

Abstract
We use a theoretical approach to reveal the electronic and structural properties of molybdenum impurities between MoS2 bilayers. We find that interstitial Mo impurities are able to reverse the well-known stability order of the pristine bilayer, because the most stable form of stacking changes from AA’ (undoped) into AB’ (doped). The occurrence of Mo impurities in different positions shows their split electronic levels in the energy gap, following octahedral and tetrahedral crystal fields. The energy stability is related to the accommodation of Mo impurities compacted in hollow sites between layers. Other less stable configurations for Mo dopants have larger interlayer distances and band gaps than those for the most stable stacking. Our findings suggest possible applications such as exciton trapping in layers around impurities, and the control of bilayer stacking by Mo impurities in the growth process.
Introduction
The recent isolation of new 2D materials, such as hexagonal boron nitride (h-BN)1, black phosphorus2, and particularly transition metal dichalcogenides (TMDCs)3 have attracted considerable attention thanks to their interesting physical, chemical, electronic, optical, and mechanical properties4–11. Among TMDCs, MoS2 is being used as prototype in several different applications, such as photovoltaic cells, photocatalysts, electronic nanodevices, and energy storage and conversion materials12. In structural terms, a layer of MoS2 has Mo centers coordinated with six sulfur ligands in a trigonal prismatic arrangement13,14, following a hexagonal lattice of alternating Mo and S atoms, as seen from above. TMDCs nanostructures can then be produced by stacking several MoS2 layers through weak van der Waals interactions. It is noteworthy that the number of layers and their stacking arrangement largely modify the electronic properties of the MoS2 semiconductor15–19. For instance, the MoS2 monolayer shows a direct band gap, compared with the indirect band gap of MoS2 bulk20–23. The stacking in the MoS2 bilayer alters the band gap, which can then be engineered not only by strain24,25, but also by sliding26 and twisting the MoS2 layers27. Thus, the control of the MoS2 bilayer stacking is relevant when considering different applications in future devices28.
The electronic and magnetic properties of MoS2 monolayers and bilayers are also tuned by defects, such as vacancies and adatoms29–31. Most of the adatom impurities considered within MoS2 layers are in groups IA and VIIA, or transition metal atoms. The adatoms in the interlayer space have interesting effects, such as n- or p-type doping, induced magnetic moments32–34, and structural phase transitions35. Although some early photoelectron spectroscopy experiments appeared to show Mo atoms embedded between MoS2 layers36, current experiments using low-temperature scanning tunneling microscopy (STM) show Mo impurities between bilayers37. However, the effects of intrinsic Mo impurity atoms on the electronic and structural properties of MoS2 bilayers with different stackings are still largely unexplored.
We present a theoretical study of the structural and electronic properties of the MoS2 bilayer considering Mo atoms as intrinsic impurities, placed at different positions within the interlayer region in a diluted regime. Using density functional with van der Waals calculations from first principles, we relax the structures and study their electronic properties. We find that the intrinsic Mo impurities in the interlayer region produce some interesting behaviors, namely (i) a change in the stability order with respect to the pristine bilayer, energetically favoring the AB’ stacking over the AA’, (ii) impurity states in the band gap region, and (iii) an increase in the distance between layers. The structural and electronic modifications induced by the impurities could be employed as electron and exciton-like traps38,39, and the change in stacking produced by the Mo impurities could also be useful to fine-tune the stacking during the growing process19,27.
Model
We first consider the most stable stackings of the pristine MoS2 bilayers, namely the 2H and the 3R phases28,40,41. These natural phases have Mo atoms in a trigonal prismatic coordination, and the MoS2 layers in different stacking orders. The bilayer in the 2H-phase has inversion symmetry7,22, and shows the AA’ and AB’ stacking related by rotating the MoS2 layers. In the AA’ stacking, the hexagons in each layer are superposed in such a way that the molybdenum atoms of the bottom layer are located just below the sulfur atoms in the top layer, and vice-versa. For AB’ stacking the hexagons in each layer are shifted with the sulfur atoms of the bottom layer beneath the hollow sites of top layer, and the molybdenum atoms in the top layer over the molybdenum atoms in the bottom layer18, as shown schematically in Fig. 1. In our calculations we found an energy difference between these stackings of EAA′ − EAB′ = 2.6 meV per atom, in good agreement with previous DFT calculations18,19,42. The AB stacking with no rotation between two layers belongs to another kind of phase, the 3R-phase. The up layer slides on the bottom layer in the armchair direction so that some Mo and S atoms in different layers match. Our results for the stability order of the pristine bilayers show that AB stacking is the most stable, nearly degenerated with the AA’ stacking with a total energy of 0.7 meV per atom, followed by the AB’ stacking with 3.3 meV per atom. Note that depending on the details of different calculations the ground energy stacking can exchanging between AB and AA’43, and the AB’ stacking remains as the third state in stability.18,19,42,43.
Figure 1.

