

Abstract
Doxorubicin (DOX) is a conventional and effective chemotherapeutic used in the treatment of hepatocellular carcinoma (HCC). However, doxorubicin administration may induce EMT, which results in the development of chemoresistance in HCC. Recent studies report that Isocorydine (ICD) selectively inhibits human cancer stem cells (CSCs), which have an important role in the development of chemoresistance. In this study, we observed that ICD co-administration enhanced DOX cytotoxicity in HCC cells, enabling the inhibition of DOX-induced epithelial-mesenchymal transition (EMT). Microarray data analysis revealed substantially decreased ERK signaling after ICD treatment. Additionally, we observed decreased IC50 for DOX upon ERK knockdown. Finally, we confirmed the enhanced efficacy of treatment with a combination of DOX and ICD in xenograft models. Collectively, the present study unveils the benefit of using DOX in combination with ICD for chemotherapy against HCC, revealing a novel potential anti-cancer strategy.
Keywords: Doxorubicin, Isocorydine, HCC, EMT
Introduction
Hepatocellular carcinoma (HCC) is the fourth most commonly diagnosed human malignancies and third cause of cancer-related death in China [1]. Despite increased health examinations, most patients have either advanced cancer or metastases at the time of diagnosis [2]. High-grade carcinomas have a larger tumor volume and vasculature and surgery alone has low success rates. Patients who undergo surgical removal of cancer and are at a high risk of relapse must be considered for adjuvant therapy [3,4]. While researchers have explored many strategies for chemotherapy in HCC treatment, the effective treatment choices remain limited. Resistance to conventional adjuvant therapy is a major problem for the effective treatment of cancer [5,6]. Hence, screening for agents that improve the chemotherapeutic efficacy of HCC is valuable to sensitize chemoresistant tumors.
Doxorubicin (DOX) is a classical antineoplastic agent that has been widely used in HCC treatment [4]. Previously, we have reported that DOX administration induces Epithelial-Mesenchymal Transition (EMT) in HCC [7]. During EMT, epithelial cells transform into motile, invasive mesenchymal cells and play an important role in the progression of malignant tumors, including tumor proliferation, invasion and migration. Previous studies have demonstrated that EMT can confer cancer stem cell-like properties [8]. EMT process in cancer cells may further promote chemoresistance in many malignancies, including pancreatic cancer [9], breast cancer [10] and HCC [7]. However, the precise mechanism of DOX-induced EMT remains ambiguous.
L-(+)-isocorydine (ICD) extract from Dicranostigma leptopodum (Maxim.) Fedde (DLF) is an active ingredient in DLF that significantly inhibits the cell proliferation of HCC side population (SP) in vitro and in vivo [11]. Side population cells, defined as a subpopulation of tumor cells with the capacity for self-renewal and tumor initiation and are an important attribute of cancer stem cells (CSC). We postulate that ICD has the ability to inhibit CSC associated development of chemoresistance in HCC cells when used in combination with DOX.
In this present study, we investigated the selective mechanisms involved in ICD mediated inhibition of DOX-induced EMT. We further report the important role of ERK signaling pathway in the inhibition of HCC cell growth after a combined treatment with DOX and ICD.
Materials and methods
Cell lines and cell culture
Human HCC cell lines (SNU-449, SNU-387, Huh-7, and Hep-G2) were procured from the Shanghai Institute for Biological Science, China. All cell lines were cultured in DMEM medium (high glucose; Gibco) containing 10% fetal bovine serum (FBS; Gibco) supplemented with 1% penicillin/streptomycin (Sigma, St. Louis, MO). Cells were maintained at 37°C in a humidified incubator with 5% CO2.
Doxorubicin was purchased from Merck KgaA (Darmstadt, Germany). Isocorydine was purchased from Herbest Company (Shanxi, China).
Cell viability assays
Cell toxicity and proliferation after treatment were determined using the cell counting kit-8 assay (CCK8; Dojindo, Kumamoto, Japan) according to the manufacturer’s specifications. Briefly, 5000 hepatoma cells were seeded per well in a 96-well microplate and exposed to different concentrations of the drug for 48 h. 10 μl of CCK8 reagent (in PBS) was added to each well and incubated for another 4 h, followed by measurements of optical density at 450 nm. The half-maximal inhibition concentration (IC50) was calculated as described previously [12]. All experiments were conducted in triplicates.
