caspase-3 was measured by Western blot after U266 cells were treated with 8-CPT-cAMP, bortezomib, H89 (PKA inhibitor), or their combination for 48 h. H929-R, H929 bortezomib-resistant cells. Epac, exchange protein directly activated by cAMP; PKA, protein kinase A. **P < 0.01. Each experiment was performed in triplicate.
Downregulation of cyclinD1 and Mcl-1 was involved in 8-CPT-cAMP- and bortezomib-induced cell death
Our previous results showed that 8-CPT-cAMP and bortezomib inhibit MM cell proliferation and induce cell apoptosis [15]. Nevertheless, the underlying mechanism is not completely understood. As illustrated in Figure 5A, 5B, the expression of cyclinD1 and Mcl-1 was significantly downregulated in both cell lines treated with the combination of 8-CPT-cAMP and bortezomib. In contrast, the expression of Bax and Bcl-2 was not significantly altered post-treatment. To clarify the mechanisms of the downregulation of cyclinD1 and Mcl-1, H929-R cells were treated with 8-CPT-cAMP in the presence or absence of the protein synthesis inhibitor cycloheximide (CHX). The half-life of cyclinD1 was reduced by the addition of CHX, whereas the half-life of Mcl-1 was not significantly changed (Figure 5C, 5D). Moreover, the expression of these two genes at the mRNA level was measured by RT-PCR, which showed that the expression of cyclinD1 at the transcriptional level was not disturbed; however, downregulation of Mcl-1 might have been caused by a decrease in mRNA levels (Figure 5E, 5F). These findings suggested that 8-CPT-cAMP-mediated cyclinD1 and Mcl-1 downregulation occurred at the protein and transcriptional level, respectively.
Figure 5.

CyclinD1 and Mcl-1 downregulation was involved in 8-CPT-cAMP- and bortezomib-induced cell death. H929 (A) and H929-R (B) cells were exposed to 8-CPT-cAMP, bortezomib, or their combination for 48 h. The expression levels of cyclinD1, Mcl-1, Bax, and Bcl-2 were measured by immunoblot analysis. Cycloheximide (CHX) was used to detect the half-life of Mcl-1 (C) and cyclinD1 (D) affected by 8-CPT-cAMP in H929-R cells. Densitometry of proteins was performed; blot density was normalized to β-actin expression levels. After the treatment with 8-CPT-cAMP, bortezomib, or their combination, the expression of cyclinD1 (E) and Mcl-1 (F) at the mRNA level was assessed by RT-PCR. H929-R, H929 bortezomib-resistant cells. **P < 0.01, ***P < 0.001. Each experiment was performed in triplicate.
8-CPT-cAMP enhanced bortezomib-induced endoplasmic reticulum stress
It has been reported that bortezomib selectively inhibits the 26S proteasome and leads to the accumulation of misfolded proteins in MM cells, resulting in endoplasmic reticulum (ER) stress, followed by a coordinated cellular unfolded protein response (UPR) [19,20]. As illustrated in Figure 6A, the expression of the ER stress markers, transcription activating factor-4 (ATF4) and CCAAT/enhancer-binding protein homologous protein (CHOP), at the mRNA level was significantly increased in H929 and H929-R cells treated with bortezomib alone. Moreover, this upregulation was further enhanced by the addition of 8-CPT-cAMP. In line with RT-PCR results, the expression of ATF4 and CHOP were dramatically increased at the protein level post-bortezomib treatment, whereas 8-CPT-cAMP enhanced the upregulation of these proteins induced by bortezomib (Figure 6B). These results indicated that ER stress was induced by bortezomib, and 8-CPT-cAMP promoted this effect to a certain extent.
Figure 6.

8-CPT-cAMP enhanced the ER stress induced by bortezomib. The expression of CHOP and ATF4 (ER stress markers) was measured at the mRNA (A, B) and protein (C, D) levels after the treatment of H929 and H929-R cells with 8-CPT-cAMP, bortezomib, or their combination for 24 h in. H929-R, H929 bortezomib-resistant cells; ER, endoplasmic reticulum. *P < 0.05, **P < 0.01. The experiments were performed in triplicate.