Stackings of MoS2 bilayers considered with low-energy: (a) AB, (b) AA’ and (c) AB’. Mo atoms are shown as cyan spheres, and sulfur atoms are shown as yellow spheres. Inset shows the Mo impurity sites relative to the bottom layer in B (bridge), H (hollow), T (top over Mo) and T’ (top over S).
We then include Mo atoms as intrinsic impurities within the interlayer region for MoS2 bilayers at different inequivalent positions, as shown in Fig. 1. The Mo impurity is labeled as Moimp. The initial absorption sites for Moimp within the MoS2 bilayer are assumed to follow Mo absorption sites as in MoS2 monolayers44,45. The Mo impurity position is then labeled relative to the bottom layer in B (bridge), H (hollow), T (top over Mo) and T’ (top over S). The MoS2 bilayer structures with the impurity in the interlayer region are fully relaxed, to allow the optimized lattice parameters and atomic coordinates to be obtained. More detailed information on the relaxed geometries is included in the Supplemental Material. The binding energy can then be calculated using Ebinding = ETotal − Ebilayer − Eimp, where ETotal is the total energy of the MoS2 bilayer with the impurity, Ebilayer is the energy of the corresponding final pristine MoS2 bilayer (either AA’ or AB’), and Eimp is the energy for the isolated Moimp atom.
Results
Energy and Geometry
Because the most stable configuration with Moimp belongs to the 2H-phase, in the main text we discuss the impurity properties between bilayers focusing on the AA’ and AB’ stacking. The discussion on the 3R-phase is reported as Supplemental Material.
Figure 2 shows the total and binding energies for the different 2H stacking and impurity positions. The binding energies are negative, which indicates that the Mo impurity atoms are indeed adsorbed in the interlayer region of the MoS2 bilayer. The binding and total energies exhibit the same trend in terms of stability. The results in increasing order of stability show that in the presence of the interlayer Moimp, the T-AB’ bilayer configuration is the most energetically favorable. This configuration has AB’ stacking with the Moimp superposed with two Mo atoms as seen from above. Note that the T-AB’ configuration is reached from the input that has the Moimp placed at the bridge (B) position in the AA’ stacking.
Figure 2.

Total and binding energy as function of the structural configurations of Mo impurities within the MoS2 bilayer. The red spheres indicate the Mo impurity in each configuration. The relaxed structures are included, grouped in octahedral (T-AB’ and H-AA’) and tetrahedral (T’-AB’ and T’-AA’) structures of sulfur atoms around Mo impurities. The zero energy point is set for the most energetically favorable structure, namely the T-AB’ configuration. Figure prepared using XCrySDen46.
The next most favorable configuration is H-AA’, with the Moimp in the hollow position, in which the bilayer structure maintains the AA’ stacking. The H-AA’ case is less stable than T-AB’ by about 1.5 eV. On the right hand side, the configurations labeled T’-AB’ for AB’ stacking and T’-AA’ for AA’ stacking are energetically close, and the least stable.
We classify the relaxed configurations according to how the Moimp is related structurally to its neighboring sulfur atoms47,48. The T-AB’ and H-AA’ configurations form octahedral sites around the Moimp. These configurations have a coordination number of six, corresponding to the six neighboring sulfur atoms. The T’-AA’ and T’-AB’ configurations for the Moimp form a tetrahedral structure with a coordination number of four, therefore, the sulfur atoms in the top and bottom layers enclose a tetrahedral site for the Moimp. These octahedral and tetrahedral environments are shown schematically at the top of Fig. 2. It is noteworthy that regardless of the final stacking, the octahedral configurations are the most stable.
Electronic Properties
Results showing the band structures and the local density of states (LDOS) projected in space, for some of the considered configurations, are presented in Fig. 3. We focus on the impurity in-gap states near the Fermi energy introduced by the Moimp atom, indicated by the areas enclosed by orange rectangles. The band structures of the most stable structures show three distinctive in-gap states, joined in two groups, labeled as regions 1 and 2 with degeneracies of one and two, respectively. Although the Moimp in these two configurations have an octahedral sulfur environment, the in-gap bands present slightly different dispersive behavior. In region 1, the band for the T-AB’ case is more dispersive than the corresponding band for the H-AA’ configuration, which is almost flat. The states in region 1 mainly have orbital character, as shown by the LDOS in panels (d) and (e). Note that in region 1, the surrounding region of the impurity for the T-AB’ case has some hybridization with bilayer orbitals, which is not observed for the H-AA’ case. In region 2 of the T-AB’ case, there are two energy bands which are mainly non-bonding Moimp d orbitals with the neighboring sulfur atoms, as shown by the LDOS in panels (d) and (e). By comparing the T-AB’ and H-AA’ configurations, the stability order can be associated with the widening of the bands in regions 1 and 2 and to the displacement of the bands in region 2 to lower energies in the T-AB’ configuration.
Figure 3.

(a–c) Band structures for the given configurations. The Fermi energy is set to 0 eV. Orange rectangles enclose the Moimp bands separated into several different energy regions, labeled as 1, 2 and 3. (d–f) Local density of states (LDOS) projected in space for the Moimp bands in the band gap region of the MoS2 bilayer. Figure prepared using XCrySDen46.