Western blotting and immunofluorescence
Cells were treated with drugs for 48 h and lysed in lysis buffer, followed by separation of proteins by 10% SDS-PAGE. The proteins were transferred electrophoretically on to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). Anti-E-cadherin, anti-Vimentin, anti-Twist1 antibodies (1:1000; Cell Signaling) were used to detect the respective proteins. HRP conjugated secondary antibodies were detected by chemiluminescence using Chemi-Doc MP system (Bio-Rad, Hercules, CA). GAPDH was used as a loading control.
Cultured cells were maintained in a chamber slide (Millipore, Billerica, MA) for 24 h. Cell were fixed with 4% paraformaldehyde for 30 min at room temperature. The slides were washed in PBS and non-specific sites were blocked with 5% BSA for 30 min. The slides were incubated overnight at 4°C with mouse anti-human Vimentin or anti-human E-cadherin primary antibody (1:100; Cell Signaling). After washing in PBS, the slides were incubated with PE or FITC conjugated secondary antibody for 1 h, followed by washing in PBS. DAPI was used to stain the nuclei. Stained slides were observed under confocal microscope (Carl Zeiss, Oberkochen, Germany).
Wound healing assay
The cells were cultured in the medium supplemented with or without the half-maximal inhibitory concentration (IC50) of indicated drugs. Same cell numbers were seeded in 12-well plates pre-coated overnight with 0.5% gelatin. Once confluent, the cells were starved in 1% FBS culture medium for 24 hours. Using a 200 µl pipette tip was used to create a scratch wound on the cell monolayer. The distance migrated by the cells was calculated as an average of three different fields observed under optical microscope. Three independent experiments were analyzed at 200x magnification.
Transwell invasion assay
The Transwell inserts were coated with Matrigel (BD Biosciences, Bedford, MA, USA). Equal number of cells in each upper chamber were cultured in medium supplemented with 1% FBS. The lower chamber was supplemented with 20% FBS medium as a chemoattractant. Cells were incubated for an additional 24 hours at 37°C in 5% CO2. Cells in the lower chamber of the Transwell filter were fixed with 95% ethanol for 15 min followed by hematoxylin and eosin staining. Once stained, the cells were evaluated under the microscope.
Flow cytometry and sorting
CD 133 staining in HCC cells was performed using fluorochrome-conjugated mAbs with parallel controls (Biolegend, San Diego, CA). Cell suspension containing 1×106 cells/mL was stained with CD133 at 4°C for 30 min. Flow cytometric analysis was performed on a Canto-II (BD Biosciences, San Jose, CA). CD133+ or CD133- cells were sorted using FACSAria II (BD Biosciences, San Jose, CA).
Xenograft experiments
Animal experiments were conducted in compliance with the Guide for the Care and Use of the Animal Ethics Committee of Wenzhou Medical University (Wenzhou, PR China). Female nude mice (Shanghai, China) aged 3-4 weeks were used for all experiments. 1×106 Huh-7 cells were suspended in 100 μl PBS and implanted subcutaneously into the right axillary fossa of each mouse. Mice were randomly assigned to four subgroups including: DOX (4 mg/kg), ICD (0.4 mg/kg) and DOX (4 mg/kg) combined with ICD (0.4 mg/kg), as well as one vehicle-treated control group (equal volume of diluents). Drugs were injected intraperitoneally every 2 days for 2 weeks.
Statistical analysis
All data are presented as mean ± standard deviation (SD). The inhibitory effects of different groups were compared using two-way (ANOVA), followed by Bonferroni post-hoc test. Student’s t-test was used to compare continuous variables and Chi square test (or Fisher’s exact test) was used to compare categorical variables. All tests are two sided and P < 0.05 was considered to be statistically significant.
Results
ICD enhances DOX sensitivity in hepatoma cells
To investigate if there was a dose-dependent effect of ICD on DOX mediated cytotoxicity in HCC cells, cell viability was measured 48 h after treatment using a CCK8 assays. As shown in Figure 1, combined treatment with ICD and DOX had a higher cytotoxicity in HCC cells in comparison to ICD or DOX alone. This is accompanied with a considerable decrease in IC50 for DOX. To further investigate the dose dependent relationship of adding ICD to DOX in HCC cells, the IC50 and combination index (CI) was calculated according to the previous study [12]. As shown in Table 1, a significant decrease in the IC50 for DOX is observed post combined treatment with ICD. The CI values were 0.605, 0.644, 0.804, and 0.707 respectively for Huh-7, Hep-G2, SNU-449 and SNU-387, indicating that the addition of ICD chemosensitizes HCC cells to DOX.
