

Summary
Asthma is a chronic inflammatory respiratory disease characterized by airway inflammation, airway hyperresponsiveness and reversible airway obstruction. Understanding the mechanisms that underlie the various endotypes of asthma could lead to novel and more personalized therapies for individuals with asthma. Using a tissue inhibitor of metalloproteinases 1 (TIMP‐1) knockout murine allergic asthma model, we previously showed that TIMP‐1 deficiency results in an asthma phenotype, exhibiting airway hyperreactivity, enhanced eosinophilic inflammation and T helper type 2 cytokine gene and protein expression following sensitization with ovalbumin. In the current study, we compared the expression of Galectins and other key cytokines in a murine allergic asthma model using wild‐type and TIMP‐1 knockout mice. We also examined the effects of Galectin‐3 (Gal‐3) inhibition on a non‐T helper type 2 cytokine interleukin‐17 (IL‐17) to evaluate the relationship between Gal‐3 and the IL‐17 axis in allergic asthma. Our results showed a significant increase in Gal‐3, IL‐17 and transforming growth factor‐β 1 gene expression in lung tissue isolated from an allergic asthma murine model using TIMP‐1 knockout. Gal‐3 gene and protein expression levels were also significantly higher in lung tissue from an allergic asthma murine model using TIMP‐1 knockout. Our data show that Gal‐3 may regulate the IL‐17 axis and play a pivotal role in the modulation of inflammation during experimental allergic asthma.
Keywords: Asthma, Galectin‐3, IL‐17, tissue inhibitor of metalloproteinases 1
Abbreviations
- BALF
bronchoalveolar lavage fluid
- COPD
chronic obstructive pulmonary disease
- ECM
extracellular matrix
- Gal‐3
Galectin‐3
- IFN‐γ
interferon‐γ
- IL‐4
interleukin‐4
- KO
knockout
- MCP‐1
monocyte chemoattractant protein 1
- MMP
matrix metalloproteinase
- OVA
ovalbumin
- siRNA
small interfering RNA
- TGF‐β
transforming growth factor‐β
- Th2
T helper type 2
- TIMP‐1
tissue inhibitors of metalloproteinase‐1
- WT
wild‐type
Introduction
Asthma is a common respiratory disease, afflicting 10% of the US population, affecting 24 million Americans,1, 2, 3 and is characterized by airway inflammation. Asthma‐related emergency room visits and hospitalizations lead to annual costs of almost $2 billion in the USA for urgent care for asthma.4 Although asthma has been traditionally defined as a T helper type 2 (Th2) ‐driven response associated with increased levels of interleukin‐4 (IL‐4), IL‐13 and IL‐55 that is responsive to corticosteroid treatment, almost 50% of asthma cases do not appear to have Th2‐driven inflammation.6 It is being recognized that asthma is a multifaceted disease with heterogeneous phenotypes and corresponding mechanistic variants, termed endotypes.7 A broad division of asthma endotypes separate eosinophilic and non‐eosinophilic asthma, including neutrophilic, paucigranulocytic and mixed granulocytic subtypes.8 Understanding the mechanisms underlying the various endotypes of asthma should lead to novel and more personalized therapies for individuals with asthma.9
Several studies have indicated that cell‐surface‐expressed glycans such as the Galectin family play an important role in trafficking, migration and recruitment of leucocytes during inflammation by virtue of their ability to bind to selectins.10, 11 The Galectin protein family are β‐galactoside‐binding lectins comprising homologous carbohydrate recognition domains, which function in a variety of biological processes including inflammation and allergic pathologies. Galectins may modulate cell adhesion by inhibiting or enhancing adhesive potential between cells or between cells and the extracellular matrix (ECM). Galectins bind glycoconjugates of the ECM via the carbohydrate recognition domains. The extracellular and intracellular concentrations and surface expression of Galectin‐3 (Gal‐3) is increased during inflammation.12, 13, 14 Gal‐3 is expressed in many airway cell types including macrophages, eosinophils, neutrophils and mast cells, and is reported to play an important role in neutrophil recruitment and activation.10, 11, 14 Expression of Gal‐3 was observed in both the nucleus and cytoplasm, which is consistent with a variable subcellular location of the protein participating in different processes in separate asthma inflammatory phenotypes based on cellular location.10 However, the role played by Galectins, specifically Gal‐3, in eosinophil and neutrophil recruitment, particularly in the context of allergic airway inflammation, is unknown.
Asthma is a chronic inflammatory respiratory disease associated with both allergic and non‐allergic mechanisms, with allergic asthma endotypes associated with Th2 elements, and non‐allergic asthma endotypes associated with non‐Th2‐associated elements, such as Th1, Th17 and neutrophilic inflammation.15 With 50% of asthma linked to non‐Th2 mechanisms associated with a variable response to corticosteroid therapy, intricate integration of animal and human studies is necessary to delineate the molecular phenotypes of this asthma type.6
We have previously shown that genetic deficiency of TIMP‐1 results in an asthma phenotype, exhibiting airway hyperreactivity, enhanced cellular peribronchial (eosinophilic) inflammation, increased Th2 cytokine gene and protein expression and significantly reduced dynamic lung compliance following sensitization with ovalbumin.15 Increased TIMP‐1 levels result in ECM accumulation, whereas loss of TIMP‐1 leads to matrix metalloproteinase (MMP) ‐mediated enhanced matrix proteolysis, hence TIMP‐1 levels can modulate ECM turnover. The MMP, MMP‐2 and MMP‐9 efficiently cleave Gal‐3 into two fragments, allowing the carbohydrate domain to bind to glycosylated ligands with higher affinity. The MMPs, however, abrogate the ability of these Gal‐3 fragments to re‐associate and develop homodimers.16, 17 As both TIMP‐1 and Gal‐3 are correlated with ECM turnover, we wanted to examine the levels of Gal‐3 in TIMP‐1 knockout (KO) and wild‐type (WT) mice in both ovalbumin (OVA) sensitized and SHAM groups. We examined the gene expression levels of key members of the Galectin family, specifically Galectin‐1 (Gal‐1), Gal‐3 and Galectin‐9 (Gal‐9) as well as levels of pro‐inflammatory cytokines such as transforming growth factor‐β 1 (TGF‐β 1), IL‐1β, interferon‐γ (IFN‐γ) and IL‐17 with a 2 × 2 factorial experiment examining the effect of OVA‐induced allergic asthma versus SHAM interventions on WT and TIMP‐1 KO mice. We also examined the effects of Gal‐3 inhibition on the key cytokine IL‐17 using Gal‐3 small interfering RNA (siRNA) transfected lung epithelial cells A549, to evaluate the relationship between Gal‐3 and the IL‐17 axis in lung epithelial cells.
Our results showed a significant increase in Gal‐3, IL‐17 and TGF‐β 1 gene expression in lung tissue isolated from OVA‐sensitized TIMP‐1 KO mice compared with WT SHAM mice. Gal‐3 gene and protein expression levels were significantly higher in lung tissue from OVA‐sensitized TIMP‐1 KO mice compared with WT OVA‐sensitized mice or TIMP‐1 KO SHAM mice. Our data supports the premise that Gal‐3 may play a role in the regulation of the IL‐17A axis during experimental allergic asthma.
