

Abstract
The α-arrestins domain-containing 1 and 3 (ARRDC1 and ARRDC3) are two members of the α-arrestins family. Yes-associated protein 1 (YAP1) is a key downstream transcription co-activator of the Hippo pathway essential for cancer initiation, progression, or metastasis in clear cell renal cell carcinoma (ccRCC). The aim of this work was to elucidate the role of the α-arrestins in ccRCC tumorigenesis by identifying molecular interacting factors and exploring potential mechanisms. In this study, we identified YAP1 as a novel ARRDC3 interacting protein in RCC cells through tandem affinity purification and mass spectrometry. We confirmed that ARRDC1 and ARRDC3, but not other α-arrestin family proteins, interact with YAP1. Binding of ARRDC1/3 to YAP1 is mediated through the WW domains of YAP1 and the PPXY motifs of ARRDC1/3. Functional analysis of ARRDC1/3 by lentiviral shRNA revealed a role for ARRDC1/3 in suppression of cell growth, migration, invasion and epithelial-mesenchymal transition in ccRCC cells, and these effects were mediated, at least in part, through YAP1. Mechanically, ARRDC1/3 negatively regulates YAP1 protein stability by facilitating E3 ubiquitin ligase Itch-mediated ubiquitination and degradation of YAP1. Moreover, ARRDC1/3 mRNA levels were significantly downregulated in ccRCC specimens. A negative correlation was identified between ARRDC3 and YAP1 expression in ccRCC specimens by immunohistochemistry. This study revealed a novel mechanism for ARRDC1/3 in the regulation of YAP1 stability and provided insight in understanding the relationship between ARRDC1/3 downregulation and aberrant Hippo-YAP1 pathway activation in ccRCC.
Keywords: ARRDC1, ARRDC3, YAP1, renal cell carcinoma, degradation, ubiquitination
Introduction
Renal cell carcinoma (RCC) is the most lethal of the common urologic cancers and constitutes 2%-3% of all adult malignant neoplasms [1,2]. Clear cell renal cell carcinoma (ccRCC) is the most common type of RCC, accounting for 70%-80% of all kidney cancers. Despite the emergence of novel targeted therapies over the last decade, such as antiangiogenetic drugs and mammalian target of rapamycin inhibitors, the prognosis of patients with metastasis or relapse remains poor with 5-year survival rates of less than 10% [3]. Therefore, better understanding of the mechanisms underlying RCC tumorigenesis is required to help develop and improve treatment options for RCC patients.
The Hippo signaling pathway impacts various cellular processes, ranging from cell cycle and metabolism to development and tumor suppression [4]. The core Hippo module consists of the tumor-suppressive MST-LATS kinases and the oncogenic transcriptional co-effectors YAP1, which contains two WW-domains [5,6]. Recent work has demonstrated that aberrant activation of Hippo signaling is involved in the maintenance and progression of various human cancers [7-10]. However, the precise role of Hippo signaling in ccRCC is not yet clear.
The mammalian α-arrestin family includes five arrestin domain-containing proteins (ARRDC1-5) and TXNIP that share similar domain homology with the mammalian β-arrestins [11]. TXNIP and ARRDC1-4 (but not ARRDC5) contain two C-terminal PPXY motifs that bind WW domain-containing proteins, including the HECT-domain-containing Nedd4-like E3 ubiquitin ligases [11]. Previous studies demonstrated that α-arrestin family members act as adaptors/scaffolds to recruit Nedd4-like E3 ubiquitin ligases to their substrates for degradation [12]. Recent studies have linked α-arrestin with cancer, and ARRDC3 and TXNIP are emerging as tumor suppressors that regulate a broad range of cellular processes [12-14]. ARRDC3 expression is either lost or suppressed in basal-like breast cancer and prostate cancer [14,15]. ARRDC3 is epigenetically silenced in basal-like breast cancer cells due to its promoter DNA methylation and deacetylation via SIRT2 [14]. ARRDC3 suppresses breast cancer progression by directly binding to a phosphorylated form of ITGβ4 (a cell surface adhesion molecule associated with aggressive tumor behavior) leading to its internalization, ubiquitination and ultimate degradation [16]. Another study showed that ARRDC3 recruits the Nedd4 ubiquitin ligase to the β2 adrenergic receptor (β2AR) to regulate the receptor ubiquitination and degradation [17].
Although the α-arrestin ARRDC3 was reported to be downregulated in a subset of human cancers, its function and expression in ccRCC are still unclear. Here we explored the functions of ARRDC3 by searching for potential interacting factors in 786-O RCC cells. In the present study, YAP1 was identified as a novel ARRDC3 interacting protein through tandem affinity purification (TAP) methods. ARRDC3 acts as an adaptor to negatively regulate YAP1 protein stability by facilitating E3 ubiquitin ligase Itch-mediated YAP1 ubiquitination and degradation. Moreover, we demonstrated that ARRDC1, but not ARRDC2, -4, or -5 or TXNIP, plays similar roles in YAP1 stability regulation as ARRDC3. Therefore, here we reveal a novel mechanism for ARRDC1/3-related YAP1 stability regulation and provide insight into understanding the relationship between ARRDC1/3 downregulation and ccRCC tumorigenesis.
Methods
Cell culture and transfection
786-O and 293T cells were obtained from the American Type Culture Collection. 786-O cells were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS). 293T cells were cultured in DMEM with 10% FBS. Cells were transiently transfected with plasmids or siRNAs using Lipofectamine 3000 or RNAiMax Transfection Reagent (Invitrogen) according to the manufacturer’s instructions.
