

Abstract
Li-Fraumeni Syndrome (LFS), which is a rare dominantly inherited cancer predisposition syndrome, is associated with germline P53 mutations. Mutations of the tumor suppressor protein P53 are associated with more than 50% of human cancers; however, almost 30% of P53 mutations occur rarely and this has raised questions about their significance. It therefore appeared of particular interest that we identified a novel mutation in a patient suffering from breast cancer and fulfilling the diagnostic criteria of LFS. In this study, a patient with remarkable family history developed breast cancer and was diagnosed with LFS. By performing next-generation sequencing on the patient and subsequent verification by Sanger sequencing among other family members, a new germ-line P53 replication error, a trinucleotide repeat mutation in the coding region, was identified in two generations of this Li-Fraumeni family.
Keywords: Li-Fraumeni syndrome, P53 mutation, breast cancer
Introduction
Li-Fraumeni syndrome (LFS; OMIM #151623) is a rare autosomal-dominant, inherited tumor predisposition genetic disease associated with heterozygous germline mutations in the P53 gene. LFS was first reported in 1969 by Li and Fraumeni and subsequently confirmed by a series of epidemiological studies [1]. LFS patients and their family members have a higher risk of developing multiple neoplasms during their lifetime, especially breast cancer (50%), soft tissue sarcomas (15%), brain tumors (6%) and osteosarcomas (5%) [2]. Other types of cancer, including lung, gastric, ovarian, colorectal cancer and early-onset melanoma have also been reported in some families. LFS is classified into two types: classic LFS and Li-Fraumeni-like (LFL) syndrome. Classic LFS is defined as a diagnosis of sarcoma before 45 years of age, a first-degree relative with cancer before 45 years of age, and another first- or second-degree relative with any cancer diagnosed by 45 years of age or with a sarcoma at any age [3]. A germline mutation in the P53 tumor-suppressor gene is present in 56-70% of families with classical LFS. Most mutations identified in LFS have been located between exons 5 and 8 of P53 gene [4].
Protein encoded by P53 gene is a transcription factor that responds to oncogenic stress by inducing cell cycle arrest or apoptosis and whose inactivation is mainly due to mutations that interfere with the DNA-binding activity of the protein [5,6]. Antagonistic cellular responses such as apoptotic cell death, senescence, reversible cell cycle arrest, DNA repair and autophagy could be triggered by P53. Cell cycle arrest and the associated DNA repair program leads to cell survival, while activation of apoptosis helps to eliminate damaged cells [7].
It is well appreciated that common P53 mutations in human cancers confer cells neoplastic properties. More than 75% of the mutations could encode a P53 protein that has lost wild-type functions and may obliterate wild-type P53 tumor suppressor functions. Furthermore, mutant P53 also acquires oncogenic functions that are entirely independent of wild-type P53 [8]. In this study, we identified a novel A Novel dysfunctional germline P53 Mutation in two generations of a LFS family.
Case report and genetic analysis
A 27-year-old woman without past history developed breast cancer and was diagnosed with LFS based on the remarkable family history. A pedigree of the family is presented in Figure 1. Notably, her identical twin sister (III-3) developed breast cancer at 24 years of age. The study was reviewed by the ethics committee of our hospital. Genetic analysis was performed after obtaining informed consent, and provided the patient with genetic counseling. All persons gave informed consent before their inclusion in the study.
Figure 1.