Material and methods
Materials
Tissue procurement: Lung tissue and serum were obtained from C57BL/6 TIMP‐1 KO and WT mice, that were both SHAM and OVA‐sensitized as previously described.15 Briefly, on days 0 and 14, TIMP‐1 KO and WT mice (age 6–8 weeks) were sensitized by 200 μl intraperitoneal injection with 10 μg chicken OVA (Grade III; Sigma, St Louis, MO) and 1 mg alum adjuvant (AlK[SO]4[H2O]12) emulsified in sterile PBS (OVA groups). SHAM mice received OVA‐free injections. On day 21, mice were challenged with 30 ml of aerosolized 1% (weight/volume) OVA or PBS (SHAM group) for 30 min on 7 consecutive days using an ultrasonic nebulizer. On day 29 mice were anaesthetized with intraperitoneal pentobarbital at a 50 mg/kg dose, and lung mechanics were measured during methacholine challenge. Following euthanasia, lungs were harvested for histopathology, protein and gene expression analyses. On days 0 and 28, blood was drawn for IgE analysis to confirm OVA sensitization. Serum OVA‐specific IgE was measured by ELISA. Lung tissue was obtained from mice anaesthetized with halothane and exsanguinated via the inferior vena cava on day 28 or 29. Animal protocols were approved by Institutional Animal Care and Use Committees of the University at Buffalo and Veterans Administration Health Care System of Western New York. Six samples/group were obtained.
Cell line: A549 (ATCC® CCL‐185™) lung epithelial cells were obtained from the American Type Culture Collection (Manassas, VA) and are grown as adherent cultures in complete Dulbecco's modified Eagle's medium (Invitrogen, Grand Island, NY), supplemented with 10% (volume/volume) fetal bovine serum (Hyclone, Logan, UT), 100 units/ml penicillin and 100 units/ml streptomycin (Gibco, Grand Island, NY). All cells were maintained in a humidified incubator with 5% CO2 at 37°.
Cell viability: A549 cells were treated with Gal‐3 siRNA (concentrations 10–20 pmol) and the Gal‐3 inhibitor (Ac‐SDKP) (concentrations 5–10 nmol) in our experiments and we tested the cell viability at those concentrations using both Trypan blue dye exclusion and MTT assay (Vybrant® MTT Cell Proliferation Assay; Thermo Fisher Scientific, Waltham, MA). Data from both assays showed no significant toxicity and > 95% cell viability in cells treated with Gal‐3 siRNA at both 10 and 20 pmol concentrations and Gal‐3 inhibitor, Ac‐SDKP, at both 5 and 10 nmol concentrations, respectively.
Small‐interfering RNA: Gal‐3‐siRNA (5′‐GGGAAUGAUGUUGCCUUCCACUUUA‐3′) and control‐siRNA (5′‐UUCUCCGAACGUGUCACGUTT‐3′) were produced by Thermo Fisher Scientific. Transient transfection was performed using the Lipofectamine RNAi MAX reagent (Invitrogen) following the manufacturer's protocols. A549 cells (1 × 105 cells/ml) were seeded into a six‐well plate at 24 hr before transfection. The lyophilized siRNAs were dissolved in diethylpyrocarbonate‐treated water according to the manufacturer's instructions and A549 cells were treated with siRNA. Cells were harvested within 48–72 hr post transfection for further experiments.
Methods
RNA extraction: Cytoplasmic RNA was extracted using Trizol reagent (Invitrogen‐Life Technologies, Carlsbad, CA) using lung tissue (entire lung) that was stored at −80° in RNAlater (Ambion, Austin, TX). The amount of RNA was quantified using a Nano‐Drop ND‐1000 spectrophotometer (Nano‐Drop Technologies, Wilmington, DE) and isolated RNA was stored at −80° until batch analysis was performed.
Real‐time quantitative PCR: Gene expression studies were performed on lung tissue obtained from OVA‐sensitized as well as SHAM‐treated groups of both TIMP‐1 KO and WT mice. Additionally, gene expression studies were performed on lung epithelial cells (A549 cell line) in which the Gal‐3 gene expression was silenced using siRNA targeting Gal‐3. Cytoplasmic RNA was extracted using Trizol, RNA was reverse transcribed to cDNA and relative abundance of Gal‐1, Gal‐3, Gal‐9, TGF‐β 1, TGF‐β 3, IL‐1β, IFN‐γ) and IL‐17(A–E) mRNA was quantified using real‐time PCR. Gene expression levels were expressed as the Transcript Accumulation Index. Untreated cells were used as controls. Five hundred nanograms of total RNA was used for the reverse transcription reaction (25 μl total volume) with the First‐Strand cDNA synthesis kit (GE Healthcare, Piscataway, NJ), according to the manufacturer's instruction. One microlitre of the resultant cDNA from the reverse transcription reaction was employed as the template in PCRs using commercially synthesized, well validated PCR primer sequences obtained from www.realtimeprimers.com. The housekeeping gene, β‐actin, was used as an internal control. The final primer concentration used in the PCR was 0·1 μm. PCR was performed using a quantitative PCR machine (Mx3005P; Stratagene, La Jolla, CA) with the following PCR conditions: 95° for 3 min, followed by 24 cycles of 95° for 40 seconds, 58° for 30 seconds and 72° for 1 min; the final extension was at 72° for 5 min. Gene expression was calculated using the comparative CT method.18 The threshold cycle (Ct) of each sample was determined, and the relative level of a transcript (2ΔCt) was calculated by obtaining ΔCt (test Ct − β‐actin Ct) and the Transcript Accumulation Index was calculated as 2−ΔΔCT.
Galectin‐3 ELISA: We measured Gal‐3 levels in serum obtained from TIMP‐1 KO and WT mice in both OVA‐sensitized and SHAM groups using a commercially available ELISA kit (RayBio® Mouse Gal‐3 ELISA Kit Cat # ELM‐Galectin‐3 RayBiotech, Inc., Norcross, GA 30092). This assay has excellent specificity for detection of Gal‐3 with an assay range of 10–10 000 pg/ml and minimum detectability/sensitivity of 6 pg/ml. No significant cross‐reactivity between analytes and analogues was observed.
Western blotting analysis: Protein was extracted from lung tissue obtained from the four groups of mice, namely the TIMP‐1 KO and WT mice, that were both SHAM‐ and OVA‐sensitized, using the T‐PER Tissue Protein Extraction Reagent (Cat #78510; Thermo Fisher Scientific). Protein concentrations were measured using the Nano‐Drop ND‐1000 spectrophotometer (Nano‐Drop Technologies).