Expression constructs
The ARRDC1, ARRDC3 and YAP1 cDNAs were purchased from Genechem. The ARRDC2, ARRDC4, ARRDC5 and TXNIP cDNAs were amplified from HeLa cDNA library. All cDNAs were subcloned into pCMV-Myc expression vectors. SFB-YAP1 constructs were purchased from Addgene. YAP1, ARRDC1 and ARRDC3 mutants were generated by the KOD-Plus Mutagenesis Kit (TOYOBO). All the constructs were verified by DNA sequencing.
RNA interference
The negative control and specific shRNAs for ARRDC1/3 and YAP1 were purchased from GenePharma. shRNAs sequence information is provided in Supplementary Table 1.
Immunoprecipitation
For immunoprecipitation of the FLAG-tagged proteins, transfected cells were lysed 24 h after transfection with BC100 buffer. The whole-cell lysates were immunoprecipitated by overnight incubation with monoclonal anti-FLAG antibody-conjugated M2 agarose beads (Sigma). After three washes with FLAG lysis buffer, followed by two washes with BC100 buffer, the bound proteins were eluted from the beads with FLAG-Peptide (Sigma)/BC100 and were subjected to Western blotting. For immunoprecipitation of the endogenous proteins, cells were lysed with cell lysis buffer (Cell Signaling), and the lysates were centrifuged. The supernatant was precleared with protein A/G beads (Sigma) and incubated with the indicated antibody overnight at 4°C. The immunocomplexes were then incubated for 2 h at 4°C with protein A/G beads. After centrifugation, the pellets were collected and washed five times with lysis buffer, resuspended in sample buffer, and further analyzed by SDS-PAGE.
Western blotting
Cell lysates or immunoprecipitates were subjected to SDS-PAGE, and then proteins were transferred onto nitrocellulose membranes (GE Healthcare). The membranes were blocked in Tris-buffered saline (TBS; pH 7.4) containing 5% nonfat milk and 0.1% Tween-20, washed three times in TBS containing 0.1% Tween-20, and incubated with the primary antibody overnight at 4°C, followed by the secondary antibody for 1 h at room temperature. Antibody binding was visualized using the ECL Chemiluminescence System (Santa Cruz). Information of primary antibodies used in this study is provided in Supplementary Table 2.
Quantitative RT-PCR
Total RNA from transiently transfected cells was extracted using the TRIzol reagent (Invitrogen), and cDNA was reverse transcribed using the Superscript RT Kit (TOYOBO), according to the manufacturer’s instructions. Primer sequence information is provided in Supplementary Table 1. PCR amplification was performed using the SYBR Green PCR Master Mix Kit (TOYOBO). Endogenous GAPDH was used for normalization.
Protein complex purification
The epitope-tagging strategy to isolate ARRDC3-containing protein complexes from human cells was performed essentially as previously described with some modifications. In brief, to obtain a FLAG-HA-ARRDC3 expressing cell line, 786-O cells were transfected with pCIN4-FLAG-HA-ARRDC3 constructs and selected for 2 weeks in 1 mg/ml G418. The tagged ARRDC3 protein levels were detected by WB analyses. The stable cell lines were chosen to expand for protein complex purification. For purification, the cells were lysed in BC100 buffer (20 mM Tris-Cl, pH 7.9, 100 mM NaCl, 0.2 mM EDTA, 20% glycerol) containing 0.2% Triton X-100 and fresh protease inhibitor on ice for 2 h. The homogenate was centrifuged for 30 min at 12000 rpm at 4°C. Cleared lysates were filtered through 0.45 μM spin filters (Millipore) and immunoprecipitated by anti-FLAG antibody-conjugated M2 agarose (Sigma). The bound polypeptides eluted with the FLAG peptide (Sigma) were further affinity purified by anti-HA antibody-conjugated agarose (Sigma). The final elutes from the HA-beads with HA peptides were resolved by SDS-PAGE on a 4%-20% gradient gel (BioRad) for Coomassie Blue staining. Gel bands were cut out from the gel and subjected to mass-spectrometric sequencing.
Immunofluorescence
Cells cultured on coverslips in 24-well plates were fixed in 4% paraformaldehyde for 10 min and permeabilized in 0.2% Triton X-100 solution for 5 min. The coverslips were blocked with 2% BSA plus 5% goat serum for 1 h, and subsequently incubated with primary antibodies against FLAG and Myc, which was followed by sequential incubation with fluorescent secondary antibodies (Alexa 488 goat anti-mouse, Alexa 488 goat anti-rabbit, or Alexa 546 goat anti-mouse; Invitrogen). Finally, cells were counterstained with DAPI to reveal the nuclei. Fluorescence images were captured and processed using a fluorescence microscope.
Cell proliferation assay
The cell proliferation rate was determined using Cell Counting Kit-8 (CCK-8) according to the manufacturer’s protocol (Dojindo Laboratories). Briefly, the cells were seeded onto 96-well plates at a density of 1000 cells per well. During 1 to 6-day culture periods, 10 μl of the CCK-8 solution was added to the cell culture every day, and incubated for 2 h. The resulting color was assayed at 450 nm using a microplate absorbance reader (Bio-Rad). Each assay was carried out in triplicate.
Cell migration and invasion assay
Cell migration and invasion were performed using a 24-well Transwell unit with polycarbonate membrane (pore size, 8 μm) (Corning). The membrane was coated with Matrigel basement membrane matrix (1 μg/μl) (BD Bioscience). Cells (0.5~2.5 × 104) were seeded into the upper chamber in a serum-free medium. The lower chamber was filled with a medium containing 10% FBS. After incubation for 24 h at 37°C, the cells on the upper side of membrane were removed. The cells invading to the underside of the membrane were fixed in methanol for 15 min and 1 mg/ml crystal violet staining for 20 min. After being washed with water three times, the membranes of chamber were covered by coverslip and observed using microscope. Six fields of three independent replicates were recorded and analyzed.
