

Abstract
In light of the declining global malaria burden attained largely due to insecticides, a deeper understanding of the factors driving insecticide resistance is needed to mitigate its growing threat to malaria vector control programs. Following evidence of microbiota-mediated insecticide resistance in agricultural pests, we undertook a comparative study of the microbiota in mosquitoes of differing insecticide resistance status. The microbiota of wild-caught Anopheles albimanus, an important Latin American malaria vector, that were resistant (FEN_Res) or susceptible (FEN_Sus) to the organophosphate (OP) insecticide fenitrothion were characterized and compared using whole metagenome sequencing. Results showed differing composition of the microbiota and its functions between FEN_Res and FEN_Sus, with significant enrichment of OP-degrading bacteria and enzymes in FEN_Res compared to FEN_Sus. Lower bacterial diversity was observed in FEN_Res compared to FEN_Sus, suggesting the enrichment of bacterial taxa with a competitive advantage in response to insecticide selection pressure. We report and characterize for the first time whole metagenomes of An. albimanus, revealing associations between the microbiota and phenotypic resistance to the insecticide fenitrothion. This study lays the groundwork for further investigation of the role of the mosquito microbiota in insecticide resistance.
Introduction
Insecticide resistance is a rapidly emerging threat to global malaria control efforts1,2, particularly in sub-Saharan Africa, where the greatest burden of disease lies3. In Latin America, regional successes in malaria control have led to a shift in focus from malaria control to malaria elimination. Vector control remains the cornerstone of malaria control and elimination programs, with a greater emphasis now being placed on understanding the role that insecticide resistance may play in compromising these elimination efforts4,5. Malaria vector control relies primarily on the use of indoor residual spraying (IRS) and long lasting insecticidal nets (LLINs) to reduce vector populations and protect people from potentially infectious mosquito bites1,6. These approaches both utilize chemical insecticides that can select for insecticide resistance in the vector populations they target7,8. Thus, the mechanisms leading to the evolution of insecticide resistance in malaria vectors are under extensive study, particularly given the limited number of insecticides approved for public health use9. To date, four key mechanisms of insecticide resistance have been described in malaria vectors: (i) target site insensitivity, where changes to the insecticide target molecules render them unsuitable for binding9; (ii) metabolic resistance, where heightened enzymatic activity leads to increased levels of insecticide detoxification9; (iii) behavioral changes resulting in the evasion of contact with insecticide treated surfaces10,11; and (iv) cuticle modification, which prevents or reduces the cuticular penetration of insecticides12.
Anopheles albimanus, the main coastal malaria vector in Latin America, has been reported to show resistance to multiple classes of insecticides, including organophosphates (OPs)7,13,14, one of the recommended classes of insecticides for IRS6. Two mechanisms have so far been documented for OP resistance in An. albimanus: a metabolic resistance mechanism involving elevated levels of nonspecific esterases, and a target site resistance mechanism involving acetylcholinesterase (AChE) insensitivity14–18. However, the mode of selection and underlying factors driving resistance to OPs and other classes of insecticides in An. albimanus remain largely unexplored.
Microbes colonize a wide variety of environments including the tissues of insects, where they undertake various metabolic functions, including the degradation of pesticides19,20. Microbes have been linked to insecticide resistance in agricultural pests21,22, but their role in conferring resistance in mosquitoes is unknown. The advent of new and affordable molecular tools has led to a plethora of microbiome studies, including studies of mosquito microbiota. Many of these studies have focused on characterizing the microbiota of medically important mosquito genera23–28, as well as identifying their role in mosquito behavior, biology, and pathogen transmission29,30, with a few studies suggesting their potential role in insecticide resistance31–34. As with most studies on insecticide resistance in malaria vectors9, studies of the microbiota of malaria vectors have mainly focused on sub- Saharan African and Southeast Asian mosquitoes35, with few such studies on Latin American vectors, particularly An. albimanus36,37. The studies of An. albimanus microbiota have largely focused on the effect of the microbiota on malaria parasite development, and have identified a limited number of bacterial species using culture dependent methods.
Based on the documented links between insecticide resistance and the presence of pesticide-degrading (particularly, OP-degrading) bacteria in agricultural pests21,22, as well as the identification of OP resistance in An. albimanus17, we hypothesized that bacteria could be contributing to insecticide resistance through increased degradation of insecticides in resistant mosquito populations. To test this, our objective was to characterize and compare the microbiota of An. albimanus with differing fenitrothion resistance phenotypes14. Specifically, bacterial compositions between fenitrothion resistant (FEN_Res) and susceptible (FEN_Sus) An. albimanus were compared using whole metagenome sequencing (WMS), a next generation sequencing (NGS) technique that captures both cultivable and non-cultivable bacteria as well as their putative functions. The findings presented here show differences between the microbiota of FEN_Res and FEN_Sus, as well as associations between the mosquito microbiota and xenobiotic degradation.
Results
Descriptive statistics
Female An. albimanus with differing fenitrothion resistance profiles were pooled (FEN_Res, 30/pool; FEN_Sus, 10/pool) and underwent WMS on the Illumina® HiSeq2500 platform. A total of 83,947,332 (FEN_Res) and 60,444,900 (FEN_Sus) raw sequencing reads were generated, with 91% and 84% of these reads passing quality control, respectively. In both samples, read lengths ranged from 60–232 bp after quality trimming, with 63% (FEN_Res) and 68% (FEN_Sus) of total reads aligning to bacterial proteins after host (An. albimanus) genome removal. Out of the reads that aligned to bacterial proteins, 0.9% and 0.8% aligned to bacterial xenobiotic degradation pathways in FEN_Res and FEN_Sus, respectively.
Differential bacterial composition between fenitrothion resistant and susceptible An. albimanus, with significant enrichment of Klebsiella pneumoniae, an OP-degrading bacterial species, in resistant An. albimanus
Based on the alignment of sequencing reads to the National Center for Biotechnology Information (NCBI) bacterial non-redundant (NR) protein database and subsequent annotation using the NCBI taxonomy tree, a total of 103 bacterial species and an uncultured β-proteobacterium (CBNPD1 BAC clone 578) were identified in FEN_Res and FEN_Sus. This comprised four phyla, six classes, 12 orders, 13 families, and 37 genera (Fig. 1 and Supplementary Table 1). Thirty-two out of these 37 genera were found in both FEN_Res and FEN_Sus, two (Bacillus and Halomonas) were unique to FEN_Res, and three (Stenotrophomonas, Microbacterium, and Lelliottia) were only identified in FEN_Sus (Table 1). The uncultured β-proteobacterium was found in both FEN_Res and FEN_Sus. Proteobacteria was the most abundant bacterial phylum, comprising 99% of reads in each sample. Likewise, Gammaproteobacteria, comprising 99% of reads, was the most predominant bacterial class. At the order level, Enterobacteriales and Pseudomonadales comprised over 98% of reads in each sample, and Enterobacteriaceae and Moraxellaceae comprised 99% of reads per sample at the family level. At the genus level, Klebsiella, Enterobacter, Acinetobacter, Escherichia, and Salmonella constituted over 93% of reads in each sample, and are considered as the predominant bacterial genera in this study (Fig. 1). The most predominant bacterial species was Klebsiella pneumoniae, comprising 74% and 49% of reads in FEN_Res and FEN_Sus, respectively (Supplementary Table 1).
Figure 1.

Differential bacterial composition between fenitrothion resistant and susceptible An. albimanus. Based on the alignment of sequencing reads (FEN_Res, n = 83,947,332; FEN_Sus, n = 60,444,900) to the NCBI-NR protein database, 37 bacterial genera and one uncultured β-proteobacterium* were identified in fenitrothion resistant and susceptible An. albimanus samples. Five genera; Klebsiella, Enterobacter, Acinetobacter, Escherichia and Salmonella comprised over 93% of identified genera in both samples and are considered as the predominant genera in this study. Plot (a) shows the proportions of each predominant genera as well as the proportions of unclassified reads in fenitrothion resistant and susceptible samples. Thirty two bacterial genera comprising the remaining proportion (1.8%) of the microbiota are grouped as ‘other genera’ and expanded in plot (b). Bacterial genera that are unique to either resistant or susceptible An. albimanus are presented in bold typeface. There were differential abundances of predominant (a) and other (b) genera between FEN_Res and FEN_Sus, with significant (p < 0.001) enrichment of Klebsiella in FEN_Res.
Table 1.
