

Abstract
Several somatic copy number variations (CNVs) have been identified in papillary thyroid cancer (PTC). However, the functional roles of CNVs and the genes responsible for the roles of CNVs are largely unknown. In this study, we identified a novel long noncoding RNA (lncRNA) CNALPTC1 (copy number amplified long noncoding RNA in papillary thyroid cancer 1). The genomic copy number of CNALPTC1 is amplified and CNALPTC1 expression level is up-regulated in PTC. Increased expression of CNALPTC1 is associated with aggressive clinicopathological characteristics. Gain-of-function and loss-of-function assays revealed that CNALPTC1 promotes proliferation and migration of PTC cells, and inhibits apoptosis of PTC cells. Mechanistically, we found that CNALPTC1 physically associates to miR-30 family and down-regulates miR-30 expression. Furthermore, CNALPTC1 up-regulates the expression of miR-30 targets, such as BCL9, SNAI1, and VIM. The mutation of miR-30 binding site on CNALPTC1 or overexpression of miR-30 abrogates the oncogenic roles of CNALPTC1 in PTC. Collectively, our results suggested that the copy number amplified lncRNA CNALPTC1 promotes PTC progression via sponging miR-30 family. Our data also implied that CNALPTC1 may be a novel therapeutic target for PTC.
Keywords: Copy number variation, papillary thyroid cancer, long noncoding RNA, proliferation, migration, apoptosis, miR-30 family
Introduction
Thyroid cancer is the most common endocrine malignancy with rapidly increased incidence over the past few decades [1,2]. Papillary thyroid cancer (PTC) accounts for more than 80% of thyroid cancer. PTC is biologically indolent and has a relatively good prognosis [3,4]. However, approximate 10%-30% of PTC cases show aggressive behaviors and progress to poor outcome [5]. Therefore, identification of molecular mechanisms contributing to the progression of PTC is critical for proper treatment of PTC.
Genetic and epigenetic alterations are associated with the initiation and progression of many cancers, including PTC [6-8]. Among these genetic and epigenetic alterations, somatic copy number variants (CNVs) are extremely common and play important roles in various cancers, including PTC [9,10]. The somatic copy number amplifications or deletions have led to the identifications of oncogenes or tumor suppressors that drive cancer initiation and development [11,12]. However, the functional roles of many CNVs and the genes responsible for the roles of CNVs are always unclear [13,14]. Although many protein-coding genes involved in cancer have been identified in aberrant chromosome regions, there are many non-protein coding genes locating at these aberrant chromosome regions and contributing considerably to cancer initiation and progression [15].
Among the RNAs encoded by these non-protein coding genes, long noncoding RNAs (lncRNAs) are a class of recently identified non-protein coding RNAs with more than 200 nucleotides in length [16-18]. Increasing evidences have revealed that many lncRNAs are frequently dysregulated in cancers, and have oncogenic or tumor suppressive roles [19-21]. LncRNAs BANCR, PTCSC3, and MEG3 are down-regulated and function as tumor suppressors in thyroid cancer [22-24]. Whereas, lncRNAs ANRIL, MALAT1, HOTAIR, and HIT000218960 are up-regulated and function as oncogenes in thyroid cancer [25-28]. However, the expressions and roles of most other lncRNAs in PTC are still unknown. Furthermore, whether the functional lncRNAs locate at copy number amplified or deleted regions, and are responsible for the roles of these aberrant chromosome regions in PTC have not been reported.
In a previous report using high-resolution comparative genomic hybridization, Passon et al. identified frequently somatic copy number amplified or depleted regions [29]. Notably, we found chromosome 1p36.22 is frequently amplified in PTC, and is markedly more frequent in the intermediate/high risk group of PTC compared with that in low risk group of PTC. Interesting, in another report using lncRNA microarray, Li et al. identified several dysregulated lncRNAs in PTC [28]. Among the aberrantly expressed lncRNAs in the microarray results, we noted that lncRNA ENST00000606790 is significantly up-regulated in PTC tissues compared with that in adjacent noncancerous thyroid tissues. Moreover, the gene encoding ENST00000606790 locates at chromosome 1p36.22-p36.21.
Therefore, we further measured the genomic copy number levels and RNA expression levels of ENST00000606790 in enlarged clinical PTC tissues, investigated the biological roles of ENST00000606790 in PTC, and explored the underlying action mechanisms of ENST00000606790 in PTC. Our results confirmed the genomic copy number amplification of the gene encoding ENST00000606790. Thus, we named this lncRNA as Copy Number Amplified Long noncoding RNA in Papillary Thyroid Cancer 1 (CNALPTC1).
Materials and methods
Tissue samples
Sixty-four pairs of PTC tissues and adjacent noncancerous thyroid tissues were obtained from PTC patients who underwent radical surgical resections at Chinese PLA General Hospital (Beijing, China). None of the patients received any anti-cancer therapy before surgery. All the tissues were diagnosed by pathological examination. All tissue samples were immediately frozen in liquid nitrogen after surgery and saved at -80°C for later experiments. This study was in strictly accordance with the guidelines and principles of the Declaration of Helsinki, and approved by the Ethical Committee of Chinese PLA General Hospital. All patients had signed the informed consents.
Cell culture
The human PTC cell lines TPC-1 and IHH-4 were obtained from Cell Bank of Chinese Academy of Sciences (Shanghai, China). The cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) in an atmosphere at 37°C with 5% CO2. The cell lines used in this study were not found in the database of commonly misidentified cell lines that is maintained by ICLAC and NCBI database. All cells were routinely tested as mycoplasma-free.
RNA isolation, DNA preparation, and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated from tissues and cells with the TRIzol Reagent (Invitrogen) in accordance with the manufacturer’s instructions. The isolated RNA was treated with DNase I to remove genomic DNA, and reverse-transcribed into the first strand complimentary DNA (cDNA) with the M-MLV Reverse Transcriptase (Invitrogen) in accordance with the manufacturer’s instructions. Genomic DNA was isolated from tissues with TIANamp Genomic DNA Kit (TIANGEN, Beijing, China) in accordance with the manufacturer’s instructions. Quantitative real-time polymerase chain reaction (qRT-PCR) was carried out with the SYBR® Premix Ex Taq™ II (Takara, Dalian, China) on the StepOnePlus™ Real-time PCR system (Applied Biosystems, Foster City, CA, USA). For microRNAs analyses, qRT-PCR was carried out as above described with TaqMan microRNA assays (Applied Biosystems) in accordance with the manufacturer’s instructions. lncRNAs and mRNAs expressions were normalized to β-actin. microRNAs expressions were normalized to U6. DNA copy number levels were normalized to long interspersed element-1 (LINE1). RNA expression levels or DNA copy number levels were calculated using the 2-ΔΔCt method. The primers sequences used were as follows: CNALPTC1 (RNA expression), 5’-CACTTTGAACTGGGGCTG-3’ (sense) and 5’-CGTCTCCTGTCCTTGGGT-3’ (anti-sense); BCL9, 5’-GGAAATGTAGAGTCAGGTG-3’ (sense) and 5’-CATCATAATAGAGAGTGGG-3’ (anti-sense); SNAI1, 5’-TGCGTCTGCGGAACCTG-3’ (sense) and 5’-GGACTCTTGGTGCTTGTGGA-3’ (anti-sense); VIM, 5’-CCTGAACCTGAGGGAAACTAA-3’ (sense) and 5’-GCAGAAAGGCACTTGAAAGC-3’ (anti-sense); β-actin, 5’-GGGAAATCGTGCGTGACATTAAG-3’ (sense) and 5’-TGTGTTGGCGTACAGGTCTTTG-3’ (anti-sense); CNALPTC1 (DNA copy number), 5’-CCCACCTTCTAACCTCCT-3’ (sense) and 5’-CTCACCTCCCAGACTTCC-3’ (anti-sense); and LINE1, 5’-AAAGCCGCTCAACTACATGG-3’ (sense) and 5’-TGCTTTGAATGCGTCCCAGAG-3’ (anti-sense).