Figure 1.
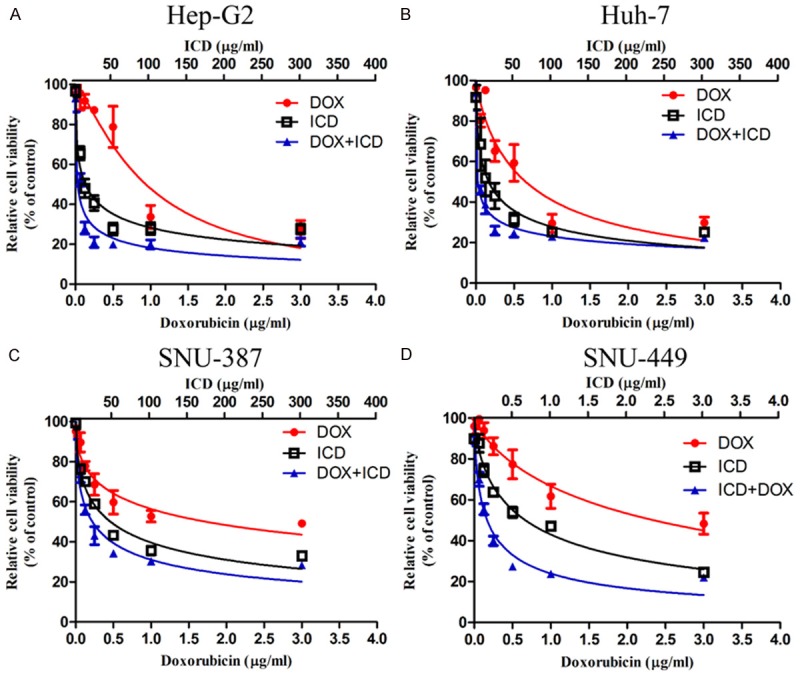
ICD enhances DOX inhibition of HCC proliferation in CCK-8 assay. Relative cell viability (mean ± SD) for ICD (black), DOX (red) and ICD + DOX (blue) in Huh-7, HepG2, SNU-449 and SNU-387 cells.
Table 1.
IC50 values for DOX and ICD in HCC cells
| Cell Line | IC50 of ICD (μg/ml)a | IC50 of Dox (μg/ml)b | Combination Index | ||
|---|---|---|---|---|---|
|
| |||||
| ICD | ICD + DOX | DOX | ICD + DOX | ||
| Huh-7 | 161.3 | 91.41** | 0.6565 | 0.02495*** | 0.605 |
| Hep-G2 | 148 | 90.45** | 0.8796 | 0.02940*** | 0.644 |
| SNU-449 | 262.2 | 178.3* | 1.739 | 0.2159*** | 0.804 |
| SNU-387 | 254.1 | 161.5* | 2.353 | 0.1684*** | 0.707 |
IC50 of ICD concentration in different treatments for 48 h;
IC50 of DOX concentration in different treatments for 48 h.
P < 0.05;
P < 0.01;
P < 0.001.
ICD inhibits DOX-induced EMT
Following treatment with DOX for 48 h, we observed an upregulation of Vimentin, β-catenin and Snail. However, protein levels of Claundin-1 and E-cadherin were downregulated (Figure 2A, 2B). The cell morphology changed upon treatment with DOX to a stretched, elongated shape. Furthermore, immunostaining experiments revealed that DOX exposure induced a strong cytoplasmic localization of Vimentin and weak membrane expression of E-cadherin (Figure 2C, 2D), indicating that treatment with DOX resulted in the acquisition of EMT-like characteristics by HCC.
Figure 2.