Clinical specimens and Immunohistochemistry
Tissue samples were obtained from patients with previously untreated, nonmetastatic ccRCC who underwent radical nephrectomy at the Department of Urology, Shanghai General Hospital from August 2012 to December 2013. The specimens were collected from a normal region (at least 5 cm from the tumor) and from a tumor for each patient. The histological diagnosis was confirmed simultaneously by examining hematoxylin and eosin (H&E) stained sections by two pathologists.
The pathological stage was determined according to the American Joint Committee on Cancer (AJCC), and the tumor grade was classified using the Fuhrman grading system. All patients were informed, and consent was given. All the specimens were fixed in formalin for up to 24 h immediately after surgery, and then dehydrated, paraffinized, and embedded in paraffin blocks. Tissue sections were cut at 3-4 μm and air-dried overnight. The sections were deparaffinized, rehydrated, and subjected to heat-induced antigen retrieval with sodium citrate buffer (10 mM sodium citrate, 0.05% Tween-20 (pH 6.0), which was followed by incubation with 3% hydrogen peroxide for 5 min to block endogenous peroxidase activity. Sections were then incubated with the appropriate primary antibody, and were sequentially incubated with biotinylated goat anti-mouse IgG. For signal detection, the VECTASTAIN ABC kit (Vector Laboratories) was used according to the manufacturer’s instructions. The slides were further counterstained with hematoxylin. Appropriate positive and negative controls were utilized for each immunostain run.
Statistical analysis
Experiments were carried out with three or more replicates unless otherwise stated. All statistical tests were two-sided and performed using GraphPad Prism (Graphpad Software). Statistical analyses were performed by Student’s t-test for most studies. The relationship between YAP1 and ARRDC3 expression was analyzed by the Spearman rank correlation. Differences between the expression of YAP1 and ARRDC3 and clinicopathological features were assessed by Fisher’s exact test. Values with P < 0.05 were considered statistically significant.
Results
ARRDC1 and ARRDC3 interact with YAP1
To investigate the cellular functions and identify molecular mediators of ARRDC3 in RCC, we isolated the ARRDC3 complex from 786-O RCC cells using TAP methods and determined the proteins present in the complex using mass spectrometry (Figure 1A, 1B). Notably, peptides of several members of the Nedd4-like ubiquitin ligases family (Nedd4 L, Nedd4, Itch and WWP1), were abundantly detected in the complex, verifying the efficiency of this approach. In addition to these known binding partners of ARRDC3, we found that several transmembrane proteins were co-purified in the ARRDC3 complex, including receptor tyrosine kinases (AXL, EPHA2), a monocarboxylate transporter (SLC16A1) and TMEM200A (Figure 1B). These results were consistent with previous studies showing that ARRDC3 interacts with transmembrane proteins β2AR and ITGβ4 [16]. We also found that YAP1 was present in the purified ARRDC3 complex (Figure 1B). Since a function for ARRDC3 in YAP1 regulation has not been previously reported, we selected YAP1 for subsequent analyses. To investigate the potential roles of ARRDC3 in the Hippo pathway through an interaction with YAP1, we first confirmed whether ARRDC3 interacts with YAP1 in cells. We co-expressed SFB-YAP1 and Myc-ARRDC3 were in 293T cells and performed co-immunoprecipitation (co-IP) with anti-FLAG antibody. As shown in Figure 1C, Myc-ARRDC3 was successfully co-immunoprecipitated by SFB-YAP1, suggesting an interaction between ARRDC3 and YAP1 proteins. We also investigated whether YAP1 interacts with other α-arrestin members. As shown in Figure 1C, co-IP assay showed that YAP1 also interacted with ARRDC1, but not ARRDC2, -4, or -5 or TXNIP. We next investigated whether endogenous ARRDC1 and ARRDC3 could interact with endogenous YAP1. Immunoprecipitation using the anti-YAP1 antibody was performed using cell lysates prepared from 786-O cells. As shown in Figure 1D, endogenous ARRDC1 and ARRDC3 were efficiently co-immunoprecipitated with endogenous YAP1. Moreover, reciprocal immunoprecipitation experiments confirmed endogenous YAP1 was co-immunoprecipitated with both endogenous ARRDC1 and ARRDC3, confirming an endogenous interaction between these proteins. To investigate whether ARRDC1 and ARRDC3 co-localize with YAP1 in vivo, 786-O cells were transfected with SFB-YAP1 along with Myc-ARRDC1 or Myc-ARRDC3. Cells were immunostained with anti-FLAG or anti-Myc antibodies and visualized by confocal microscopy (Figure 1E). We found that YAP1 was present in both the cytoplasm and nucleus in the majority of 786-O cells. When ARRDC1 or ARRDC3 was co-expressed, YAP1 was co-localized with ARRDC1 or ARRDC3 in the cytoplasm. Taken together, these results indicate that ARRDC1 and ARRDC3 associate with YAP1 in 786-O cells.
Figure 1.