5’ and 3’ rapid amplification of cDNA Ends (RACE)
5’ and 3’ RACE analyses were carried out to determine the transcriptional initiation and termination sites of CNALPTC1 with the 5’/3’ RACE Kit (Roche, Mannheim, Germany) in accordance with the manufacturer’s instructions. The primers sequences used for the PCR of the RACE analyses were as follows: SP1, 5’-CAAAAGCCTGTAAACAAAACG-3’; SP2, 5’-GGCACCCACCTTCTAACCTC-3’; SP3, 5’-TAACCTCCTGCCCTTCCCGAC-3’; SP5, 5’-AGGAGGTTAGAAGGTGGGTGCC-3’.
Plasmids and stable cell lines construction
Two independent oligonucleotides for shRNAs targeting CNALPTC1 were synthesized and inserted into the shRNA expression vector pGPH1/Neo (GenePharma, Shanghai, China). The two shRNAs target sequences were: 5’-GCAACAGACAACTCTAAATAG-3’ and 5’-GAAGGGCAGGAGGTTAGAAGG-3’. A scrambled non-silencing shRNA was used as negative control for constructed shRNAs. To obtain CNALPTC1 stably depleted TPC-1 and IHH-4 cells, CNALPTC1 specific or negative control shRNAs were transfected into TPC-1 and IHH-4 cells with Lipofectamine 3000 (Invitrogen) in accordance with the manufacturer’s instructions, and then the cells were selected with 800 μg/ml neomycin for four weeks.
The full-length CNALPTC1 sequences were PCR amplified with the Thermo Scientific Phusion Flash High-Fidelity PCR Master Mix (Thermo-Fisher Scientific, Waltham, MA, USA) and the primers 5’-CCCAAGCTTGCTCCTCCGGCCCCGGGAA-3’ (sense) and 5’-CGGGATCCAGCTGGGAGCATATTTATTGCTCAT-3’ (anti-sense) in accordance with the manufacturer’s instructions. Then, the PCR products were subcloned into the Hind III and BamH I sites of pcDNA3.1 vector (Invitrogen), named as pcDNA3.1-CNALPTC1. Full-length CNALPTC1 sequences with the mutation of miR-30 binding sites were synthesized by GenScript (Nanjing, Jiangsu, China) and subcloned into the Hind III and BamH I sites of pcDNA3.1 vector, named as pcDNA3.1-CNALPTC1-mut. miR-30a, miR-30b, miR-30c, miR-30d, and miR-30e mimics were obtained from GenePharma. The transfections of plasmids and microRNAs mimics were carried out with Lipofectamine 3000 (Invitrogen) in accordance with the manufacturer’s instructions.
Cell proliferation assay
Cell proliferation was determined using Cell Counting Kit-8 (CCK-8) assays and Ethynyl deoxyuridine (EdU) incorporation assays. For CCK-8 assays, 4000 indicated PTC cells were plated in 96-well plates per well. After culture for 0, 24, 48, and 72 hours, the cell viability was determined with the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). The absorbance values at 450 nm at each time point were used to plot cell growth curves. EdU incorporation assays were carried out with an EdU kit (RiboBio, Guangzhou, Guangdong, China) in accordance with the manufacturer’s instructions. The results were determined with Zeiss photomicroscope (Carl Zeiss, Oberkochen, Germany) and quantified by counting ten random fields.
Cell apoptosis assay
Cell apoptosis was determined using the TdT-mediated dUTP nick end labeling (TUNEL) assays with the Dead EndTM Fluorometric TUNEL System (Promega, Madison, WI, USA) in accordance with the manufacturer’s instructions. The results were determined with Zeiss photomicroscope (Carl Zeiss) and quantified by counting ten random fields.
Cell migration assay
Cell migration was determined using transwell assays. Briefly, 50,000 indicated PTC cells suspended in FBS-free DMEM with 1 μg/ml mitomycin C to inhibit cell proliferation were plated into the upper well of a 24-well poly-carbonate transwell filters (Millipore, Bedford, MA, USA). DMEM supplemented with 10% FBS was added to the lower well. After incubation for 48 hours, cells on the upper surface of filters were scraped off, and cells on the lower surface were fixed and stained. The results were determined with Zeiss photomicroscope (Carl Zeiss) and quantified by counting ten random fields.
RNA immunoprecipitation (RIP) assay
pSL-MS2-12X (Addgene, Cambridge, MA, USA) was double digested using EcoR I and Not I, and the MS2-12X fragment was inserted into pcDNA3.1, pcDNA3.1-CNALPTC1, or pcDNA3.1-CNALPTC1-mut, named as pcDNA3.1-MS2, pcDNA3.1-CNALPTC1-MS2, or pcDNA3.1-CNALPTC1-mut-MS2, respectively. pcDNA3.1-MS2, pcDNA3.1-CNALPTC1-MS2, or pcDNA3.1-CNALPTC1-mut-MS2 was co-transfected with pMS2-GFP (Addgene) into TPC-1 cells. Forty-eight hours later, RNA Immunoprecipitation (RIP) assays were carried out with these cells, the Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore), and a GFP antibody (Roche) in accordance with the manufacturer’s instructions. The retrieved RNAs were determined by qRT-PCR as above described.
RNA pull-down assay
pcDNA3.1-CNALPTC1 and pcDNA3.1-CNALPTC1-mut were double digested using Hind III and BamH I. Then the CNALPTC1 or CNALPTC1-mut fragment was inserted into the pSPT19 vector (Roche), named as pSPT19-CNALPTC1 or pSPT19-CNALPTC1-mut, respectively. CNALPTC1 or CNALPTC1-mut was in vitro transcribed and biotinylated from pSPT19-CNALPTC1 or pSPT19-CNALPTC1-mut with the T7 RNA polymerase (Roche) and Biotin RNA Labeling Mix (Roche) in accordance with the manufacturer’s instructions. After being treated with RNase-free DNase I (Roche), the in vitro transcribed RNA was purified with an RNeasy Mini Kit (Qiagen, Valencia, CA, USA) in accordance with the manufacturer’s instructions. Three micrograms of purified biotinylated CNALPTC1 or CNALPTC1-mut were incubated with one microgram of whole-cell lysates from TPC-1 cells for one hour at 25°C, followed by being retrieved with streptavidin agarose beads (Invitrogen). The retrieved RNAs were determined by qRT-PCR as above described.
Luciferase reporter assay
3’ untranslated region (UTR) of BCL9, SNAI1, and VIM mRNA containing miR-30 binding sites were PCR amplified with the Thermo Scientific Phusion Flash High-Fidelity PCR Master Mix (Thermo-Fisher) in accordance with the manufacturer’s instructions, and subcloned into the Sac I and Xba I sites of pmirGLO vector (Promega), named as pmirGLO-BCL9, pmirGLO-SNAI1, or pmirGLO-VIM, respectively. The primers sequences used were as follows: for BCL9, 5’-CGAGCTCTTTTTGTGGACTTGGGTATC-3’ (sense) and 5’-GCTCTAGATCTGAGGTCGTAGTTTGGTT-3’ (anti-sense); for SNAI1, 5’-CGAGCTCGCTGACAGACTCACTGGG-3’ (sense) and 5’-GCTCTAGAGTTTGAAATATAAATACCAGTG-3’ (anti-sense); and for VIM, 5’-CGAGCTCGGAATAGGAATAAGCTCTAG-3’ (sense) and 5’-GCTCTAGAGCTGGAAAAAAAAAAGCAGT-3’ (anti-sense). pmirGLO, pmirGLO-BCL9, pmirGLO-SNAI1, or pmirGLO-VIM was co-transfected with pcDNA3.1, pcDNA3.1-CNALPTC1, or pcDNA3.1-CNALPTC1-mut into TPC-1 cells. The luciferase activity was determined with the Dual-Luciferase® Reporter Assay System (Promega) 48 hours after transfection.