The band structures in the T-AB’ and H-AA’ configurations are spin-compensated and related to the impurity in an octahedral sulfur environment. We now focus on the T’-AA’ and T’-AB’ cases. These two configurations have similar energy band structures and are found to have close energies. Both systems exhibit spin polarized behavior with a total magnetic moment of 2 μB, which is determined by a similar Moimp tetrahedral environment. In particular for the T’-AA’ configuration, the spin up and spin down components of the spatial resolved LDOS are shown in Fig. 3 panel (f). The state in region 1 has a orbital character; however, it is above the Fermi level. The LDOS of the lower impurity states, the up states in region 3 and the spin down component in region 2 are nearly equal, depleting the LDOS in the Moimp-S bond direction as in the T-AB’ case. However, the states responsible for the spin polarization in the T’-AB’ and T’-AA’ configurations are the spin up d-orbitals in region 2, which are along the Moimp-S bonds.
We also find that the LDOS is localized not only on the Moimp, but also on one of the MoS2 layers. This layer asymmetry indicates that doping by electrons or holes could spatially differentiate between the two layers in the MoS2 bilayer, a finding that could be of use in optoelectronic applications22,49.
The electronic structure for the impurity level states is best understood using crystal field theory. We analyze the ligand field splitting for the Moimp d-orbitals produced by the interactions with the sulfur ligands for the octahedral and tetrahedral sites. The bonding and non-bonding interactions of d-orbitals for octahedral and tetrahedral sites are in agreement with the energy level scheme shown in Fig. 4. We consider the z-axis perpendicular to the layers and the x and y axes in the in-plane layer. In the octahedral environment for the T-AB’ and H-AA’ configurations, the sulfur ligands overlap less with the in-plane dxy and orbitals, these orbitals are therefore non-bonding and have the lowest energy. The orbital remains non-bonding at an intermediate energy, interacting less with the sulfur atoms. We next find that the dxz and dyz orbitals are more strongly directed and interact with the sulfur atoms along Moimp-S bonds, lying at higher energies, as is showed in Fig. 4(a). In the case of tetrahedral environment for T’-AB’ and T’-AA’, shown in Fig. 4(b), the dxy and orbitals behave similarly to the octahedral structure; however, the dxz and dyz orbitals exchange roles with the orbital. Thus, the orbital in the tetrahedral environment interacts more with the sulfur ligands increasing its energy, as shown for region 1 of Fig. 3(c) and (f) in the T’-AA’ case. The level filling help to explain why the tetrahedral cases have spin polarization, and a total magnetization of 2 μB, as shown by the arrow counting.
Figure 4.

Ligand field splitting for Moimp d-orbitals produced by the neighboring sulfur atoms in (a) octahedral and (b) tetrahedral environments. Figure prepared using VESTA50.
Molybdenum impurities in the 3R-phase follow the same general trends as the previously discussed for the 2H stackings. The octahedral Mo environment is the most stable and the tetrahedral environment is less stable and presents a non-zero magnetic moment. A more detailed discussion of the stacking configurations for the 3R-phase with Mo impurities is included in Supplemental Material.
The main effect in our calculations is the interaction of the Mo impurity with the MoS2 layers in the order of eVs. The effect of spin-orbit coupling (SOC) in the stacking stability is expected to be minimal although it would split bands near the Fermi level7,51. Since the spin-orbit interaction is particularly noticeable in materials without inversion symmetry52, we can expect the following effects. On the one hand, the pristine bilayers in the 2H-phase presents point-center inversion symmetry, and the band structure remains spin degenerate even in the presence of SOC14,40. SOC can thus change the pristine bilayer bands in the 3R-phase because the lack of inversion symmetry would breaks the spin degeneracy and could lead to valley dependent spin polarization53. On the other hand, we can expect splitting of the Moimp-bands due to the presence of spin-orbit and the lack of inversion symmetry in the tetrahedral configurations (in Fig. 4). However, the impurity in the tetrahedral arrangement is not the most stable configuration by an energy difference of 1.7 eV, much large than values typically associated to the spin-orbit coupling energies.