Forskolin and bortezomib synergistically inhibits MM cell death in vivo
Finally, we investigated the in vivo efficacy of forskolin (a cAMP-elevating compound) and bortezomib in the MOPC-315 xenograft model. The combination of forskolin and bortezomib inhibited more the significantly tumor growth than the treatment with forskolin or bortezomib alone (Figure 7A-C). Furthermore, no significant body weight loss was observed in the groups, indicating adequate tolerance to the combination of forskolin and bortezomib (Figure 7D). Importantly, the forskolin and bortezomib combination significantly inhibited the proliferation and enhanced the cell death of tumor cells, as indicated by PCNA and TUNEL staining (Figure 7E). Furthermore, the expression of cyclinD1 and Mcl-1 in tumor tissues was significantly attenuated by the combination of forskolin and bortezomib (Figure 7E), which was in accordance with results obtained from cells experiments. These data further supported the notion that cAMP synergized with bortezomib to induce cell death of MM cells both in vitro and in vivo.
Figure 7.
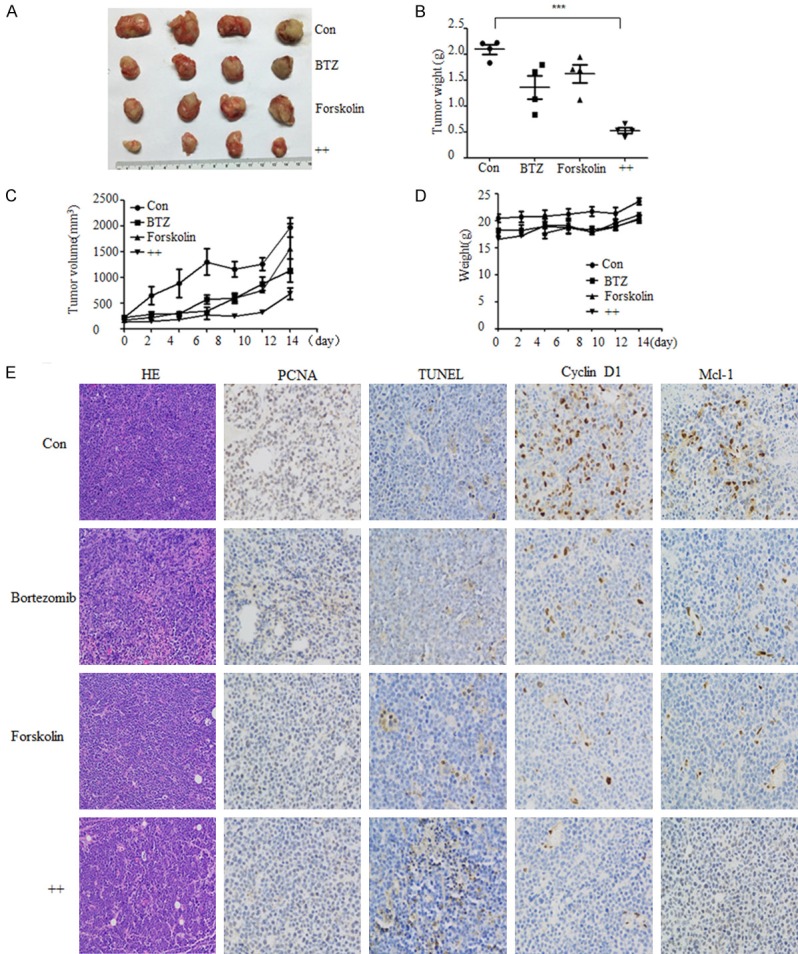
The combination of forskolin and bortezomib inhibits myeloma tumor growth in vivo. A xenograft MOPC315 MM mouse model was established, in which mice were administered with forskolin (5 mg/kg/d, i.p.), bortezomib (0.8 mg/kg, twice a week, i.v.), or their combination for two weeks. A. Tumor volume; B. Tumor weight; C. Consecutive measurement of tumor volume; D. Body weight of mice; E. Hematoxylin-eosin staining for histology, the expression of PCNA, TUNEL, cyclinD1, and Mcl-1 were analyzed by immunohistochemistry (IHC). ***P < 0.001. Scale bar = 50 μm.