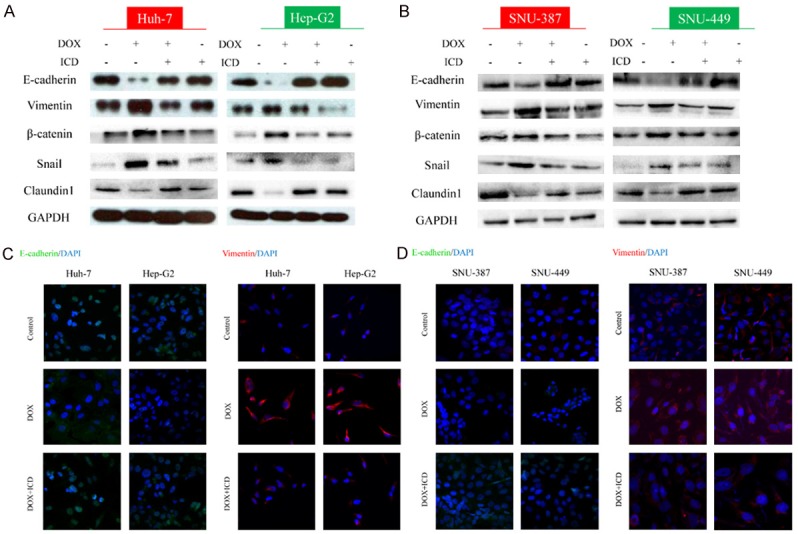
ICD alters the expression of DOX-induced EMT in HCC cells. Expression of EMT markers E-cadherin, Vimentin, β-catenin, Snail and Claundin1, as examined by western blot in Huh-7, Hep-G2 (A) and SNU-387, SNU-449 (B) cells, or treated with DOX, DOX + ICD, or ICD alone for 48 h. Immunofluorescence staining of E-cadherin and Vimentin in Huh-7, Hep-G2 (C) and SNU-387, SNU-449 (D) cells after DOX or DOX + ICD treatment (200x magnification).
We next investigated the effect of ICD on DOX-induced EMT after co-treatment of HCC with DOX and ICD or DOX alone for 48 h. ICD abrogated DOX-induced upregulation of mesenchymal markers and the downregulation of epithelial markers (Figure 2A, 2B). Furthermore, immunofluorescence staining showed a weaker signal for Vimentin and strong E-cadherin staining in co-treated cells (Figure 2C, 2D). These results demonstrate that DOX-induced EMT in epithelial cells was inhibited by ICD.
ICD inhibits the invasive capacity of DOX-induced EMT in HCC cells
Transwell assays showed remarkably less hepatoma cells passing through the matrigel coated membranes when compared with the corresponding cells under single drug condition (Figure 3A, 3B). Wound healing assays also showed decreased migration in the cells exposed to combined treatment with ICD and DOX, when compared to the cells treated with DOX or ICD alone (Figure 3C, 3D). These results suggest that addition of ICD decreased the invasiveness of DOX-induce EMT in HCC cells.
Figure 3.

ICD inhibits the DOX-induced tumor invasive capacity in HCC cells. (A) Transwell assays showing the invasion of Huh-7 and Hep-G2 cells for control, DOX, DOX + ICD or ICD group (100x magnification). (B) Number of cells per field that crossed the transwell chamber under the different treatment conditions (*P < 0.05, **P < 0.01, ***P < 0.001). (C) Wound healing assay for migration of Huh-7 and Hep-G2 cells in the control, DOX, DOX + ICD or ICD group (100x magnification). The broken white line indicates the wound areas without migrating cells. (D) The gap between the migrating cells is expressed as a percentage of the initial area. (E) Levels of CD133 in HCC cells (Huh-7 and Hep-G2), or HCC cells treated with DOX, DOX + ICD, or ICD alone for 48 h. Sorting CD133 + HCC cells (F) and the relative cell viability for ICD (black), DOX (red) and ICD + DOX (blue) in Huh-7 and Hep-G2 (G). One of three representative images are shown in (A, C).
The proportion of CD133 positive cells was markedly elevated in the cells treated with DOX. This phenotype disappeared after the combined treatment with DOX and ICD (Figure 3E). To further test the inhibition of CD133+ HCC cells, we sorted CD133+ cells (Figure 3F). CCK8 assay confirmed higher resistance of CD133+ cells to chemotherapeutic agents (Figure 3G). However, this subpopulation of CD133+ cells was sensitive to the combined treatment with ICD and DOX. The CI value were 0.456 and 0.556 respectively for Huh-7 and Hep-G2 (Table 2). These results suggest that the stem cell-like subpopulation of CD133+ cells was sensitive to the combined treatment with ICD and DOX.
Table 2.
IC50 values for DOX and ICD in CD133+ HCC cells
| Cell Line | IC50 of ICD (μg/ml)a | IC50 of Dox (μg/ml)b | Combination Index | ||
|---|---|---|---|---|---|
|
| |||||
| ICD | ICD + DOX | DOX | ICD + DOX | ||
| Huh-7 | 207.1 | 69.72** | 6.147 | 0.735*** | 0.456 |
| Hep-G2 | 370.4 | 129.9** | 7.199 | 1.540*** | 0.556 |
IC50 of ICD concentration in different treatments for 48 h;
IC50 of DOX concentration in different treatments for 48 h.