Fifty micrograms of protein was loaded on a 10–20% Novex™ electrophoresis gel (Cat # XP00100BOX, Tris–Glycine, 10‐well mini gel; Thermo Fisher Scientific). Protein separation by gel electrophoresis was followed by electro‐blotting and protein transfer onto a PVDF membrane using the iBlot™ system (Original iBlot® Gel Transfer Device; Thermo Fisher Scientific). Protein blots were blocked with Blocking buffer (3% PBS in 1× PBST) and incubated overnight with the following primary mouse antibodies: anti‐Gal‐3 antibody (Cat # ab2785; Abcam, Cambridge, MA) (1 : 500) or anti‐β‐actin (Cat # ab8226; Abcam) (1 : 1000). After an overnight incubation with primary antibodies, blots were washed three times in 1× PBST followed by treatment with anti‐mouse alkaline phosphatase‐conjugated secondary antibody at a dilution of 1 : 5000 (in 2·5% bovine serum albumin in PBST buffer) for 1·5 hr at room temperature. All antibodies were obtained from Abcam. β‐Actin expression was used to monitor equal protein loading in each lane. Immunoreactivity was detected using an enhanced chemiluminescence detection using the 1‐Step™ NBT/BCIP Substrate Solution (Cat #34042; Thermo Fisher Scientific). For densitometric analysis of Western blots, alpha image software (Alpha Innotech, San Leandro, CA) was used.
Statistical analysis: Data are expressed as mean ± SD. Statistical comparisons were made between genotypes using a two‐tailed t‐test. A P‐value of < 0·05 was considered statistically significant. Analyses were performed using prism (GraphPad Software, Inc., San Diego, CA) software.
Results
Expression levels of Galectins and key cytokines in TIMP‐1 KO mice in comparison with WT SHAM mice
We examined the expression levels of key Galectins and cytokines that are relevant to allergic asthma using real‐time PCR. Comparative analysis was done to examine the gene expression levels of Gal‐1, Gal‐3, ‐9, TGF‐β 1, TGF‐β 3, IL‐1β, IFN‐γ and IL‐17 between OVA‐sensitized TIMP‐1 KO OVA (Group III) and WT SHAM (Group II) (Fig. 1a). Our data showed a significant increase in the Gal‐3 (130% increase; P < 0·005), TGF‐β 1 (394% increase; P < 0·004) and IL‐17 (56% increase; P < 0·003) and a decrease in Gal‐9 (47% decrease, P < 0·004) gene expression between the OVA‐sensitized TIMP‐1 KO group and the WT SHAM controls. Comparative analysis to examine gene expression of Galectins and inflammatory cytokines above were also performed in lung tissue obtained from OVA‐sensitized TIMP‐1 KO mice versus OVA‐sensitized WT mice. Our data (Fig. 1b) showed trends that were similar to those observed between Group II and Group III. Specifically, we observed significant increase in the Gal‐3 (95% increase; P < 0·01), TGF‐β 1 (750% increase; P < 0·00001) and IL‐17 (41% increase; P < 0·05) and a decrease in Gal‐9 (42% decrease, P < 0·05) gene expression between the OVA‐sensitized TIMP‐1 KO mice and the OVA‐sensitized WT mice.
Figure 1.
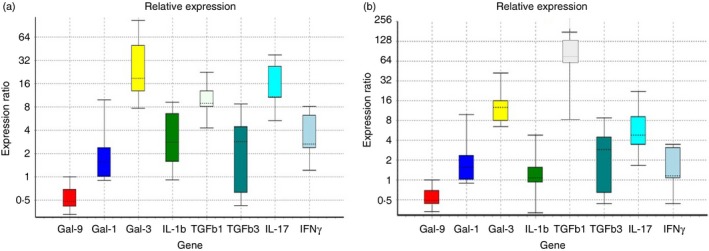
Gene expression comparison of Galectins and inflammatory cytokines in lung tissue obtained from (a) ovalbumin (OVA) ‐sensitized tissue inhibitor of metalloproteinases 1 knockout (TIMP‐1 KO) mice versus wild‐type (WT) SHAM mice and (b) OVA‐sensitized TIMP‐1 KO mice versus OVA‐sensitized WT mice. Comparative real‐time quantitative PCR analysis showing relative gene expression of Galectin‐1 (Gal‐1), Gal‐3, ‐9, transforming growth factor‐β 1 (TGF‐β 1), TGF‐β 3, interleukin‐1β (IL1β), interferon‐γ (IFN‐γ) and IL‐17 in lung tissue obtained from OVA‐sensitized TIMP‐1 KO mice versus WT SHAM mice (a) and OVA‐sensitized TIMP‐1 KO mice versus OVA‐sensitized WT mice (b), respectively. Data normalized to the housekeeping gene β‐actin. Gene expression was calculated using the comparative CT method. Results are expressed as the mean ± SD, n = 3 separate experiments. [Colour figure can be viewed at wileyonlinelibrary.com]
Serum Gal‐3 levels in the four study groups
We evaluated the Gal‐3 protein levels via ELISA in the serum obtained from mice in the following four study groups: WT OVA (Group I), WT SHAM (Group II), TIMP‐1 KO OVA (Group III), and TIMP‐1 KO SHAM (Group IV). Our results (Fig. 2) show a significant increase in Gal‐3 levels in OVA‐sensitized animals both in the WT OVA and TIMP‐1 KO OVA mice compared with the SHAM groups. However, on comparisons between OVA‐sensitized TIMP‐1 KO mice and OVA‐sensitized WT mice, the Gal‐3 levels were significantly higher in the OVA‐sensitized TIMP‐1 KO mice (45·68 ± 12·52 versus 36·76 ± 4·93 pg/ml; P < 0·05). No significant differences in the Gal‐3 levels were observed between the WT SHAM and the TIMP‐1 KO SHAM mice (7·15 ± 5·56 versus 8·31 ± 1·1 pg/ml; P = NS). Comparison of Gal‐3 levels between WT SHAM and the TIMP‐1 KO OVA mice (7·15 ± 5·56 versus 45·68 ± 12·52 pg/ml; P < 0·001) showed a significant increase in Gal‐3 levels in the TIMP‐1 KO OVA mice.
Figure 2.
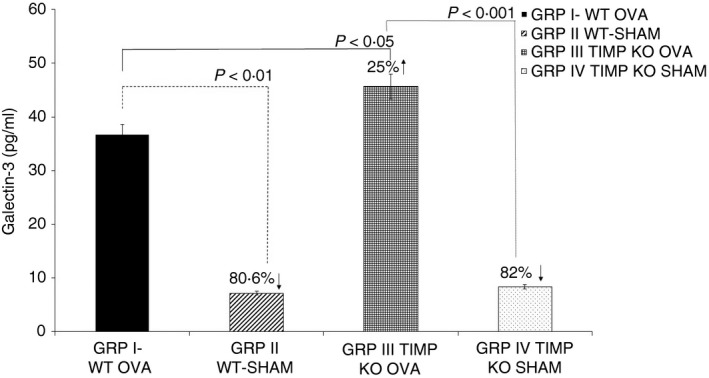
Increased Galectin‐3 levels in tissue inhibitor of metalloproteinases 1 knockout (TIMP‐1 KO) ovalbumin (OVA) ‐sensitized mice serum. Galectin‐3 levels were measured by ELISA in serum samples from mice in the four study groups: wild‐type (WT) OVA (Group I), WT SHAM (Group II), TIMP‐1 KO OVA (Group III), and TIMP‐1 KO SHAM (Group IV). Data are presented as mean ± SD from n = 6 samples per group.