Discussion
The cAMP is a multifunctional second messenger that has been used in the treatment of hematological malignancies and solid tumors. In the present study, we demonstrated that cAMP synergizes with bortezomib to induce MM cell death and overcomes bortezomib resistance both in vitro and in vivo. Therefore, targeting cAMP may enhance the anti-proliferative and pro-apoptotic activities of bortezomib.
Currently, the combination chemotherapy based on proteasome inhibitors (PI) or immunomodulatory drugs (IMiDs) is the backbone of MM therapy. Bortezomib-containing regimens, including CyBorD (cyclophosphamide, bortezomib, and dexamethasone) and RVD (lenalidomide, bortezomib, and dexamethasone), have been highlighted as frontline regimens for newly diagnosed or relapsed/refractory MM [21,22]. However, the majority of MM patients relapse due to drug resistance [2,23]. The combinations of bortezomib and novel agents, including monoclonal antibodies, inhibitors of deacetylases, and phosphatidylinositol 3-kinase (PI3K) inhibitors, have yielded promising results [24,25]. The cAMP signaling pathway has been identified as a key regulator of hematopoietic cell proliferation, differentiation, and apoptosis [10,26-28]. In our previous study, we reported that a cAMP-modulating agent induced cell cycle arrest and apoptosis in MM cells [15]. In the present investigation, we found that cAMP synergized with bortezomib to trigger apoptosis in MM cells. Strikingly, cAMP substantially enhanced the apoptosis induced by low-dose bortezomib treatment not only in bortezomib-sensitive U266 and H929 parental cells, but also in U266-R and H929-R cells. Moreover, this ability of cAMP was also observed in isolated primary CD138-positive cells from recurrent MM patients as well as in in vivo studies. These results were in accordance with previously published data showing synergistic inhibition of bortezomib and forskolin on MM cells apoptosis [16].
Previous reports have indicated that cAMP functions via Epac and PKA, which are the main downstream effectors of cAMP in lymphoid cells [29-31]. The present study demonstrated that the activation of cAMP/Epac pathways did not affect cell growth and apoptosis of MM cells, whereas the activation of cAMP/PKA pathway did. Besides, the knockdown of PKA or the administration of an antagonist of PKA significantly abrogated the inhibition of cell growth and apoptosis, induced by the combination of cAMP and bortezomib, which confirmed that PKA was involved in cAMP- and bortezomib-induced cell death. In contrast, the role of the cAMP/PKA pathway in MM tumor cell pathophysiology and bortezomib resistance remains to be further studied.
It has long been recognized that interventions resulting in growth inhibition or apoptosis of cells influence the central regulators of cell cycle or apoptosis [18,32]. This study showed that the exposure of MM cells to 8-CPT-cAMP led to dramatic downregulation of the expression of cyclinD1, which is in agreement with the findings of previous studies [33,34]. The accumulation of Mcl-1 (one of the antiapoptotic Bcl-2 family proteins) has been shown to induce MM cell resistance to bortezomib-induced lethality [35], which was associated with the selective activation of the ATF4 signaling branch of the UPR [36,37]. Our examination revealed that the activation of the cAMP signaling pathway led to rapid downregulation of Mcl-1 at the transcriptional and protein levels. This observation is in line with the results obtained by Virginie et al. that cAMP induced the downregulation of Mcl-1 in MM cells [13]. Interestingly, no significant expression modulation of cell cycle or apoptosis regulatory proteins was observed in the presence of a low dose of bortezomib alone, and 8-CPT-cAMP the applied alone exhibited lower apoptotic activity than drug combinations, which suggests that additional signals are involved in the synergistic induction of apoptosis.
In the case of MM, extensive immunoglobulin production sensitizes MM cells for ER stress-induced apoptosis. Thus, one approach for MM cell apoptosis is to trigger ER stress signaling pathways [38,39]. In this context, two strategies may be taken into account: (1) interference with the ER function and (2) target inhibition of ER-associated degradation pathway to dispose misfolded proteins [40-42]. In this study, we found that bortezomib exposure triggered ER stress signaling, which was confirmed by the upregulation of the ER stress markers ATF4 and CHOP [37,43]. Moreover, we observed that 8-CPT-cAMP synergized with bortezomib to enhance ER stress, which might be associated with cAMP signaling-mediated suppression of the homeostasis of the ER.