Western blot
Proteins were extracted from indicated PTC cells using RIPA Lysis Buffer and PMSF (Beyotime, Jiangsu, China) in accordance with the manufacturer’s instructions. Equal amount of proteins was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by being transferred to PVDF membranes. After being blocked using bovine serum albumin, the membranes were incubated with primary antibodies specific for BCL9 (Cell Signaling Technology, Boston, MA, USA), SNAI1 (Cell Signaling Technology), VIM (Cell Signaling Technology), or β-actin (Proteintech, Rosemont, IL, USA). After being washed three times, the membranes were further incubated with IRDye 800CW goat anti-rabbit IgG or IRDye 700CW goat anti-mouse IgG (Li-Cor, Lincoln, NE, USA), and detected with an Odyssey infrared scanner (Li-Cor). Proteins expression was normalized to β-actin.
Statistical analysis
All statistical analyses were carried out with the GraphPad Prism Software. For comparisons, Wilcoxon signed-rank test, Pearson correlation analysis, Pearson chi-square test, and Student’s t-test were performed as indicated. P < 0.05 was considered as statistically significant.
Results
CNALPTC1 expression pattern in PTC
To investigate CNALPTC1 expression pattern and clinical significances in PTC, genomic copy number of CNALPTC1 was measured by qRT-PCR in 64 pairs of PTC tissues and adjacent noncancerous thyroid tissues. The results showed that the genomic copy number level of CNALPTC1 was markedly increased in PTC tissues compared with that in noncancerous thyroid tissues (Figure 1A). The RNA expression level of CNALPTC1 was also measured in the same 64 pairs of PTC tissues and adjacent noncancerous thyroid tissues. The results showed that the RNA expression level of CNALPTC1 was also markedly up-regulated in PTC tissues compared with that in noncancerous thyroid tissues (Figure 1B). Furthermore, the RNA expression level of CNALPTC1 was significantly correlated with the genomic copy number level of CNALPTC1 in PTC tissues (Figure 1C). These data implied that the genomic copy number amplification contributed to the up-regulation of CNALPTC1 in PTC. Correlation regression analyses of the association between CNALPTC1 expression and clinicopathological characteristics of PTC patients showed that increased expression of CNALPTC1 was associated with large tumor size (P = 0.008), lymph node metastasis (P = 0.005), and advanced TNM stage (P = 0.017) (Table 1). These data implied that CNALPTC1 may be involved in the progression of PTC.
Figure 1.
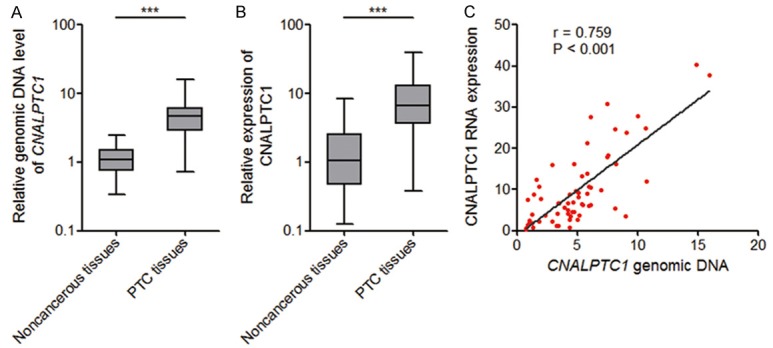
The expression pattern of CNALPTC1 in PTC. A. Genomic copy number levels of CNALPTC1 were determined by qRT-PCR in 64 pairs of PTC tissues and adjacent noncancerous thyroid tissues. The horizontal lines in the box plots represent the medians, the boxes represent the interquartile range, and the whiskers represent the minimum and maximum values. ***P < 0.001 by Wilcoxon signed-rank test. B. CNALPTC1 RNA expression levels were determined by qRT-PCR in 64 pairs of PTC tissues and adjacent noncancerous thyroid tissues. The horizontal lines in the box plots represent the medians, the boxes represent the interquartile range, and the whiskers represent the minimum and maximum values. ***P < 0.001 by Wilcoxon signed-rank test. C. Correlation between CNALPTC1 RNA expression levels and CNALPTC1 genomic copy number levels in PTC tissues (n = 64). x, the relative CNALPTC1 genomic copy number levels. y, the relative CNALPTC1 RNA expression levels. P < 0.001, r = 0.759 by Pearson correlation analysis. PTC, papillary thyroid cancer; qRT-PCR, quantitative real-time PCR.
Table 1.
Association between CNALPTC1 expression and clinicopathological characteristics of PTC patients
| Clinicopathologic characteristics | CNALPTC1 | P-value | |
|---|---|---|---|
|
| |||
| Low | High | ||
| All cases | 32 | 32 | |
| Gender | 0.412 | ||
| Male | 8 | 11 | |
| Female | 24 | 21 | |
| Age (years) | 0.211 | ||
| < 45 | 18 | 13 | |
| ≥45 | 14 | 19 | |
| Focality | 1.000 | ||
| Unifocal | 19 | 19 | |
| Multifocal | 13 | 13 | |
| Primary tumor | 0.008 | ||
| T1-T2 | 26 | 16 | |
| T3-T4 | 6 | 16 | |
| Lymph node metastasis | 0.005 | ||
| N0 | 18 | 7 | |
| N1 | 14 | 25 | |
| TNM stage | 0.017 | ||
| I-II | 26 | 17 | |
| III-IV | 6 | 15 | |
Median expression level of CNALPTC1 was used as the cutoff. P-value was acquired by Pearson chi-square test.
Effects of CNALPTC1 on the proliferation of PTC cells
To explore whether CNALPTC1 genuinely has biological effects on PTC, we first established the transcription initiation and termination sites of CNALPTC1 using 5’ and 3’ RACE analyses (Supplementary Figure 1). Then, we constructed CNALPTC1 stably depleted TPC-1 cells using two independent CNALPTC1 specific shRNAs (Figure 2A). CCK-8 assays showed that depletion of CNALPTC1 significantly repressed TPC-1 cell proliferation (Figure 2B). EdU incorporation assays also revealed that depletion of CNALPTC1 markedly inhibited TPC-1 cell proliferation (Figure 2C). To further confirm the effects of CNALPTC1 on the proliferation of PTC cells, we stably depleted CNALPTC1 in another PTC cell line IHH-4 (Figure 2D). Consistently, CCK-8 assays and EdU incorporation assays showed that depletion of CNALPTC1 markedly repressed IHH-4 cell proliferation (Figure 2E and 2F). These data suggested that depletion of CNALPTC1 represses the proliferation of PTC cells.
Figure 2.
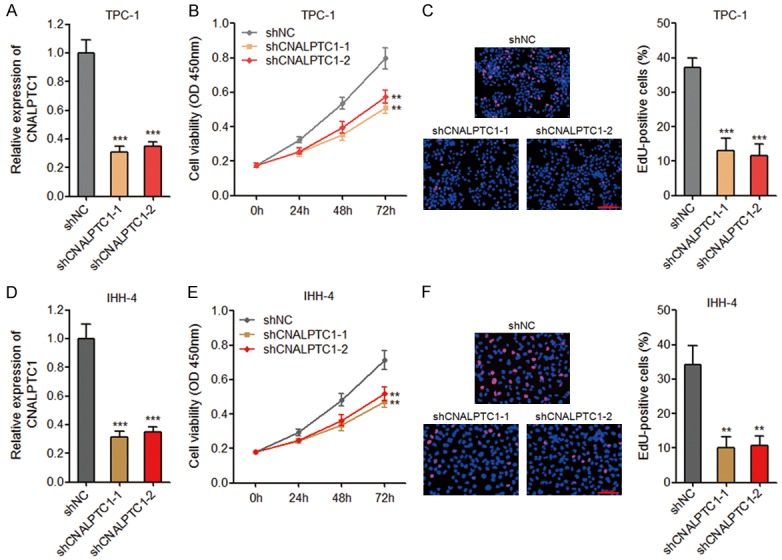
Depletion of CNALPTC1 inhibits proliferation of PTC cells. A. CNALPTC1 RNA expression levels were determined by qRT-PCR in CNALPTC1 stably depleted and control TPC-1 cells. B. Cell proliferation of CNALPTC1 stably depleted and control TPC-1 cells were determined by CCK-8 assays. C. Cell proliferation of CNALPTC1 stably depleted and control TPC-1 cells were determined by EdU incorporation assays. The red colour indicates EdU-positive nuclei. Scale bars = 100 µm. D. CNALPTC1 RNA expression levels were determined by qRT-PCR in CNALPTC1 stably depleted and control IHH-4 cells. E. Cell proliferation of CNALPTC1 stably depleted and control IHH-4 cells were determined by CCK-8 assays. F. Cell proliferation of CNALPTC1 stably depleted and control IHH-4 cells were determined by EdU incorporation assays. The red colour indicates EdU-positive nuclei. Scale bars = 100 µm. Results are shown as mean ± s.d. of 3 independent experiments. **P < 0.01, ***P < 0.001 by Student’s t-test. PTC, papillary thyroid cancer; qRT-PCR, quantitative real-time PCR; CCK-8, Cell Counting Kit-8; EdU, Ethynyl deoxyuridine.