Galectin‐3 gene and protein expression in the four study groups
We evaluated the Gal‐3 gene and protein expression in lung tissue obtained from mice in the four study groups: WT OVA (Group I), WT SHAM (Group II), TIMP‐1 KO OVA (Group III) and TIMP‐1 KO SHAM (Group IV). Gal‐3 gene expression levels were quantified using real time quantitative PCR. Our results (Fig. 3a) show an increase in Gal‐3 expression levels in the WT OVA (0·88 ± 0·04; P = NS) and TIMP‐1 KO OVA (0·98 ± 0·03; P < 0·05) mice compared with the WT SHAM (0·78 ± 0·005) mice. No significant difference in Gal‐3 gene expression was observed on comparison between OVA‐sensitized TIMP‐1 KO (0·98 ± 0·03; P < 0·05) mice and OVA‐sensitized SHAM (0·91 ± 0·02; P = NS) mice. Figure 3(b) shows representative data of Gal‐3 protein expression levels measured by Western blot analysis in the four study groups. Our cumulative Gal‐3 protein expression results (Fig. 3c) show significantly higher Gal‐3 levels in both the OVA‐sensitized TIMP‐1 KO and OVA‐sensitized SHAM mice compared with respective non‐OVA‐sensitized controls, indicating that OVA sensitization significantly increases Gal‐3 protein expression. Comparison of Gal‐3 protein levels between WT SHAM versus the TIMP‐1 KO OVA mice showed a significant increase (72%; P < 0·0001) in Gal‐3 protein levels in the TIMP‐1 KO OVA mice.
Figure 3.

Increased Galectin‐3 (Gal‐3) levels in tissue inhibitor of metalloproteinases 1 knockout (TIMP KO) ovalbumin (OVA) ‐sensitized mouse lungs. Galectin‐3 gene and protein expression in lung tissue obtained from our study groups: wild‐type (WT) OVA (Group I), WT SHAM (Group II), TIMP‐1 KO OVA (Group III) and TIMP‐1 KO SHAM (Group IV). (a) Relative Gal‐3 gene expression in lung tissue among the four groups. (b) Gal‐3 protein expression in lung tissue via Western blot. (c) Gal‐3 protein levels in lung tissue via ELISA. Data are presented as mean ± SD from n = 6 samples per group. [Colour figure can be viewed at wileyonlinelibrary.com]
Galectin‐3 gene silencing modulates levels of IL‐17 in lung epithelial A549 cells
We observed a significant increase in IL‐17 levels in the OVA‐sensitized TIMP‐1 KO mice. There is evidence that IL‐17 attenuates allergic responses in asthma, and is important for neutrophil recruitment.19, 20, 21 Interleukin‐17 has been detected in bronchial biopsies, bronchoalveolar lavage fluid and sputum from asthma patients.11, 22, 23, 24, 25, 26, 27, 28 We posit that Gal‐3 plays a key role in asthma immunopathogenesis by modulating the inflammatory response via the steroid‐insensitive Th17 pathway. To investigate the role of Gal‐3 in the immunomodulatory response in lung epithelial cells, we transfected the lung epithelial A549 cells using Gal‐3 siRNA and 48 hr post transfection, evaluated the Gal‐3 gene expression levels (Fig. 4) as well as the gene expression levels of several members of the IL‐17 family, specifically IL‐17A‐E (Fig. 6). A known Gal‐3 inhibitor N‐acetyl‐seryl‐aspartyl‐lysyl‐proline (Ac‐SDKP),29 a naturally occurring tetrapeptide was used as a control, in addition to an siRNA with scrambled sequences as a transfection control. Our results (Fig. 4) showed a 61% (P < 0·01) and 65% (P < 0·001) decrease in Gal‐3 gene expression on treatment with 10 and 20 pmol of Gal‐3 siRNA, respectively. Further, treatment of A549 cells with the Gal‐3 inhibitor peptide Ac‐SDKP resulted in a 35% (P < 0·05) and 78% (P < 0·001) decrease in Gal‐3 gene expression at concentrations of 5 and 10 nmol of the inhibitor, respectively.
Figure 4.
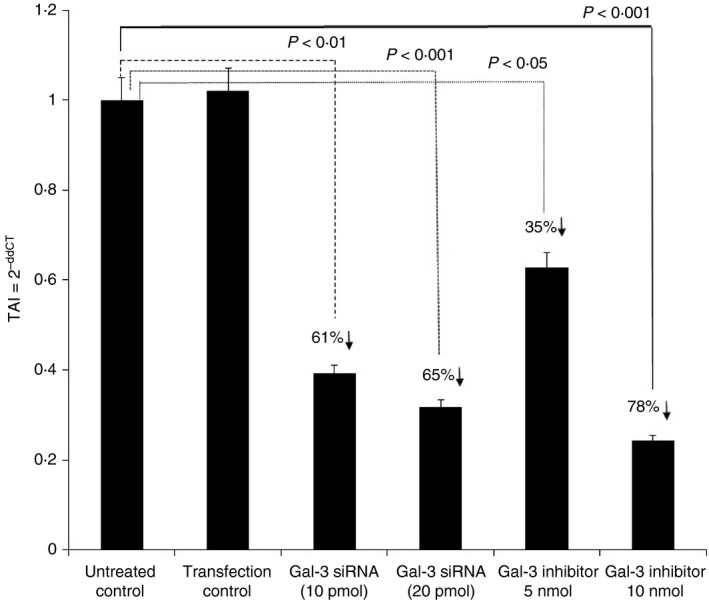
Effect of Galectin‐3 (Gal‐3) inhibition in lung epithelial cell line A549 on Gal‐3 expression. Gal‐3 gene expression in A549 cells treated with Gal‐3 small interfering RNA (siRNA) and Gal‐3 inhibitor (Ac‐SDKP) Data are presented as mean ± SD from n = 3 separate experiments.
Figure 6.
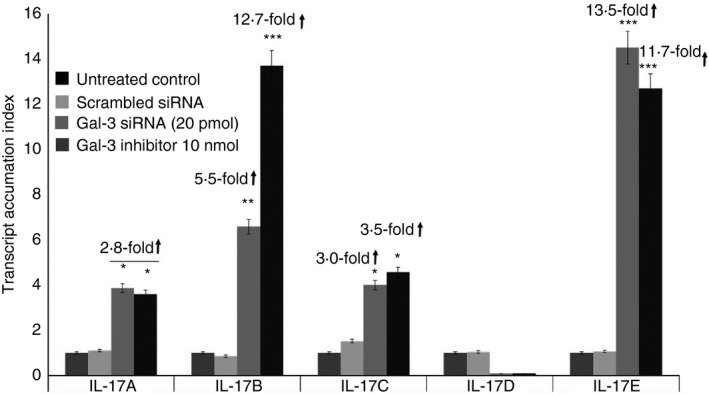
Effect of Galectin‐3 (Gal‐3) gene silencing on interleukin‐17 (IL‐17) gene expression in lung epithelial cell line A549. Gene expression levels of IL‐17 subtypes A–E in A549 cells treated with Gal‐3 small interfering RNA (siRNA) and Gal‐3 inhibitor (Ac‐SDKP). Data are presented as mean ± SD from n = 3 separate experiments. *P < 0.05, **P < 0.01, ***P < 0.0001.
