

Summary
Current cancer therapies target the bulk of the tumour, while a population of highly resistant tumour cells may be able to repopulate the tumour and metastasize to new sites. Cancer cells with such stem cell‐like characteristics can be identified based on their phenotypical and/or functional features which may open up ways for their targeted elimination. In this review we discuss potential off‐target effects of inhibiting cancer stem‐cell self‐renewal pathways on immune cells, and summarize some recent immunological studies specifically targeting cancer stem cells based on their unique antigen expression.
Keywords: antigens/peptides/epitopes, immunotherapy, stem cell, T‐cell, tumour immunology
Introduction
Cancer stem cells (CSCs), also called stem‐like cells or tumour‐initiating cells (TICs), are a distinct subpopulation of tumour cells. They may arise in a variable and unpredictable manner due to genetic and epigenetic changes during tumour development (stochastic theory), or they are cells that possess a unique intrinsic ability to initiate tumour growth and self‐renewal (hierarchical theory). The existence of CSCs had been postulated for some time before experimental evidence was first provided in 1994. A subpopulation of acute myeloid leukaemia cells, with CD34high CD32low phenotype, was shown to be highly capable of engrafting leukaemia in severe combined immunodeficiency (SCID) mice.1 These cells were called leukaemia‐initiating cells. Solid tumour TICs were first described in breast cancer (CD44+ CD24−/low/linage− cells) in 2003,2 followed by further discoveries in a variety of malignancies. Regarding using the correct designation, CSCs are perceived as immature progenitors of tumour cells, residing at the top of a hierarchical organization of tumour cell differentiation. Conversely, the definition of TICs is based on the function of these cells, as they are uniquely capable, at very low cell numbers, to initiate heterogeneous, complex tumours in vivo. In this review, we use the combined CSCs/TICs abbreviation, as suggested by Maccalli et al.3 reflecting the variety of the references.
While traditional cancer therapies, such as radio‐ or chemotherapy, may eliminate the bulk of the tumours, treatment resistance in CSCs/TICs is thought to be responsible for relapse. In order to prevent or significantly delay relapse, these cells should be specifically targeted and eliminated. There are numerous ongoing trials targeting CSCs/TICs (for the latest review see Qureshi‐Baig et al.4); however, in order to design efficient novel treatment approaches we need a clearer understanding of the biology of these cells. In this review we summarize the current state of knowledge concerning the feasibility of immune targeting CSCs/TICs in solid tumours.
We also point out how some of the biological targeting of CSCs/TICs may act as a double‐edged sword by also affecting immune responses.
Developmental pathways in CSC/TIC signalling
Developmental pathways play important roles in normal stem cell function. The core stem cell pathways, Notch, Wnt/β‐catenin, Hedgehog (discussed in more detail below) and some other crucial pathways, such as Janus kinase/signal transducer and activator of transcription/phosphoinositide 3‐kinase/phosphatase and tensin homologue (JAK/STAT, PI3K/PTEN and nuclear factor kappa B (NF‐κB), promote cell proliferation and the formation of CSC‐like colonies.5, 6 They are frequently altered in cancers, such as becoming deregulated or persistently activated, and are suggested to be responsible for CSC/TIC regulation.7, 8, 9, 10
Notch
Notch ligands, located in the plasma membrane of adjacent cells are transactivated, triggering the transcription of Notch target genes, such as the hairy and enhancer of split (HES)‐related family, c‐myc, PI3K, protein kinase B (AKT), NF‐κB, peroxisome proliferator‐activated receptor (PPAR), cyclin D1, p21 and p27. The activation of these downstream targets regulates cell fate leading to differentiation, cell‐cycle progression and survival, depending on the particular signalling context. In stem‐like cells Notch may delay differentiation and promote cell survival.11, 12 There are numerous ongoing Phase I and II clinical trials in cancer with a range of targets and mechanisms investigating the usefulness of Notch targeting, alone or in combination with other therapies.11 However, Notch signalling has also been linked to peripheral T‐cell maturation into effector cells, such as developing cytotoxic T‐cell function or cytokine production.13 T‐cell activity has been shown to be impaired by Notch‐inhibition with a γ‐secretase inhibitor.14
Wnt