Effects of CNALPTC1 on apoptosis of PTC cells
The effects of CNALPTC1 on apoptosis of PTC cells were evaluated by TUNEL assays. As shown in Figure 3A, depletion of CNALPTC1 markedly promoted apoptosis of TPC-1 cells. Consistently, depletion of CNALPTC1 significantly enhanced apoptosis of IHH-4 cells (Figure 3B). These data suggested that depletion of CNALPTC1 promotes apoptosis of PTC cells.
Figure 3.
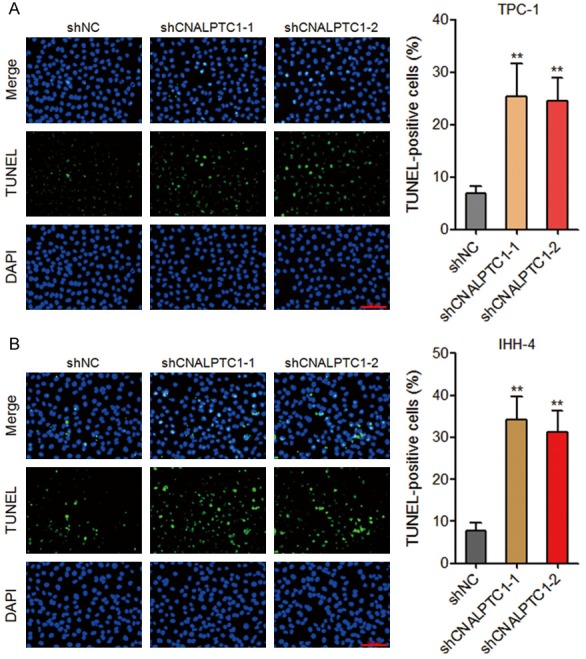
Depletion of CNALPTC1 promotes apoptosis of PTC cells. A. Apoptosis of CNALPTC1 stably depleted and control TPC-1 cells were determined by TUNEL assays. The green colour indicates TUNEL-positive and apoptotic cells. Scale bars = 100 µm. B. Apoptosis of CNALPTC1 stably depleted and control IHH-4 cells were determined by TUNEL assays. The green colour indicates TUNEL-positive and apoptotic cells. Scale bars = 100 µm. Results are shown as mean ± s.d. of 3 independent experiments. **P < 0.01 by Student’s t-test. PTC, papillary thyroid cancer; TUNEL, TdT-mediated dUTP nick end labeling.
Effects of CNALPTC1 on migration of PTC cells
Transwell assays were performed to evaluate the effects of CNALPTC1 on migration of PTC cells. As shown in Figure 4A, depletion of CNALPTC1 markedly inhibited migration of TPC-1 cells. Consistently, depletion of CNALPTC1 also markedly repressed migration of IHH-4 cells (Figure 4B). These data suggested that depletion of CNALPTC1 inhibits migration of PTC cells.
Figure 4.
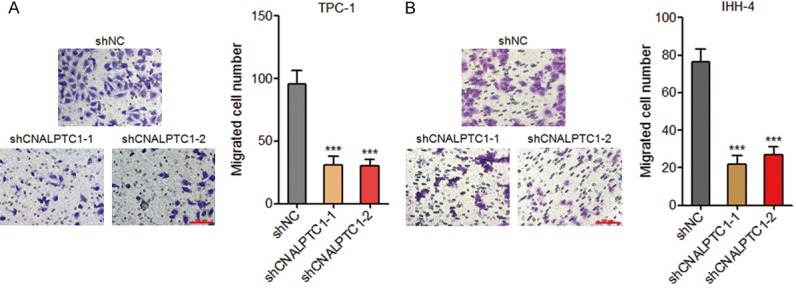
Depletion of CNALPTC1 represses migration of PTC cells. A. Migration of CNALPTC1 stably depleted and control TPC-1 cells were determined by transwell assays. Represent images are shown. Scale bars = 100 µm. B. Migration of CNALPTC1 stably depleted and control IHH-4 cells were determined by transwell assays. Represent images are shown. Scale bars = 100 µm. Results are shown as mean ± s.d. of 3 independent experiments. ***P < 0.001 by Student’s t-test. PTC, papillary thyroid cancer.
CNALPTC1 sponges and down-regulates miR-30 family
Recently, many lncRNAs have been regarded as competing endogenous RNAs (ceRNA) via competitively binding common microRNAs [30-32]. Using bioinformatics analysis by TargetScan and Miranda, we found a putative microRNA-30 (miR-30) family response element on CNALPTC1 (Figure 5A). To investigate whether CNALPTC1 genuinely sponges miR-30 family, MS2 based RIP assays were performed to pull down endogenous microRNAs bound to CNALPTC1. As shown in Figure 5B, miR-30 family members, including miR-30a, miR-30b, miR-30c, miR-30d, and miR-30e were all markedly enriched in RNAs retrieved from MS2-CNALPTC1 compared with that from control MS2 and miR-30 binding site mutated MS2-CNALPTC1 (termed as CNALPTC1-mut). In addition, in vitro transcribed biotin-labeled CNALPTC1 was used to pull down endogenous microRNAs interacting with CNALPTC1. The results showed that all five miR-30 family members were significantly enriched in RNAs retrieved from CNALPTC1 compared with that from beads control and CNALPTC1-mut (Figure 5C). These data suggested the specific interaction between CNALPTC1 and miR-30 family. Moreover, transfection of CNALPTC1 overexpression plasmids markedly inhibited the expression of miR-30 family in PTC cells, which was abolished by the mutation of miR-30 binding site (Figure 5D). On the other hand, the expression of miR-30 family was markedly increased in CNALPTC1 stably depleted TPC-1 cells (Figure 5E). Collectively, these data suggested that CNALPTC1 sponges and down-regulates miR-30 family.
Figure 5.