We evaluated the effect of Gal‐3 inhibition on the gene expression of some key pro‐inflammatory cytokines namely, monocyte chemoattractant protein (MCP‐1), IL‐8 and IL‐6 by treating the A549 cells with 10 nmol Gal‐3 inhibitor peptide Ac‐SDKP. Statistical comparisons were performed between Ac‐SDKP‐treated A549 cells and untreated A549 cells used as controls. Our results (Fig. 5) showed that treatment with Gal‐3 inhibitor peptide (10 nmol) resulted in a 45% decrease (P < 0·05) in MCP‐1 gene expression; a 65% increase (P < 0·01) in IL‐8 gene expression and a 16% increase (P = NS) in IL‐6 gene expression levels compared with the respective untreated controls (Fig. 5).
Figure 5.
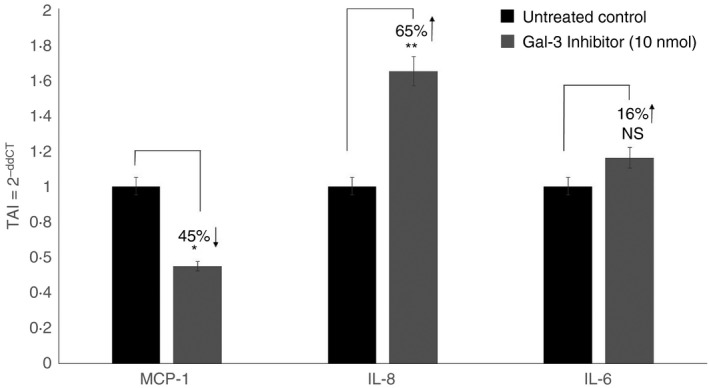
Effect of Galectin‐3 (Gal‐3) inhibition on gene expression of interleukin‐8 (IL‐8), IL‐6 and monocyte chemoattractant protein 1 (MCP‐1) in lung epithelial cell line A549. A549 cells were treated with Gal‐3 inhibitor (Ac‐SDKP) and gene expression of IL‐8, IL‐6 and MCP‐1 was evaluated using real‐time PCR. Data are presented as mean ± SD from n = 3 separate experiments. *P < 0.05, **P < 0.01.
Galectin‐3 siRNA (20 pmol) treatment resulted in a 2·8‐fold increase (P < 0·05); 5·5‐fold increase (P < 0·01); 3·0‐fold increase (P < 0·05); and 13·5‐fold increase (P < 0·0001) in the gene expression levels of IL‐17A, IL‐17B, IL‐17C and IL‐17E (Fig. 6), respectively. Treatment with Gal‐3 inhibitor peptide (10 nmol) resulted in a 2·8‐fold increase (P < 0·05); 12·7‐fold increase (P < 0·0001); 3·5‐fold increase (P < 0·05); and 11·7‐fold increase (P < 0·0001) in the gene expression levels of IL‐17A, IL‐17B, IL‐17C and IL‐17E, respectively (Fig. 5). Both Gal‐3 siRNA silencing and Gal‐3 inhibition with Ac‐SDKP resulted in a decrease in IL‐17D gene expression but the effect was not statistically significant. These data suggest that Gal‐3 may play a pivotal role in the balance between a pro‐inflammatory response and a protective anti‐inflammatory response by modulation of IL‐17 levels that either exacerbate or attenuate the allergic response in asthma.
Discussion
Asthma appears to be due to allergic and non‐allergic mechanisms, with allergic asthma associated with Th2‐associated elements, and non‐allergic asthma associated with non‐Th2‐associated elements, such as Th1, Th17 and neutrophilic inflammation.30 With 50% of asthma linked to non‐Th2 mechanisms, that are associated with variable response to corticosteroid therapy, intricate integration of animal and human studies is urgently needed to delineate the molecular phenotypes of this asthma type.6
One non‐Th2‐mediated endotype of asthma associated with steroid resistance is a subtype with high IL‐17 and high IFN‐γ levels, which may be possible therapeutic targets.31 Another study by McKinley et al.,32 also notes Th17 cells linked to steroid resistance in a murine model of asthma. Investigators examining expression of IL‐17A and IL‐17F in the airways of asthmatic patients detected increased IL‐17A and IL‐17F immunoreactivity in lung tissue sections obtained via bronchoscopy, and measured L‐17A and IL‐17F cytokine mRNA expression that increased with asthma severity.33
The airway smooth muscle hypertrophy associated with asthma may interact with components of the ECM, the enzymes that degrade the ECM, MMP and the inhibitors of MMP enzymes, TIMP.34 Similar to the observations of our TIMP‐1 KO study on murine asthma, Gal‐3 is associated with ECM turnover and is also reported to be involved in many aspects in asthma, such as eosinophil recruitment airway remodelling, development of a Th2 phenotype as well as increased expression of inflammatory mediators.11, 35, 36 The imbalance between MMPs (particularly MMP‐2 and MMP‐9) and TIMPs (particularly TIMP‐1) plays an active role in airway remodelling in asthma, with the MMP‐9/TIMP‐1 ratio elevated in individuals with asthma compared with non‐asthmatic control participants.37, 38, 39, 40
In a previous study, Sands et al. employed TIMP‐1 KO mice to assess the allergic response of a murine model of OVA‐induced allergic asthma, which suggests that TIMP‐1 plays a protective role by preventing airway hyperreactivity and modulating inflammation, remodelling and cytokine expression in this animal model of asthma.15 Galectins are β‐galactoside binding lectins that regulate biological and cellular processes such as production of inflammatory mediators, cell adhesion, migration and apoptosis.41, 42 Galectins are expressed by various cell types, specifically eosinophils, which are present in large numbers in the lungs and airways in allergic asthma. Various members of the Galectin family differentially regulate eosinophil recruitment, activation and apoptosis and therefore exert a pro‐ or anti‐inflammatory outcome.
To determine the impact that MMP‐2 and MMP‐9 may have on Gal‐3 and components of the ECM, we examined the expression of Gal‐3 in the TIMP1 KO mice exposed to OVA challenges compared with WT controls. MMP‐2 and MMP‐9 efficiently cleave Gal‐3 into two fragments, allowing the carbohydrate domain to bind to glycosylated ligands with higher affinity, but abolish the ability of the molecules to self‐associate and develop homodimers.16, 17 Given that TIMP‐1 KO mice had increased levels of MMP‐2 and MMP‐9, we expected less intact Gal‐3 leading to a reduced Th2 response. Acute allergen exposure is believed to result in increased recruitment of Gal‐3‐expressing inflammatory cells such as macrophages and eosinophils to the airways resulting in elevated levels of Gal‐3 in the lung.11
A comparative analysis of the gene expression levels of key Galectins and cytokines in lung tissue, between OVA‐sensitized TIMP‐1 KO and WT SHAM controls showed a significant increase in Gal‐3, TGF‐β 1 and IL‐17 expression in OVA‐sensitized TIMP‐1 KO mice (Fig. 1a). Similar gene expression trends were observed when gene expression levels of Gal‐1, Gal‐3, Gal‐9, TGF‐β 1, TGF‐β 3, IL‐1β, IFN‐γ and IL‐17 were compared in OVA‐sensitized TIMP‐1 KO and OVA‐sensitized WT mice (Fig. 1b). However significantly higher TGF‐β 1 gene expression levels were observed in OVA‐sensitized TIMP‐1 KO mice when compared with the OVA‐sensitized WT mice (Fig. 1b). TGF‐β 1 is believed to be a neutrophil chemoattractant and may promote Th17 differentiation,33 so increased expression of TGF‐β 1 may also contribute the increased IL‐17 gene expression in these OVA‐sensitized mice.