Two pathways have been identified: the canonical pathway is β‐catenin‐dependent and involved in cell fate determination. The non‐canonical pathway is β‐catenin independent and involved in cell movement and polarity. Wnt signalling is initiated by soluble ligands released by neighbouring cells. In cancer, gain of function and loss of function mutations, regulation by methylation and histone modification of this pathway have all been observed.9, 15, 16 Its inhibition in CSCs is a main area of cancer therapy research.17 However, the Wnt/β‐catenin pathway is also a key regulator of T‐cell development and activation. Wnt/β‐catenin signalling is crucial for CD8+ memory T‐cell development,18 while agonists of the pathway improve immunotherapy outcomes.19
The Hedgehog (Hh) signalling pathway has been implicated in tissue homeostasis and repair and epithelial to mesenchymal transition (EMT) in normal tissues. In cancer, aberrant Hh‐signalling, such as over‐expression of its ligands, loss of function of the receptor and dysregulation of transcription factors, promote tumorigenesis and tumour progression. Hh signalling can also be canonical and non‐canonical, and triggered by a variety of factors in the tumour microenvironment, such as transforming growth factor (TGF)‐β, tumour necrosis factor (TNF)‐α and interleukin (IL)‐6. Inhibition of Hh signalling is also undergoing intense investigations for cancer treatment.20 Hh signalling is relevant in immune cell development and function, although its effect on peripheral T‐cell function is controversial.21, 22, 23, 24 Because it is also involved in myeloid‐derived suppressor cell (MDSC) function,25 Hh inhibitors may deliver additional benefits.
As there is a considerable overlap between these pathways, single targeting is unlikely to achieve a physiologically relevant level of inhibition. Furthermore, the fact that they are also involved in normal tissue homeostasis and development, including immune cell behaviour and peripheral effector function, makes their targeting a difficult challenge.
Identification and isolation of CSCs/TICs
Surface marker‐based identification
CSCs/TICs are typically isolated based on their expression of proteins shared in common with healthy stem cells. The markers most commonly used in solid tumours to identify CSCs/TICs are CD133, CD44, IL‐6R, CD24, epithelial cell adhesion molecule (EpCAM), leucine‐rich repeat‐containing G‐protein coupled receptor 5 (Lgr5), CD166 and CD29, alone or in combination. The use of these markers is relatively conserved across the spectrum of solid cancers. However, there are technical considerations which may give rise to false positives or inconsistencies in the results, including subjectivity in flow cytometry gating, the use of cell lines versus primary cells, confirmation of function in clonogenic cultures and animal models. For some of these markers there is evidence for direct stem cell‐like function, while recently the validity of some, as bona fide CSC/TIC markers, has been called into question, as discussed later. A few common markers are discussed below.
CD133
CD133 (Prominin‐1) is a five‐transmembrane glycoprotein used to identify CSCs/TICs in prostate, pancreatic, colon and liver cancer and glioblastoma.5 Although the precise function of CD133 has not been elucidated, it is known to bind cholesterol and is localized in protrusions of the membrane, e.g. in villi and cilia. Despite its initial acceptance as a CSC/TIC marker, in some instances cells expressing this marker have not demonstrated exclusive tumour‐initiating ability.26, 27 CD133 is also present in a number of adult tissues, including the kidneys, pancreas and colon28, 29 and is used as a marker for haematopoietic stem cells. Thus it is important to acknowledge that it is not a universal CSC marker, nor is it a cancer cell‐specific antigen.
Some of the inconsistencies observed in the application of CD133 as a CSC/TIC marker may be associated with its pattern of expression and the antibodies used to detect it.30 The most commonly used antibodies for CD133 detection are mouse monoclonal antibodies CD133/1 and CD133/2, which detect the epitopes AC133 and AC141, respectively. These epitopes are distinct from each other and both are glycosylated. The different glycosylation status of CD133 across different tissues may give rise to false negatives. Glycosylation status is also suggested to change as a result of differentiation in some lineages,31, 32 although this may be advantageous in the specific detection of early progenitor cells. However, a number of studies have shown that AC133 epitope expression (as detected by the CD133/1 antibody) does not correlate with CD133 protein or mRNA levels.32 The functional outcome of the loss of this epitope upon differentiation is unclear.