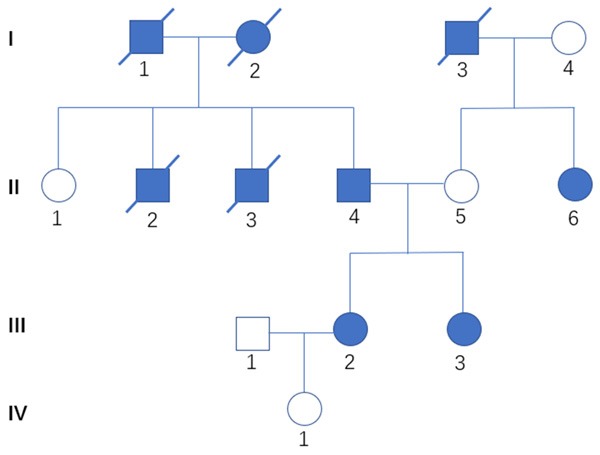
A pedigree of the family. III-2 represents the patient presented in this article. Three of her grandparents died of pancreatic cancer (I-3), Liver cancer (I-1) and breast cancer (I-2), respectively. Her father (II-4) was diagnosed with rhabdomyosarcoma at the age of 28. One of her uncle (II-2) was diagnosed with leukaemia at the age of 18. Her another uncle (II-3) developed lung cancer when he was 42 years old. Her two uncles have already died. Her mother (II-5) has not developed any types of tumor so far. But her aunt (II-6) has suffered from breast cancer since 56 years old. Her identical twin sister (III-3) developed breast cancer at 24 years of age. Square, male; circle, female. slash mark, deceased. Solid symbols represent cancer patients.
One painless lump was accidently found in her right breast. Physical examination showed a 2 cm lump in the upper inner quadrant of her left breast and a 2.5 cm lump in the right armpit near the chest wall. Ultrasonography showed low echogenic position in the right breast (7*17*17 mm, BI-RADS: 4A) and lymphadenectasis of the right axillary lymph node (10*26*24). Calcifications in the right breast were detected by molybdenum target mammography. Right breast cancer with axillary lymph node metastases was suspected. Her complete blood count, liver function test, serum electrolytes, and creatinine were all within normal limits. Without operative taboo, right mammary lobectomy was performed to excise the lump. While the Intraoperative frozen section examination identified the mass as breast infiltrating carcinoma, thus she underwent right breast modified radical mastectomy as a standard procedure. Histopathological examination of the specimen showed invasive ductal carcinoma (Figure 2A) and mamillary Paget’s Disease. Immunohistochemical examination showed (detected by immunocytochemistry): negative results for progesterone receptor (Figure 2B), estrogen receptor (Figure 2D), P53 (Figure 2E), CK14 and EGFR; positive results for HER2 (Figure 2C) and CerbB-2 (3+); the Ki67 index was 80%.
Figure 2.
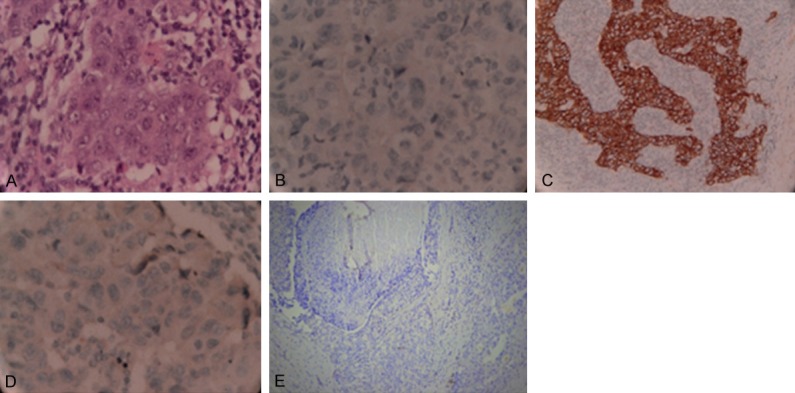
H&E and ImmunohistochemicaI results of the present breast cancer patient. H&E stain showed invasive ductal carcinoma (A); Immunohistochemistry results: progesterone receptor (B), HER2 (C), estrogen receptor (D), P53 (E).
Considering the complicated family history of this patient, to further clarify the genetic causes, the patient accepted genetic testing. Target area capture combined with next-generation sequencing was adopted to analysis the variation of related exons and their adjacent ± 10 bp intron regions. Next-generation sequencing based panel testing for 21 known representative breast cancer & ovarian cancer susceptibility genes (BRCA1/2, CHEK2, PALB2, BRIP1, P53, PTEN, STK11, CDH1, ATM, BARD1, MLH1, MRE11A, MSH2/6, MUTYH, NBN, PMS1/2, RAD50 and RAD51C) on a high-throughput platform was performed at the BGI Clinical Laboratories (Shenzhen, China). A suspected pathogenic heterozygous frameshift mutation c.685dup (p.Cys229Leufs*11) in the P53 gene was detected. This mutation inserted a thymidine (T) between 685th and 686th nucleotides of P53 gene, which result in a truncated polypeptide chain harbouring only 238 amino acids. We have not found any related research about the function and clinical significance of this mutation.