In conclusion, the present study demonstrated that synergism between bortezomib and cAMP triggers cell apoptosis in bortezomib-sensitive and -resistant MM cells, which might, to some extent, provide rational support for the clinical evaluation of the combination of bortezomib and cAMP signaling modulator in relapsed MM patients who are resistant to bortezomib.
Acknowledgements
This work was supported by the Shanghai Committee of Science and Technology (Grant no. 12ZR1416800, 13ZR1423800).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Yu W, Chen Y, Xiang R, Xu W, Wang Y, Tong J, Zhang N, Wu Y, Yan H. Novel phosphatidylinositol 3-kinase inhibitor BKM120 enhances the sensitivity of multiple myeloma to bortezomib and overcomes resistance. Leuk Lymphoma. 2017;58:428–437. doi: 10.1080/10428194.2016.1190968. [DOI] [PubMed] [Google Scholar]
- 2.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7:585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 3.Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010;15:243–249. doi: 10.1016/j.drudis.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 4.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14:1649–1657. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 5.Muz B, Kusdono HD, Azab F, de la Puente P, Federico C, Fiala M, Vij R, Salama NN, Azab AK. Tariquidar sensitizes multiple myeloma cells to proteasome inhibitors via reduction of hypoxia-induced P-gp-mediated drug resistance. Leuk Lymphoma. 2017;58:2916–2925. doi: 10.1080/10428194.2017.1319052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richardson PG, Xie W, Jagannath S, Jakubowiak A, Lonial S, Raje NS, Alsina M, Ghobrial IM, Schlossman RL, Munshi NC, Mazumder A, Vesole DH, Kaufman JL, Colson K, McKenney M, Lunde LE, Feather J, Maglio ME, Warren D, Francis D, Hideshima T, Knight R, Esseltine DL, Mitsiades CS, Weller E, Anderson KC. A phase 2 trial of lenalidomide, bortezomib, and dexamethasone in patients with relapsed and relapsed/refractory myeloma. Blood. 2014;123:1461–1469. doi: 10.1182/blood-2013-07-517276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nikesitch N, Ling SC. Molecular mechanisms in multiple myeloma drug resistance. J Clin Pathol. 2016;69:97–101. doi: 10.1136/jclinpath-2015-203414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Podar K, Hideshima T, Chauhan D, Anderson KC. Targeting signalling pathways for the treatment of multiple myeloma. Expert Opin Ther Targets. 2005;9:359–381. doi: 10.1517/14728222.9.2.359. [DOI] [PubMed] [Google Scholar]
- 9.Dolloff NG. Emerging therapeutic strategies for overcoming proteasome inhibitor resistance. Adv Cancer Res. 2015;127:191–226. doi: 10.1016/bs.acr.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Lerner A, Epstein PM. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological malignancies. Biochem J. 2006;393:21–41. doi: 10.1042/BJ20051368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong H, Carlton ME, Lerner A, Epstein PM. Effect of cAMP signaling on expression of glucocorticoid receptor, Bim and Bad in glucocorticoid-sensitive and resistant leukemic and multiple myeloma cells. Front Pharmacol. 2015;6:230. doi: 10.3389/fphar.2015.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halgren RG, Traynor AE, Pillay S, Zell JL, Heller KF, Krett NL, Rosen ST. 8Cl-cAMP cytotoxicity in both steroid sensitive and insensitive multiple myeloma cell lines is mediated by 8Cladenosine. Blood. 1998;92:2893–2898. [PubMed] [Google Scholar]
- 13.Follin-Arbelet V, Torgersen ML, Naderi EH, Misund K, Sundan A, Blomhoff HK. Death of multiple myeloma cells induced by cAMP-signaling involves downregulation of Mcl-1 via the JAK/STAT pathway. Cancer Lett. 2013;335:323–331. doi: 10.1016/j.canlet.2013.02.042. [DOI] [PubMed] [Google Scholar]