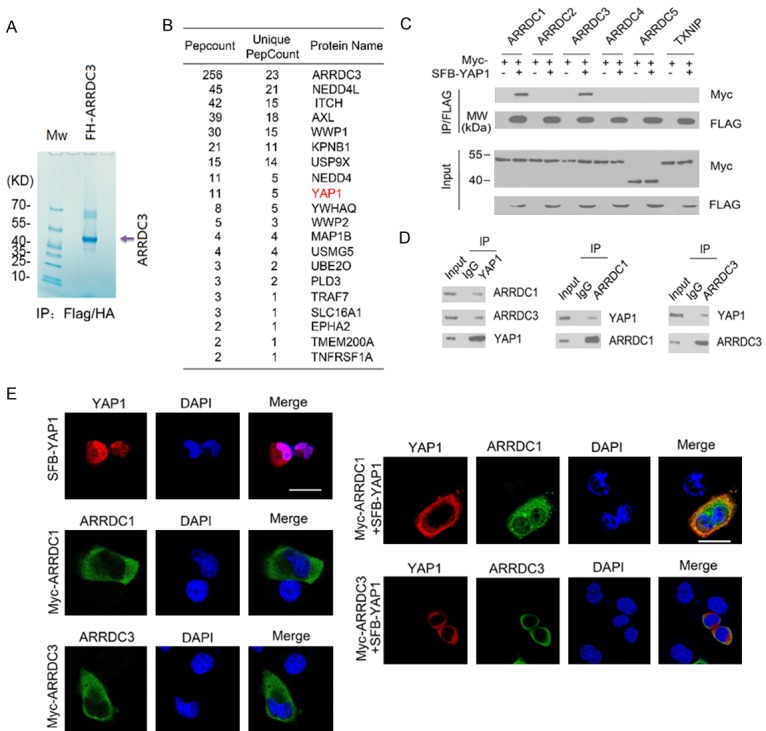
ARRDC1 and ARRDC3 interact with YAP1. (A, B) Tandem affinity purification of the ARRDC3-containing protein complex was conducted using 786-O cells stably expressing FH-ARRDC3. Associated proteins were separated by SDS-PAGE and visualized by Coomassie Blue (CB) staining (A). The number of total/unique peptides identified by mass spectrometry analysis is shown in the table (B). (C) 293T cells were co-transfected with the indicated constructs. Cell lysates were prepared and subjected to immunoprecipitation with anti-FLAG antibody followed by western blotting (WB) with indicated antibodies. (D) Immunoprecipitation using anti-YAP1, anti-ARRDC1, or anti-ARRDC3 antibodies in cell lysates prepared from 786-O cells followed by WB with the indicated antibodies. (E) 786-O cells were transiently transfected with SFB-YAP1, Myc-ARRDC1 or Myc-ARRDC3 constructs either alone or in combination. YAP1 was immunostained in paraformaldehyde-fixed cells with anti-FLAG antibody (red). ARRDC1 or ARRDC3 was immunostained with anti-Myc antibody (green). Nuclei were stained with DAPI.
Identification of the mutual-binding regions of ARRDC1/3 and YAP1
YAP1 contains two WW domains that mediate the interaction with various PPXY motif-containing proteins (Figure 2A). ARRDC1 and ARRDC3 have two C-terminal PPXY motifs that mediate the interaction with WW domains in Nedd4-like ubiquitin ligases (Figure 2B, 2C). To gain more insight into the ARRDC1/3-YAP1 interactions, we determined which region in YAP1 mediated its interaction with ARRDC1/3. As shown in Figure 2D and 2E, all YAP1 fragments could immunoprecipitate ARRDC1 and ARRDC3 except for YAP1-ΔWW1+2, in which both WW domains were deleted. Moreover, deletion of a single WW domain of YAP1 decreased the interaction with ARRDC1/3 compared with wild-type YAP1, suggesting that both WW domains of YAP1 are involved the interaction with ARRDC1/3. We next determined the regions in ARRDC1/3 that bind to YAP1. We generated PPXY motif deletion mutants of ARRDC1/3 and tested their ability to interact with YAP1. Co-IP results showed that only the wild-type ARRDC1/3, but not the deletion mutants, could be immunoprecipitated by SFB-YAP1 (Figure 2F and 2G), suggesting that the two PPXY motifs of ARRDC1/3 were responsible for binding to YAP1.
Figure 2.
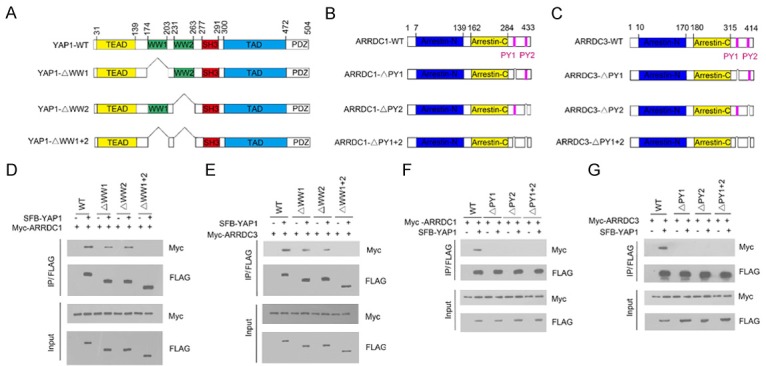
Identification of the mutual-binding regions of ARRDC1/3 and YAP1. (A) Schematic representation of YAP1 deletion mutants used in the study. (B, C) Schematic representation of ARRDC1 (B) and ARRDC3 (C) mutants used in the study. (D) 293T cells were co-transfected with Myc-ARRDC1 and the indicated full length SFB-YAP1 or deletion mutants. Cell lysates were prepared and subjected to immunoprecipitation using the anti-FLAG antibody, followed by WB analyses with the indicated antibodies. (E) 293T cells were co-transfected with Myc-ARRDC3 and the indicated full length SFB-YAP1 or deletion mutants. Cell lysates were prepared and subjected to immunoprecipitation using the anti-FLAG antibody, followed by WB analyses with the indicated antibodies. (F) 293T cells were co-transfected with SFB-YAP1 and Myc-ARRDC1 or the indicated mutants. Cell lysates were prepared and subjected to immunoprecipitation using the anti-FLAG antibody, followed by WB analyses with the indicated antibodies. (G) 293T cells were co-transfected with SFB-YAP1 and Myc-ARRDC3 or the indicated mutants. Cell lysates were prepared and subjected to immunoprecipitation using the anti-FLAG antibody, followed by WB analyses with the indicated antibodies.