In order to test whether other family members carry this identified mutation, peripheral blood samples from family members (II1, 4, 5, 6; III2, 3; IV1) of the patient were collected to detect the existence of this P53 mutation. DNA was extracted from peripheral blood lymphocytes. Sanger sequencing targeting P53 genes were performed using P53 gene fragments amplified by PCR. Genome version GRCh37/hg19 was used for sequence alignment. The reference sequence of P53 gene is NM_000546.5. It turned out that her father and her sister carried the gene mutation among the included family members (Figure 3).
Figure 3.
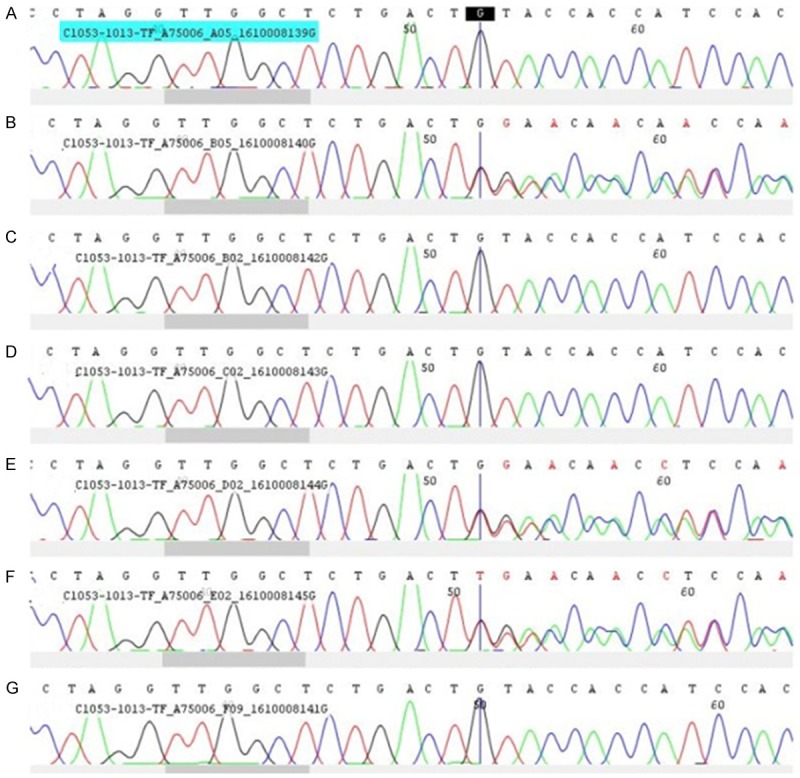
Sanger Sequencing analysis of the P53 gene in the present patient and her family members. A-G. Represent relative P53 gene sequence of II6, III3, II5, IV1, II4, III2, II1, respectively, consistent with the identifier in Figure 1.
Discussion
The majority of P53 mutations are missense substitutions (73.6%). Other alterations include frameshift insertions and deletions (8.65%), nonsense mutations (7.67%), silent mutations (4.37%), splice site mutations (1.84%) and other infrequent alterations (IARC P53 Database, http://www-P53.iarc.fr/). Most germ line P53 mutations have been found in families with strong histories of cancer. Wild-type P53 has been demonstrated to induce apoptosis within the context of DNA damage; loss of wild-type P53 is thought to result in abrogation of apoptosis in DNA-damaged cells, leading to propagation of DNA lesions that could result in neoplastic transformation [9].
The P53 protein consists of 393 residues and can be divided into three functional regions: (i) an N-terminal domain (1-93) containing a transcriptional activation domain and a proline-rich domain; (ii) a core DNA-binding domain (102-292), which contains most of the inactivating mutations found in human tumors; and (iii) a C-terminal domain (CTD) consisting of a tetramerization domain (320-356) and a regulatory domain (363-393) [10]. The extreme CTD, which binds to non-specific DNA sequences, is in fact of major importance for the regulation of the protein [11]. It seems to have a negative effect on specific DNA-binding activity of the core domain or by altering the conformation of P53 or by interfering by steric hindrance with the ability of the full-length protein to bind DNA [12,13]. Deletion of this regulatory region, binding of antibodies, phosphorylation and acetylation abolish the negative effect on DNA binding [14].