Differential abundance of identified bacterial genera in fenitrothion resistant and susceptible An. albimanus. The table shows the relative abundance (%) of bacterial genera identified in each sample, and the difference in relative abundance of each genera with 95% confidence intervals, calculated using two-sided Fisher’s exact test with Benjamini Hochberg’s false discovery rate p-value corrections. The level of significance was set to p < 0.05. Bacterial genera unique to either sample are presented in bold typeface. The relative abundance of 21 of these bacterial genera was significantly higher in FEN_Res compared to FEN_Sus, with the proportion of Klebsiella showing the greatest enrichment (Diff 25.1%, p < 0.0001). Conversely, the proportion of Enterobacter was the most reduced in FEN_Res compared to FEN_Sus (Diff −21.5% p < 0.0001).
| S/N | Bacterial genera | FEN_Res Rel. abundance (%) | FEN_Sus Rel. abundance (%) | Diff. Rel abundance (%) | 95% lower CI | 95% upper CI | p-values | Corrected p-values |
|---|---|---|---|---|---|---|---|---|
| 1 | Klebsiella †,‡ | 77.571 | 52.424 | 25.148 | 25.129 | 25.167 | <1E-301 | <1E-301 |
| 2 | Bacillus † | 0.456 | — | 0.456 | 0.454 | 0.458 | <1E-301 | <1E-301 |
| 3 | Escherichia †,‡ | 2.239 | 1.846 | 0.393 | 0.387 | 0.398 | <1E-301 | <1E-301 |
| 4 | Pantoea †,‡ | 0.196 | 0.100 | 0.096 | 0.094 | 0.097 | <1E-301 | <1E-301 |
| 5 | Raoultella † | 0.103 | 0.076 | 0.028 | 0.026 | 0.029 | <1E-301 | <1E-301 |
| 6 | Salmonella † | 0.738 | 0.718 | 0.020 | 0.016 | 0.023 | 1.70E-28 | 3.82E-28 |
| 7 | Rahnella † | 0.065 | 0.046 | 0.018 | 0.018 | 0.019 | 1.54E-301 | 6.04E-301 |
| 8 | Pluralibacter ♦ | 0.044 | 0.032 | 0.012 | 0.011 | 0.012 | 3.40E-173 | 1.23E-172 |
| 9 | Halomonas ±,c | 0.011 | — | 0.011 | 0.011 | 0.012 | <1E-301 | <1E-301 |
| 10 | Edwardsiella † | 0.035 | 0.024 | 0.011 | 0.010 | 0.012 | 7.82E-208 | 2.94E-207 |
| 11 | Pseudomonas †,‡ | 0.037 | 0.028 | 0.009 | 0.008 | 0.010 | 1.37E-124 | 4.44E-124 |
| 12 | Providencia † | 0.027 | 0.022 | 0.006 | 0.005 | 0.006 | 3.26E-69 | 9.28E-69 |
| 13 | Erwinia †,‡†,‡ | 0.031 | 0.026 | 0.005 | 0.005 | 0.006 | 3.80E-51 | 9.65E-51 |
| 14 | Haemophilus † | 0.017 | 0.012 | 0.005 | 0.005 | 0.006 | 2.40E-94 | 7.53E-94 |
| 15 | Photorhabdus † | 0.023 | 0.018 | 0.005 | 0.004 | 0.006 | 3.75E-60 | 9.78E-60 |
| 16 | Photobacterium ♦ | 0.016 | 0.012 | 0.003 | 0.003 | 0.004 | 6.02E-43 | 1.49E-42 |
| 17 | Shigella †,‡ | 0.097 | 0.094 | 0.003 | 0.002 | 0.004 | 1.10E-06 | 1.85E-06 |
| 18 | Staphylococcus † | 0.015 | 0.012 | 0.003 | 0.003 | 0.004 | 2.53E-36 | 5.94E-36 |
| 19 | Kosakonia ♦ | 0.049 | 0.046 | 0.003 | 0.002 | 0.004 | 5.99E-10 | 1.10E-09 |
| 20 | *CBNPD1 BAC clone 578♦ | 0.016 | 0.013 | 0.003 | 0.002 | 0.003 | 3.61E-24 | 7.72E-24 |
| 21 | Vibrio † | 0.017 | 0.016 | 0.001 | 0.000 | 0.001 | 0.02556166 | 0.039390101 |
| 22 | Morganella † | 0.015 | 0.015 | −0.001 | −0.001 | 0.000 | 0.02829376 | 0.042896987 |
| 23 | Pectobacterium ♦ | 0.021 | 0.022 | −0.001 | −0.002 | −0.001 | 1.01E-05 | 1.66E-05 |
| 24 | Xenorhabdus † | 0.017 | 0.019 | −0.002 | −0.002 | −0.001 | 2.97E-10 | 5.58E-10 |
| 25 | Yokenella ±a | 0.026 | 0.028 | −0.002 | −0.002 | −0.001 | 2.62E-07 | 4.56E-07 |
| 26 | Yersinia †,‡ | 0.014 | 0.016 | −0.002 | −0.003 | −0.002 | 7.66E-18 | 1.56E-17 |
| 27 | Cedecea † | 0.012 | 0.014 | −0.002 | −0.003 | −0.002 | 3.05E-25 | 6.66E-25 |
| 28 | Cronobacter † | 0.052 | 0.055 | −0.003 | −0.004 | −0.002 | 1.03E-10 | 2.02E-10 |
| 29 | Kluyvera † | 0.036 | 0.039 | −0.003 | −0.004 | −0.002 | 1.47E-15 | 2.95E-15 |
| 30 | Stenotrophomonas † | — | 0.016 | −0.016 | −0.016 | −0.015 | <1E-301 | <1E-301 |
| 31 | Bradyrhizobium † | 0.027 | 0.044 | −0.017 | −0.017 | −0.016 | <1E-301 | <1E-301 |
| 32 | Microbacterium † | — | 0.017 | −0.017 | −0.017 | −0.016 | <1E-301 | <1E-301 |
| 33 | Leclercia ♦ | 0.013 | 0.042 | −0.029 | −0.030 | −0.028 | <1E-301 | <1E-301 |
| 34 | Lelliottia ♦ | — | 0.039 | −0.039 | −0.040 | −0.038 | <1E-301 | <1E-301 |
| 35 | Citrobacter †,‡ | 0.227 | 0.276 | −0.049 | −0.051 | −0.047 | <1E-301 | <1E-301 |
| 36 | Serratia †,‡ | 0.052 | 0.115 | −0.063 | −0.064 | −0.061 | <1E-301 | <1E-301 |
| 37 | Acinetobacter †,‡ | 7.742 | 11.913 | −4.172 | −4.184 | −4.159 | <1E-301 | <1E-301 |
| 38 | Enterobacter †,‡ | 5.563 | 27.091 | −21.528 | −21.543 | −21.513 | <1E-301 | <1E-301 |
| 39 | Unclassified Bacteria | 4.381509301 | 4.674589901 | −0.2930806 |
†Previously identified in Anopheles.
‡Previously identified in Latin American Anopheles.
*Uncultured β-proteobacteria.
±Reported in Anopheles for the first time in this study.
aOnly reported in Aedes previously.
cOnly reported in Culex previously.
♦Reported in mosquitoes for the first time in this study.
There were significant differences in the relative abundance of identified bacterial taxa between FEN_Res and FEN_Sus, with lower Simpson’s reciprocal index (a measure of taxonomic richness and evenness) in FEN_Res (1.4) compared to FEN_Sus (2.5). Out of the four bacterial phyla identified in both samples, the relative abundances of Firmicutes and Actinobacteria was significantly (p < 0.0001) higher in FEN_Res compared to FEN_Sus, with a corresponding lower (p < 0.0001) relative abundance of Proteobacteria in FEN_Res. There was no significant difference in the relative abundance (Diff) of Bacteroidetes between both samples (p = 0.43) (Supplementary Table 1). However, since Proteobacteria was the predominant bacterial phylum in both samples (Supplementary Table 1), bacteria belonging to this phylum showed differential compositions between FEN_Res and FEN_Sus at lower taxonomic levels (Supplementary Table 3). The relative abundances of eighteen of the 32 bacterial genera as well as the uncultured β-proteobacterium identified in both samples were significantly (p < 0.001) higher in FEN_Res compared to FEN_Sus (Table 1). These enriched bacterial genera comprised three of the predominant genera; Klebsiella, Escherichia and Salmonella, with Klebsiella being the most enriched (Diff 25.2%, p < 0.0001) in FEN_Res overall (Fig. 2). There was a significantly reduced relative abundance of the remaining 14 genera in FEN_Res (Table 1), with Enterobacter showing the greatest reduction (Diff −21.5%, p < 0.0001) (Fig. 2). At the species level, 29 bacterial species were significantly enriched (p < 0.01) in FEN_Res compared to FEN_Sus (Supplementary Table 3), with Klebsiella pneumoniae being the most enriched (Diff 24.3%, p < 0.0001). Conversely, the relative abundance of 34 bacterial species was significantly reduced in FEN_Res compared to FEN_Sus (p < 0.001), with Enterobacter cloacae showing the greatest reduction (Diff −13.3%, p < 0.0001) (Supplementary Table 3). Out of the remaining 40 bacterial species, seven were unique to FEN_Res, while 33 were only identified in FEN_Sus (Supplementary Table 1).
Figure 2.

Differential abundance of predominant bacterial genera in fenitrothion resistant and susceptible An. albimanus. Plots show the difference in relative abundance (%) of each bacterial genera between fenitrothion resistant and susceptible samples (p < 0.0001).
Differential abundance of bacterial xenobiotic-degrading enzymes between fenitrothion resistant and susceptible An. albimanus, with significant enrichment of OP-degrading carboxylesterases and phosphomonoesterases in resistant An. albimanus
A total of 88 bacterial xenobiotic-degrading enzymes were identified in FEN_Res and FEN_Sus based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) annotations of aligned sequencing reads. This comprised all six major classes of enzymes: oxidoreductases, transferases, hydrolases, isomerases, lyases, and ligases, with oxidoreductases, transferases and hydrolases constituting over 80% of identified enzymes in each sample. Within each enzyme class, the most abundant enzymes in both samples were consistent: fumarate reductase (oxidoreductase); glutathione S-transferase and acetyl-CoA C-acetyltransferase (transferase); carboxymethylenebutenolidase (hydrolase); muconate cycloisomerase (isomerase) 4-carboxymuconolactone decarboxylase (lyase); guanosine monophosphate synthase (ligase); and enoyl-CoA isomerase (isomerase) (Supplementary Table 2).