P < 0.01;
P < 0.001.
ICD inhibits EMT induced by DOX by inhibiting ERK signaling
Next, we used microarray analysis to gain specific insight into the mechanism of inhibition of EMT by ICD. Genes whose expression levels showed a ≥ 2-fold change in control vs. DOX-treated and DOX-treated vs. DOX + ICD-treated group were identified. Data showed that ICD significantly inhibits the DOX-induced EMT (Figure 4A). Following an overlay analysis, 502 genes were identified and clustered (Figure 4B). 101 genes were associated with specific signaling pathway highlighted by GO analysis (Figure 4C). A relevant percentage of these genes i.e. ELK1, ELK4 and MYC (Figure 4D) were correlated with mitogen-activated protein kinase (MAPK) signaling, indicating the role of MAPKs in ICD mediated reversal of DOX-induced EMT.
Figure 4.
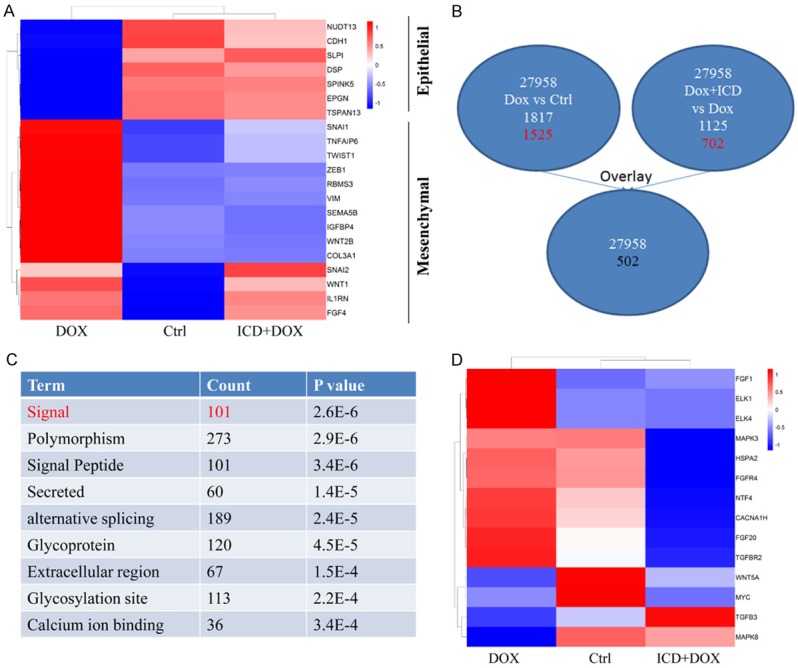
MAPK cascade is operative in the ICD and DOX treated HCC cells. A. Heat map of the EMT-related genes in control, DOX and ICD + DOX samples. B. Schematic representation of comparing gene expression profiles in Huh-7 cells (2-fold difference, DOX vs. Ctrl; DOX + ICD vs. DOX) and overlay analysis. C. Hierarchical clustering of the above-mention genes. D. Heat map of the EMT-related genes in control, DOX and ICD + DOX.
We further investigated the involvement of the MAPK pathway in DOX induced EMT and found that treatment with DOX increased the levels of p-ERK expression in comparison to the other groups. However, the levels of p-ERK decreased after the co-treatment with ICD (Figure 5A). To confirm the involvement of the ERK pathway, we used siRNA to knockdown p-ERK1/2 expression. Knockdown of p-ERK significantly attenuated the expression of Vimentin, β-catenin and ZEB-1, while the level of epithelial markers was upregulated (Figure 5B). Immunofluorescence staining also showed the similar trend for E-cadherin and Vimentin expression (Figure 5C). In addition, IC50 concentration for DOX significantly decreased after ERK knockdown (Figure 5D). Our findings confirm the importance of the ERK signaling pathway in ICD mediated suppression of DOX induced EMT.
Figure 5.