We now consider the gap changes in the bulk bands, shown in light-color in Fig. 3, and induced by the Moimp. It is important to mention that the indirect gap along Γ − K direction of 1 × 1 unit cell becomes a direct band gap at the Γ-point due to the k-space folding of the large 3 × 3 unit cell. Our calculated pristine bands are in agreement with the literature, and are included in Supplemental Material. It is well known that band gaps calculated using GGA/LDA infra-estimate the values produced in experiments, so we discuss differences in magnitude gaps. The layer-gap is indicated by the energy difference between the HOMO-LUMO bulk bands at the Γ-point. The energy gaps of the pristine MoS2 layers are correlated with the interlayer distances. We check that the gap and distances for the AB’ pristine stacking are 0.09 eV smaller and 0.03 Å shorter than the values for the AA’ pristine case. The layer-gaps and the interlayer separation including Moimp show larger values in comparison with the pristine cases. Among the Mo doped systems, the most stable T-AB’ case has the smaller layer-gap and the shortest interlayer separation. The layer-gap is 0.2 eV above the AB’-pristine, and the interlayer separation is 0.03 Å larger than that of AB’-pristine. The layer-gap values for the H-AA’ and the T’-AB’ (T’-AA’) increase from the T-AB’ case by 0.2 eV and by 0.5 eV respectively. These gap differences are somewhat correlated with the difference between layer-layer distances, ∼0.5 Å, between the T-AB’ and H-AA’ cases, a value that increases up to 0.7 Å for the T’-AB’ and T’-AA’ configurations. The increase in the layer-band-gap with interlayer distance is explained by a weaker interlayer coupling. Detailed information is provided in the Supplemental Material.
The interlayer distances in the proximity of the impurity are between 0.1 and 0.16 Å larger than those far from it, which indicates the role of local strain. Furthermore, experiments prove than the band gap of bilayer TMDCs can be controlled by strain39,54,55. We propose that the electronic and structural modifications around the impurity could be used in a similar way to electronic confinement for embedded quantum dots. Current experimental techniques employing cross-sectional scanning transmission electron microscope analysis in encapsulated TMDC materials can provide evidence of impurity species being trapped in the interstitial region56. This effect thus has potential applications for optoelectronic devices as exciton traps around Mo-doped bilayers. A number of different experimental techniques can be used to corroborate our theoretical predictions, for instance angle-resolved photoemission spectroscopy and cross-sectional scanning transmission electron microscope analysis56,57.
Stacking change
Another possible implication of our results is that transition metal ions could be used to engineer the stacking between TMDC bilayers and to tune their electronic properties. In the T-AB’ and H-AA’ configurations, the Moimp is located within sulfur ligands forming octahedral sites. In these two configurations, the Moimp presents structural differences in the relative position respect to the nearest Mo atoms, belonging to the top and bottom MoS2 layers. The Moimp bonding produces a stacking change in T-AB’ related to the total energy gain. The interlayer Mo-Mo distance around the impurity is smaller around 0.1 Å than the interlayer distance away from the impurity. The shorter distance promotes the hybridization of the impurity states with the layer states increasing the dispersion of the in-gap impurity states.
A scheme for the stacking change is shown in Fig. 5. Starting from the AB’ stacking, the AA’ stacking is found by shifting a layer in the armchair direction. The maximum sliding coordinate corresponds to the distance of the Mo-atom to the center of the hexagon, a0 ≈ 1.86 Å. The energy profile along the sliding coordinate is calculated shifting the top layer, then fixing the in-plane coordinates for the Moimp and a MoS2 unit far from the impurity and relaxing. Figure 5(a) shows the total energy along the above discussed sliding route. Away from the T-AB’ configuration the energy increases smoothly passing through an energy maximum before reaching the final T’-AA’ configuration. The maximum energy is found for a displacement of 1.76 Å (≈0.95 a0) being 5 meV above the T’-AA’ value. It is noteworthy that the total energy as a function the sliding coordinate shows an inflexion point corresponding to the point where the slip force is maximum. That point corresponds to a displacement of 0.69 Å (≈0.37 a0) in the armchair direction. The slipping force calculated as the derivative of the total energy as a function of displacement shows a maximum force around 2.1 eV/Å, similar to the forces calculated with DFT.
Figure 5.
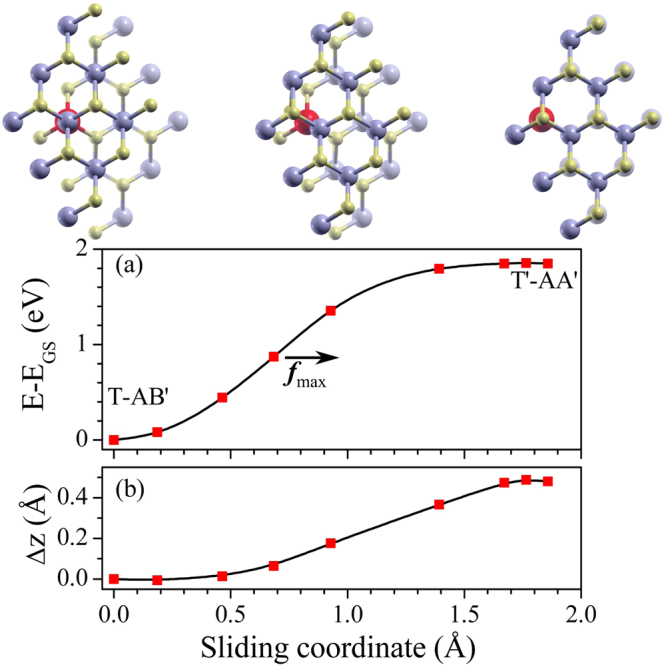
(a) Energy profile and (b) interlayer distance (Δz) by sliding between the 2H stackings. The sliding coordinate goes along the armchair direction. Full lines are just guiding the eyes. Energies and distances shown are referenced with respect to to the most stable configuration.