We evaluated the gene and protein expression levels of Gal‐3 in lung tissue as well as soluble Gal‐3 levels in serum obtained from OVA‐sensitized TIMP‐1 KO and WT mice and compared the levels to those obtained from WT SHAM and TIMP‐1 KO SHAM mice. Our data showed a significant increase in Gal‐3 expression in OVA‐sensitized animals from both the WT and TIMP‐1 KO groups compared with the SHAM controls, corroborating reports from previous studies suggesting that Gal‐3 plays a pro‐inflammatory role in allergic asthma by promoting eosinophil trafficking and migration. In contrast to its pro‐inflammatory role in allergic asthma, some studies have shown that Gal‐3 can suppress eosinophil infiltration and normalize pulmonary function in acute as well as chronic settings of allergic asthma by negatively regulating gene expression of suppressors of cytokine signalling, which play an important role in controlling the Th1—Th2 balance.43, 44, 45
In the context of asthma, several studies have highlighted the role of Gal‐3 as a key mediator of the Th1–Th2 balance. In a murine model of OVA‐induced asthma phenotype, Gal‐3 was expressed by airway immune cells, particularly macrophages in bronchoalveolar lavage fluid (BALF), with increased Gal‐3 expressed in the BALF of OVA‐challenged mice compared with the control mice.11 In Gal‐3 KO mice (gal3 −/−) compared with WT mice there were fewer eosinophils; there was lower goblet cell metaplasia and Th2 response but a higher Th1 response after OVA challenge.11 Examination of the bronchial epithelium samples noted increased Gal‐3, and neutrophils in the small airways of individuals of chronic obstructive pulmonary disease (COPD) compared with individuals without COPD who smoke.46 In gal3 −/− mice challenged with chronic OVA exposure of 12 weeks, examination of the lung tissue and BALF reveals decreased airway remodelling, decreased subepithelial fibrosis, decreased airway smooth muscle thickness, and decreased peribronchial angiogenesis, compared with wild‐type mice chronically exposed to OVA.35 Gal3 −/− mice chronically exposed to OVA also had lower levels of IL‐5, IL‐13 and TGF‐β compared with WT mice with chronic OVA exposure.35 In a mouse model of pulmonary infection with inhaled Francisella novicida, WT mice had increased Gal‐3 expression and extracellular release after pulmonary infection, as opposed to gal3 −/− mice with reduced inflammatory response (tumour necrosis factor‐α, IL‐10, IL‐1β), decreased neutrophil activity in the lung tissue, and improved survival.47
Further, Gal‐3 was decreased in BALF from individuals with COPD compared with smokers without COPD and non‐smoking controls, and addition of exogenous Gal‐3 to human alveolar macrophages obtained from BALF led to increased efferocytosis.48 These studies in Gal‐3‐deficient mice and data from the current study suggest a putative role for Gal‐3 in modulating allergic asthma.
We observed a significant increase in IL‐8 and a decrease in MCP‐1 gene expression levels in A549 cells treated with Gal‐3 inhibitor (Fig. 5), suggesting that Gal‐3 may regulate the balance between pro‐inflammatory and anti‐inflammatory responses that modulates the progression of allergic asthma. Further, we observed that Gal‐3 silencing resulted in a significant increase in IL‐17A, ‐B, ‐C and E gene expression levels (Fig. 6) in A549 lung epithelial cells, indicating that Gal‐3 is a key mediator of inflammation and exerts its effects through the modulation of other cytokine/chemokine regulators such as TGF‐β. Interleukin‐17 induces MMPs, which aids collagen destruction, and neutrophil recruitment could further sustain pro‐inflammatory responses that might be particularly damaging in asthma. Th17 cell differentiation, characterized by IL‐17 production, depends on the presence of TGF‐β and IL‐6. The presence of TGF‐β drives Th17 cell differentiation and down‐regulates Th1 cell differentiation through the inhibition of IFN‐γ, whereas IL‐12 inhibits the development of Th17 cells. Traditionally, TGF‐β is an anti‐inflammatory cytokine, a paradox given its role as a crucial factor for the differentiation of the IL‐17‐producing T‐cell subset Th17. This highlights the fact that it is a cytokine with numerous functions and that TGF‐β regulates a plethora of biological processes including inflammation and tissue repair. Studies showed that IL‐17 monoclonal antibody was moderately effective only with individuals with good responses to bronchodilator use, suggesting that IL‐17 contributes to less severe disease.49, 50, 51
Pro‐inflammatory cytokines such as IL‐17 are also potent inducers of MUC5B and also regulate the expression of Gal‐3 in allergic asthma, as observed in our study. It is speculated, that protein–protein interactions between MUC5B and Gal‐3 could alter the biophysical properties of mucus – Gal‐3 via carbohydrate‐dependent interactions with cell‐surface mucins maintaining mucosal barrier function in the eye.52 Mucin–Gal‐3 interactions may contribute substantially to the integrity of mucosal epithelial barrier function and may regulate the transcellular flux of extracellular components into epithelial cells.53, 54 Additional studies to evaluate associations between Gal‐3 and the Mucin family and their role in allergic asthma need to be investigated.
In summary, our study suggests that Gal‐3 may be a key regulator of the fine balance between pro‐inflammatory and anti‐inflammatory responses in the progression of allergic asthma. These studies have enormous clinical translational significance, as administration of exogenous Gal‐3 could potentially serve as a therapeutic tool for allergic asthma. Increased Gal‐3 levels in the bronchial tissues, derived from asthmatic individuals who are refractory to corticosteroid therapy, can predict the response or non‐response of the patient to the treatment regimen before treatment with the anti‐IgE multiclonal antibody, omalizumab,.55 The authors posit that the rationale for this action is the ability of Gal‐3 to disrupt IgE and FcεR1 interactions, improving anti‐IgE efficacy.
Conclusion
Our results showed a significant increase in Gal‐3 gene and protein expression in OVA‐treated TIMP‐1 KO mice compared with the WT controls, which is associated with increased expression of inflammatory mediators such as IL‐17 that further contribute to asthma progression. This study confirms that Gal‐3 plays a significant role in the pathophysiological mechanisms in asthma and may be a candidate biomarker or therapeutic target for specific asthma endotypes.
Funding
University at Buffalo, Faculty start‐up funds to MJM.