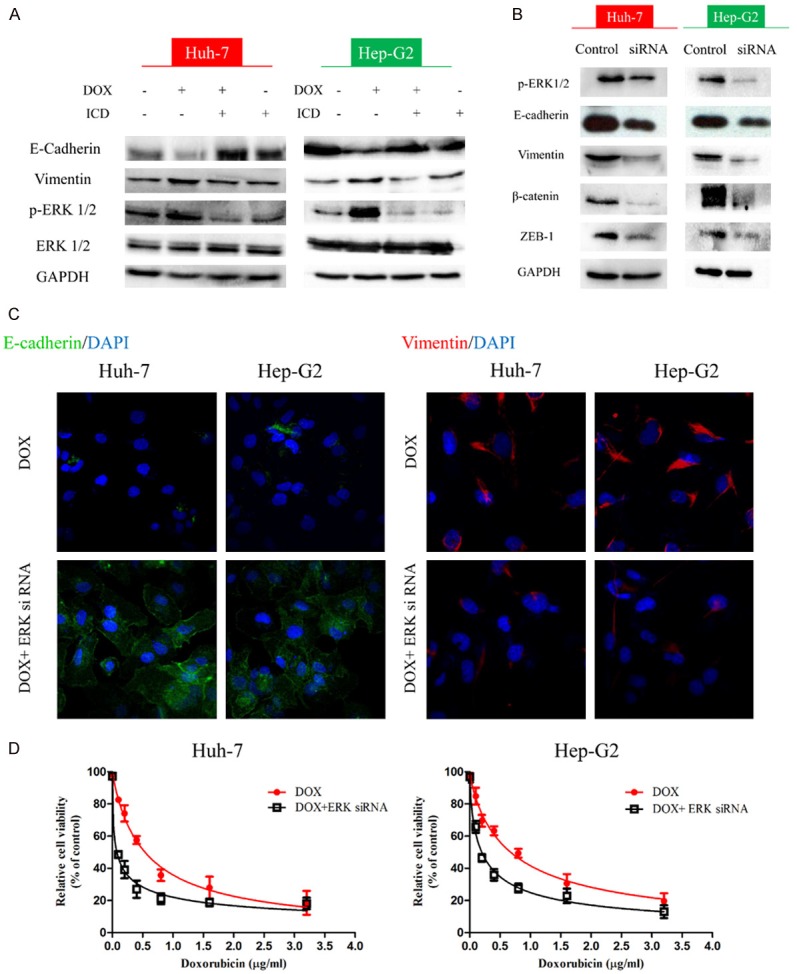
ICD reverses DOX-induced EMT through ERK signaling. A. Expression of ERK1/2 examined by western blot in Huh-7, Hep-G2, or treated with DOX, DOX + ICD, or ICD alone for 48 h. B. Detection of EMT markers upon treatment with ERK1/2 siRNA. D. Immunofluorescence staining of E-cadherin and Vimentin in DOX and DOX + ERK siRNA in Huh-7 and Hep-G2 (200x magnification). C. The relative cell viability for DOX (red) and DOX + ERK siRNA (black) in Huh-7 and Hep-G2.
ICD enhances the effect of doxorubicin in vivo
To validate the in vitro results, Huh-7 cells were inoculated subcutaneously into nude mice. Once a visible tumor mass was observed (in approximately 3 weeks), the animals were randomly divided into treatment groups and a control group. The treatment group was administered ICD, DOX or ICD + DOX intraperitoneally on alternate days, 6 times. Subsequently, tumor size and body weight of the mice were measured every second. The control group was administered PBS. After 4 weeks of treatment, the mice were euthanized and subjected to analysis. Administration of DOX or ICD alone retarded the tumor growth, but the combined treatment of DOX or ICD significantly inhibited tumor growth (P < 0.01 vs. DOX or ICD group) (Figure 6A, 6B). Importantly, there was no significant change in the body weight of mice (Figure 6C). Taken together with the data of in vitro experiments, combined treatment of ICD and DOX show a promising potential to eradicate HCC.
Figure 6.
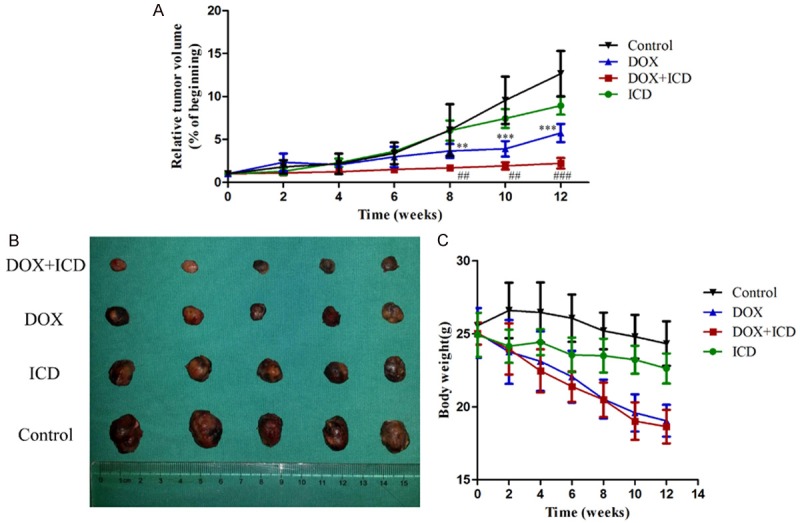
Effect of ICD, DOX, and their combination in nude mice implanted with subcutaneous HCC xenografts. A. Tumor volume over time in the control group (black), groups treated with DOX (blue), ICD (green) or DOX + ICD (red). Relative tumor volume ratios (% of original volume after initiation of therapy) are presented as the mean ± SD, (**P < 0.01, ***P < 0.001, for control vs. DOX group; ##P < 0.01, ###P < 0.001, for DOX + ICD vs. DOX alone). B. Resected tumors in the different groups. C. The body weight of mice after treatment.