CD44
CD44 is used to identify CSCs/TICs in breast, prostate, colon, head and neck and pancreatic cancer. CD44 is a transmembrane glycoprotein that functions as a receptor for hyaluronic acid. It has a multitude of physiological and pathological functions, including adhesion and migration, proliferation, growth and survival. However, CD44 is widely expressed in healthy tissues and in multiple cell types in the cancer microenvironment, making it difficult to apply as a specific CSC/TIC marker. CD44 is subject to alternate splicing and it has been suggested that CD44 splice variants (CD44v) specifically identify cells with greater tumorigenic potential compared to cells expressing CD44s, the standard isoform.33, 34 Additionally, certain splice variants have been suggested to have pathological functions in colon and pancreatic cancer35 and have prognostic utility in other cancers, including non‐small‐cell lung carcinoma (NSCLC), acute myeloid leukaemia (AML) and gastric cancer.34 CD44high cells were shown to be less immunogenic than CD44low tumour cells in head and neck cancer, partially via enhanced expression of the programmed cell death ligand‐1 (PD‐L1).36 A Phase I trial of a humanized IgG1 antibody, targeting the extracellular hyaluronic acid binding domain of all CD44 isoforms37 in heavily pretreated cancer patients, proved to be safe, but had only modest clinical effects. Conversely, however, CD44 also regulates T helper type 1 (Th1) cell survival, memory function,38 T‐cell IL‐17 and IFN‐γ production, thus its targeting may impair anti‐tumour immune responses.39
IL‐6R
IL‐6 has been shown to enhance stemness markers [Notch, Lgr5 and octamer‐binding transcription factor 4 (Oct‐4)] in colon cancer40 and the survival and tumorigenicity of cancer stem cells identified as aldehyde dehydrogenases (ALDH)high/CD44high in head and neck carcinoma.41 Inhibition of IL‐6 signalling with an IL‐6R inhibitor antibody (tocilizumab) prevents human CSC‐mediated tumour initiation.41 Similar observations were made in an NSCLC cell line, where inhibition of IL‐6 or IL‐6R, separately or in combination, significantly inhibited CSC proliferation and growth.42 In colon cancer, blocking the IL‐6 receptor with a monoclonal antibody reduced spheroid formation, stem‐cell‐related gene expression and enhanced resistance to chemotherapy by 5‐fluorouracil.40 However, IL‐6R also plays an important role in naive and central memory T‐cells, regulating their survival, proliferation and effector function while also blocking regulatory T‐cell (Treg) function.43
Further markers
There is a huge array of further markers that have been used alone or in combination in a variety of cancers to identify CSCs/TICs. They include CD24, EpCAM, Lgr5, CD90, CD117, CD166, CD29, CD177, epithelial‐specific antigen (ESA), alpha‐fetoprotein (AFP), nestin, chemokine receptor 4 (CXCR4) and stage‐specific embryonic antigen 4 (SSEA4). However, the plasticity of these markers means that they can be up‐regulated on cells originally not expressing them.44 Their heterogeneous expression throughout the tumour tissue45 has also been observed, making their isolation more difficult due to being a moving target. From our review viewpoint, some of these markers, when unique or over‐expressed, can serve as CSC/TIC‐associated antigens for T‐cell recognition, as discussed later.
Metabolism‐based identification
Aldehyde dehydrogenase
The aldehyde dehydrogenase (ALDH) family consists of 19 genes (in humans) that express enzymes which catalyse the oxidation of aldehyde.46 Aldehyde oxidation is required to metabolize many physiological substrates, such as vitamins, lipids and amino acids.46 These enzymes also catabolize aldehydes derived from pharmacological substrates; overall, they have a protective detoxifying effect. High ALDH activity is associated with both normal stem cells and CSCs. ALDH activity has been used to identify CSCs in breast, colon, head and neck, pancreatic and liver cancer. It is measured using the flow cytometry‐based ALDEFLUOR assay. An advantage this assay has over phenotypical detection methods is the direct readout of a fluorescent signal, based on enzyme activity. This may be less susceptible to inconsistencies encountered by antibody‐based staining, such as epitope down‐regulation or masking, or expression of splice variants. The ALDEFLUOR assay mainly measures the activity of the ALDH1 family, as the inhibitor N,N‐diethylaminobenzaldehyde (DEAB) used in the assay is a specific inhibitor of ALDH1.47 However, this may mean that the frequency of ALDH high cells is underestimated in tissues in which the predominant ALDH isozyme is not of the ALDH1 family.