Author contributions
MJM conceived the study. MJM, SDM, MS, initiated the study design and EAJ and RA helped with implementation. MJM, SDM, SAS NUP and JLR participated in data analysis. All authors contributed to refinement of the study protocol and approved the final manuscript.
Disclosure
The authors of this manuscript do not have any competing interests.
Acknowledgements
Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR001412 to the University at Buffalo. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Contributor Information
Manoj J. Mammen, Email: mammen@buffalo.edu
Supriya D. Mahajan, Email: smahajan@buffalo.edu.
References
- 1. Bateman ED, Hurd SS, Barnes PJ, Bousquet J, Drazen JM, FitzGerald M et al Global strategy for asthma management and prevention: GINA executive summary. Eur Respir J 2008; 31:143–78. [DOI] [PubMed] [Google Scholar]
- 2. Akinbami LJ, Moorman JE, Liu X, National Center for Health Statistics (U.S.) . Asthma Prevalence, Health Care Use, and Mortality: United States, 2005–2009. Hyattsville, MD: U.S. Dept. of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics, 2011:16 pp. [PubMed] [Google Scholar]
- 3. Masoli M, Fabian D, Holt S, Beasley R. Global Initiative for Asthma P. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy 2004; 59:469–78. [DOI] [PubMed] [Google Scholar]
- 4. Bender BG, Rand C. Medication non‐adherence and asthma treatment cost. Curr Opin Allergy Clin Immunol 2004; 4:191–5. [DOI] [PubMed] [Google Scholar]
- 5. Venkayya R, Lam M, Willkom M, Grunig G, Corry DB, Erle DJ. The Th2 lymphocyte products IL‐4 and IL‐13 rapidly induce airway hyperresponsiveness through direct effects on resident airway cells. Am J Respir Cell Mol Biol 2002; 26:202–8. [DOI] [PubMed] [Google Scholar]
- 6. Wenzel SE. Emergence of biomolecular pathways to define novel asthma phenotypes. Type‐2 immunity and beyond. Am J Respir Cell Mol Biol 2016; 55:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lotvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A et al Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol 2011; 127:355–60. [DOI] [PubMed] [Google Scholar]
- 8. Simpson JL, Scott R, Boyle MJ, Gibson PG. Inflammatory subtypes in asthma: assessment and identification using induced sputum. Respirology 2006; 11:54–61. [DOI] [PubMed] [Google Scholar]
- 9. Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet 2008; 372:1107–19. [DOI] [PubMed] [Google Scholar]
- 10. Gil CD, La M, Perretti M, Oliani SM. Interaction of human neutrophils with endothelial cells regulates the expression of endogenous proteins annexin 1, galectin‐1 and galectin‐3. Cell Biol Int 2006; 30:338–44. [DOI] [PubMed] [Google Scholar]
- 11. Zuberi RI, Hsu DK, Kalayci O, Chen HY, Sheldon HK, Yu L et al Critical role for galectin‐3 in airway inflammation and bronchial hyperresponsiveness in a murine model of asthma. Am J Pathol 2004; 165:2045–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao P, Gibson PG, Baines KJ, Yang IA, Upham JW, Reynolds PN et al Anti‐inflammatory deficiencies in neutrophilic asthma: reduced galectin‐3 and IL‐1RA/IL‐1β . Respir Res 2015; 16:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nieminen J, St‐Pierre C, Bhaumik P, Poirier F, Sato S. Role of galectin‐3 in leukocyte recruitment in a murine model of lung infection by Streptococcus pneumoniae . J Immunol 2008; 180:2466–73. [DOI] [PubMed] [Google Scholar]
- 14. Nieminen J, St‐Pierre C, Sato S. Galectin‐3 interacts with naive and primed neutrophils, inducing innate immune responses. J Leukoc Biol 2005; 78:1127–35. [DOI] [PubMed] [Google Scholar]
- 15. Sands MF, Ohtake PJ, Mahajan SD, Takyar SS, Aalinkeel R, Fang YV et al Tissue inhibitor of metalloproteinase‐1 modulates allergic lung inflammation in murine asthma. Clin Immunol 2009; 130:186–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ochieng J, Fridman R, Nangia‐Makker P, Kleiner DE, Liotta LA, Stetler‐Stevenson WG et al Galectin‐3 is a novel substrate for human matrix metalloproteinases‐2 and ‐9. Biochemistry 1994; 33:14109–14. [DOI] [PubMed] [Google Scholar]
- 17. Ochieng J, Green B, Evans S, James O, Warfield P. Modulation of the biological functions of galectin‐3 by matrix metalloproteinases. Biochim Biophys Acta 1998; 1379:97–106. [DOI] [PubMed] [Google Scholar]
- 18. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative CT method. Nat Protoc 2008; 3:1101. [DOI] [PubMed] [Google Scholar]
- 19. Laan M, Cui ZH, Hoshino H, Lotvall J, Sjostrand M, Gruenert DC et al Neutrophil recruitment by human IL‐17 via C‐X‐C chemokine release in the airways. J Immunol 1999; 162:2347–52. [PubMed] [Google Scholar]
- 20. Miyamoto M, Prause O, Sjostrand M, Laan M, Lotvall J, Linden A. Endogenous IL‐17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. J Immunol 2003; 170:4665–72. [DOI] [PubMed] [Google Scholar]
- 21. Roussel L, Houle F, Chan C, Yao Y, Berube J, Olivenstein R et al IL‐17 promotes p38 MAPK‐dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J Immunol 2010; 184:4531–7. [DOI] [PubMed] [Google Scholar]
- 22. Agache I, Ciobanu C, Agache C, Anghel M. Increased serum IL‐17 is an independent risk factor for severe asthma. Respir Med 2010; 104:1131–7. [DOI] [PubMed] [Google Scholar]
- 23. Barczyk A, Pierzchala W, Sozanska E. Interleukin‐17 in sputum correlates with airway hyperresponsiveness to methacholine. Respir Med 2003; 97:726–33. [DOI] [PubMed] [Google Scholar]
- 24. Doe C, Bafadhel M, Siddiqui S, Desai D, Mistry V, Rugman P et al Expression of the T helper 17‐associated cytokines IL‐17A and IL‐17F in asthma and COPD. Chest 2010; 138:1140–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mantel PY, Schmidt‐Weber CB. Transforming growth factor‐β: recent advances on its role in immune tolerance. Methods Mol Biol 2011; 677:303–38. [DOI] [PubMed] [Google Scholar]
- 26. Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Page N et al IL‐17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol 2001; 108:430–8. [DOI] [PubMed] [Google Scholar]
- 27. Sun YC, Zhou QT, Yao WZ. Sputum interleukin‐17 is increased and associated with airway neutrophilia in patients with severe asthma. Chin Med J 2005; 118:953–6. [PubMed] [Google Scholar]
- 28. Zhao Y, Yang J, Gao YD, Guo W. Th17 immunity in patients with allergic asthma. Int Arch Allergy Immunol 2010; 151:297–307. [DOI] [PubMed] [Google Scholar]