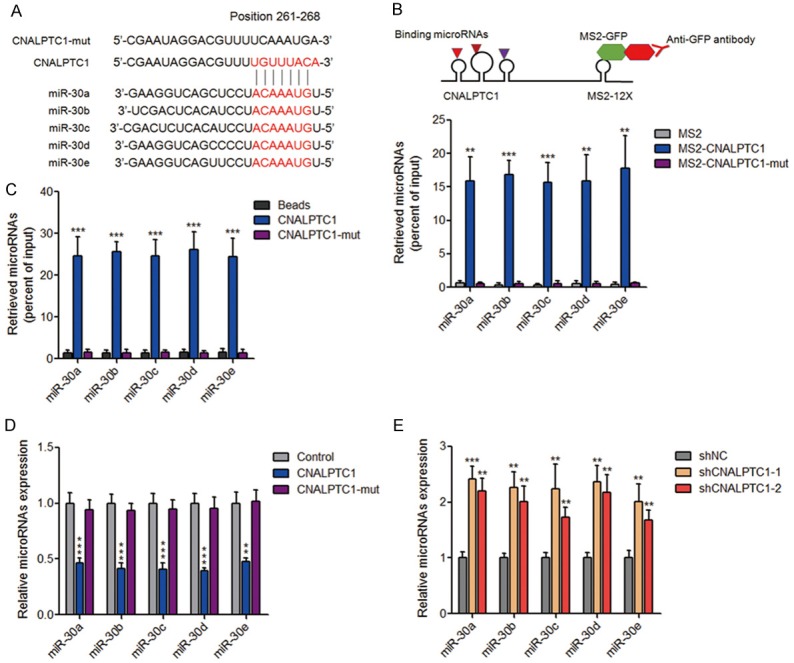
CNALPTC1 sponges and down-regulates miR-30 family. A. Schematic outlining the predicted binding site of miR-30 family on CNALPTC1. B. The specific bindings of miR-30 family to CNALPTC1 or miR-30 binding site mutated CNALPTC1 (CNALPTC1-mut) were determined by MS2 based RIP assays, followed by qRT-PCR. C. TPC-1 cell lysates were incubated with biotin-labeled CNALPTC1 or CNALPTC1-mut; after pull-down, microRNAs were extracted and determined by qRT-PCR. D. CNALPTC1 or miR-30 binding site mutated CNALPTC1 (CNALPTC1-mut) overexpression plasmids were transfected into TPC-1 cells. Forty-eight hours later, miR-30 family expression levels were determined by qRT-PCR. E. Expression levels of miR-30 family were determined by qRT-PCR in CNALPTC1 stably depleted and control TPC-1 cells. Results are shown as mean ± s.d. of 3 independent experiments. **P < 0.01, ***P < 0.001 by Student’s t-test. RIP, RNA Immunoprecipitation; qRT-PCR, quantitative real-time PCR.
CNALPTC1 up-regulates BCL9, SNAI1, and VIM expression via competitively sponging miR-30 family
miR-30 family has been shown to function as tumor suppressors via directly targeting BCL9, SNAI1, VIM, and et al. in multiple myeloma, breast cancer, hepatocellular carcinoma, gastric cancer, non-small cell lung cancer, and et al. [33-37]. Therefore, we next investigated whether CNALPTC1 regulates the expression of miR-30 targets in PTC. 3’-UTR of BCL9, SNAI1, and VIM were inserted into the luciferase reporter pmirGLO. Dual luciferase reporter assays showed that enhanced expression of CNALPTC1 significantly up-regulated the luciferase activity of the reporters containing BCL9, SNAI1, or VIM 3’-UTR (Figure 6A). Mutation of miR-30 binding site on CNALPTC1 or overexpression of miR-30 family mix abrogated the up-regulation of the luciferase activity (Figure 6A). On the other hand, depletion of CNALPTC1 markedly reduced the luciferase activity of the reporters containing BCL9, SNAI1, or VIM 3’-UTR (Figure 6B). Transfection of CNALPTC1 overexpression plasmids markedly up-regulated the RNA expression levels of miR-30 targets BCL9, SNAI1, and VIM, which was abolished by the mutation of miR-30 binding site or concurrent overexpression of miR-30 family mix (Figure 6C). On the other hand, the RNA expression levels of miR-30 targets BCL9, SNAI1, and VIM were markedly decreased in CNALPTC1 stably depleted TPC-1 cells (Figure 6D). Western blot assays showed that transfection of CNALPTC1 overexpression plasmids significantly up-regulated the protein levels of miR-30 targets BCL9, SNAI1, and VIM, which was abolished by the mutation of miR-30 binding site or concurrent overexpression of miR-30 family mix (Figure 6E). On the other hand, the protein levels of miR-30 targets BCL9, SNAI1, and VIM were significantly reduced in CNALPTC1 stably depleted TPC-1 cells (Figure 6F). Collectively, these data suggested that CNALPTC1 up-regulates BCL9, SNAI1, and VIM expression via competitively sponging miR-30 family.
Figure 6.
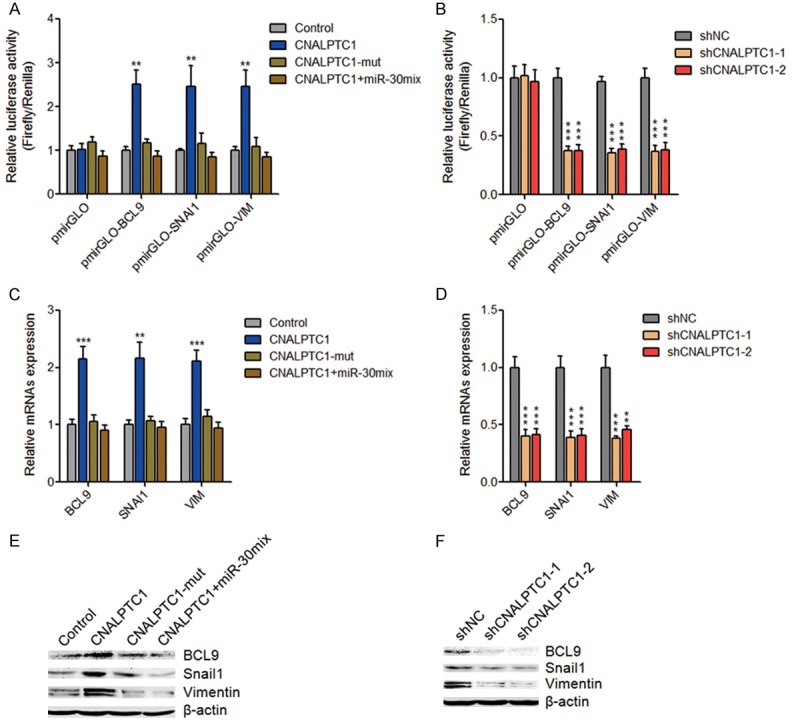
CNALPTC1 up-regulates miR-30 targets BCL9, SNAI1, and VIM expression. A. Luciferase activities of pmirGLO, pmirGLO-BCL9, pmirGLO-SNAI1, or pmirGLO-VIM upon transfection of CNALPTC1 or miR-30 binding site mutated CNALPTC1 (CNALPTC1-mut) overexpression plasmids, or co-transfection of CNALPTC1 overexpression plasmids and miR-30 mimics mix. Results are presented as the relative ratio of firefly luciferase activity to renilla luciferase activity. B. Luciferase activities of pmirGLO, pmirGLO-BCL9, pmirGLO-SNAI1, or pmirGLO-VIM in CNALPTC1 stably depleted and control TPC-1 cells. Results are presented as the relative ratio of firefly luciferase activity to renilla luciferase activity. C. After transfection of CNALPTC1 or CNALPTC1-mut overexpression plasmids, or co-transfection of CNALPTC1 overexpression plasmids and miR-30 mimics mix into TPC-1 cells, BCL9, SNAI1, and VIM RNA expression levels were determined by qRT-PCR. D. BCL9, SNAI1, and VIM RNA expression levels were determined by qRT-PCR in CNALPTC1 stably depleted and control TPC-1 cells. E. After transfection of CNALPTC1 or CNALPTC1-mut overexpression plasmids, or co-transfection of CNALPTC1 overexpression plasmids and miR-30 mimics mix into TPC-1 cells, BCL9, SNAI1, and VIM protein expression levels were determined by western blot. F. BCL9, SNAI1, and VIM protein expression levels were determined by western blot in CNALPTC1 stably depleted and control TPC-1 cells. Results are shown as mean ± s.d. of 3 independent experiments. **P < 0.01, ***P < 0.001 by Student’s t-test. qRT-PCR, quantitative real-time PCR.