- 14.Follin-Arbelet V, Hofgaard PO, Hauglin H, Naderi S, Sundan A, Blomhoff R, Bogen B, Blomhoff HK. Cyclic AMP induces apoptosis in multiple myeloma cells and inhibits tumor development in a mouse myeloma model. BMC Cancer. 2011;11:301. doi: 10.1186/1471-2407-11-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng YM, Zhu Q, Yao YY, Tang Y, Wang MM, Zou LF. 8-Chloroadenosine 3’,5’-monophosphate induces cell cycle arrest and apoptosis in multiple myeloma cells through multiple mechanisms. Oncol Lett. 2012;4:1384–1388. doi: 10.3892/ol.2012.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Follin-Arbelet V, Misund K, Naderi EH, Ugland H, Sundan A, Blomhoff HK. The natural compound forskolin synergizes with dexamethasone to induce cell death in myeloma cells via BIM. Sci Rep. 2015;5:13001. doi: 10.1038/srep13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.General Assembly of the World Medical Association. World medical association declaration of helsinki: ethical principles for medical research involving human subjects. J Am Coll Dent. 2014;81:14–18. [PubMed] [Google Scholar]
- 18.Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. doi: 10.1038/nrm1496. [DOI] [PubMed] [Google Scholar]
- 19.Nicolao MC, Loos JA, Rodriguez Rodrigues C, Beas V, Cumino AC. Bortezomib initiates endoplasmic reticulum stress, elicits autophagy and death in Echinococcus granulosus larval stage. PLoS One. 2017;12:e0181528. doi: 10.1371/journal.pone.0181528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong X, Liao Y, Liu N, Hua X, Cai J, Liu J, Huang H. Combined therapeutic effects of bortezomib and anacardic acid on multiple myeloma cells via activation of the endoplasmic reticulum stress response. Mol Med Rep. 2016;14:2679–2684. doi: 10.3892/mmr.2016.5533. [DOI] [PubMed] [Google Scholar]
- 21.Morgan GJ, Rasche L. Haematological cancer: Where are we now with the treatment of multiple myeloma? Nat Rev Clin Oncol. 2017;14:461–462. doi: 10.1038/nrclinonc.2017.82. [DOI] [PubMed] [Google Scholar]
- 22.Mimura N, Hideshima T, Anderson KC. Novel therapeutic strategies for multiple myeloma. Exp Hematol. 2015;43:732–741. doi: 10.1016/j.exphem.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapoor P, Ramakrishnan V, Rajkumar SV. Bortezomib combination therapy in multiple myeloma. Semin Hematol. 2012;49:228–242. doi: 10.1053/j.seminhematol.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J. Clin. Oncol. 2005;23:6333–6338. doi: 10.1200/JCO.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 25.Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel PL, Chesi M, Davies FE, Drach J, Greipp PR, Kirsch IR, Kuehl WM, Hernandez JM, Minvielle S, Pilarski LM, Shaughnessy JD Jr, Stewart AK, Avet-Loiseau H. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 2004;64:1546–1558. doi: 10.1158/0008-5472.can-03-2876. [DOI] [PubMed] [Google Scholar]
- 26.Naderi EH, Skah S, Ugland H, Myklebost O, Sandnes DL, Torgersen ML, Josefsen D, Ruud E, Naderi S, Blomhoff HK. Bone marrow stroma-derived PGE2 protects BCP-ALL cells from DNA damage-induced p53 accumulation and cell death. Mol Cancer. 2015;14:14. doi: 10.1186/s12943-014-0278-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medina EA, Oberheu K, Polusani SR, Ortega V, Velagaleti GV, Oyajobi BO. PKA/AMPK signaling in relation to adiponectin’s antiproliferative effect on multiple myeloma cells. Leukemia. 2014;28:2080–2089. doi: 10.1038/leu.2014.112. [DOI] [PubMed] [Google Scholar]
- 28.Dumaz N, Marais R. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the gesellschaft fur Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the special FEBS meeting in Brussels. FEBS J. 2005;272:3491–3504. doi: 10.1111/j.1742-4658.2005.04763.x. [DOI] [PubMed] [Google Scholar]