In this study, we found a novel inherited germline P53 mutation in a breast cancer patient with a thymidine (T) insertion between 685th and 686th nucleotide, which result in a truncated polypeptide chain harbouring only 238 amino acids. Lacking part of core DNA-binding domain and full length of C-terminal domain, it was speculated this frame-shifted mutant P53 protein may have disrupted tumor suppressor function by causing misfolding, aggregation or degradation.
Considering the patients’ identical twin sister carrying the same mutation developed breast cancer at young age, we believe that this mutation played a role in breast tumorigenesis in this case. This P53 mutation was also detected in the twin sisters’ father, indicating it a hereditable heterozygous mutation. What’s more, the mutation we present here has never been described in the International Agency for Research on Cancer (IARC) P53 mutation database (www.iarc.fr). No related research about the function and clinical significance of this mutation has been found.
Mutations of the P53 gene have been associated with resistance to chemotherapy as well as a poor prognosis in many different malignancies [4], the present patient is being carefully monitored to ensure early identification of recurrent breast cancer or any second unrelated tumors.
Disclosure of conflict of interest
None.
References
- 1.Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat. 2003;21:313–320. doi: 10.1002/humu.10185. [DOI] [PubMed] [Google Scholar]
- 2.Mai PL, Best AF, Peters JA, DeCastro RM, Khincha PP, Loud JT, Bremer RC, Rosenberg PS, Savage SA. Risks of first and subsequent cancers among TP53 mutation carriers in the national cancer institute Li-Fraumeni syndrome cohort. Cancer. 2016;122:3673–3681. doi: 10.1002/cncr.30248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, Bouvignies E, Caron O, Bressac-de Paillerets B, Berthet P, Dugast C, Bonaiti-Pellie C, Stoppa-Lyonnet D, Frebourg T. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J. Clin. Oncol. 2009;27:e108–109. doi: 10.1200/JCO.2009.22.7967. author reply e110. [DOI] [PubMed] [Google Scholar]
- 4.Sakurai N, Iwamoto S, Miura Y, Nakamura T, Matsumine A, Nishioka J, Nakatani K, Komada Y. Novel p53 splicing site mutation in Li-Fraumeni-like syndrome with osteosarcoma. Pediatr Int. 2013;55:107–111. doi: 10.1111/j.1442-200X.2012.03641.x. [DOI] [PubMed] [Google Scholar]
- 5.Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brachmann RK, Yu K, Eby Y, Pavletich NP, Boeke JD. Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J. 1998;17:1847–1859. doi: 10.1093/emboj/17.7.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carvajal LA, Manfredi JJ. Another fork in the road-life or death decisions by the tumour suppressor p53. EMBO Rep. 2013;14:414–421. doi: 10.1038/embor.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 9.Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- 10.Pirolli D, Carelli Alinovi C, Capoluongo E, Satta MA, Concolino P, Giardina B, De Rosa MC. Insight into a novel p53 single point mutation (G389E) by molecular dynamics simulations. Int J Mol Sci. 2010;12:128–140. doi: 10.3390/ijms12010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn J, Prives C. The C-terminus of p53: the more you learn the less you know. Nat Struct Biol. 2001;8:730–732. doi: 10.1038/nsb0901-730. [DOI] [PubMed] [Google Scholar]
- 12.Anderson ME, Woelker B, Reed M, Wang P, Tegtmeyer P. Reciprocal interference between the sequence-specific core and nonspecific Cterminal DNA binding domains of p53: implications for regulation. Mol Cell Biol. 1997;17:6255–6264. doi: 10.1128/mcb.17.11.6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hupp TR, Lane DP. Allosteric activation of latent p53 tetramers. Curr Biol. 1994;4:865–875. doi: 10.1016/s0960-9822(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 14.Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53. Cell. 1992;71:875–886. doi: 10.1016/0092-8674(92)90562-q. [DOI] [PubMed] [Google Scholar]