The Mo-Mo interlayer distance denoted as Δz follows a similar trend as the total energy (Fig. 5 (b)). For the T’-AA’ configuration, the interlayer distance is 0.48 Å larger than the value for the most stable configuration T-AB’. The Δz maximum is found for a displacement of 1.76 Å ≈ 0.95 a0 where the Δz separation is 10 mÅ higher than in T’-AA’ . The maximum force and change in z along sliding seems interesting parameters to describe tribology between MoS2 layers26,58.
Conclusion
We studied the structural and electronic properties of a MoS2 bilayer with intrinsic Mo impurities within the interlayer region.We find that the most stable configuration is T-AB’, with an energy gain above the van der Waals interaction because the Mo impurity levels strongly hybridize with the nearest atoms. A change in the stacking stability order from AA’ to AB’ is observed to be induced by impurities, with the corresponding change in energy gap. Thus, it is possible to engineer the stacking between TMDC bilayers during the growth process, enabling their electronic properties to be fine-tuned. The states and deformations induced by impurities could also be used for electronic confinement applications in optoelectronic devices, based on exciton/electron trapping.
Simulation Details
The MoS2 bilayer systems was described using density functional of van der Waals (vdW-DF) calculations with the SIESTA method59. To describe the core electrons, we consider norm-conserving relativistic ab-initio pseudopotentials in the Troullier Martins form60, including nonlinear core corrections for inner d-electrons61. The exchange and correlation energy are calculated by the non-local vdW-DF, using the parametrization proposed by Dion et al.62, taking into account the exchange energy modification included by Cooper (C09)63. In the C09 parametrization, the long-range dispersion effects are included as a perturbation to the local-density approximation correlation term. The van der Waals parameterization was chosen after comparing the band structure of the AA’ system for the 1 × 1 unit cell with the existing literature22, in particular the presence of an indirect band gap between the Γ and K points. Note that the bottom of the conduction band can move away from point K-point depending on the chosen van der Waals functional.
To include impurities, the structures of the MoS2 bilayers was extended to a 3 × 3 MoS2 supercell, using periodic boundary conditions. We are dealing with a diluted regime corresponding to 1 impurity every 54 atoms in the unit cell, around 2% doping reasonable in experimental setups. Basis set is double-ζ polarized (DZP) with numerical atomic orbital with an energy shift of 30 meV, converged to have an extended basis good to describe long van der Waals bonds. The mesh cutoff energy for the integration grid was well converged using 230 Ry. A k-grid of 10 × 10 × 1 Monkhorst-Pack is used to sample the Brillouin zone. A vacuum region in the z-direction of at least 20 Å avoids interactions with periodic images. The structures were relaxed until the force in each atom was less than 10−2 eV/Å. Further technical details are included in Supplemental Material.
Electronic supplementary material
Acknowledgements
This work was part financed by a Fondecyt grant 1140388 and Anillo Bicentenario de Ciencia y Tecnología, Conicyt grant Act-1204. J.W. González and A. Ayuela acknowledge the financial support of the Spanish Ministry of Economy and Competitiveness MINECO projects FIS2016-76617-P, the Basque Government under the ELKARTEK project(SUPER), and the University of the Basque Country grant No. IT-756-13. N. Cortés acknowledge support from the FSM1204 project, Conicyt grant No 21160844 and the hospitality of CFM-MPC and DIPC. The authors are indebted to Prof. B. Harmon, L. Chico and T. Alonso-Lanza for their helpful discussions, we also acknowledge the technical support of the DIPC computer center.