- 29.Naderi EH, Ugland HK, Diep PP, Josefsen D, Ruud E, Naderi S, Blomhoff HK. Selective inhibition of cell death in malignant vs normal B-cell precursors: implications for cAMP in development and treatment of BCP-ALL. Blood. 2013;121:1805–1813. doi: 10.1182/blood-2012-08-452698. [DOI] [PubMed] [Google Scholar]
- 30.Safa M, Kazemi A, Zaker F, Razmkhah F. Cyclic AMP-induced p53 destabilization is independent of EPAC in pre-B acute lymphoblastic leukemia cells in vitro. J Recept Signal Transduct Res. 2011;31:256–263. doi: 10.3109/10799893.2011.578140. [DOI] [PubMed] [Google Scholar]
- 31.Tiwari S, Dong H, Kim EJ, Weintraub L, Epstein PM, Lerner A. Type 4 cAMP phosphodiesterase (PDE4) inhibitors augment glucocorticoid-mediated apoptosis in B cell chronic lymphocytic leukemia (B-CLL) in the absence of exogenous adenylyl cyclase stimulation. Biochem Pharmacol. 2005;69:473–483. doi: 10.1016/j.bcp.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 32.Condorelli F, Salomoni P, Cotteret S, Cesi V, Srinivasula SM, Alnemri ES, Calabretta B. Caspase cleavage enhances the apoptosis-inducing effects of BAD. Mol Cell Biol. 2001;21:3025–3036. doi: 10.1128/MCB.21.9.3025-3036.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ely S, Di Liberto M, Niesvizky R, Baughn LB, Cho HJ, Hatada EN, Knowles DM, Lane J, Chen-Kiang S. Mutually exclusive cyclin-dependent kinase 4/cyclinD1 and cyclin-dependent kinase 6/cyclin D2 pairing inactivates retinoblastoma protein and promotes cell cycle dysregulation in multiple myeloma. Cancer Res. 2005;65:11345–11353. doi: 10.1158/0008-5472.CAN-05-2159. [DOI] [PubMed] [Google Scholar]
- 34.Chesi M, Bergsagel PL, Brents LA, Smith CM, Gerhard DS, Kuehl WM. Dysregulation of cyclinD1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood. 1996;88:674–681. [PubMed] [Google Scholar]
- 35.Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, Harousseau JL, Amiot M, Bataille R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005;19:1248–1252. doi: 10.1038/sj.leu.2403784. [DOI] [PubMed] [Google Scholar]
- 36.Nencioni A, Hua F, Dillon CP, Yokoo R, Scheiermann C, Cardone MH, Barbieri E, Rocco I, Garuti A, Wesselborg S, Belka C, Brossart P, Patrone F, Ballestrero A. Evidence for a protective role of Mcl-1 in proteasome inhibitor-induced apoptosis. Blood. 2005;105:3255–3262. doi: 10.1182/blood-2004-10-3984. [DOI] [PubMed] [Google Scholar]
- 37.Hu J, Dang N, Menu E, De Bruyne E, Xu D, Van Camp B, Van Valckenborgh E, Vanderkerken K. Activation of ATF4 mediates unwanted Mcl-1 accumulation by proteasome inhibition. Blood. 2012;119:826–837. doi: 10.1182/blood-2011-07-366492. [DOI] [PubMed] [Google Scholar]
- 38.Hershko A, Ciechanover A, Varshavsky A. Basic medical research award. The ubiquitin system. Nat Med. 2000;6:1073–1081. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]
- 39.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 40.Mevissen TET, Komander D. Mechanisms of deubiquitinase specificity and regulation. Annu Rev Biochem. 2017;86:159–192. doi: 10.1146/annurev-biochem-061516-044916. [DOI] [PubMed] [Google Scholar]
- 41.Imai J, Yashiroda H, Maruya M, Yahara I, Tanaka K. Proteasomes and molecular chaperones: cellular machinery responsible for folding and destruction of unfolded proteins. Cell Cycle. 2003;2:585–590. [PubMed] [Google Scholar]
- 42.Benbrook DM, Long A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp Oncol. 2012;34:286–297. [PubMed] [Google Scholar]
- 43.Ghosh AP, Klocke BJ, Ballestas ME, Roth KA. CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS One. 2012;7:e39586. doi: 10.1371/journal.pone.0039586. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.