The relative abundance of hydrolases (Diff 2.55%), isomerases (Diff 1.15%), and lyases (Diff 0.39%) was higher in FEN_Res compared to FEN_Sus. This corresponded to a reduced relative abundance of oxidoreductases (Diff −3.12%), ligases (Diff −0.44%), transferases (Diff −0.34%) and isomerases (Diff −0.22%) in FEN_Res (Fig. 3). Hydrolases, the most enriched enzyme class, comprised of two significantly (p < 0.0001) enriched carboxylesterases; carboxymethylenebutenolidase (Diff 1.31%) and gluconolactonase (Diff 0.99%), and two significantly (p < 0.0001) enriched phosphomonoesterases; alkaline phosphatase (Diff 0.43%) and acid phosphatase (Diff 0.36%) in FEN_Res compared to FEN_Sus, with the carboxylesterases being the most enriched overall (Fig. 4 and Supplementary Table 2). With the exception of ligase, six other enzymes in the remaining classes were also significantly (p < 0.0001) enriched in FEN_Res compared to FEN_Sus: transferases (acetyl-CoA acyltransferase, Diff 0.48% and glutathione S-transferase, Diff 0.58%); lyase (4-carboxymuconolactone decarboxylase, Diff 0.47%); oxidoreductase (p-hydroxybenzoate 3-monooxygenase, Diff 0.48%); and isomerases (3-hydroxybutyryl-CoA epimerase, Diff 0.35%, and enoyl-CoA isomerase, Diff 0.80%) (Fig. 4 and Supplementary Table 2).
Figure 3.
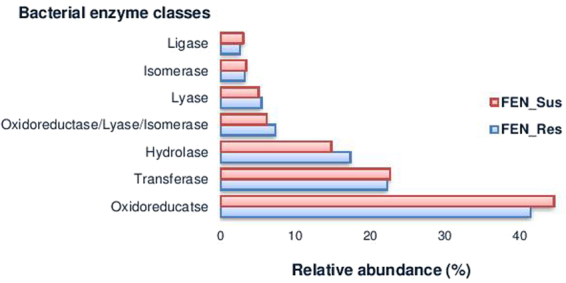
Relative abundance of bacterial enzyme classes associated with xenobiotic degradation pathways in fenitrothion resistant and susceptible An. albimanus. Bar plot shows the relative abundance (%) of reads assigned to each enzyme class in FEN_Res (n = 458,614) and FEN_Sus (n = 311,919).
Figure 4.

Putative bacterial enzymes involved in microbial xenobiotic degradation in fenitrothion resistant and susceptible An. albimanus. The plot shows the proportion (%) of sequencing reads (FEN_Res, n = 458,614; and FEN_Sus, n = 311,919) aligned to bacterial enzymes in the microbial xenobiotic degradation pathway, and the difference in relative abundance of identified enzymes between fenitrothion resistant and susceptible An. albimanus (Diff.). Only enzymes with Diff. ≥ 0.2% at p < 0.0001 are shown, and significantly enriched enzymes in FEN_Res are presented in bold typeface.
The microbial xenobiotic degradation pathways in fenitrothion resistant An. albimanus are comprised of fenitrothion-degrading and other OP-degrading bacterial species that were also confirmed via in vitro insecticide treatments
Sequencing reads associated with annotated microbial xenobiotic degradation pathways in FEN_Res mapped to 21 bacterial species belonging to 11 genera (Table 2). Of these, 13 bacterial species belonged to the predominant genera – Klebsiella, Enterobacter, Acinetobacter, Escherichia, and Salmonella. Bacteria belonging to the genus Bacillus were only identified in FEN_Res (Table 2 and Supplementary Table 1). With the exception of Pluribacter, each bacterial genus associated with xenobiotic degradation in FEN_Res contained bacterial species that have been documented to degrade OP compounds, including fenitrothion, as well as other classes of pesticides (Table 2). The presence of B. cereus and A. baumannii, two OP-degrading bacteria identified by WMS in this study (Table 2), was confirmed by PCR following in vitro insecticide treatments of the microbiota of FEN_Res.
Table 2.
Bacterial species associated with microbial xenobiotic degradation in fenitrothion resistant An. albimanus. The table lists identified bacterial species, documented OP compounds, and other pesticides metabolized by each associated bacteria.
| S/N | Bacterial species | OPs metabolized | Other pesticides metabolized | Source |
|---|---|---|---|---|
| 1 | Acinetobacter baumannii | Fenitrothion, Diazinon, Chlorpyrifos, Malathion, Methyl parathion | Diclofop-methyl | 80,91,92 |
| 2 | Acinetobacter calcoaceticus | Chlorpiryfos, Biological phosphorus in activated sludge | — | 93–97 |
| 3 | Acinetobacter pittii | — | — | — |
| 4 | Bacillus anthracis † | — | — | — |
| 5 | Bacillus cereus † | Dimethoate, Malathion, Chlorpiryfos, Malaoxon, Monocrotophos, Acephate, Phosphonates | DDT, Cypermethrin, Fenvalerate | 98–105 |
| 6 | Bacillus thuringiensis † | Malathion | Fipronil | 106–108 |
| 7 | Citrobacter koseri | Glyphosphate, Profenofos | — | 109,110 |
| 8 | Enterobacter aerogenes | Malathion | Bifenthrin, Fenpropathrin, Cypermethrin, DDT | 103,104,106,111 |
| 9 | Enterobacter asburiae | Acephate, Glyphosphate | Cypermethrin, Endosulfan, Quizalofop-p-ethyl, Clodinafop, Metribuzin | 109,118,113 |
| 10 | Enterobacter cloacae | Dimethoate, Glyphosphate | DDT, Endosulfan | 98,103,114–116 |
| 11 | Enterobacter hormaechei | — | — | — |
| 12 | Escherichia coli | Fenitrothion, Phosphonates | Cypermethrin, DDT, Aldicarb | 103,104,117–119 |
| 13 | Klebsiella oxytoca | Dimethoate, Chlorpiryfos, Phosphonates | Endosulfan | 120–123 |
| 14 | Klebsiella pneumoniae | Fensulfothion, Tributyl phosphate, Methyl-parathion, Phosphonates | DDT, Endosulfan, Triazines, Bromoxynil, Imidacloprid | 103,114,123–129 |
| 15 | Klebsiella quasipneumoniae | — | — | — |
| 17 | Klebsiella variicola | — | — | — |
| 18 | Kluyvera cryocrescens | Phosphonates | — | 123 |
| 19 | Pluralibacter gergoviae | — | — | — |
| 20 | Salmonella enterica | Phosphonates | — | 130 |
| 21 | Serratia marcescens | Fenitrothion, Chlorpiryfos, Diazinon, Coumaphos, Parathion, Isazofos, Monocrotophos | — | 100,114,131–133 |
†Only identified in fenitrothion resistant An. albimanus.
Discussion
Mosquito microbiota has been shown to impact several key host characteristics including growth, nutrition, reproduction, parasite interactions, and vector competence33, and the present study provides the first comprehensive characterization of mosquito microbiota in relation to insecticide resistance. The findings presented here show differential composition of the microbiota and its functions between fenitrothion resistant and susceptible An. albimanus, with significant enrichment of OP degrading bacteria and putative enzymes in FEN_Res compared to FEN_Sus. Bacteria belonging to the genera Serratia, Enterobacter, Flavobacterium, Pseudomonas and Acinetobacter36,37 have previously been identified in An. albimanus. In the present study, 103 bacterial species belonging to 37 bacterial genera (including four of the previously identified genera and unclassified Flavobacteriaceae) were identified, providing a comprehensive update to the composition of An. albimanus microbiota. Bacteria belonging to the phylum Proteobacteria were predominant in the present study, indicating that the composition at the phylum level is similar to that of Anopheles described previously from Asia38–42, Africa43–46 and the Americas36,37,47. The predominant bacterial genera identified in the present study were Klebsiella, Enterobacter, Acinetobacter, Escherichia, and Salmonella. These have all been previously identified in Anopheles, and all, except Salmonella, have previously been documented in Latin American Anopheles48. The majority (29 out of 37) of all bacterial genera identified in the current study have previously been identified in Anopheles29,35,48, with 11 of these documented in Latin American Anopheles48. Out of the eight remaining genera, Yokenella and Halomonas have so far been reported in Aedes49 and Culex26 respectively, while Kosakonia, Leclercia, Photobacterium, Pluralibacter, Pectobacterium and Lelliottia are documented herein for the first time in Anopheles. Two of these newly documented genera have been identified in other insect vectors of human disease, Pluralibacter in sandflies (Phlebotomus chinensis, a primary vector of leishmaniasis in China)50 and Pectobacterium in triatomine bugs (Rhodnius prolixus, an important vector of Chagas disease in Latin America)51. The remaining genera have been identified in insects of agricultural importance: Kosakonia in the coffee berry borer (Hypothenemus hampei), Photobacterium in the date palm borer (Oryctes Agamemnon)52, Lelliotia in the Asian honeybee (Apis dorsata)53, and Leclercia in several classes of agricultural pests54–57. Bacterial species within two of these genera have been shown to degrade xenobiotics, such as caffeine by K. cowanii within the coffee berry borer58, and clorpyrifos ethyl, an OP pesticide, by L. adecarboxylata isolated from the fall armyworm (Spodoptera frugiperda)55.
The lower bacterial diversity observed in FEN_Res compared to FEN_Sus could be a consequence of insecticide exposure, whereby bacteria with the ability to effectively utilize fenitrothion as a nutrient source dominate the microbiota, displacing other bacteria with a resulting decrease in bacterial diversity. Such shifts in microbial composition due to insecticide exposure have previously been documented59. For example, the application of fenitrothion to soil in an experimental setting resulted in a significant enrichment of fenitrothion degrading bacteria in the treated soil, from previously undetectable levels to >80% of identified bacteria21. Furthermore, the exposure of the diamondback moth, Plutella xylostella, to two different organophosphate insecticides resulted in significant enrichment of Lactobacillales in insecticide treated moths60. In the present study, Klebsiella was significantly enriched in FEN_Res compared to FEN_Sus, with a corresponding reduction in the relative abundance of Enterobacter. This was also evident in other genera, as well as at the species level. While this could have occurred as a consequence of insecticide exposure, this may not have been the principal factor due to the short time of insecticide exposure during the bioassays (30 minutes)14. This posits the presence of other factors such as a previous selection for OP-degrading bacteria that might have developed alongside resistance to fenitrothion in the mosquitoes. This is exemplified in the diamondback moth, where susceptible and resistant moths from a generation with no insecticide exposure showed differential bacterial compositions. The resistant moths from the unexposed generation showed significant enrichment of Lactobacillales, a bacterial taxa that was also enriched in resistant moths from a different generation that had been exposed to insecticides60. These together suggest the enrichment of bacterial taxa with a competitive advantage over other taxa in response to selection pressure. This could also explain finding certain bacterial taxa in one sample and not the other, for instance, the identification of Bacillus and Halomonas only in FEN_Res, and Stenotrophomonas, Microbacterium, and Lelliottia only in FEN_Sus. It is also possible that rather than arising from selective pressures, the mere presence of certain bacteria could be mediating insecticide resistance in mosquitoes, and both possibilities merit further exploration.