Author Contributions
N.C. and J.W.G. performed the calculations. N.C., L.R., P.O., A.A. and J.W.G. contributed to conceptual developments and manuscript preparation.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-20289-1.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Novoselov K, et al. Two-dimensional atomic crystals. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10451–10453. doi: 10.1073/pnas.0502848102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castellanos-Gomez A, et al. Isolation and characterization of few-layer black phosphorus. 2D Materials. 2014;1:025001. doi: 10.1088/2053-1583/1/2/025001. [DOI] [Google Scholar]
- 3.Yang D, Sandoval SJ, Divigalpitiya W, Irwin J, Frindt R. Structure of single-molecular-layer MoS2. Physical Review B. 1991;43:12053. doi: 10.1103/PhysRevB.43.12053. [DOI] [PubMed] [Google Scholar]
- 4.Splendiani A, et al. Emerging photoluminescence in monolayer MoS2. Nano Letters. 2010;10:1271–1275. doi: 10.1021/nl903868w. [DOI] [PubMed] [Google Scholar]
- 5.Chhowalla M, et al. The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets. Nature Chemistry. 2013;5:263–275. doi: 10.1038/nchem.1589. [DOI] [PubMed] [Google Scholar]
- 6.Wang QH, Kalantar-Zadeh K, Kis A, Coleman JN, Strano MS. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nature Nanotechnology. 2012;7:699–712. doi: 10.1038/nnano.2012.193. [DOI] [PubMed] [Google Scholar]
- 7.Kośmider K, González JW, Fernández-Rossier J. Large spin splitting in the conduction band of transition metal dichalcogenide monolayers. Physical Review B. 2013;88:245436. doi: 10.1103/PhysRevB.88.245436. [DOI] [Google Scholar]
- 8.Radisavljevic B, Radenovic A, Brivio J, Giacometti iV, Kis A. Single-layer MoS2 transistors. Nature Nanotechnology. 2011;6:147–150. doi: 10.1038/nnano.2010.279. [DOI] [PubMed] [Google Scholar]
- 9.Lopez-Sanchez O, Lembke D, Kayci M, Radenovic A, Kis A. Ultrasensitive photodetectors based on monolayer MoS2. Nature nanotechnology. 2013;8:497–501. doi: 10.1038/nnano.2013.100. [DOI] [PubMed] [Google Scholar]
- 10.Zeng H, Dai J, Yao W, Xiao D, Cui X. Valley polarization in MoS2 monolayers by optical pumping. Nature nanotechnology. 2012;7:490–493. doi: 10.1038/nnano.2012.95. [DOI] [PubMed] [Google Scholar]
- 11.Castellanos-Gomez A, et al. Elastic properties of freely suspended MoS2 nanosheets. Advanced Materials. 2012;24:772–775. doi: 10.1002/adma.201103965. [DOI] [PubMed] [Google Scholar]
- 12.Zhiming, M. & Wang, E. MoS2Materials, Physics, and Devices, vol. 1 of 21, 1 edn (Springer International Publishing, Springer International Publishing Switzerland, 2014).
- 13.Liu G-B, Xiao D, Yao Y, Xu X, Yao W. Electronic structures and theoretical modelling of two-dimensional group-VIB transition metal dichalcogenides. Chemical Society Reviews. 2015;44:2643. doi: 10.1039/C4CS00301B. [DOI] [PubMed] [Google Scholar]
- 14.Roldán R, et al. Electronic properties of single-layer and multilayer transition metal dichalcogenides MX2(M = Mo, W and X = S, Se) Annalen der Physik. 2014;526:347–357. doi: 10.1002/andp.201400128. [DOI] [Google Scholar]
- 15.Ataca C, Topsakal M, Akturk E, Ciraci S. A comparative study of lattice dynamics of three-and two-dimensional MoS2. The Journal of Physical Chemistry C. 2011;115:16354–16361. doi: 10.1021/jp205116x. [DOI] [Google Scholar]
- 16.López-Suárez M, Neri I, Rurali R. Band gap engineering of MoS2 upon compression. Journal of Applied Physics. 2016;119:165105. doi: 10.1063/1.4948376. [DOI] [Google Scholar]
- 17.Yuan H, et al. Evolution of the Valley Position in Bulk Transition-Metal Chalcogenides and Their Monolayer Limit. Nano Letters. 2016;16:4738–4745. doi: 10.1021/acs.nanolett.5b05107. [DOI] [PubMed] [Google Scholar]
- 18.He J, Hummer K, Franchini C. Stacking effects on the electronic and optical properties of bilayer transition metal dichalcogenides MoS2, MoSe2, WS2, and Wse2. Physical Review B. 2014;89:075409. doi: 10.1103/PhysRevB.89.075409. [DOI] [Google Scholar]
- 19.Yan A, et al. Identifying different stacking sequences in few-layer CVD-grown MoS2 by low-energy atomic-resolution scanning transmission electron microscopy. Physical Review B. 2016;93:041420. doi: 10.1103/PhysRevB.93.041420. [DOI] [Google Scholar]
- 20.Mahatha S, Patel K, Menon KS. Electronic structure investigation of MoS2 and MoSe2 using angle-resolved photoemission spectroscopy and ab initio band structure studies. Journal of Physics: Condensed Matter. 2012;24:475504. doi: 10.1088/0953-8984/24/47/475504. [DOI] [PubMed] [Google Scholar]