Discussion
Carcinoma cells lose their epithelial characteristics upon EMT and acquire mesenchymal attributes. The EMT transition also results in the increased capacity for migration and invasion. The EMT process may be responsible for chemoresistance and invasion in hepatoma, leading to metastatic events [13-15]. Doxorubicin is an effective chemotherapeutic agent for TACE, and has been shown to induce EMT in HCC in our previous study [7]. Doxorubicin-induced EMT may substantially decrease the chemotherapeutic efficacy and lead to distant metastasis. However, there is no systematic study investigating the role of signaling events resulting in DOX resistance.
Accumulating studies indicate that activation of the EMT is necessary, not only for the physical dissemination of tumor cells, but also for the transformation to the CSC state, enabling metastasis of the cells disseminated from the primary tumor, and development of resistance to the chemotherapeutic agents [16-18]. EMT also results in the production of several autocrine signaling loops, including the transforming growth factor β (TGFβ) and Wnt pathways. These positive signaling loops contribute further to the acquisition of CSC properties in the cells [13]. We found that DOX-treated cells substantially increase the expression level of CD133 and therefore presumed that a combination of CSC-target drug may provide a viable strategy to tackle DOX-resistance.
Isocorydine (ICD) is an alkaloid monomer derived from Papaveraceae spp. plant, including Dactylicapnos scandens Hutchins and Dicranostigma leptopodum (Maxim) Fedde (DLF). ICD, which is the active ingredient in DLF, is known to induce apoptosis in HCC cell lines and the side population (SP) cells [11]. Previous results indicate that ICD may target CSC-like cells [11]. In the present study, we evaluated the synergistic inhibitory effect of ICD in HCC cell lines when co-administered with DOX. ICD remarkably decreased the expression of DOX induced CD133. Furthermore, ICD reversed DOX-induced changes in the expression of EMT-markers and attenuated the invasion and migration ability after treatment with DOX.
The transcription factors involved in EMT can typically be classified into three protein families: the Snail family (including Snail and Slug), ZEB family (including ZEB1 and ZEB2), and basic helix-loop-helix (including TWIST1, TWIST2, and TCF3) family [19]. The transcription factor ZEB1 is involved in the EMT process, repressing the expression of E-cadherin, which can be activated by the TGF-β, TNF-α and IGF1 signaling pathways [20]. A correlation between ZEB1 expression and loss of E-cadherin has been confirmed in lung adenocarcinomas [21]. ERK induces EMT via regulation of ZEB protein expression [22]. Our data from the microarray assay and western blot analysis suggest that MAPK cascade is also involved in EMT. In addition, we found that the knockdown of ERK1/2 significantly inhibits ZEB-1 levels. Based on our results, we propose that ICD inhibits the phosphorylation of ERK1/2 and ZEB-1 expression resulting in the reversal of DOX-induced EMT.
In conclusion, our study provides evidence for the development of a potential DOX based antitumor chemotherapy to counter the issue of chemoresistance in HCC. However, further studies evaluating the role of specific transcription factor(s) regulated by the ERK cascade in ICD treatment are mandated for the comprehensive understanding of the development of chemoresistance in HCC.
Acknowledgements
This study was financially supported by Zhejiang Traditional Chinese Medicine Foundation (2017ZQ020, JX Pan), Zhejiang Provincial Medical and Health Research Project (2017KY462, JX Pan; 2018KY126, LY Ye), Wenzhou Municipal Science and Technology Bureau (Y20160037, JX Pan; Y20170095, LY Ye), National Natural Science Foundation of China (81701630, LY Ye), Zhejiang Provincial Natural Science Foundation (LQ18H040005, JX Pan).