Other factors such as a mosquito’s physiological status61 and age24, have been shown to affect the composition of the mosquito microbiota. We recognize that such factors may also be at play in the present study, particularly because wild-caught An. albimanus with unknown age and physiological status were utilized14. Nonetheless, the findings in this study showed significant enrichment of bacterial carboxylesterases and phosphomonoesterases (Fig. 4) – critical enzyme families involved in bacterial OP-degradation62–64 – in FEN_Res compared to FEN_Sus, suggesting that the microbiota in FEN_Res may be involved in OP-degradation. The present study, which reports the first description of putative microbial enzymes associated with xenobiotic degradation in mosquitoes (Supplementary Table 2), detected all major enzyme classes in FEN_Res and FEN_Sus, with oxidoreductases, transferases and hydrolases constituting the majority of identified enzymes (Fig. 3). These three classes of enzymes catalyze the biodegradation of various xenobiotics65,66 including OPs, whose catabolism is initiated by hydrolysis – the first and most critical step in OP degradation, which is primarily catalyzed by hydrolases67. The most abundant enzyme families (fumarate reductase68,69, glutathione S-transferase70–72, acetyl-CoA C-acetyltransferase73,74, carboxymethylenebutenolidase75, muconate cycloisomerase76, 4-carboxymuconolactone decarboxylase77, guanosine monophosphate synthase78, and enoyl-CoA isomerase79) within these three classes have all been documented in bacterial xenobiotic degradation. This suggests the potential involvement of An. albimanus microbiota in xenobiotic degradation within the mosquito host. Interestingly, hydrolases, the most important class of enzymes involved in bacterial degradation of OPs, were the most significantly enriched in FEN_Res compared to FEN_Sus (Figs 3 and 4). These enriched hydrolases63,64,67, along with other enriched enzymes such as glutathione S-transferase80 have been documented in bacterial degradation of OPs, further suggesting the involvement of the microbiota in host xenobiotic degradation. The identification of OP-degrading bacterial species, including fenitrothion-degrading species, in association with FEN_Res microbial degradation pathways (Table 2), plus the validation via in vitro assays also suggests the involvement of the microbiota in host xenobiotic degradation. It should be noted that although microbial xenobiotic degradation has been studied extensively (particularly in light of increasing interests in bioremediation), the focus has been on cultivable bacteria, which constitute <1% of environmental bacteria59. Thus, the xenobiotic degradation profiles of the majority of known bacteria, as well as their corresponding genes and gene products are yet to be characterized. In the present study, all but six of the identified bacterial species associated with microbial xenobiotic degradation in FEN_Res (A. pittii, B. anthracis, E. hormaechei, K. quasipneumoniae, K. variicola, and P. gergoviae) have been documented in OP-metabolism, as well as in the metabolism of other pesticides (Table 2). For these six bacterial species, this may be the first documentation of their association with xenobiotic degradation.
The findings presented here demonstrate differences between the microbiota of FEN_Res and FEN_Sus, as well as associations between the mosquito microbiota and xenobiotic degradation. These initial findings lay the groundwork for future research that will characterize and compare the expression levels of specific microbial genes involved in insecticide degradation, and further elucidate the role of the mosquito microbiota in conferring resistance to insecticides.
Methods
Sample collection and determination of resistance profiles
Female An. albimanus samples collected from La Jota, Tumbes, Peru in 2014, were screened for fenitrothion resistance using the CDC bottle bioassay81. Briefly, samples were exposed to bottles coated with one to five times the diagnostic dose (50 µg/bottle) of fenitrothion for 30 mins. These were subsequently removed and classified as susceptible (FEN_Sus), if they were knocked down at the diagnostic dose, or resistant (FEN_Res), if they survived five times the diagnostic dose. The sample collection and bioassay procedures have previously been described in detail14. Immediately following the bioassays, samples were stored at −20 °C until processed.
Preparation of genomic DNA
Pools of 10 (FEN_Sus) and 30 (FEN_Res) whole mosquito samples were processed to digest mosquito host DNA using the MolYsis™ Complete 5 kit (Molzym, Bremen, Germany) following manufacturer’s instructions. We processed available samples as pools to reduce the effect of the variation found in the microbiota among individual mosquitoes of the same species44. Prior to host DNA removal, each pool of mosquitoes was surface sterilized by suspending in 70% ethanol and agitating with a vortex mixer for 15–20 seconds, then re-suspending in nuclease free water with agitation for another 15–20 seconds, followed by a final rinse with nuclease free water. Each pool was subsequently suspended in 50 µL of MolYsis chaotrophic buffer, CM, and homogenized to begin the host DNA removal process.
Following the removal of mosquito DNA, genomic DNA was extracted from FEN_Sus using DNeasy Blood and Tissue Kit (QIAGEN), and from FEN_Res using MolYsis™ Complete 5 kit, per manufacturers’ instructions. In processing FEN_Sus, which had less mosquitoes/pool, the DNeasy blood and Tissue Kit was substituted for MolYsis to optimize the yield of metagenomic DNA. Negative controls for the extractions (tubes without mosquitoes) were included with each extraction process. For all extractions, DNA was eluted to a final volume of 60 µL and stored in −20 °C until library preparation and sequencing. Prior to library preparation and sequencing, each extraction product was tested for the presence of the 16S rRNA gene using the HDA1 and HDA2 primers23. Both DNA samples from FEN_Res and FEN_Sus were positive, while both extraction controls were negative.
Library preparation and whole metagenome sequencing
Genomic DNA was sheared to a mean size of 600 bp using a Covaris LE220 focused ultrasonicator (Covaris Inc., Woburn, MA). DNA fragments were cleaned with Ampure (Beckman Coulter Inc., Indianapolis, IN) and used to prepare dual-indexed sequencing libraries using the NEBNext Ultra library prep reagents (New England Biolabs Inc., Ipswich, MA), and barcoding indices synthesized at the CDC Biotechnology Core Facility. Libraries were analyzed for size and concentration, pooled and denatured for loading onto flowcells for cluster generation. Sequencing was performed on an Illumina HiSeq2500 platform using HiSeq Rapid SBS 250 × 250 cycle paired-end sequencing kits. Each library pool was loaded onto both lanes of the HiSeq Rapid flowcell, and on completion, sequence reads were filtered for read quality, basecalled and demultiplexed using Casava (v1.8.2). There was no significant difference in resulting reads from either lane of the flowcell. Thus, only outcomes from one lane of the flowcell are reported.
Sequencing data quality control and removal of mosquito genome
Demultiplexed reads from each sample were examined for quality using FastQC v0.11.582. Adapters and low quality reads were removed using Trimmomatic v0.3583. A custom adapter file containing TruSeq universal and index primers, as well as each primers’ reverse complement, was used with the ILLUMINACLIP command in Trimmomatic to remove adapters. Next, the first 14 and last five bases were removed from each read, and the resulting reads were scanned using a sliding window of four nucleotides. Nucleotides within each sliding window were removed until the average Phred score across the window was >20. Finally, sequences along with their mate-pairs with length less than 60 bp were removed.
The quality trimmed sequences for each sample were aligned to the host reference genome (An. albimanus STECLA strain, AalbS2 assembly84) using the BWA-MEM algorithm of the Burrows-Wheeler aligner (BWA v0.77)85 with default settings for paired-end reads (BWA alignments have been deposited in the NCBI Sequence Read Archive, SRA; SRR5630719 and SRR5630720). Reads that aligned to An. albimanus genome were removed, and resulting non-host reads were checked for quality with FastQC and used in downstream analysis.
Taxonomic and functional annotations
To assess taxonomic and functional composition, non-host reads were aligned to the NCBI-NR database (downloaded June, 2016) using the BLASTx algorithm in DIAMOND v0.8.6v86, with the following adjusted parameters –e 0.00001,–top 3, –f 6, and –c 1. Each read pair was analyzed separately, then subsequent aligned reads were merged, imported into the MEtaGenome ANalyzer (MEGAN) v.6.5.787 using the paired-read mode, and parsed using default parameters of the lowest common ancestor (LCA) algorithm. NCBI taxonomy (prot-acc2taxid-August2016.bin) and the Kyoto Encyclopedia of Genes and Genomes, KEGG (gi2kegg-Feb2015X.bin) mapping files were used to map aligned reads to the NCBI taxonomy tree and KEGG pathways, respectively. One final check for host genome contamination was performed at this stage, and reads that mapped to the Eukaryota domain were removed from downstream analysis. To identify bacteria associated with bacterial xenobiotic degradation pathways, reads that mapped to the KEGG bacterial xenobiotic degradation pathways were extracted and processed for taxonomic annotation as described above.