ARRDC1/3 negatively regulate YAP1 protein stability
Previous studies demonstrated ARRDC1 and ARRDC3 act as adaptors to recruit Nedd4-like ubiquitin ligases to ubiquitinate a set of substrates, such as β2AR, ALIX and Notch [11,15,16]. Thus, we next assessed whether ARRDC1/3 promote YAP1 ubiquitination and degradation by recruiting Nedd4-like ubiquitin ligases. As shown in Figure 3A, ARRDC1/3, but not ARRDC2, -4, or -5 or TXNIP, decreased the level of co-expressed YAP1 in a dose-dependent manner. Moreover, expression of wild-type ARRDC1/3, but not the ARRDC1-ΔPY1+2 or ARRDC3-ΔPY1+2 mutants, decreased the co-expressed YAP1 protein levels in a dose-dependent manner (Figure 3B). Next, we depleted endogenous ARRDC1/3 by specific shRNAs in 786-O cells and observed that YAP1 protein level was markedly elevated compared with controls (Figure 3C). Moreover, co-depletion of ARRDC1/3 caused more YAP1 elevation than ARRDC1 or ARRDC3 single depletion (Figure 3C).
Figure 3.

ARRDC1/3 negatively regulates YAP1 protein stability. A. 786-O cells were transfected with the indicated constructs. Cells were harvested for WB analyses with the indicated antibodies. B. 786-O cells were co-transfected with the indicated constructs. Cells were harvested for WB analyses. C. 786-O cells were infected with lentivirus expressing control or ARRDC1/3-specific small hairpin RNAs. After 48 h, cells were harvested for WB analyses. D. qRT-PCR measurement of the mRNA levels of ARRDC1 and YAP1 in ARRDC1-depleted cells. GAPDH mRNA was used for normalization. The mean values (S.D.) of three independent experiments are shown. *P < 0.05. E. qRT-PCR measurement of the mRNA levels of ARRDC3 and YAP1 in ARRDC3-depleted cells. F, G. 786-O cells were infected with the indicated shRNA lentivirus. After 48 h, cells were collected at various times after cycloheximide (CHX) treatment and then subjected to WB analyses. The relative intensities of YAP1 were first normalized to the intensities of actin and then to the value of the 0-h time point. H, I. 293T cells were co-transfected with the indicated constructs. Cells were harvested for WB analyses. After 24 h, cells were treated with 20 μM MG132 for 6 h. Flag-YAP1 protein was immunoprecipitated with anti-FLAG antibody. The ubiquitinated forms of YAP1 were analyzed by WB with anti-HA antibody. J, K. 786-O cells were transfected with control, ARRDC1 or ARRDC3 constructs. After 48 h, cells were harvested for qRT-PCR measurement of the mRNA levels of CYR61 and CTGF. GAPDH mRNA was used for normalization. The mean values (S.D.) of three independent experiments are shown. *P < 0.05.
To exclude the possibility that YAP1 protein upregulation resulted from transcriptional upregulation, we performed qRT-PCR to measure ARRDC1, ARRDC3 and YAP1 mRNA levels in ARRDC1/3-depleted 786-O cells. In contrast to the significant decrease in ARRDC1/3 mRNA levels upon shRNA transfection, YAP1 mRNA levels in ARRDC1 or ARRDC3-depleted 786-O cells stayed at a level similar to that of the control cells (Figure 3D and 3E), suggesting that the effects of ARRDC1/3 on YAP1 levels are not mediated through the regulation of YAP1 mRNA expression. To determine whether ARRDC1/3 decreased YAP1 protein level by shortening its half-life, the protein level of YAP1 was monitored after treatment with the protein synthesis inhibitor cycloheximide. In the absence of de novo protein synthesis, the half-life of endogenous YAP1 protein was much longer in the ARRDC1/3 co-depleted cells than that in control cells (Figure 3F and 3G), further suggesting that ARRDC1/3 regulate YAP1 at the posttranslational level. To further determine whether ARRDC1/3 promote YAP1 degradation through the regulation of YAP1 ubiquitination, HA-ubiquitin and FLAG-YAP1 constructs were co-expressed with ARRDC1-WT or the ΔPY1+2 mutant. As shown in Figure 3H, ectopic expression of ARRDC1-WT, but not the ΔPY1+2 mutant, enhanced YAP1 poly-ubiquitination (Figure 3H). Moreover, when Myc-Itch was co-expressed, YAP1 ubiquitination was significantly increased. Similar results were obtained with the ARRDC3 constructs (Figure 3I). Moreover, overexpression of ARRDC1 or ARRDC3 downregulated the mRNA expression of CTGF and Cyr61, two well-known YAP1 transcriptional targets (Figure 3J and 3K). Taken together, these data demonstrate that ARRDC1/3 negatively regulate the Hippo pathway through promoting Itch-mediated YAP1 ubiquitination.