Disclosure of conflict of interest
None.
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia XF, Sun X, Li GG, Hu QD, Fu QH, Liang TB. Hypoxia-Induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res. 2016;76:818–830. doi: 10.1158/0008-5472.CAN-15-0977. [DOI] [PubMed] [Google Scholar]
- 3.Bruix J, Sherman M American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53:1020–1022. doi: 10.1002/hep.24199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang QH, Li AJ, Yang GM, Lai EC, Zhou WP, Jiang ZH, Lau WY, Wu MC. Surgical resection versus conformal radiotherapy combined with TACE for resectable hepatocellular carcinoma with portal vein tumor thrombus: a comparative study. World J Surg. 2013;37:1362–1370. doi: 10.1007/s00268-013-1969-x. [DOI] [PubMed] [Google Scholar]
- 5.Moncharmont C, Levy A, Gilormini M, Bertrand G, Chargari C, Alphonse G, Ardail D, Rodriguez-Lafrasse C, Magne N. Targeting a cornerstone of radiation resistance: cancer stem cell. Cancer Lett. 2012;322:139–147. doi: 10.1016/j.canlet.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 6.Ramos P, Bentires-Alj M. Mechanism-based cancer therapy: resistance to therapy, therapy for resistance. Oncogene. 2015;34:3617–3626. doi: 10.1038/onc.2014.314. [DOI] [PubMed] [Google Scholar]
- 7.Pan JX, Wang F, Ye LY. Doxorubicin-induced epithelial-mesenchymal transition through SEMA 4A in hepatocellular carcinoma. Biochem Biophys Res Commun. 2016;479:610–614. doi: 10.1016/j.bbrc.2016.09.167. [DOI] [PubMed] [Google Scholar]
- 8.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asiedu MK, Beauchamp-Perez FD, Ingle JN, Behrens MD, Radisky DC, Knutson KL. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene. 2014;33:1316–1324. doi: 10.1038/onc.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu P, Sun H, Zhang L, Hou H, Zhang L, Zhao F, Ge C, Yao M, Wang T, Li J. Isocorydine targets the drug-resistant cellular side population through PDCD4-related apoptosis in hepatocellular carcinoma. Mol Med. 2012;18:1136–1146. doi: 10.2119/molmed.2012.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Y, Liang C, Xue F, Chen W, Zhi X, Feng X, Bai X, Liang T. Salinomycin decreases doxorubicin resistance in hepatocellular carcinoma cells by inhibiting the beta-catenin/TCF complex association via FOXO3a activation. Oncotarget. 2015;6:10350–10365. doi: 10.18632/oncotarget.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC, Wang SB, Wang YZ, Yang Y, Yang N, Zhou WP, Yang GS, Sun SH. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666–681. doi: 10.1016/j.ccr.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 14.Giannelli G, Bergamini C, Fransvea E, Sgarra C, Antonaci S. Laminin-5 with transforming growth factor-beta1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005;129:1375–1383. doi: 10.1053/j.gastro.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 15.Niu RF, Zhang L, Xi GM, Wei XY, Yang Y, Shi YR, Hao XS. Up-regulation of Twist induces angiogenesis and correlates with metastasis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2007;26:385–394. [PubMed] [Google Scholar]
- 16.Bao B, Azmi AS, Ali S, Ahmad A, Li Y, Banerjee S, Kong D, Sarkar FH. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim Biophys Acta. 2012;1826:272–296. doi: 10.1016/j.bbcan.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, Kearsley JH, Li Y. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875. doi: 10.1038/cddis.2013.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16:488–494. doi: 10.1038/ncb2976. [DOI] [PubMed] [Google Scholar]
- 20.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 21.Bae GY, Choi SJ, Lee JS, Jo J, Lee J, Kim J, Cha HJ. Loss of E-cadherin activates EGFR-MEK/ERK signaling, which promotes invasion via the ZEB1/MMP2 axis in non-small cell lung cancer. Oncotarget. 2013;4:2512–2522. doi: 10.18632/oncotarget.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu LY, Hsin IL, Yang TY, Sung WW, Chi JY, Chang JT, Ko JL, Sheu GT. The ERK-ZEB1 pathway mediates epithelial-mesenchymal transition in pemetrexed resistant lung cancer cells with suppression by vinca alkaloids. Oncogene. 2017;36:242–253. doi: 10.1038/onc.2016.195. [DOI] [PMC free article] [PubMed] [Google Scholar]