CNALPTC1 exerts oncogenic activity in PTC via sponging miR-30 family
To explore whether the interaction with miR-30 family contributes to the biological roles of CNALPTC1 in PTC, CNALPTC1 overexpression plasmids, miR-30 binding site mutated CNALPTC1 overexpression plasmids, or control plasmids were transfected into TPC-1 cells. Furthermore, CNALPTC1 overexpression plasmids were co-transfected with miR-30 mix into TPC-1 cells. CCK-8 assays showed that enhanced expression of CNALPTC1 significantly promoted proliferation of TPC-1 cells (Figure 7A). The mutation of miR-30 binding site or concurrent overexpression of miR-30 mix abolished the proliferation promoting roles of CNALPTC1 (Figure 7A). In addition, EdU incorporation assays also showed that enhanced expression of CNALPTC1 markedly promoted proliferation of TPC-1 cells, which was abolished by the mutation of miR-30 binding site or concurrent overexpression of miR-30 mix (Figure 7B). TUNEL assays showed that enhanced expression of CNALPTC1 significantly inhibited apoptosis of TPC-1 cells (Figure 7C). The mutation of miR-30 binding site or concurrent overexpression of miR-30 mix abolished the apoptosis inhibiting roles of CNALPTC1 (Figure 7C). Transwell assays showed that enhanced expression of CNALPTC1 markedly promoted migration of TPC-1 cells, which was abrogated by the mutation of miR-30 binding site or concurrent overexpression of miR-30 mix (Figure 7D). Collectively, these data suggested that CNALPTC1 promotes cell proliferation and migration, and inhibits cell apoptosis of PTC cells in a miR-30 dependent manner.
Figure 7.
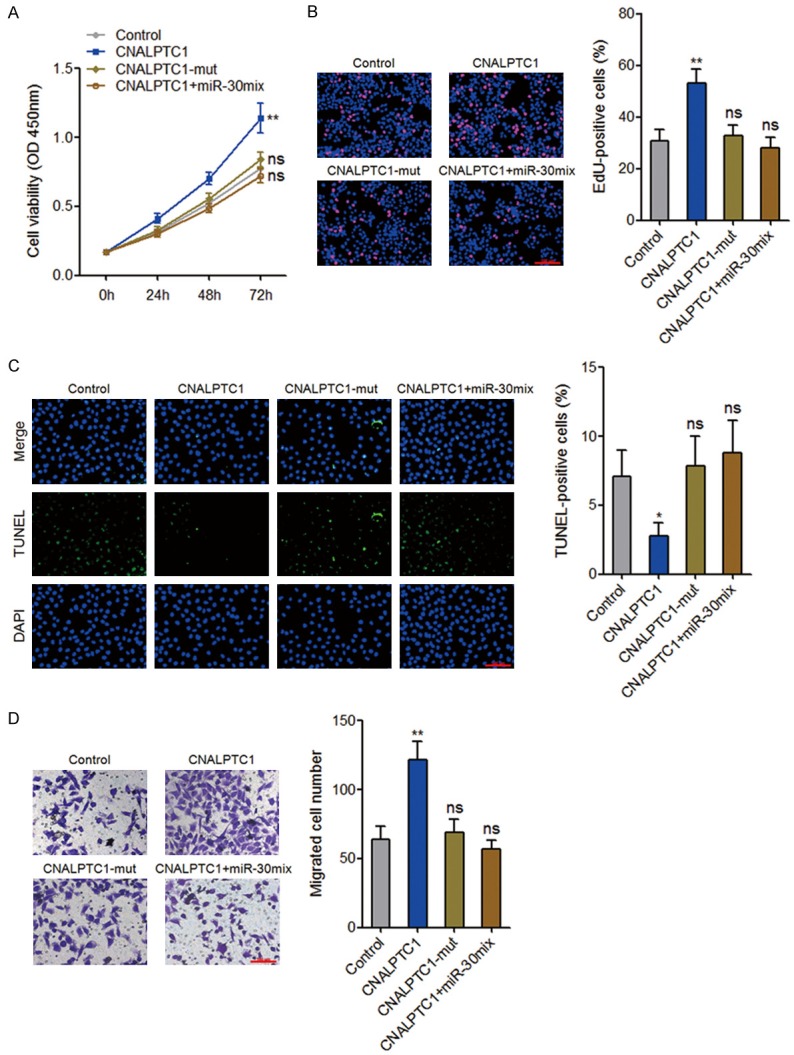
CNALPTC1 exerts oncogenic activity in PTC via sponging miR-30 family. A. After transfection of CNALPTC1 or CNALPTC1-mut overexpression plasmids, or co-transfection of CNALPTC1 overexpression plasmids and miR-30 mimics mix into TPC-1 cells, cell proliferation was determined by CCK-8 assays. B. After transfection of CNALPTC1 or CNALPTC1-mut overexpression plasmids, or co-transfection of CNALPTC1 overexpression plasmids and miR-30 mimics mix into TPC-1 cells, cell proliferation was determined by EdU incorporation assays. The red colour indicates EdU-positive nuclei. Scale bars = 100 µm. C. After transfection of CNALPTC1 or CNALPTC1-mut overexpression plasmids, or co-transfection of CNALPTC1 overexpression plasmids and miR-30 mimics mix into TPC-1 cells, cell apoptosis was determined by TUNEL assays. The green colour indicates TUNEL-positive or apoptotic cells. Scale bars = 100 µm. D. After transfection of CNALPTC1 or CNALPTC1-mut overexpression plasmids, or co-transfection of CNALPTC1 overexpression plasmids and miR-30 mimics mix into TPC-1 cells, cell migration was determined by transwell assays. Represent images are shown. Scale bars = 100 µm. Results are shown as mean ± s.d. of 3 independent experiments. *P < 0.05, **P < 0.01, ns, not significant, by Student’s t-test. PTC, papillary thyroid cancer; CCK-8, Cell Counting Kit-8; EdU, Ethynyl deoxyuridine; TUNEL, TdT-mediated dUTP nick end labeling.
Discussion
With the great progresses of genetic researches, many somatic CNVs have been identified in cancers [14]. In most cases, the protein-coding genes locating at these abnormal chromosome regions contribute to the functional roles of these somatic CNVs [38]. However, somatic CNVs often encompass unknown genes, which may also play important roles in cancers [39]. Several CNVs-related lncRNAs have been reported, such as the copy number deletion-related lncRNA PRAL in hepatocellular carcinoma [40]. As to PTC, although several abnormal chromosome regions have been identified, whether these regions encompass lncRNAs and whether these lncRNAs have functional roles in PTC are still unknown [29,41].
In this study, we identified a novel lncRNA CNALPTC1, whose genomic copy number level is amplified in PTC tissues compared with that in noncancerous thyroid tissues. Furthermore, the RNA expression level of CNALPTC1 is also up-regulated in PTC tissues compared with that in noncancerous thyroid tissues. Our data also revealed that the genomic copy number amplification contributes to the up-regulation of CNALPTC1 in PTC. Clinical correlation analyses revealed that increased expression of CNALPTC1 is associated with large tumor size, lymph node metastasis, and advanced TNM stage. Gain-of-function and loss-of-function assays revealed that CNALPTC1 significantly promotes proliferation and migration of PTC cells, and inhibits apoptosis of PTC cells. Collectively, our data suggest that the copy number amplification-related lncRNA CNALPTC1 is up-regulated and functions as an oncogene in PTC.
The most complex and difficult section of the researches on lncRNAs is exploring the action mechanisms of lncRNAs. lncRNAs could directly bind to proteins, microRNAs, or DNAs, and change the expression, localization, and functions of the interaction partners [42]. Recently, many lncRNAs were regarded as competing endogenous RNAs (ceRNAs) via competitively binding common microRNAs, and further relieved the repression of targets caused by bound microRNAs [31]. microRNA is another class of non-protein coding transcript with 20-25 nucleotides in length [43-46]. Similar to lncRNAs, microRNAs are also demonstrated to be dysregulated and have critical roles in various cancers [47-52]. miR-30 family, including miR-30a, miR-30b, miR-30c, miR-30d, and miR-30e, are well known tumor suppressors and directly target BCL9, SNAI1, VIM, and et al. in many cancers [33-36]. In this study, combining bioinformatic analysis and experimental verification, we found that CNALPTC1 directly binds and down-regulates miR-30 family expression. Via competitively binding miR-30 family, CNALPTC1 up-regulates the expression of miR-30 targets BCL9, SNAI1, and VIM. Functional assays also showed that the oncogenic roles of CNALPTC1 in PTC are dependent on the sponging of miR-30 family.