Comparisons and statistical analysis
A single MEGAN comparison file was generated using both FEN_Res and FEN_Sus individual MEGAN files. Simpson’s reciprocal indices were calculated for each sample using MEGAN, to determine species diversity (the number of identified bacteria taxa) and evenness (the relative abundance of each identified taxa) within each sample. Taxonomic profiles at each level of classification and corresponding read abundances were exported from the comparison file into the Statistical Analysis of Metagenomic Profiles (STAMP) software v2.1.388. Likewise, functional (KEGG) profiles were exported into STAMP for statistical analysis. For each sample, the relative abundance of reads assigned to each identified taxon/function were calculated. The resulting relative abundances were compared between samples using two-sided Fisher’s exact test with Benjamini-Hochberg False Discovery Rate (FDR) correction. The level of significance was set to p < 0.05, and any taxa/function with < 100 assigned reads in both samples were grouped as unclassified.
Identification of OP-degrading bacteria in in vitro insecticide treatments of An. albimanus microbiota
The microbiota of mosquitoes that survived 10 times the diagnostic dose of fenitrothion81 from the same mosquito population were cultured on insecticide treated bacterial media. Three pools of three female mosquitoes each were surface sterilized as described above and homogenized in nuclease free ultra-purified water. One hundred microliters of each homogenate was spread on Luria-Bertani (LB) agar plates that were surface-treated with either 1 mL of the diagnostic dose (50 µg/mL) or five times the diagnostic dose of fenitrothion and incubated overnight at 37 ± 0.5 °C. Untreated LB agar plates and plates treated with 1 mL of absolute ethanol (solvent) were included as controls. Following incubation, bacterial colonies were collected for DNA extraction. Genomic DNA was extracted from bacterial colonies using the Extracta™ DNA Prep for PCR kit (Quanta BioSciences, USA) following manufacturer’s instructions. Using PCR, bacterial genomic DNA was screened for two bacterial species/groups known to degrade organophosphate insecticides including fenitrothion – A. baumannii, and B. cereus – that were also associated with putative microbial xenobiotic degradation pathways in FEN_Res. Following a previously described method89 with slight modifications, the PCR for A. baumannii identification was conducted using A. baumannii specific primers P-Ab-ITSF (CATTATCACGGTAATTAGTG) and P-Ab-ITSB (AGAGCACTGTGCACTTAAG), along with internal control primers P-rA1 (CCTGAATCTTCTGGTAAAAC) and P-rA2 (GTTTCTGGGCTGCCAAACATTAC). The specific primer pair (P-Ab-ITSF and P-Ab-ITSB) amplifies a 208 bp fragment of the ITS region in A. baumannii, while the internal control primers (P-rA1 and P-rA2) target a 425 bp region of the recA gene in all Acinetobacter species. To screen for B. cereus, the BCFomp1 (ATCGCCTCGTTGGATGACGA) and BCRomp1 (CTGCATATCCTACCGCAGCTA) primer set89 which targets a 575 bp region of the motB gene in B. cereus was used. PCR was performed in a total reaction volume of 20 µL (A. baumannii) and 25 µL (B. cereus), each containing 60–160ng/µL DNA template, 1 µM of each respective primer, 12.5 µL of 2 × KAPA HiFi HotStart PCR mix (Roche, Switzerland), and PCR grade water to final volume. Reactions were conducted using a T100™ Thermal Cycler (Bio-Rad, USA) following the previously described conditions89,90.
Data Accessibility
The WMS reads obtained from this study (SRP108310) have been deposited in NCBI under the BioProject PRJNA388280.
Electronic supplementary material
Acknowledgements
This work was supported by the US Centers for Disease Control and Prevention (CDC) through the American Society for Microbiology’s (ASM) Infectious disease and Public Health Microbiology Postdoctoral Fellowship program, and the CDC’s Advanced Molecular Detection (AMD) program. We thank Yvonne Qvarnstrom for providing the host DNA removal kit, Gregory Dasch and Mike Frace for inputs on the design, the CDC’s AMD scientific computing team for bioinformatics support, Lucrecia Vizcaino for assisting with in vitro assays, and William Brogdon for useful discussions throughout the development of the project and preparation of the manuscript. The findings and conclusions in this paper are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Author Contributions
Conceptualization and design: N.D. & A.L.; Sample collection and bioassays: K.L. & J.P.; Molecular analysis and sequencing: N.D. & M.S.; Bioinformatics: N.D.; Manuscript preparation: N.D. & A.L.; All authors read and approved the final version of the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-20367-4.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hemingway, J. The role of vector control in stopping the transmission of malaria: threats and opportunities. Philosophical Transactions of the Royal Society B: Biological Sciences369 (2014). [DOI] [PMC free article] [PubMed]
- 2.Barnes KG, et al. Genomic Footprints of Selective Sweeps from Metabolic Resistance to Pyrethroids in African Malaria Vectors Are Driven by Scale up of Insecticide-Based Vector Control. PLOS Genetics. 2017;13:e1006539. doi: 10.1371/journal.pgen.1006539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO. Fact Sheet: World Malaria Day 2016 (2016).
- 4.Herrera S, et al. Prospects for malaria elimination in non-Amazonian regions of Latin America. Acta Tropica. 2012;121:315–323. doi: 10.1016/j.actatropica.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alimi TO, et al. Prospects and recommendations for risk mapping to improve strategies for effective malaria vector control interventions in Latin America. Malaria Journal. 2015;14:519. doi: 10.1186/s12936-015-1052-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organisation. Global Plan for insecticide resistance management in malaria vectors (GPIRM). Geneva, Switzerland: WHO (2012).
- 7.Quiñones ML, et al. Insecticide Resistance in Areas under Investigation by the International Centers of Excellence for Malaria Research: A Challenge for Malaria Control and Elimination. The American Journal of Tropical Medicine and Hygiene. 2015;93:69–78. doi: 10.4269/ajtmh.14-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chanda E. Optimizing Strategic Insecticide Resistance Management Planning in Malaria Vectors. In: Insecticides Resistance (ed Trdan S.). InTech (2016).
- 9.Corbel V., N’Guessan R. D, Mechanisms, Impact and Management of Insecticide Resistance in Malaria Vectors: A Pragmatic Review. In: Anopheles mosquitoes - New insights into malaria vectors (ed Manguin S.). InTech (2013).
- 10.Sougoufara S, Doucoure S, Backe Sembene PM, Harry M, Sokhna C. Challenges for malaria vector control in sub-SaharanAfrica: Resistance and behavioral adaptations in Anopheles populations. Journal of vector borne diseases. 2017;54:4–15. [PubMed] [Google Scholar]
- 11.Stone C, Chitnis N, Gross K. Environmental influences on mosquito foraging and integrated vector management can delay the evolution of behavioral resistance. Evolutionary Applications. 2016;9:502–517. doi: 10.1111/eva.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balabanidou V, et al. Cytochrome P450 associated with insecticide resistance catalyzes cuticular hydrocarbon production in Anopheles gambiae. Proceedings of the National Academy of Sciences. 2016;113:9268–9273. doi: 10.1073/pnas.1608295113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Penilla RP, et al. Resistance management strategies in malaria vector mosquito control. Baseline data for a large-scale field trial against Anopheles albimanus in Mexico. Med Vet Entomol. 1998;12:217–233. doi: 10.1046/j.1365-2915.1998.00123.x. [DOI] [PubMed] [Google Scholar]
- 14.Liebman KA, et al. Novel mutations on the ace-1 gene of the malaria vector Anopheles albimanus provide evidence for balancing selection in an area of high insecticide resistance in Peru. Malaria Journal. 2015;14:74. doi: 10.1186/s12936-015-0599-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hemingway J, Georghiou GP. Studies on the acetylcholinesterase of Anopheles albimanus resistant and susceptible to organophosphate and carbamate insecticides. Pesticide Biochemistry and Physiology. 1983;19:167–171. doi: 10.1016/0048-3575(83)90136-0. [DOI] [Google Scholar]
- 16.Ayad H, Georghiou GP. Resistance to Organophosphates and Carbamates in Anopheles albimanus Based on Reduced Sensitivity of Acetylcholinesterase. Journal of Economic Entomology. 1975;68:295–297. doi: 10.1093/jee/68.3.295. [DOI] [PubMed] [Google Scholar]
- 17.Brogdon WG, Barber AM. Fenitrothion-deltamethrin cross-resistance conferred by esterases in Guatemalan Anopheles albimanus. Pesticide Biochemistry and Physiology. 1990;37:130–139. doi: 10.1016/0048-3575(90)90118-L. [DOI] [Google Scholar]
- 18.Brogdon WG, Beach RF, Stewart JM, Castanaza L. Microplate assay analysis of the distribution of organophosphate and carbamate resistance in Guatemalan Anopheles albimanus. Bulletin of the World Health Organization. 1988;66:339–346. [PMC free article] [PubMed] [Google Scholar]
- 19.van den Bosch TJM, Welte CU. Detoxifying symbionts in agriculturally important pest insects. Microbial Biotechnology. 2017;10:531–540. doi: 10.1111/1751-7915.12483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Werren JH. Symbionts provide pesticide detoxification. Proceedings of the National Academy of Sciences. 2012;109:8364–8365. doi: 10.1073/pnas.1206194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kikuchi Y, et al. Symbiont-mediated insecticide resistance. Proceedings of the National Academy of Sciences. 2012;109:8618–8622. doi: 10.1073/pnas.1200231109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia X, et al. DNA Sequencing Reveals the Midgut Microbiota of Diamondback Moth, Plutella xylostella (L.) and a Possible Relationship with Insecticide Resistance. PLOS ONE. 2013;8:e68852. doi: 10.1371/journal.pone.0068852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dada N, et al. Comparative assessment of the bacterial communities associated with Aedes aegypti larvae and water from domestic water storage containers. Parasites & Vectors. 2014;7:391. doi: 10.1186/1756-3305-7-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.David MR, Santos LM, Vicente ACP, Maciel-de-Freitas R. Effects of environment, dietary regime and ageing on the dengue vector microbiota: evidence of a core microbiota throughout Aedes aegypti lifespan. Memórias do Instituto Oswaldo Cruz. 2016;111:577–587. doi: 10.1590/0074-02760160238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yadav KK, et al. Molecular characterization of midgut microbiota of Aedes albopictus and Aedes aegypti from Arunachal Pradesh, India. Parasites & Vectors. 2015;8:641. doi: 10.1186/s13071-015-1252-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duguma D, et al. Developmental succession of the microbiome of Culex mosquitoes. BMC Microbiology. 2015;15:140. doi: 10.1186/s12866-015-0475-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muturi EJ, Kim C-H, Bara J, Bach EM, Siddappaji MH. Culex pipiens and Culex restuans mosquitoes harbor distinct microbiota dominated by few bacterial taxa. Parasites & Vectors. 2016;9:18. doi: 10.1186/s13071-016-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gimonneau G, et al. Composition of Anopheles coluzzii and Anopheles gambiae microbiota from larval to adult stages. Infection, Genetics and Evolution. 2014;28:715–724. doi: 10.1016/j.meegid.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 29.Minard G, Mavingui P, Moro CV. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasites & Vectors. 2013;6:146. doi: 10.1186/1756-3305-6-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Tol, S. & Dimopoulos, G. Chapter Nine - Influences of the Mosquito Microbiota on Vector Competence. In: Advances in Insect Physiology (ed Alexander S. R.). Academic Press (2016).