In prostate cancer (PCa), ALDHhigh population frequency varies from 1·2 to 8·3% in the classic prostate cancer cell lines (DU145, PC3, LNCaP and 22RV.1), while cell lines derived from metastatic PCa cells had an even higher ALDHhigh frequency; up to 30%.48, 49 ALDHhigh PCa CSCs have demonstrated enhanced clonogenicity and migration in vitro, expression of stemness‐associated genes, in‐vivo tumour initiation49, 50, 51 and induction of metastasis.48 ALDH activity has been correlated with the expression of other CSC or clinical markers; CD44, EpCaM and integrin expression were significantly higher in ALDHhigh compared to ALDHlow cells in one report,48 while CD44 and α 2‐integrin levels did not differ in another PCa cell line.50 CD133 was not detectable in both these studies; in primary samples it was detected in freshly isolated cells but was greatly reduced upon passaging of the cells.52 In ovarian cancer, both ALDH activity and CD133 expression were detected in primary specimens. Upon passage of the primary cells, CD133 expression was reduced but could be rescued by CSC culture conditions, i.e. sphere culture and serum‐free conditions.53 ALDHhigh ovarian cancer cells, which range in frequency from 0·1 to 7·9% in cell lines and 1–7% in primary samples, demonstrated tumorigenicity in vivo.53, 54 High ALDH activity has been used to identify both stem and cancer stem cells in breast cancer with frequencies of 8 and 4% ALDHhigh cells, respectively.55 ALDHhigh CD44+ CD24− and ALDHhigh CD44+ CD133+ breast cancer cells demonstrated greater tumorigenicity than the corresponding low/negative populations.56 Conflicting evidence exists for the utility of ALDH activity as a CSC marker in lung cancer.57 In one study, both ALDHhigh and ALDHlow cells (from a single cell line) were capable of initiating tumours in vivo,58 while in another, ALDHhigh but not ALDHlow cells from two different cell lines demonstrated tumorigenicity in vivo.59 One further study using eight lung cancer cell lines identified STAT‐3 signalling as a mediator of ALDH3A1 activity and demonstrated tumorigenicity in two cell lines.60 This suggests that variability in cell lines could lead to rejection of a potentially applicable CSC marker and highlights the importance of testing in multiple cell lines/primary tissues.
ALDH expression has also been correlated with clinical outcome in a number of cancers. High ALDH expression (as well as the presence of other CSC markers) correlates with poor prognosis in pancreatic cancer61 and in both serous (n = 62) and clear cell (n = 37) ovarian carcinoma cases54 and in 112 serous carcinoma cases in a further study.62 In prostate cancer, ALDH expression correlates with more advanced stage, compared to localized cancer and BPH,63 while there seems to be no consensus on the utility of ALDH activity for the selection of tumorigenic melanoma cells.64
ALDH has been shown to contribute to resistance mechanisms to radiation therapy and chemotherapy, thus a different, more specific, therapeutic approach must be undertaken. Some groups have investigated chemical inhibitors47 such drug development requires the design of highly specific inhibitors, as the widespread expression of ALDH in healthy stem cells may result in off‐target effects. It is also possible that functional redundancy within the large isozyme family could compensate for inhibition of one ALDH target. In the immunotherapy setting, ALDHhigh CSC‐loaded dendritic cells (DC) have been used successfully in two in‐vivo melanoma models.65, 66 ALDH activity has been demonstrated in the Treg cell immune subset in the transplantation setting67; thus, ALDH targeting may also have an anti‐tumour effect via affecting the Treg subset.
Glycolytic activity
CSCs/TICs have fewer mitochondria, thus they are more glycolytic than differentiated tumour cells, as shown in melanoma, breast, lung and liver carcinomas.68, 69, 70, 71 This can be detected by reduced mitochondrial activity, perinuclear mitochondrial distribution, lower intracellular concentrations of ROS and ATP and lower amounts of mitochondrial DNA in CSCs/TICs.72
CSC/TIC inhibitors targeting either self‐renewal pathways, surface markers or enzymes have potential off‐target effects on other cell types. Figure 1 illustrates the potential side effects of some of these inhibitors on immune cells.
Figure 1.