Knockdown of ARRDC1/3 promote cell growth, migration, invasion, and epithelial-mesenchymal transition (EMT) in ccRCC cells
To determine the functional significance of ARRDC1/3-mediated YAP1 protein destabilization, we examined the effect of reduced ARRDC1/3 levels on cell proliferation in 786-O cells infected with lentivirus expressing control shRNAs or shRNAs towards ARRDC1 or ARRDC3. As shown in Figure 4A, ARRDC1 or ARRDC3 depletion resulted in an increase of 786-O cell growth. Moreover, co-depletion of ARRDC1/3 resulted in a stronger increase of 786-O cell growth compared with depletion of ARRDC1 or ARRDC3 alone. Notably, we found that co-depletion of YAP1 reversed the ARRDC1/3 knockdown-mediated acceleration of 786-O cell growth (Figure 4A). Previous studies demonstrated that YAP1 not only promotes cell proliferation but also leads to EMT, which lessens cell contact inhibition and thus allows tumorigenesis [6-9]. We found that ARRDC1 or ARRDC3 depletion resulted in a marked increase of N-cadherin and vimentin and decrease of E-cadherin (Figure 4B and 4C). However, these effects were reversed by co-depletion of YAP1. Finally, cell migration and invasion were determined by transwell assay. Similar to the cell growth assay, we found that ARRDC1/3 co-depletion resulted in a marked increase of 786-O cell migration and invasion, but these effects were reversed by co-depletion of YAP1 (Figure 4D and 4E). Taken together, these results suggest that ARRDC1/3 play negative roles in cell growth, migration and invasion and EMT, at least in part by regulating the Hippo-YAP1 pathway.
Figure 4.

Knockdown of ARRDC1/3 promotes cell growth, migration and invasion, and epithelial-mesenchymal transition in ccRCC cells. A. 786-O cells were infected with the indicated lentivirus. After 48 h, cell growth was measured by CCK-8 assays at indicated days. The mean values (S.D.) of three independent experiments are shown. B, C. 786-O cells were infected with the indicated lentivirus. After 48 h, cells were harvested for WB analyses with the indicated antibodies. D, E. 786-O cells were infected with indicated lentivirus. After 48 h, cells were seeded in transwell chambers and incubated for 24 h. The migrating or invading cells were counted. *P < 0.05.
ARRDC3 protein expression is correlated with YAP1 expression in ccRCC patients
ARRDC3 has been recognized as a tumor suppressor and its expression is downregulated in breast and prostate cancers [14,15]. However, no study has explored whether ARRDC1/3 expression is dysregulated in ccRCC. We first determined the levels of ARRDC1/3 mRNA in 40 ccRCC tissues paired with their corresponding neighboring non-cancerous tissues. The clinical and pathologic characteristics of the patients are shown in Supplementary Table 3. Figure 5A shows the log2-transformed fold changes of ARRDC1 mRNA expression ratio of T/N (cancer tissues/non-cancerous tissues), and a two-fold threshold was set for significant changes in expression. The expression of ARRDC1 was significantly downregulated in 30 of 40 (75.0%) cases compared with adjacent non-cancerous tissues. Six of 40 (15.0%) cases showed no significant alteration, and only 4 of 40 (10.0%) cases showed upregulation of ARRDC1 in ccRCC. Similarly, we found that the expression of ARRDC3 was significantly downregulated in 30 of 40 (75.0%) cases compared with adjacent non-cancerous tissues. Five of 40 (12.5%) cases showed no significant alteration, and only 5 of 40 (12.5%) cases showed upregulation of ARRDC3 in ccRCC (Figure 5B). These results suggested that ARRDC1/3 is downregulated in a large proportion of ccRCC tissues.
Figure 5.

ARRDC3 protein expression is correlated with YAP1 expression in ccRCC patients. A. ARRDC1 mRNA expression levels were analyzed by qRT-PCR in 40 paired ccRCCs with the corresponding non-cancerous tissues. Log2-transformed fold changes of ARRDC1 mRNAs with respect to non-cancerous tissues were normalized to GAPDH mRNA. B. ARRDC3 mRNA expression levels were analyzed by qRT-PCR in the same 40 paired ccRCCs with the corresponding non-cancerous tissues. Log2-transformed fold changes of ARRDC3 mRNAs with respect to non-cancerous tissues were normalized to GAPDH mRNA. C. ARRDC3 and YAP1 staining patterns in 40 paired ccRCC cases. D. ARRDC3 and YAP1 expression were negatively correlated (r=-0.3720, P < 0.001). Staining intensities were quantified by morphometry. The x/y axis represents the expression ratio of the indicated proteins between paired tumor and normal tissues. E. Representative immunohistochemistry of normal and tumor tissues with positive or negative staining of ARRDC3 and YAP1. Scale bar =50 μm.
We next investigated whether YAP1 is upregulated in ARRDC1/3-downregulated ccRCC tissues by immunohistochemistry (IHC) analysis. Since an ARRDC1 antibody suitable for IHC analysis is not available, we were not able to perform IHC analysis of ARRDC1. Instead, IHC analysis of ARRDC3 and YAP1 was performed. We scored the staining of ARRDC3 and YAP1 from 0 to 3 and designated scores 0-1 as negative and scores 2-3 as positive, as described in Methods. As shown in Figure 5C, among the 40 paired ccRCC cases, 4/40 (10%) tumor ti-ssues were YAP1 negative, and 14/40 (35%) tumor tissues were ARRDC3 negative. The expressions of ARRDC3 and YAP1 were strongly correlated (Figure 5D, r=-0.3720, P < 0.001). Representative images are shown in Figure 5E; case 1 shows ccRCC tissues with ARRDC3 loss and strong staining of YAP1 and case 2 shows ccRCC tissues with strong staining of ARRDC3 and weak staining of YAP1.