- 31.Nkya TE, Akhouayri I, Kisinza W, David J-P. Impact of environment on mosquito response to pyrethroid insecticides: Facts, evidences and prospects. Insect Biochemistry and Molecular Biology. 2013;43:407–416. doi: 10.1016/j.ibmb.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Berticat C, Rousset F, Raymond M, Berthomieu A, Weill M. High Wolbachia density in insecticide-resistant mosquitoes. Proceedings of the Royal Society B: Biological Sciences. 2002;269:1413–1416. doi: 10.1098/rspb.2002.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minard G, Mavingui P, Moro CV. Diversity and function of bacterial microbiota in the mosquito holobiont. Parasites & Vectors. 2013;6:1–12. doi: 10.1186/1756-3305-6-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soltani A, Vatandoost H, Oshaghi M, Enayati A, Chavshin AR. The role of midgut symbiotic bacteria in resistance of Anopheles stephensi (Diptera: Culicidae) to organophosphate insecticides. Pathogens and Global Health. 2017;111:289–296. doi: 10.1080/20477724.2017.1356052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villegas LM, Pimenta PFP. Metagenomics, paratransgenesis and the Anopheles microbiome: a portrait of the geographical distribution of the anopheline microbiota based on a meta-analysis of reported taxa. Memórias do Instituto Oswaldo Cruz. 2014;109:672–684. doi: 10.1590/0074-0276140194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez-Ceron L, Santillan F, Rodriguez MH, Mendez D, Hernandez-Avila JE. Bacteria in Midguts of Field-Collected Anopheles albimanus Block Plasmodium vivax Sporogonic Development. Journal of Medical Entomology. 2003;40:371–374. doi: 10.1603/0022-2585-40.3.371. [DOI] [PubMed] [Google Scholar]
- 37.Pumpuni CB, Demaio J, Kent M, Davis JR, Beier JC. Bacterial Population Dynamics in Three Anopheline Species: The Impact on Plasmodium Sporogonic Development. The American Journal of Tropical Medicine and Hygiene. 1996;54:214–218. doi: 10.4269/ajtmh.1996.54.214. [DOI] [PubMed] [Google Scholar]
- 38.Ngo CT, Aujoulat F, Veas F, Jumas-Bilak E, Manguin S. Bacterial Diversity Associated with Wild Caught Anopheles Mosquitoes from Dak Nong Province, Vietnam Using Culture and DNA Fingerprint. PLoS ONE. 2015;10:e0118634. doi: 10.1371/journal.pone.0118634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ngo, C. T., Romano-Bertrand, S., Manguin, S., Jumas-Bilak, E. Diversity of the Bacterial Microbiota of Anopheles Mosquitoes from Binh Phuoc Province, Vietnam. Frontiers in microbiology7 (2016). [DOI] [PMC free article] [PubMed]
- 40.Favia G, et al. Bacteria of the genus Asaia stably associate with Anopheles stephensi, an Asian malarial mosquito vector. Proceedings of the National Academy of Sciences. 2007;104:9047–9051. doi: 10.1073/pnas.0610451104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rani A, Sharma A, Rajagopal R, Adak T, Bhatnagar RK. Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi-an Asian malarial vector. BMC Microbiology. 2009;9:96. doi: 10.1186/1471-2180-9-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manguin S. et al. Bacterial Biodiversity in Midguts of Anopheles Mosquitoes, Malaria Vectors in Southeast Asia. In: Anopheles mosquitoes -New insights into malaria vectors (ed Manguin S.). InTech (2013).
- 43.Boissière A, et al. Midgut Microbiota of the Malaria Mosquito Vector Anopheles gambiae and Interactions with Plasmodium falciparum Infection. PLoS Pathog. 2012;8:e1002742. doi: 10.1371/journal.ppat.1002742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osei-Poku J, Mbogo CM, Palmer WJ, Jiggins FM. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol Ecol. 2012;21:5138–5150. doi: 10.1111/j.1365-294X.2012.05759.x. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Gilbreath TM, III, Kukutla P, Yan G, Xu J. Dynamic Gut Microbiome across Life History of the Malaria Mosquito Anopheles gambiae in Kenya. PLOS ONE. 2011;6:e24767. doi: 10.1371/journal.pone.0024767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akorli J, et al. Seasonality and Locality Affect the Diversity of Anopheles gambiae and Anopheles coluzzii Midgut Microbiota from Ghana. PLOS ONE. 2016;11:e0157529. doi: 10.1371/journal.pone.0157529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terenius O, et al. 16S rRNA Gene Sequences from Bacteria Associated with Adult Anopheles darlingi (Diptera: Culicidae) Mosquitoes. Journal of Medical Entomology. 2008;45:172–175. doi: 10.1093/jmedent/45.1.172. [DOI] [PubMed] [Google Scholar]
- 48.Gendrin M., Christophides G.K. The Anopheles Mosquito Microbiota and Their Impact on Pathogen Transmission. In: Anopheles mosquitoes - New insights into malaria vectors (ed. (eds Manguin S.). InTech (2013).
- 49.Zouache K, et al. Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Aedes aegypti, from different geographic regions of Madagascar. FEMS Microbiology Ecology. 2011;75:377–389. doi: 10.1111/j.1574-6941.2010.01012.x. [DOI] [PubMed] [Google Scholar]
- 50.Li K, et al. Diversity of bacteriome associated with Phlebotomus chinensis (Diptera: Psychodidae) sand flies in two wild populations from China. Scientific Reports. 2016;6:36406. doi: 10.1038/srep36406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Díaz S, Villavicencio B, Correia N, Costa J, Haag KL. Triatomine bugs, their microbiota and Trypanosoma cruzi: asymmetric responses of bacteria to an infected blood meal. Parasites & Vectors. 2016;9:636. doi: 10.1186/s13071-016-1926-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El-Sayed WS, Ibrahim RA. Diversity and phylogenetic analysis of endosymbiotic bacteria of the date palm root borer Oryctes agamemnon (Coleoptera: Scarabaeidae) BMC Microbiology. 2015;15:88. doi: 10.1186/s12866-015-0422-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saraithong P, Li Y, Saenphet K, Chen Z, Chantawannakul P. Midgut bacterial communities in the giant Asian honeybee (Apis dorsata) across 4 developmental stages: A comparative study. Insect Science. 2017;24:81–92. doi: 10.1111/1744-7917.12271. [DOI] [PubMed] [Google Scholar]
- 54.Su L, et al. Comparative Gut Microbiomes of Four Species Representing the Higher and the Lower Termites. Journal of Insect Science. 2016;16:97. doi: 10.1093/jisesa/iew081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Almeida LG, Moraes LA, Trigo JR, Omoto C, Cônsoli FL. The gut microbiota of insecticide-resistant insects houses insecticide-degrading bacteria: A potential source for biotechnological exploitation. PLOS ONE. 2017;12:e0174754. doi: 10.1371/journal.pone.0174754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramya SL, Venkatesan T, Srinivasa Murthy K, Jalali SK, Verghese A. Detection of carboxylesterase and esterase activity in culturable gut bacterial flora isolated from diamondback moth, Plutella xylostella (Linnaeus), from India and its possible role in indoxacarb degradation. Brazilian Journal of Microbiology. 2016;47:327–336. doi: 10.1016/j.bjm.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pekas A, et al. Comparison of bacterial microbiota of the predatory mite Neoseiulus cucumeris (Acari: Phytoseiidae) and its factitious prey Tyrophagus putrescentiae (Acari: Acaridae) Scientific Reports. 2017;7:2. doi: 10.1038/s41598-017-00046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ceja-Navarro JA, et al. Gut microbiota mediate caffeine detoxification in the primary insect pest of coffee. Nature Communications. 2015;6:7618. doi: 10.1038/ncomms8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Singh BK. Organophosphorus-degrading bacteria: ecology and industrial applications. Nat Rev Micro. 2009;7:156–164. doi: 10.1038/nrmicro2050. [DOI] [PubMed] [Google Scholar]
- 60.Xia, X., Zheng, D., Zhong, H., Qin, B., Gurr, G. M. DNA sequencing reveals the midgut microbiota of diamondback moth, Plutella xylostella (L.) and a possible relationship with insecticide resistance. PLoS One8 (2013). [DOI] [PMC free article] [PubMed]
- 61.Tchioffo MT, et al. Dynamics of Bacterial Community Composition in the Malaria Mosquito’s Epithelia. Frontiers in microbiology. 2015;6:1500. doi: 10.3389/fmicb.2015.01500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh B. Review on microbial carboxylesterase: general properties and role in organophosphate pesticides degradation. Biochem Mol Biol. 2014;2:1–6. doi: 10.12966/bmb.03.01.2014. [DOI] [Google Scholar]
- 63.Singh B, Kaur J, Singh K. Microbial degradation of an organophosphate pesticide, malathion. Critical Reviews in Microbiology. 2014;40:146–154. doi: 10.3109/1040841X.2013.763222. [DOI] [PubMed] [Google Scholar]
- 64.Singh BK, Walker A, Morgan JAW, Wright DJ. Biodegradation of Chlorpyrifos by Enterobacter Strain B-14 and Its Use in Bioremediation of Contaminated Soils. Applied and Environmental Microbiology. 2004;70:4855–4863. doi: 10.1128/AEM.70.8.4855-4863.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karigar CS, Rao SS. Role of Microbial Enzymes in the Bioremediation of Pollutants: A Review. Enzyme Research. 2011;2011:11. doi: 10.4061/2011/805187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Allocati N, Federici L, Masulli M, Di Ilio C. Glutathione transferases in bacteria. The FEBS journal. 2009;276:58–75. doi: 10.1111/j.1742-4658.2008.06743.x. [DOI] [PubMed] [Google Scholar]
- 67.Singh BK, Walker A. Microbial degradation of organophosphorus compounds. FEMS microbiology reviews. 2006;30:428–471. doi: 10.1111/j.1574-6976.2006.00018.x. [DOI] [PubMed] [Google Scholar]
- 68.Sierra-Garcia I. N. & Oliveira, V. M. d. Microbial Hydrocarbon Degradation: Efforts to Understand Biodegradation in Petroleum Reservoirs. In: Biodegradation - Engineering and Technology (eds Chamy, R., Rosenkranz, F.). InTech (2013).