- 21.Zhao W, et al. Origin of indirect optical transitions in few-layer MoS2, WS2, and Wse2. Nano Letters. 2013;13:5627–5634. doi: 10.1021/nl403270k. [DOI] [PubMed] [Google Scholar]
- 22.Kośmider K, Fernández-Rossier J. Electronic properties of the MoS2-WS2 heterojunction. Physical Review B. 2013;87:075451. doi: 10.1103/PhysRevB.87.075451. [DOI] [Google Scholar]
- 23.Padilha J, Peelaers H, Janotti A. & Van de Walle, C. Nature and evolution of the band-edge states in MoS2: From monolayer to bulk. Physical Review B. 2014;90:205420. doi: 10.1103/PhysRevB.90.205420. [DOI] [Google Scholar]
- 24.Dong L, Dongare AM, Namburu RR, O’Regan TP, Dubey M. Theoretical study on strain induced variations in electronic properties of 2H-MoS2 bilayer sheets. Applied Physics Letters. 2014;104:053107. doi: 10.1063/1.4863827. [DOI] [Google Scholar]
- 25.Sharma M, Kumar A, Ahluwalia P, Pandey R. Strain and electric field induced electronic properties of two-dimensional hybrid bilayers of transition-metal dichalcogenides. Journal of Applied Physics. 2014;116:063711. doi: 10.1063/1.4892798. [DOI] [Google Scholar]
- 26.Levita G, Cavaleiro A, Molinari E, Polcar T, Righi M. Sliding properties of MoS2 layers: load and interlayer orientation effects. The Journal of Physical Chemistry C. 2014;118:13809–13816. doi: 10.1021/jp4098099. [DOI] [Google Scholar]
- 27.Liu K, et al. Evolution of interlayer coupling in twisted molybdenum disulfide bilayers. Nature Communications. 2014;5:4966. doi: 10.1038/ncomms5966. [DOI] [PubMed] [Google Scholar]
- 28.Xia J, et al. Current rectification and asymmetric photoresponse in MoS2 stacking-induced homojunctions. 2D Materials. 2017;4:035011. doi: 10.1088/2053-1583/aa79db. [DOI] [Google Scholar]
- 29.Chang J, Larentis S, Tutuc E, Register LF, Banerjee SK. Atomistic simulation of the electronic states of adatoms in monolayer MoS2. Applied Physics Letters. 2014;104:141603. doi: 10.1063/1.4870767. [DOI] [Google Scholar]
- 30.Huang Z, et al. Density functional theory study of Fe adatoms adsorbed monolayer and bilayer MoS2 sheets. Journal of Applied Physics. 2013;114:083706. doi: 10.1063/1.4818952. [DOI] [Google Scholar]
- 31.Wang X-Q, Chen W-G, Zhu Z-L, Jia Y. Electronic and Magnetic Properties Modulated by Adsorption of 3d Transition Metal Atoms in Monolayer and Bilayer MoS2 Sheets. Acta Metallurgica Sinica (English Letters) 2015;28:793–798. doi: 10.1007/s40195-015-0265-1. [DOI] [Google Scholar]
- 32.Wang Y, et al. First-principles study of transition-metal atoms adsorption on MoS2 monolayer. Physica E: Low-dimensional Systems and Nanostructures. 2014;63:276–282. doi: 10.1016/j.physe.2014.06.017. [DOI] [Google Scholar]
- 33.He J, et al. Magnetic properties of nonmetal atoms absorbed MoS2 monolayers. Applied Physics Letters. 2010;96:082504. doi: 10.1063/1.3318254. [DOI] [Google Scholar]
- 34.Lu S-C, Leburton J-P. Electronic structures of defects and magnetic impurities in MoS2 monolayers. Nanoscale Research Letters. 2014;9:1–9. doi: 10.1186/1556-276X-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pandey M, Bothra P, Pati SK. Phase transition of MoS2 bilayer structures. The Journal of Physical Chemistry C. 2016;120:3776–3780. doi: 10.1021/acs.jpcc.5b10904. [DOI] [Google Scholar]
- 36.Fives K, et al. The photoelectron bandstructure of molybdenum disulphide. Journal of Physics: Condensed Matter. 1992;4:5639. [Google Scholar]
- 37.Wan W, et al. Incorporating isolated molybdenum (Mo) atoms into bilayer epitaxial graphene on 4H-SiC (0001) ACS nano. 2013;8:970–976. doi: 10.1021/nn4057929. [DOI] [PubMed] [Google Scholar]
- 38.He K, Poole C, Mak KF, Shan J. Experimental demonstration of continuous electronic structure tuning via strain in atomically thin MoS2. Nano letters. 2013;13:2931–2936. doi: 10.1021/nl4013166. [DOI] [PubMed] [Google Scholar]
- 39.Castellanos-Gomez A, et al. Local strain engineering in atomically thin MoS2. Nano letters. 2013;13:5361–5366. doi: 10.1021/nl402875m. [DOI] [PubMed] [Google Scholar]
- 40.Yan J, et al. Stacking-dependent interlayer coupling in trilayer MoS2 with broken inversion symmetry. Nano letters. 2015;15:8155–8161. doi: 10.1021/acs.nanolett.5b03597. [DOI] [PubMed] [Google Scholar]
- 41.Xia J, Yan J, Shen ZX. Transition metal dichalcogenides: structural, optical and electronic property tuning via thickness and stacking. FlatChem. 2017;4:1–19. doi: 10.1016/j.flatc.2017.06.007. [DOI] [Google Scholar]