Discussion
Previous studies demonstrated that YAP1 is ubiquitinated and degraded by the Skp1/Cul1/β-TRCP E3 ligase complex in the cytoplasm, and this process requires coordinated phosphorylation of YAP1 by Lats and CK1 kinases [6]. In this study, we revealed another YAP1 degradation pathway that is regulated by ARRDC1 and ARRDC3 and demonstrated that this pathway is dysregulated in ccRCC. ARRDC1/3 act as adaptors to negatively regulate YAP1 protein stability by facilitating E3 ubiquitin ligase Itch-mediated ubiquitination and degradation. Interestingly, this regulatory mechanism may be conserved between mammalian cells and Drosophila, as a previous study showed that Leash (CG4674), an ancestral α-arrestin protein in Drosophila, interacts with Yki (the YAP1 ortholog in Drosophila) in Drosophila S2R+ cells through TAP methods [18]. A subsequent study demonstrated that depletion of Leash by RNAi increased Yki-reporter activity and overexpression had the opposite effect, suggesting that Leash restrains Yki activity [18]. The authors investigated whether human ARRDCs could downregulate Yki and found that expression of ARRDC1 or ARRDC3, but not ARRDC2, ARRDC4 or TXNIP, reduced Yki-reporter activity and Yki protein abundance [18]. These results were very consistent with our finding that only ARRDC1 and ARRDC3 interact with YAP1 and promote its degradation in human cells.
Although our findings implicate a role for ARRDC1/3 in ccRCC suppression via interactions with YAP1 and promoting its degradation, it is also possible that ARRDC1/3 participate in ccRCC suppression through regulating other proteins and pathways. Previous reports demonstrated that ARRDC3 can suppress breast cancer progression by negatively regulating ITGβ4 [16]. We also found that two receptor tyrosine kinases, AXL and EPHA2, were co-purified with the ARRDC3 protein complex (Figure 1B). Since ARRDC3 promotes internalization, ubiquitination and degradation of membrane proteins ITGβ4 and β2AR, it is possible that ARRDC3 exerts similar effects on AXL and EPHA2. AXL has been shown to be overexpressed and to have mitogenic and prosurvival roles in a broad spectrum of human malignancies [19]. High expression of AXL is associated with poor prognosis in ccRCC patients [20,21]. Moreover, AXL affects multiple cellular behaviors required for neovascularization, such as endothelial proliferation, migration, survival, and tube formation in vitro and regulates angiogenesis in vivo [21-23]. Cabozantinib, a recently approved inhibitor of multiple tyrosine kinase receptors, including AXL, MET and VEGFRs, has proven to increase progression-free survival and overall survival compared with everolimus in advanced ccRCC patients who had progressed after prior VEGFR-tyrosine kinase inhibitor therapy [24]. Similar to AXL, high EphA2 protein expression in ccRCC is associated with a poor disease outcome. EPHA2 signaling pathway plays a critical role in the malignant cellular behavior of ccRCC and appears to be functional particularly in the early stage of malignant progression of non-metastatic ccRCC [25]. ARRDC1 acts as a negative regulator of Notch pathway by promoting Notch receptor degradation [26]. Collectively, our studies suggest that ARRDC1/3 may be implicated in tumor suppression by modulating the levels of YAP1 and other membrane receptors.
Acknowledgements
This work was in part supported by the National Natural Science Foundation of China (81372753 to J.F., 81672558 and 81201533 to C.W.; 31400753 to K.G.).
Disclosure of conflict of interest
None.
Abbreviations
- ARRDC1
arrestin domain containing 1
- ARRDC3
arrestin domain containing 3
- YAP1
Yes-associated protein 1
- ccRCC
clear cell renal cell carcinoma
- UB
ubiquitination enzymes
- Co-IP
co-immunoprecipitation
- EMT
epithelial-mesenchymal transition
- TXNIP
Thioredoxin interacting protein
- IHC
Immunohistochemical
- WB
Western Blotting
Supporting Information
References
- 1.Flanigan RC, Campbell SC, Clark JI, Picken MM. Metastatic renal cell carcinoma. Curr Treat Options Oncol. 2003;4:385–390. doi: 10.1007/s11864-003-0039-2. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Ljungberg B, Cowan NC, Hanbury DC, Hora M, Kuczyk MA, Merseburger AS, Patard JJ, Mulders PF, Sinescu IC European Association of Urology Guideline Group. EAU guidelines on renal cell carcinoma: the 2010 update. Eur Urol. 2010;58:398–406. doi: 10.1016/j.eururo.2010.06.032. [DOI] [PubMed] [Google Scholar]
- 4.Mo JS, Park HW, Guan KL. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014;15:642–656. doi: 10.15252/embr.201438638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee MJ, Ran Byun M, Furutani-Seiki M, Hong JH, Jung HS. YAP and TAZ regulate skin wound healing. J Invest Dermatol. 2014;134:518–525. doi: 10.1038/jid.2013.339. [DOI] [PubMed] [Google Scholar]