- 69.Zhang C, Bennett GN. Biodegradation of xenobiotics by anaerobic bacteria. Applied Microbiology and Biotechnology. 2005;67:600–618. doi: 10.1007/s00253-004-1864-3. [DOI] [PubMed] [Google Scholar]
- 70.Van Eerd LL, Hoagland RE, Zablotowicz RM, Hall JC. Pesticide metabolism in plants and microorganisms. Weed Science. 2003;51:472–495. doi: 10.1614/0043-1745(2003)051[0472:PMIPAM]2.0.CO;2. [DOI] [Google Scholar]
- 71.McGuinness M, Dowling D. Plant-Associated Bacterial Degradation of Toxic Organic Compounds in Soil. International Journal of Environmental Research and Public Health. 2009;6:2226–2247. doi: 10.3390/ijerph6082226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vuilleumier S, Pagni M. The elusive roles of bacterial glutathione S-transferases: new lessons from genomes. Applied Microbiology and Biotechnology. 2002;58:138–146. doi: 10.1007/s00253-001-0836-0. [DOI] [PubMed] [Google Scholar]
- 73.Zhu D, et al. Biodegradation of alkaline lignin by Bacillus ligniniphilus L1. Biotechnology for Biofuels. 2017;10:44. doi: 10.1186/s13068-017-0735-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guazzaroni M-E, et al. Metaproteogenomic insights beyond bacterial response to naphthalene exposure and bio-stimulation. ISME J. 2013;7:122–136. doi: 10.1038/ismej.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bers K, et al. A Novel Hydrolase Identified by Genomic-Proteomic Analysis of Phenylurea Herbicide Mineralization by Variovorax sp. Strain SRS16. Applied and Environmental Microbiology. 2011;77:8754–8764. doi: 10.1128/AEM.06162-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arora PK, Bae H. Bacterial degradation of chlorophenols and their derivatives. Microbial Cell Factories. 2014;13:31. doi: 10.1186/1475-2859-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Eulberg D, Lakner S, Golovleva LA, Schlömann M. Characterization of a Protocatechuate Catabolic Gene Cluster from Rhodococcus opacus 1CP: Evidence for a Merged Enzyme with 4-Carboxymuconolactone-Decarboxylating and 3-Oxoadipate Enol-Lactone-Hydrolyzing Activity. Journal of Bacteriology. 1998;180:1072–1081. doi: 10.1128/jb.180.5.1072-1081.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu Y, Ding Y, Cohen Y, Cao B. Elevated level of the second messenger c-di-GMP in Comamonas testosteroni enhances biofilm formation and biofilm-based biodegradation of 3-chloroaniline. Applied Microbiology and Biotechnology. 2015;99:1967–1976. doi: 10.1007/s00253-014-6107-7. [DOI] [PubMed] [Google Scholar]
- 79.Hiessl S, et al. Involvement of Two Latex-Clearing Proteins during Rubber Degradation and Insights into the Subsequent Degradation Pathway Revealed by the Genome Sequence of Gordonia polyisoprenivorans Strain VH2. Applied and Environmental Microbiology. 2012;78:2874–2887. doi: 10.1128/AEM.07969-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Longkumer T, Parthasarathy S, Vemuri SG, Siddavattam D. OxyR-dependent expression of a novel glutathione S-transferase (Abgst01) gene in Acinetobacter baumannii DS002 and its role in biotransformation of organophosphate insecticides. Microbiology. 2014;160:102–112. doi: 10.1099/mic.0.070664-0. [DOI] [PubMed] [Google Scholar]
- 81.Brogdon, W. G., Chan, A. Guideline for evaluating insecticide resistance in vectors using the CDC bottle bioassay. CDC Atlanta (2010).
- 82.Andrews, S. FastQC: A quality control tool for high throughput sequence data BabrahamBioinformatics, (2016).
- 83.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neafsey DE, et al. Highly evolvable malaria vectors: the genomes of 16 Anopheles mosquitoes. Science (New York, NY) 2015;347:1258522–1258522. doi: 10.1126/science.1258522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment usingDIAMOND . Nat Meth. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 87.Huson DH, et al. MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS computational biology. 2016;12:e1004957. doi: 10.1371/journal.pcbi.1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oliwa-Stasiak K, Molnar CI, Arshak K, Bartoszcze M, Adley CC. Development of a PCR assay for identification of the Bacillus cereus group species. J Appl Microbiol. 2010;108:266–273. doi: 10.1111/j.1365-2672.2009.04419.x. [DOI] [PubMed] [Google Scholar]
- 90.Chen TL, et al. Comparison of one-tube multiplex PCR, automated ribotyping and intergenic spacer (ITS) sequencing for rapid identification of Acinetobacter baumannii. Clinical Microbiology and Infection. 2007;13:801–806. doi: 10.1111/j.1469-0691.2007.01744.x. [DOI] [PubMed] [Google Scholar]
- 91.Azmy AF, Saafan AE, Essam TM, Amin MA, Ahmed SH. Biodegradation of Malathion by Acinetobacter baumannii Strain AFA Isolated from Domestic Sewage in Egypt. International Journal of Biological, Biomolecular, Agricultural, Food and Biotechnological Engineering. 2015;9:54–65. [Google Scholar]
- 92.Smith-Grenier LL, Adkins A. Isolation and characterization of soil microorganisms capable of utilizing the herbicide diclofop-methyl as a sole source of carbon and energy. Canadian Journal of Microbiology. 1996;42:221–226. doi: 10.1139/m96-033. [DOI] [PubMed] [Google Scholar]
- 93.Zhao L, Wang F, Zhao J. Identification and functional characteristics of chlorpyrifos-degrading and plant growth promoting bacterium Acinetobacter calcoaceticus. Journal of Basic Microbiology. 2014;54:457–463. doi: 10.1002/jobm.201200639. [DOI] [PubMed] [Google Scholar]
- 94.Akbar S, Sultan S, Kertesz M. Bacterial community analysis in chlorpyrifos enrichment cultures via DGGE and use of bacterial consortium for CP biodegradation. World Journal of Microbiology and Biotechnology. 2014;30:2755–2766. doi: 10.1007/s11274-014-1699-8. [DOI] [PubMed] [Google Scholar]
- 95.Murphy M, Lötter LH. The effect of acetate and succinate on polyphosphate formation and degradation in activated sludge, with particular reference to Acinetobacter calcoaceticus. Applied Microbiology and Biotechnology. 1986;24:512–517. doi: 10.1007/BF00250333. [DOI] [Google Scholar]
- 96.Ohtake H, Takahashi K, Tsuzuki Y, Toda K. Uptake and release of phosphate by a pure culture of Acinetobacter calcoaceticus. Water Research. 1985;19:1587–1594. doi: 10.1016/0043-1354(85)90404-X. [DOI] [Google Scholar]
- 97.Auling G, et al. Analysis of the polyphosphate-accumulating microflora in phosphorus-eliminating, anaerobic-aerobic activated sludge systems by using diaminopropane as a biomarker for rapid estimation of Acinetobacter spp. Applied and Environmental Microbiology. 1991;57:3585–3592. doi: 10.1128/aem.57.12.3585-3592.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Begum SFM, Rajesh G, Narendran RR. Isolation, Characterization and Identification of Dimethoate Degrading Bacteria from Soil Series of Tamil Nadu. International Journal of Advanced Scientific and Technical Research. 2016;3:220–230. [Google Scholar]
- 99.Chen S, et al. Enhancement of cypermethrin degradation by a coculture of Bacillus cereus ZH-3 and Streptomyces aureus HP-S-01. Bioresource Technology. 2012;110:97–104. doi: 10.1016/j.biortech.2012.01.106. [DOI] [PubMed] [Google Scholar]
- 100.Vidya Lakshmi C, Kumar M, Khanna S. Biotransformation of chlorpyrifos and bioremediation of contaminated soil. International Biodeterioration & Biodegradation. 2008;62:204–209. doi: 10.1016/j.ibiod.2007.12.005. [DOI] [Google Scholar]
- 101.Singh B, Kaur J, Singh K. Biodegradation of malathion by Brevibacillus sp. strain KB2 and Bacillus cereus strain PU. World Journal of Microbiology and Biotechnology. 2012;28:1133–1141. doi: 10.1007/s11274-011-0916-y. [DOI] [PubMed] [Google Scholar]
- 102.Bhadbhade BJ, Sarnaik SS, Kanekar PP. Biomineralization of an organophosphorus pesticide, Monocrotophos, by soil bacteria. Journal of Applied Microbiology. 2002;93:224–234. doi: 10.1046/j.1365-2672.2002.01680.x. [DOI] [PubMed] [Google Scholar]
- 103.Sharma, A., Pankaj, P. K., Gangola, S. & Kumar, G. Microbial Degradation of Pesticides for Environmental Cleanup. In: Bioremediation ofIndustrial pollutants(eds Saxena, R. N. B. G.). Write & Print Publications (2016).