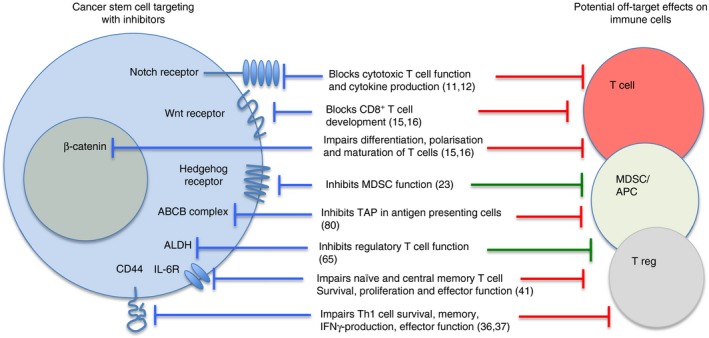
Targeting stem cell pathways, functions or markers may have off‐target effects on immune cells. The red blocking symbols on the right represent negative effects by inhibiting immune responses, while the green symbols indicate positive effects by blocking immunosuppressive cells.
Function‐based identification
Slow cell division
Slow cell division of CSCs/TICs, that exist mainly in G0 phase, can be detected by their characteristic of retaining dyes that normally become diluted during proliferation, such as PKH, bromodeoxyuridine (BrdU) or carboxyfluorescein succinimidyl ester (CFSE). Dye‐retaining cells that give progeny to xenotransplants have been observed in glioma, melanoma, breast and pancreatic cancer.73, 74, 75, 76, 77
Enhanced drug resistance and detoxifying pathways
The ATP‐binding cassette (ABC) transporter family of proteins, especially ABCB1, ABCC1, ABCG2 and ABCB5, are active in cancer stem cells, but switched off during differentiation.78 These transporter proteins are extremely efficient in pumping out complex molecules from the cytoplasm, thus protecting the cells from exogenous toxins, including numerous chemotherapeutic drugs. They have a wide range of substrates, such as peptides, lipids, hydrophobic drugs, polysaccharides and proteins.79 Their targeting with specific inhibitors is one of the active areas of drug development in cancer.80 As CSCs/TICs also exclude hydrophobic Hoescht dyes via this mechanism, they can be identified by forming a side population (SP) based on low dye levels.81 However, ABCB proteins, such as transporter associated with antigen processing (TAP), also have an important role in intracellular peptide trafficking across membranes, with crucial involvement in major histocompatibility complex (MHC) class I antigen presentation and DC function.82 Thus, the off‐target effect of tumour ABCB targeting may be deleterious for generating efficient anti‐tumour T‐cell responses.
Enhanced resistance to radiotherapy
It has been speculated that the failure of radiation therapy correlates with the survival of at least some of the CSCs/TICs in the tumour, although the underlying protective mechanisms are incompletely understood. Radiation causes direct damage to the DNA, such as single‐ or double‐strand breaks or acts indirectly via reactive oxygen species (ROS). The consequence of radiation damage can be temporary or permanent cell cycle arrest, mainly via checkpoint kinase activity (Chk‐1 and Chk‐2), which allows time for DNA damage repair (DDR) to take place. If the damage is irreparable, DDR mediates senescence or cell death.83 ROS mediates oxidative stress which, if beyond the capacity of the cell's anti‐oxidant defence mechanism, may also lead to cell death. Both DDR and the ROS scavenging system have been reported to be highly efficient in CSCs/TICs in numerous solid cancers,84 contributing to their intrinsic radioresistance. Extrinsic radioresistance may be supported by the localization of CSCs. It has been suggested that CSCs/TICs residing in hypoxic areas are more resistant to radiation‐induced damage than cells in normoxic areas. Hypoxia‐inducing factors (HIF)1α and HIF2α may lead to the activation of the Notch, Wnt and Hedgehog signalling pathways, which are essential for CSC/TIC maintenance. There is clinical evidence that radiotherapy is generally less successful in CSC‐rich laryngeal cancer where CSCs were identified as a residing subset within the CD44high population.85
However, the effects of hypoxia are far from inhibitory on immune cell function. Activated DC become glycolytic and their long‐term survival is regulated by HIF1α.86 Effector T‐cells are also resistant to hypoxia – even more, their cytolytic machinery is turned on by HIF2α.87 As effector T‐cells also display glycolytic characteristics, they can exert their effector function even in the depth of tissues with poor vasculature, maybe where CSCs/TICs reside. Furthermore, we have shown that low dose radiation spares effector and memory T‐cells compared to naive T‐cells.88 Thus, combination of radiation with T‐cell targeting of stem cells sounds like a two‐pronged attack with potential synergistic effects.