- 6.Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhi X, Zhao D, Zhou Z, Liu R, Chen C. YAP promotes breast cell proliferation and survival partially through stabilizing the KLF5 transcription factor. Am J Pathol. 2012;180:2452–2461. doi: 10.1016/j.ajpath.2012.02.025. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Dong Q, Zhang Q, Li Z, Wang E, Qiu X. Overexpression of yes-associated protein contributes to progression and poor prognosis of non-small-cell lung cancer. Cancer Sci. 2010;101:1279–1285. doi: 10.1111/j.1349-7006.2010.01511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pei T, Li Y, Wang J, Wang H, Liang Y, Shi H, Sun B, Yin D, Sun J, Song R, Pan S, Sun Y, Jiang H, Zheng T, Liu L. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget. 2015;6:17206–17220. doi: 10.18632/oncotarget.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schütte U, Bisht S, Heukamp LC, Kebschull M, Florin A, Haarmann J, Hoffmann P, Bendas G, Buettner R, Brossart P, Feldmann G. Hippo signaling mediates proliferation, invasiveness, and metastatic potential of clear cell renal cell carcinoma. Transl Oncol. 2014;7:309–321. doi: 10.1016/j.tranon.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tian X, Irannejad R, Bowman SL, Du Y, Puthenveedu MA, von Zastrow M, Benovic JL. The α-Arrestin ARRDC3 regulates the endosomal residence time and intracellular signaling of the β-Adrenergic receptor. J Biol Chem. 2016;28:14510–14525. doi: 10.1074/jbc.M116.716589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masutani H, Yoshihara E, Masaki S, Chen Z, Yodoi J. Thioredoxin binding protein (TBP)-2/Txnip and α-arrestin proteins in cancer and diabetes mellitus. J Clin Biochem Nutr. 2012;50:23–34. doi: 10.3164/jcbn.11-36SR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison JA, Pike LA, Sams SB, Sharma V, Zhou Q, Severson JJ, Tan AC, Wood WM, Haugen BR. Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol Cancer. 2014;13:62. doi: 10.1186/1476-4598-13-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soung YH, Pruitt K, Chung J. Epigenetic silencing of ARRDC3 expression in basal-like breast cancer cells. Sci Rep. 2014;24:3846. doi: 10.1038/srep03846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng Y, Lin ZY, Xie JJ, Jiang FN, Chen CJ, Li JX, Zhou X, Zhong WD. ARRDC3 inhibits the progression of human prostate cancer through ARRDC3-ITGβ4 pathway. Curr Mol Med. 2017;17:221–229. doi: 10.2174/1566524017666170807144711. [DOI] [PubMed] [Google Scholar]
- 16.Draheim KM, Chen HB, Tao Q, Moore N, Roche M, Lyle S. ARRDC3 suppresses breast cancer progression by negatively regulating integrin beta4. Oncogene. 2010;29:5032–5047. doi: 10.1038/onc.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nabhan JF, Pan H, Lu Q. Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the β-adrenergic receptor. EMBO Rep. 2010;11:605–611. doi: 10.1038/embor.2010.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwon Y, Vinayagam A, Sun X, Dephoure N, Gygi SP, Hong P, Perrimon N. The hippo signaling pathway interactome. science. 2013;342:737–740. doi: 10.1126/science.1243971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mudduluru G, Ceppi P, Kumarswamy R, Scagliotti GV, Papotti M, Allgayer H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene. 2011;30:2888–2899. doi: 10.1038/onc.2011.13. [DOI] [PubMed] [Google Scholar]
- 20.Zhou L, Liu XD, Sun M, Zhang X, German P, Bai S, Ding Z, Tannir N, Wood CG, Matin SF, Karam JA, Tamboli P, Sircar K, Rao P, Rankin EB, Laird DA, Hoang AG, Walker CL, Giaccia AJ, Jonasch E. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene. 2016;35:2687–2697. doi: 10.1038/onc.2015.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y, Chen W, Liu F, Sun W, Li XF, Wang X, Wang Y, Xu ZY, Gao L, Yang Q, Xu B, Li YM, Fang ZY, Xu ZP, Bao Y, Wu DS, Miao X, Sun HY, Sun YH, Wang HY, Wang LH. Exosome-transmitted lncarsr promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell. 2016;29:653–668. doi: 10.1016/j.ccell.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Fridell YW, Jin Y, Quilliam LA, Burchert A, Mc-Closkey P, Spizz G, Varnum B, Der C, Liu ET. Differential activation of the Ras/extracellularsignal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol Cell Biol. 1996;16:135–145. doi: 10.1128/mcb.16.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braunger J, Schleithoff L, Schulz AS, Kessler H, Lammers R, Ullrich A, Bartram CR, Janssen JW. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multisubstrate docking-site. Oncogene. 1997;14:2619–2631. doi: 10.1038/sj.onc.1201123. [DOI] [PubMed] [Google Scholar]
- 24.Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, Hammers H, Hutson TE, Lee JL, Peltola K, Roth BJ, Bjarnason GA, Géczi L, Keam B, Maroto P, Heng DY, Schmidinger M, Kantoff PW, Borgman-Hagey A, Hessel C, Scheffold C, Schwab GM, Tannir NM, Motzer RJ METEOR Investigators. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1814–1823. doi: 10.1056/NEJMoa1510016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herrem CJ, Tatsumi T, Olson KS, Shirai K, Finke JH, Bukowski RM, Zhou M, Richmond AL, Derweesh I, Kinch MS, Storkus WJ. Expression of EphA2 is prognostic of disease-free interval and overall survival in surgically treated patients with renal cell carcinoma. Clin Cancer Res. 2005;11:226–231. [PubMed] [Google Scholar]
- 26.Puca L, Chastagner P, Meas-Yedid V, Israël A, Brou C. Α-arrestin 1 (ARRDC1) and β-arrestins cooperate to mediate Notch degradation in mammals. J Cell Sci. 2013;126:4457–4468. doi: 10.1242/jcs.130500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.