In conclusion, our results demonstrated that the genomic copy number of CNALPTC1 is amplified and the expression of CNALPTC1 is up-regulated in PTC. Increased expression of CNALPTC1 is associated with aggressive clinical characteristics. CNALPTC1 promotes proliferation and migration of PTC cells, and inhibits apoptosis of PTC cells via sponging miR-30 family. These finding suggested that CNALPTC1 could be a novel therapeutic target for PTC.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81403424).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Burns WR, Zeiger MA. Differentiated thyroid cancer. Semin Oncol. 2010;37:557–566. doi: 10.1053/j.seminoncol.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 4.Aschebrook-Kilfoy B, Ward MH, Sabra MM, Devesa SS. Thyroid cancer incidence patterns in the united states by histologic type, 1992-2006. Thyroid. 2011;21:125–134. doi: 10.1089/thy.2010.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundgren CI, Hall P, Dickman PW, Zedenius J. Clinically significant prognostic factors for differentiated thyroid carcinoma: a populationbased, nested case-control study. Cancer. 2006;106:524–531. doi: 10.1002/cncr.21653. [DOI] [PubMed] [Google Scholar]
- 6.Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13:184–199. doi: 10.1038/nrc3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Roder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigo R, Gingeras TR. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tartari CJ, Donadoni C, Manieri E, Mologni L, Mina PD, Villa A, Gambacorti-Passerini C. Dissection of the RET/beta-catenin interaction in the TPC1 thyroid cancer cell line. Am J Cancer Res. 2011;1:716–725. [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang N, Wang M, Zhang P, Huang T. Classification of cancers based on copy number variation landscapes. Biochim Biophys Acta. 2016;1860:2750–2755. doi: 10.1016/j.bbagen.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676–690. doi: 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pongtheerat T, Saelee P. Role of GSTM1 Copy Number Variant in the Prognosis of Thai Colorectal Cancer Patients Treated with 5-FUbased Chemotherapy. Asian Pac J Cancer Prev. 2016;17:4719–4722. doi: 10.22034/APJCP.2016.17.10.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poniah P, Mohd Zain S, Abdul Razack AH, Kuppusamy S, Karuppayah S, Sian Eng H, Mohamed Z. Genome-wide copy number analysis reveals candidate gene loci that confer susceptibility to high-grade prostate cancer. Urol Oncol. 2017;35:545.e1–545.e11. doi: 10.1016/j.urolonc.2017.04.017. [DOI] [PubMed] [Google Scholar]
- 13.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park RW, Kim TM, Kasif S, Park PJ. Identification of rare germline copy number variations over-represented in five human cancer types. Mol Cancer. 2015;14:25. doi: 10.1186/s12943-015-0292-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirano T, Yoshikawa R, Harada H, Harada Y, Ishida A, Yamazaki T. Long noncoding RNA, CCDC26, controls myeloid leukemia cell growth through regulation of KIT expression. Mol Cancer. 2015;14:90. doi: 10.1186/s12943-015-0364-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC, Wang SB, Wang YZ, Yang Y, Yang N, Zhou WP, Yang GS, Sun SH. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666–681. doi: 10.1016/j.ccr.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Liu X, Xiao ZD, Han L, Zhang J, Lee SW, Wang W, Lee H, Zhuang L, Chen J, Lin HK, Wang J, Liang H, Gan B. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat Cell Biol. 2016;18:431–442. doi: 10.1038/ncb3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartonicek N, Maag JL, Dinger ME. Long noncoding RNAs in cancer: mechanisms of action and technological advancements. Mol Cancer. 2016;15:43. doi: 10.1186/s12943-016-0530-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin A, Li C, Xing Z, Hu Q, Liang K, Han L, Wang C, Hawke DH, Wang S, Zhang Y, Wei Y, Ma G, Park PK, Zhou J, Zhou Y, Hu Z, Zhou Y, Marks JR, Liang H, Hung MC, Lin C, Yang L. The LINK-A lncRNA activates normoxic HIF1alpha signalling in triple-negative breast cancer. Nat Cell Biol. 2016;18:213–224. doi: 10.1038/ncb3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu X, Tian X, Yu C, Shen C, Yan T, Hong J, Wang Z, Fang JY, Chen H. A long non-coding RNA signature to improve prognosis prediction of gastric cancer. Mol Cancer. 2016;15:60. doi: 10.1186/s12943-016-0544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Y, Li J, Liu Y, Ding J, Fan Y, Tian Y, Wang L, Lian Y, Wang K, Shu Y. The long noncoding RNA SPRY4-IT1 increases the proliferation of human breast cancer cells by upregulating ZNF703 expression. Mol Cancer. 2015;14:51. doi: 10.1186/s12943-015-0318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao T, Qu N, Shi RL, Guo K, Ma B, Cao YM, Xiang J, Lu ZW, Zhu YX, Li DS, Ji QH. BRAFactivated LncRNA functions as a tumor suppressor in papillary thyroid cancer. Oncotarget. 2017;8:238–247. doi: 10.18632/oncotarget.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Lu X, Geng Z, Yang G, Shi Y. LncRNA PTCSC3/miR-574-5p governs cell proliferation and migration of papillary thyroid carcinoma via Wnt/beta-catenin signaling. J Cell Biochem. 2017;118:4745–4752. doi: 10.1002/jcb.26142. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Yan G, Zhang Y, Jia X, Bu P. Long non-coding RNA MEG3 suppresses migration and invasion of thyroid carcinoma by targeting of Rac1. Neoplasma. 2015;62:541–549. doi: 10.4149/neo_2015_065. [DOI] [PubMed] [Google Scholar]
- 25.Zhao JJ, Hao S, Wang LL, Hu CY, Zhang S, Guo LJ, Zhang G, Gao B, Jiang Y, Tian WG, Luo DL. Long non-coding RNA ANRIL promotes the invasion and metastasis of thyroid cancer cells through TGF-beta/Smad signaling pathway. Oncotarget. 2016;7:57903–57918. doi: 10.18632/oncotarget.11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang JK, Ma L, Song WH, Lu BY, Huang YB, Dong HM, Ma XK, Zhu ZZ, Zhou R. LncRNAMALAT1 promotes angiogenesis of thyroid cancer by modulating tumor-associated macrophage FGF2 protein secretion. J Cell Biochem. 2017;118:4821–4830. doi: 10.1002/jcb.26153. [DOI] [PubMed] [Google Scholar]
- 27.Zhu H, Lv Z, An C, Shi M, Pan W, Zhou L, Yang W, Yang M. Onco-lncRNA HOTAIR and its functional genetic variants in papillary thyroid carcinoma. Sci Rep. 2016;6:31969. doi: 10.1038/srep31969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li T, Yang XD, Ye CX, Shen ZL, Yang Y, Wang B, Guo P, Gao ZD, Ye YJ, Jiang KW, Wang S. Long noncoding RNA HIT000218960 promotes papillary thyroid cancer oncogenesis and tumor progression by upregulating the expression of high mobility group AT-hook 2 (HMGA2) gene. Cell Cycle. 2017;16:224–231. doi: 10.1080/15384101.2016.1261768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Passon N, Bregant E, Sponziello M, Dima M, Rosignolo F, Durante C, Celano M, Russo D, Filetti S, Damante G. Somatic amplifications and deletions in genome of papillary thyroid carcinomas. Endocrine. 2015;50:453–464. doi: 10.1007/s12020-015-0592-z. [DOI] [PubMed] [Google Scholar]