High‐dose radiation alone may eliminate CSCs/TICs, as early prostate and lung cancers can be cured by radiotherapy alone. It may happen partly via generating immunogenic cell death, which then initiates tumour antigen uptake and antigen cross‐presentation by DC.89 One could speculate whether radiation‐induced CSC death is also immunogenic and weather radiation would generate CSC/TIC‐specific effector and memory T‐cells, significantly contributing to an abscopal effect and subsequent protection from relapse. Despite its obvious clinical importance, this question has not been studied previously.
Immunological characteristics of CIC/CSC
Immunosuppression
Proliferative T‐cell responses and IL‐2 production were inhibited by CSCs/TICs in gliobastoma90 and melanoma91 in vitro. Treg frequencies were also increased in melanoma91 but not in glioblastoma CSC–T‐cell co‐cultures.90 Secretion of TGF‐β, IL‐10, IL‐4 and IL‐13 by CSCs/TICs has been shown to have immunosuppressive effects on NK‐cells, T‐cells and antigen‐presenting cells.90, 92, 93 Cell surface molecules, such as CD200, expressed on CSCs/TICs,94 can also dampen immune responses. CD200 over‐expression has been associated with the suppression of Th1 responses, decreased neutrophil infiltration and increased IL‐10 production induced by the tumour, as shown in a breast carcinoma model.95 PD‐L1 is often over‐expressed on tumour cells, with a function of promoting tumour glycolysis. Up‐regulation on CSCs/TICs is probably tumour‐type or localization‐dependent as hypoxia, for example, is one of the triggers that can up‐regulate PD‐L1.96 High expression on CSCs/TICs has been reported on head and neck carcinoma,36 on CD133+ colorectal97 and gastric,98 but not on melanoma CSCs/TICs.91
Immune resistance
Some extent of MHC class I down‐regulation has been shown on glioblastoma CSCs/TICs compared to non‐CSCs. Nevertheless, these CSCs/TICs were still able to induce autologous T‐cell responses in vitro. Furthermore, IFN‐γ‐treatment enhanced the susceptibility of CSCs to T‐cell‐mediated immune responses.90
Cancer vaccines that generate T‐cell and antibody‐responses against tumour‐associated antigens (TAA) work well in preclinical preventative models; however, the results obtained with therapeutic vaccines in the clinic are somewhat disappointing. Focusing on CSCs, it is feasible that stem‐like cells are inherently resistant to T‐cell attack, although lack of vaccine specificity for CSC/TIC‐antigens is also a possibility. Development of stem‐like features, such as Nanog, in surviving tumour cells following vaccination has been observed.99 Resistance to T‐cell killing was successfully abolished by silencing Nanog in these cells. This work suggests that stem‐like features may develop in a population of cells under immunological pressure; however, it does not show inherent stem cell resistance.
The argument that cancer stem‐like cells may not be inherently resistant to immune attack is also supported by using purified CSCs/TICs as a vaccine.66 This treatment generated T‐cells in immunocompetent mice that were highly efficient at killing CSCs/TICs and provided greater protection in the D5 melanoma model against pulmonary metastasis than vaccination with unseparated cells. As a further example to prove T‐cell susceptibility, CD33+ brain tumour CSCs/TICs, transfected with the pp65 antigen of human cytomegalovirus, were efficiently killed by virus‐specific memory T‐cells,100 indicating the feasibility of CSC/TIC targeting by T‐cells.
Potential immune targeting of CSCs/TICs
Antigen non‐specific immune targeting
NK cells
As MHC class I molecule expression is often lower on CSCs/TICs than on the bulk of tumour cells, CSCs/TICs are more likely to be susceptible to NK cell‐mediated killing. However, they also often lack NK‐activating ligands, such as NKG2D, as shown in brain and breast cancers.101, 102 Conversely, glioma, oral squamous cell carcinoma and colorectal cancer CSCs/TICs 103, 104, 105 have been reported to express various ligands for NK cells [most frequently poliovirus receptor (PVR), which is recognized by DNAX accessory molecule‐1 (DNAM‐1)] and are highly susceptible to NK cell killing. However, while cytokine‐activated NK cells have efficient CSC/TIC killing ability, freshly isolated NK cells from the same patient do not kill CSCs.103 This points towards microenvironmental regulation of NK cell activity in cancer patients.