- 42.Yang S, Kang J, Yue Q, Yao K. Vapor phase growth and imaging stacking order of bilayer molybdenum disulfide. The Journal of Physical Chemistry C. 2014;118:9203–9208. doi: 10.1021/jp500050r. [DOI] [Google Scholar]
- 43.Liu Q, et al. Tuning electronic structure of bilayer MoS2 by vertical electric field: a first-principles investigation. The Journal of Physical Chemistry C. 2012;116:21556–21562. doi: 10.1021/jp307124d. [DOI] [Google Scholar]
- 44.Komsa H-P, Krasheninnikov AV. Native defects in bulk and monolayer MoS2 from first principles. Physical Review B. 2015;91:125304. doi: 10.1103/PhysRevB.91.125304. [DOI] [Google Scholar]
- 45.Hong J, et al. Direct imaging of kinetic pathways of atomic diffusion in monolayer molybdenum disulphide. Nano Letters. 2017;17:3383. doi: 10.1021/acs.nanolett.6b05342. [DOI] [PubMed] [Google Scholar]
- 46.Kokalj A. XCrySDen–a new program for displaying crystalline structures and electron densities. Journal of Molecular Graphics and Modelling. 1999;17:176–179. doi: 10.1016/S1093-3263(99)00028-5. [DOI] [PubMed] [Google Scholar]
- 47.Kertesz M, Hoffmann R. Octahedral vs. trigonal-prismatic coordination and clustering in transition-metal dichalcogenides. Journal of the American Chemical Society. 1984;106:3453–3460. doi: 10.1021/ja00324a012. [DOI] [Google Scholar]
- 48.Benavente E, Santa Ana M, Mendizábal F, González G. Intercalation chemistry of molybdenum disulfide. Coordination Chemistry Reviews. 2002;224:87–109. doi: 10.1016/S0010-8545(01)00392-7. [DOI] [Google Scholar]
- 49.Hong X, et al. Ultrafast charge transfer in atomically thin MoS2/WS2 heterostructures. Nature Nanotechnology. 2014;9:682–686. doi: 10.1038/nnano.2014.167. [DOI] [PubMed] [Google Scholar]
- 50.Momma K, Izumi F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. Journal of Applied Crystallography. 2011;44:1272–1276. doi: 10.1107/S0021889811038970. [DOI] [Google Scholar]
- 51.Xiao D, Liu G-B, Feng W, Xu X, Yao W. Coupled spin and valley physics in monolayers of MoS2 and other group-vi dichalcogenides. Physical Review Letters. 2012;108:196802. doi: 10.1103/PhysRevLett.108.196802. [DOI] [PubMed] [Google Scholar]
- 52.Xiao D, Chang M-C, Niu Q. Berry phase effects on electronic properties. Reviews of modern physics. 2010;82:1959. doi: 10.1103/RevModPhys.82.1959. [DOI] [Google Scholar]
- 53.Fan X, et al. Modulation of electronic properties from stacking orders and spin-orbit coupling for 3R-type MoS2. Scientific reports. 2016;6:24140. doi: 10.1038/srep24140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.San-Jose P, Parente V, Guinea F, Roldán R, Prada E. Inverse Tunnel Effect of Excitons in Strained BlackPhosphorus. Physical Review X. 2016;6:031046. doi: 10.1103/PhysRevX.6.031046. [DOI] [Google Scholar]
- 55.Feng J, Qian X, Huang C-W, Li J. Strain-engineered artificial atom as a broad-spectrum solar energy funnel. Nature Photonics. 2012;6:866–872. doi: 10.1038/nphoton.2012.285. [DOI] [Google Scholar]
- 56.Rooney, A. P. et al. Observing imperfection in atomic interfaces for van der Waals heterostructures. Nano Letters (2017). [DOI] [PubMed]
- 57.Brauer H, Starnberg H, Holleboom L, Hughes H, Strocov V. Modifying the electronic structure of TiS2 by alkali metal intercalation. Journal of Physics: Condensed Matter. 1999;11:8957. [Google Scholar]
- 58.Miura K, Kamiya S. Observation of the amontons-coulomb law on the nanoscale: frictional forces between MoS2 flakes and MoS2 surfaces. EPL (Europhysics Letters) 2002;58:610. doi: 10.1209/epl/i2002-00439-9. [DOI] [Google Scholar]
- 59.Soler JM, et al. The SIESTA method for ab initio order-N materials simulation. Journal of Physics: Condensed Matter. 2002;14:2745. [Google Scholar]
- 60.Troullier N, Martins JL. Efficient pseudopotentials for plane-wave calculations. Physical Review B. 1991;43:1993–2006. doi: 10.1103/PhysRevB.43.1993. [DOI] [PubMed] [Google Scholar]
- 61.Louie SG, Froyen S, Cohen ML. Nonlinear ionic pseudopotentials in spin-density-functional calculations. Physical Review B. 1982;26:1738–1742. doi: 10.1103/PhysRevB.26.1738. [DOI] [Google Scholar]
- 62.Dion M, Rydberg H, Schröder E, Langreth DC, Lundqvist BI. Van der Waals Density Functional for General Geometries. Physical Review Letters. 2004;92:246401. doi: 10.1103/PhysRevLett.92.246401. [DOI] [PubMed] [Google Scholar]
- 63.Cooper VR. Van der waals density functional: An appropriate exchange functional. Physical Review B. 2010;81:161104. doi: 10.1103/PhysRevB.81.161104. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.