- 30.Deng L, Yang SB, Xu FF, Zhang JH. Long noncoding RNA CCAT1 promotes hepatocellular carcinoma progression by functioning as let-7 sponge. J Exp Clin Cancer Res. 2015;34:18. doi: 10.1186/s13046-015-0136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li RK, Gao J, Guo LH, Huang GQ, Luo WH. PTENP1 acts as a ceRNA to regulate PTEN by sponging miR-19b and explores the biological role of PTENP1 in breast cancer. Cancer Gene Ther. 2017;24:309–315. doi: 10.1038/cgt.2017.29. [DOI] [PubMed] [Google Scholar]
- 33.Zhao JJ, Lin J, Zhu D, Wang X, Brooks D, Chen M, Chu ZB, Takada K, Ciccarelli B, Admin S, Tao J, Tai YT, Treon S, Pinkus G, Kuo WP, Hideshima T, Bouxsein M, Munshi N, Anderson K, Carrasco R. miR-30-5p functions as a tumor suppressor and novel therapeutic tool by targeting the oncogenic Wnt/beta-catenin/BCL9 pathway. Cancer Res. 2014;74:1801–1813. doi: 10.1158/0008-5472.CAN-13-3311-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng CW, Wang HW, Chang CW, Chu HW, Chen CY, Yu JC, Chao JI, Liu HF, Ding SL, Shen CY. MicroRNA-30a inhibits cell migration and invasion by downregulating vimentin expression and is a potential prognostic marker in breast cancer. Breast Cancer Res Treat. 2012;134:1081–1093. doi: 10.1007/s10549-012-2034-4. [DOI] [PubMed] [Google Scholar]
- 35.Kumarswamy R, Mudduluru G, Ceppi P, Muppala S, Kozlowski M, Niklinski J, Papotti M, Allgayer H. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer. 2012;130:2044–2053. doi: 10.1002/ijc.26218. [DOI] [PubMed] [Google Scholar]
- 36.Liu Z, Tu K, Liu Q. Effects of microRNA-30a on migration, invasion and prognosis of hepatocellular carcinoma. FEBS Lett. 2014;588:3089–3097. doi: 10.1016/j.febslet.2014.06.037. [DOI] [PubMed] [Google Scholar]
- 37.Liu Z, Chen L, Zhang X, Xu X, Xing H, Zhang Y, Li W, Yu H, Zeng J, Jia J. RUNX3 regulates vimentin expression via miR-30a during epithelial-mesenchymal transition in gastric cancer cells. J Cell Mol Med. 2014;18:610–623. doi: 10.1111/jcmm.12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, Ronning P, Reuland B, Guenther K, Beadnell TC, Essig J, Otto GM, O’Sullivan MG, Largaespada DA, Schwertfeger KL, Marahrens Y, Kawakami Y, Bagchi A. PVT1 dependence in cancer with MYC copynumber increase. Nature. 2014;512:82–86. doi: 10.1038/nature13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bailey SD, Desai K, Kron KJ, Mazrooei P, Sinnott-Armstrong NA, Treloar AE, Dowar M, Thu KL, Cescon DW, Silvester J, Yang SY, Wu X, Pezo RC, Haibe-Kains B, Mak TW, Bedard PL, Pugh TJ, Sallari RC, Lupien M. Noncoding somatic and inherited single-nucleotide variants converge to promote ESR1 expression in breast cancer. Nat Genet. 2016;48:1260–1266. doi: 10.1038/ng.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou CC, Yang F, Yuan SX, Ma JZ, Liu F, Yuan JH, Bi FR, Lin KY, Yin JH, Cao GW, Zhou WP, Wang F, Sun SH. Systemic genome screening identifies the outcome associated focal loss of long noncoding RNA PRAL in hepatocellular carcinoma. Hepatology. 2016;63:850–863. doi: 10.1002/hep.28393. [DOI] [PubMed] [Google Scholar]
- 41.Stein L, Rothschild J, Luce J, Cowell JK, Thomas G, Bogdanova TI, Tronko MD, Hawthorn L. Copy number and gene expression alterations in radiation-induced papillary thyroid carcinoma from chernobyl pediatric patients. Thyroid. 2010;20:475–487. doi: 10.1089/thy.2009.0008. [DOI] [PubMed] [Google Scholar]
- 42.Yuan JH, Liu XN, Wang TT, Pan W, Tao QF, Zhou WP, Wang F, Sun SH. The MBNL3 splicing factor promotes hepatocellular carcinoma by increasing PXN expression through the alternative splicing of lncRNA-PXN-AS1. Nat Cell Biol. 2017;19:820–832. doi: 10.1038/ncb3538. [DOI] [PubMed] [Google Scholar]
- 43.Klimenko OV, Shtilman MI. Transfection of Kasumi-1 cells with a new type of polymer carriers loaded with miR-155 and antagomiR-155. Cancer Gene Ther. 2013;20:237–241. doi: 10.1038/cgt.2013.11. [DOI] [PubMed] [Google Scholar]
- 44.Najafi Z, Sharifi M, Javadi G. Degradation of miR-21 induces apoptosis and inhibits cell proliferation in human hepatocellular carcinoma. Cancer Gene Ther. 2015;22:530–535. doi: 10.1038/cgt.2015.51. [DOI] [PubMed] [Google Scholar]
- 45.Fong MY, Zhou W, Liu L, Alontaga AY, Chandra M, Ashby J, Chow A, O’Connor ST, Li S, Chin AR, Somlo G, Palomares M, Li Z, Tremblay JR, Tsuyada A, Sun G, Reid MA, Wu X, Swiderski P, Ren X, Shi Y, Kong M, Zhong W, Chen Y, Wang SE. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. 2015;17:183–194. doi: 10.1038/ncb3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliverio M, Schmidt E, Mauer J, Baitzel C, Hansmeier N, Khani S, Konieczka S, Pradas-Juni M, Brodesser S, Van TM, Bartsch D, Bronneke HS, Heine M, Hilpert H, Tarcitano E, Garinis GA, Frommolt P, Heeren J, Mori MA, Bruning JC, Kornfeld JW. Dicer1-miR-328-Bace1 signalling controls brown adipose tissue differentiation and function. Nat Cell Biol. 2016;18:328–336. doi: 10.1038/ncb3316. [DOI] [PubMed] [Google Scholar]
- 47.Reddi HV, Driscoll CB, Madde P, Milosevic D, Hurley RM, McDonough SJ, Hallanger-Johnson J, McIver B, Eberhardt NL. Redifferentiation and induction of tumor suppressors miR-122 and miR-375 by the PAX8/PPARgamma fusion protein inhibits anaplastic thyroid cancer: a novel therapeutic strategy. Cancer Gene Ther. 2013;20:267–275. doi: 10.1038/cgt.2013.16. [DOI] [PubMed] [Google Scholar]
- 48.Ahmadi S, Sharifi M, Salehi R. Locked nucleic acid inhibits miR-92a-3p in human colorectal cancer, induces apoptosis and inhibits cell proliferation. Cancer Gene Ther. 2016;23:199–205. doi: 10.1038/cgt.2016.10. [DOI] [PubMed] [Google Scholar]
- 49.Shoshan E, Mobley AK, Braeuer RR, Kamiya T, Huang L, Vasquez ME, Salameh A, Lee HJ, Kim SJ, Ivan C, Velazquez-Torres G, Nip KM, Zhu K, Brooks D, Jones SJ, Birol I, Mosqueda M, Wen YY, Eterovic AK, Sood AK, Hwu P, Gershenwald JE, Robertson AG, Calin GA, Markel G, Fidler IJ, Bar-Eli M. Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat Cell Biol. 2015;17:311–321. doi: 10.1038/ncb3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ge Y, Zhang L, Nikolova M, Reva B, Fuchs E. Strand-specific in vivo screen of cancer-associated miRNAs unveils a role for miR-21(*) in SCC progression. Nat Cell Biol. 2016;18:111–121. doi: 10.1038/ncb3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Usmani A, Shoro AA, Memon Z, Hussain M, Rehman R. Diagnostic, prognostic and predictive value of MicroRNA-21 in breast cancer patients, their daughters and healthy individuals. Am J Cancer Res. 2015;5:2484–2490. [PMC free article] [PubMed] [Google Scholar]
- 52.Giunti L, da Ros M, Vinci S, Gelmini S, Iorio AL, Buccoliero AM, Cardellicchio S, Castiglione F, Genitori L, de Martino M, Giglio S, Genuardi M, Sardi I. Anti-miR21 oligonucleotide enhances chemosensitivity of T98G cell line to doxorubicin by inducing apoptosis. Am J Cancer Res. 2015;5:231–242. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.