γδ T‐cells
γδ T‐cells also exhibit MHC‐unrestricted lysis of targets, including tumour cells. Vγ9Vδ2 T‐cells can be activated with phosphoantigens or aminobisphosphonates and have been shown to efficiently kill CSCs/TICs in colon, ovarian and breast cancer models, especially after treatment with Zoledronate.106, 107, 108 They have also been observed infiltrating the tumour tissue, emphasizing their physiological relevance.
Antigen‐specific targeting by T‐cells
Solid cancer cells and their CSCs/TICs express human leucocyte antigen (HLA) class I but not HLA class II molecules. Their efficient targeting by CD8+ T‐cells depends upon a sufficient level of HLA class‐I molecule expression and intact antigen presenting machinery in these cells. CSC/TIC resistance or susceptibility to T‐cell killing has both been reported, depending on tumour type, origin of cells and culture conditions. Tissue‐derived CSCs/TICs from colon cancer were shown to express lower levels of MHC class I molecules than non‐stem‐like cells.105
Some TAA, such as mucin 1 (MUC‐1) or centrosomal protein 55 (CEP55), are expressed equally in both CSCs/TICs and non‐CSCs/TICs,109, 110 while others, such as the olfactory receptor family 7 subfamily C member 1 (OR7C1) are dominantly expressed in CSCs/TICs.111 The former TAA are classified as shared antigens and the latter are functionally linked to cancer stemness, because their expression ceases after CSC differentiation into non‐CSCs. T‐cell responses to shared TAA could achieve temporary tumour control, but it may lead to tumour escape by inducing loss of antigens that are not necessary for cellular fitness (immunoediting).112 Multiple antigen targeting, incorporating those TAA that are specifically expressed in CSCs/TICs, is much more likely to result in therapeutic success in clinical settings,113 as indicated in Fig. 2. Although it may be crucial to have high frequencies of CSC/TIC‐specific T‐cells present in the tumour tissue, breaking the localized immunosuppressive milieu is likely be helped by activated infiltrating T‐cells of multiple specificity.
Figure 2.
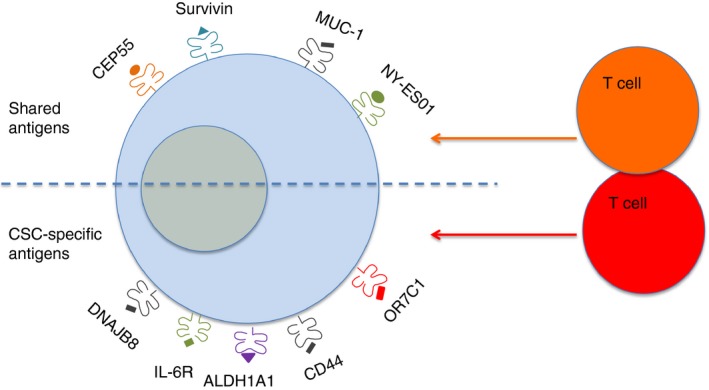
Simultaneous targeting of shared and cancer stem cell/tumour‐initiating cell (CSC/TIC)‐specific antigens by antigen‐specific T‐cells can eliminate CSC/TIC‐like cells and differentiated tumour cells alike.
The main types of TAA, expressed in CSCs/TICs and can be targeted by T‐cells, are summarized below.