- 104.Langlois BE, Collins JA, Sides KG. Some Factors Affecting Degradation of Organochlorine Pesticides by Bacteria1. Journal of dairy science. 1970;53:1671–1675. doi: 10.3168/jds.S0022-0302(70)86461-X. [DOI] [PubMed] [Google Scholar]
- 105.Lee KS, Metcalf WW, Wanner BL. Evidence for two phosphonate degradative pathways in Enterobacter aerogenes. Journal of Bacteriology. 1992;174:2501–2510. doi: 10.1128/jb.174.8.2501-2510.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mohamed ZK, Ahmed MA, Fetyan NA, Elnagdy SM. Isolation and molecular characterisation of malathion-degrading bacterial strains from waste water in Egypt. Journal of Advanced Research. 2010;1:145–149. doi: 10.1016/j.jare.2010.03.007. [DOI] [Google Scholar]
- 107.Mandal K, Singh B, Jariyal M, Gupta VK. Microbial degradation of fipronil by Bacillus thuringiensis. Ecotoxicology and Environmental Safety. 2013;93:87–92. doi: 10.1016/j.ecoenv.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 108.Zeinat Kamal M, Nashwa AH, Fetyan A, Ibrahim MA, El-Nagdy S. Biodegradation and Detoxification of Malathion by of Bacillus thuringiensis MOS-5. Australian Journal of Basic and Applied Sciences. 2008;2:724–732. [Google Scholar]
- 109.Abubacker MN, Visvanathan M, Srinivasan S. Biodegradation of glyphosate herbicide by bacterial isolates from Banana (Musa spp.) Plantation soil Biolife. Journal. 2016;4:243–250. [Google Scholar]
- 110.Jabeen H, Iqbal S, Anwar S, Parales RE. Optimization of profenofos degradation by a novel bacterial consortium PBAC using response surface methodology. International Biodeterioration & Biodegradation. 2015;100:89–97. doi: 10.1016/j.ibiod.2015.02.022. [DOI] [Google Scholar]
- 111.Liao M, Zhang HJ, Xie XM. [Isolation and identification of degradation bacteria Enterobacter aerogenes for pyrethriods pesticide residues and its degradation characteristics] Huan jing ke xue = Huanjing kexue/[bian ji, Zhongguo ke xue yuan huan jing ke xue wei yuan hui “Huan jing ke xue” bian ji wei yuan hui] 2009;30:2445–2451. [PubMed] [Google Scholar]
- 112.Ramya SL, Venkatesan T, Murthy KS, Jalali SK, Varghese A. Degradation of acephate by Enterobacter asburiae, Bacillus cereus and Pantoea agglomerans isolated from diamondback moth Plutella xylostella (L), a pest of cruciferous crops. Journal of environmental biology. 2016;37:611–618. [PubMed] [Google Scholar]
- 113.Ahemad M, Khan MS. Influence of Selective Herbicides on Plant Growth Promoting Traits of Phosphate Solubilizing Enterobacter asburiae Strain PS2. Research Journal of Microbiology. 2010;5:849–857. doi: 10.3923/jm.2010.849.857. [DOI] [Google Scholar]
- 114.Abraham J, Silambarasan S, Logeswari P. Simultaneous degradation of organophosphorus and organochlorine pesticides by bacterial consortium. Journal of the Taiwan Institute of Chemical Engineers. 2014;45:2590–2596. doi: 10.1016/j.jtice.2014.06.014. [DOI] [Google Scholar]
- 115.Kryuchkova YV, et al. Isolation and characterization of a glyphosate-degrading rhizosphere strain, Enterobacter cloacae K7. Microbiological Research. 2014;169:99–105. doi: 10.1016/j.micres.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 116.Beunink J, Rehm H-J. Synchronous anaerobic and aerobic degradation of DDT by an immobilized mixed culture system. Applied Microbiology and Biotechnology. 1988;29:72–80. doi: 10.1007/BF00258354. [DOI] [PubMed] [Google Scholar]
- 117.Gangolli, S. & Chemistry, R. S. O. The Dictionary of Substances and Their Effects: E-J. Royal Society of Chemistry (1999).
- 118.Schowanek D, Verstraete W. Phosphonate utilization by bacterial cultures and enrichments from environmental samples. Appl Environ Microbiol. 1990;56:895–903. doi: 10.1128/aem.56.4.895-903.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lawrence KS, Feng Y, Lawrence GW, Burmester CH, Norwood SH. Accelerated Degradation of Aldicarb and Its Metabolites in Cotton Field Soils. Journal of Nematology. 2005;37:190–197. [PMC free article] [PubMed] [Google Scholar]
- 120.Kwon G-S, Sohn H-Y, Shin K-S, Kim E, Seo B-I. Biodegradation of the organochlorine insecticide, endosulfan, and the toxic metabolite, endosulfan sulfate, by Klebsiella oxytoca KE-8. Applied Microbiology and Biotechnology. 2005;67:845–850. doi: 10.1007/s00253-004-1879-9. [DOI] [PubMed] [Google Scholar]
- 121.Surekha Rani M, et al. Isolation and characterization of a chlorpyrifos-degrading bacterium from agricultural soil and its growth response. African Journal of Microbiology Research. 2008;2:26–31. [Google Scholar]
- 122.Ifediegwu MC, et al. Isolation, Growth and Identification of Chlorpyrifos Degrading Bacteria from Agricultural Soil in Anambra State, Nigeria. Universal Journal of Microbiology Research. 2015;3:46–52. doi: 10.13189/ujer.2015.030107. [DOI] [Google Scholar]
- 123.Wackett LP, Shames SL, Venditti CP, Walsh CT. Bacterial carbon-phosphorus lyase: products, rates, and regulation of phosphonic and phosphinic acid metabolism. Journal of Bacteriology. 1987;169:710–717. doi: 10.1128/jb.169.2.710-717.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Timms P, MacRae IC. Conversion of fensulfothion by Klebsiella pneumoniae to fensulfothion sulfide and its accumulation. Australian journal of biological sciences. 1982;35:661–667. doi: 10.1071/BI9820661. [DOI] [PubMed] [Google Scholar]
- 125.Cook AM, Huetter R. s-Triazines as nitrogen sources for bacteria. Journal of Agricultural and Food Chemistry. 1981;29:1135–1143. doi: 10.1021/jf00108a009. [DOI] [Google Scholar]
- 126.McBride KE, Kenny JW, Stalker DM. Metabolism of the herbicide bromoxynil by Klebsiella pneumoniae subsp. ozaenae. Appl Environ Microbiol. 1986;52:325–330. doi: 10.1128/aem.52.2.325-330.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kulkarni SV, Markad VL, Melo JS, D’Souza SF, Kodam KM. Biodegradation of tributyl phosphate using Klebsiella pneumoniae sp. S3. Applied Microbiology and Biotechnology. 2014;98:919–929. doi: 10.1007/s00253-013-4938-2. [DOI] [PubMed] [Google Scholar]
- 128.Ortiz-Hernandez ML, Monterosas-Brisson M, Yanez-Ocampo G, Sanchez-Salinas E. Biodegradation of methyl-parathion by bacteria isolated of agricultural soil. Rev Int Contam Ambient. 2001;17:147–155. [Google Scholar]
- 129.Phugare SS, Kalyani DC, Gaikwad YB, Jadhav JP. Microbial degradation of imidacloprid and toxicological analysis of its biodegradation metabolites in silkworm (Bombyx mori) Chemical Engineering Journal. 2013;230:27–35. doi: 10.1016/j.cej.2013.06.042. [DOI] [Google Scholar]
- 130.Jiang W, Metcalf WW, Lee KS, Wanner BL. Molecular cloning, mapping, and regulation of Pho regulon genes for phosphonate breakdown by the phosphonatase pathway of Salmonella typhimurium LT2. J Bacteriol. 1995;177:6411–6421. doi: 10.1128/jb.177.22.6411-6421.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Abo-Amer A. Biodegradation of diazinon by Serratia marcescens DI101 and its use in bioremediation of contaminated environment. Journal of microbiology and biotechnology. 2011;21:71–80. doi: 10.4014/jmb.1007.07024. [DOI] [PubMed] [Google Scholar]
- 132.Cycon M, Wojcik M, Piotrowska-Seget Z. Biodegradation of the organophosphorus insecticide diazinon by Serratia sp. and Pseudomonas sp. and their use in bioremediation of contaminated soil. Chemosphere. 2009;76:494–501. doi: 10.1016/j.chemosphere.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 133.Cycoń M, Żmijowska A, Wójcik M, Piotrowska-Seget Z. Biodegradation and bioremediation potential of diazinon-degrading Serratia marcescens to remove other organophosphorus pesticides from soils. Journal of Environmental Management. 2013;117:7–16. doi: 10.1016/j.jenvman.2012.12.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The WMS reads obtained from this study (SRP108310) have been deposited in NCBI under the BioProject PRJNA388280.