- 29. Liu YH, D'Ambrosio M, Liao TD, Peng H, Rhaleb NE, Sharma U et al N‐acetyl‐seryl‐aspartyl‐lysyl‐proline prevents cardiac remodeling and dysfunction induced by galectin‐3, a mammalian adhesion/growth‐regulatory lectin. Am J Physiol Heart Circ Physiol 2009; 296:H404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med 2012; 18:716–25. [DOI] [PubMed] [Google Scholar]
- 31. Chambers ES, Nanzer AM, Pfeffer PE, Richards DF, Timms PM, Martineau AR et al Distinct endotypes of steroid‐resistant asthma characterized by IL‐17Ahigh and IFN‐γ high immunophenotypes: potential benefits of calcitriol. J Allergy Clin Immunol 2015; 136:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A et al TH17 cells mediate steroid‐resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol 2008; 181:4089–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Al‐Ramli W, Prefontaine D, Chouiali F, Martin JG, Olivenstein R, Lemiere C et al TH17‐associated cytokines (IL‐17A and IL‐17F) in severe asthma. J Allergy Clin Immunol 2009; 123:1185–7. [DOI] [PubMed] [Google Scholar]
- 34. Johnson PR. Role of human airway smooth muscle in altered extracellular matrix production in asthma. Clin Exp Pharmacol Physiol 2001; 28:233–6. [DOI] [PubMed] [Google Scholar]
- 35. Ge XN, Bahaie NS, Kang BN, Hosseinkhani MR, Ha SG, Frenzel EM et al Allergen‐induced airway remodeling is impaired in galectin‐3‐deficient mice. J Immunol 2010; 185:1205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saegusa J, Hsu DK, Chen HY, Yu L, Fermin A, Fung MA et al Galectin‐3 is critical for the development of the allergic inflammatory response in a mouse model of atopic dermatitis. Am J Pathol 2009; 174:922–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Atkinson JJ, Senior RM. Matrix metalloproteinase‐9 in lung remodeling. Am J Respir Cell Mol Biol 2003; 28:12–24. [DOI] [PubMed] [Google Scholar]
- 38. Cataldo DD, Bettiol J, Noel A, Bartsch P, Foidart JM, Louis R. Matrix metalloproteinase‐9, but not tissue inhibitor of matrix metalloproteinase‐1, increases in the sputum from allergic asthmatic patients after allergen challenge. Chest 2002; 122:1553–9. [DOI] [PubMed] [Google Scholar]
- 39. Mattos W, Lim S, Russell R, Jatakanon A, Chung KF, Barnes PJ. Matrix metalloproteinase‐9 expression in asthma: effect of asthma severity, allergen challenge, and inhaled corticosteroids. Chest 2002; 122:1543–52. [DOI] [PubMed] [Google Scholar]
- 40. Ohbayashi H, Shimokata K. Matrix metalloproteinase‐9 and airway remodeling in asthma. Curr Drug Targets Inflamm Allergy 2005; 4:177–81. [DOI] [PubMed] [Google Scholar]
- 41. Cooper D, Iqbal AJ, Gittens BR, Cervone C, Perretti M. The effect of galectins on leukocyte trafficking in inflammation: sweet or sour? Ann N Y Acad Sci 2012; 1253:181–92. [DOI] [PubMed] [Google Scholar]
- 42. Liu FT, Yang RY, Hsu DK. Galectins in acute and chronic inflammation. Ann N Y Acad Sci 2012; 1253:80–91. [DOI] [PubMed] [Google Scholar]
- 43. del Pozo V, Rojo M, Rubio ML, Cortegano I, Cardaba B, Gallardo S et al Gene therapy with galectin‐3 inhibits bronchial obstruction and inflammation in antigen‐challenged rats through interleukin‐5 gene downregulation. Am J Respir Crit Care Med 2002; 166:732–7. [DOI] [PubMed] [Google Scholar]
- 44. Lopez E, del Pozo V, Miguel T, Sastre B, Seoane C, Civantos E et al Inhibition of chronic airway inflammation and remodeling by galectin‐3 gene therapy in a murine model. J Immunol 2006; 176:1943–50. [DOI] [PubMed] [Google Scholar]
- 45. Lopez E, Zafra MP, Sastre B, Gamez C, Lahoz C, del Pozo V. Gene expression profiling in lungs of chronic asthmatic mice treated with galectin‐3: downregulation of inflammatory and regulatory genes. Mediators Inflamm 2011; 2011:823279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pilette C, Colinet B, Kiss R, Andre S, Kaltner H, Gabius HJ et al Increased galectin‐3 expression and intra‐epithelial neutrophils in small airways in severe COPD. Eur Respir J 2007; 29:914–22. [DOI] [PubMed] [Google Scholar]
- 47. Mishra BB, Li Q, Steichen AL, Binstock BJ, Metzger DW, Teale JM et al Galectin‐3 functions as an alarmin: pathogenic role for sepsis development in murine respiratory tularemia. PLoS One 2013; 8:e59616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mukaro VR, Bylund J, Hodge G, Holmes M, Jersmann H, Reynolds PN et al Lectins offer new perspectives in the development of macrophage‐targeted therapies for COPD/emphysema. PLoS One 2013; 8:e56147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Busse WW, Holgate S, Kerwin E, Chon Y, Feng J, Lin J et al Randomized, double‐blind, placebo‐controlled study of brodalumab, a human anti‐IL‐17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med 2013; 188:1294–302. [DOI] [PubMed] [Google Scholar]
- 50. Manni ML, Trudeau JB, Scheller EV, Mandalapu S, Elloso MM, Kolls JK et al The complex relationship between inflammation and lung function in severe asthma. Mucosal Immunol 2014; 7:1186–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin RA, Ather JL, Daggett R, Hoyt L, Alcorn JF, Suratt BT et al The endogenous Th17 response in NO2‐promoted allergic airway disease is dispensable for airway hyperresponsiveness and distinct from Th17 adoptive transfer. PLoS One 2013; 8:e74730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Argueso P, Guzman‐Aranguez A, Mantelli F, Cao Z, Ricciuto J, Panjwani N. Association of cell surface mucins with galectin‐3 contributes to the ocular surface epithelial barrier. J Biol Chem 2009; 284:23037–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bernacki SH, Nelson AL, Abdullah L, Sheehan JK, Harris A, Davis CW et al Mucin gene expression during differentiation of human airway epithelia in vitro. Muc4 and muc5b are strongly induced. Am J Respir Cell Mol Biol 1999; 20:595–604. [DOI] [PubMed] [Google Scholar]
- 54. Radicioni G, Cao R, Carpenter J, Ford AA, Wang TT, Li Y et al The innate immune properties of airway mucosal surfaces are regulated by dynamic interactions between mucins and interacting proteins: the mucin interactome. Mucosal Immunol 2016; 9:1442–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mauri P, Riccio AM, Rossi R, Di Silvestre D, Benazzi L, De Ferrari L et al Proteomics of bronchial biopsies: galectin‐3 as a predictive biomarker of airway remodelling modulation in omalizumab‐treated severe asthma patients. Immunol Lett 2014;162(Pt A):2–10. [DOI] [PubMed] [Google Scholar]