Cancer/testis (CT) antigens
CT antigens are expressed only in germ cells; however, they have been shown to be re‐expressed in some malignancies. This cancer‐specific expression makes these, and the onco‐fetal antigen group (discussed in the next paragraph), therapeutically highly relevant compared to other types of TAA that are discussed later. Germ cells do not express MHC class‐I molecules, thus deletional tolerance may not occur during the negative selection phase of T‐cell development against these antigens. CT antigens have oncogenic functions, such as supporting tumour growth, enhancing treatment resistance and facilitating metastasis.114 There are more than 100 gene families of CT antigens listed on the Ludwig Institute of Cancer Research's website (http://www.cta.lncc.br/), such as MAGE, BAGE, GAGE, XAGE, SPANX, NY‐ESO1, etc.115 Because of their unique expression pattern, they often serve as TAA for immunotherapy trials. Interestingly, a transcriptome analysis of SP versus main population (MP) cells from colon, breast and lung cancer cell lines revealed that 18 of 74 of these antigens are preferentially expressed on CSCs/TICs.116 Novel CT antigens have recently been identified by transcriptome analysis, such as the DNAJ Hsp40 homologue, subfamily B, member 8 (DNAJB8), in SP cells of renal cancer cells.117 OLF7C1 is also a novel cancer‐testis antigen, observed on SP cells of colorectal cancer. It has been successfully targeted by HLA A24‐restricted T‐cells.111 Another CT antigen, the brother of the regulator of the imprinted site (BORIS), subfamily 6, was found preferentially expressed on cervical cancer CSCs/TICs. It has a role in maintaining CSC function and serves as a target for BORIS‐specific cytotoxic T‐cells.118
Oncofetal antigens
These antigens are typically only expressed during embryonic development in fetal tissues, but similarly to CT antigens, they can be re‐expressed in some cancers. A typical example, carcinoembryonic antigen (CEA), is expressed in numerous solid cancers but not associated with stem‐like cells. Other antigens, such as stage‐specific embryonic antigen 3 and Globo‐H, are expressed on stem‐like and non‐stem‐like breast cancer cells at similar rates,119 while 5T4 is preferentially expressed on CSC‐like cells in lung cancer and head and neck cancer.120, 121 5T4 expression, in both malignancies, is associated with poor prognosis. Xenograft models indicated that the CSC‐fraction can be reduced, tumour progression halted and local recurrence inhibited by treatment with an antibody‐drug conjugate targeting 5T4.120, 121
Over‐expressed antigens
This group of antigens has recently been reviewed by Hirohashi et al.122 and includes apoptosis‐resistance genes, such as survivin, proto‐oncogenes, such as human epidermal growth factor receptor 2 (HER‐2), centrosomal protein 55 (CEP55), sex‐determining region Y‐box 2 (SOX‐2) and cytochrome C oxidase assembly factor 1 homologue (COA‐190, and stress‐response related genes, such as heat shock protein (Hsp) or HOX genes.123 Some of the over‐expressed antigens are general stem cell‐ and not exclusively cancer stem‐cell‐specific. In PCa, Numb has been identified as a potential target for controlling tumorigenesis.124 Numb is lost in differentiating tumour cells, due to exaggerated ubiquitination and subsequent proteasomal degradation, but its expression is maintained in stem‐like cells. Its close homologue, Numb‐like (NumbL), is able to regulate the CSC/TIC pool by inhibiting the Notch pathway.125 Numb‐1‐ and Notch‐specific T‐cells eliminated luminal CSC/TIC‐like cells in a breast cancer model.126 In a gynaecological cancer (endometrioid adenocarcinoma), ALDHhigh cells preferentially expressed Hsp27, the over‐expression of which is associated with poor prognosis.127 Mitogen‐activated kinase (MAPK)13, pituitary tumour‐transforming gene protein‐binding factor (PTTG1IP), calpain 1 (CAPN1) and ubiquilin 2 (UBQLN2) were also found highly expressed in these cells compared with that in ALDHlow cells.128 SOX2, a transcription factor that regulates the Wnt/β‐catenin pathway, is amplified in numerous solid tumours and expressed predominantly in CSCs/TICs.129, 130 Interestingly, class I histone deacetylase (HDAC) inhibitors increase the frequency of SP cells with CSC/TIC markers, due to de‐differentiation of cancer cells. HOXA5 was shown to be the main transcription factor responsible for de‐differentiation and induction of SOX2 in lung cancer cells.131
Differentiation antigens
Many differentiation antigens are expressed in primary cancer cells as well as in CSC, such as MUC‐1 in breast cancer, tyrosinase and gp100 in melanoma. The human telomerase reverse transcriptase (hTERT) is also a target in primary cancer cells and in stem cells. T‐cells have been generated against a HLA‐A3‐restricted epitope: peptide K973 (KLFGVLRLK).132 Successful targeting of these antigens has the potential of eliminating CSCs and non‐CSCs alike.
Neoantigens
Although CT and onco‐fetal antigens can also be considered as neoantigens, this category typically includes mutated tumour antigens. The mutations may generate entirely new T‐cell epitopes for which high‐affinity T‐cell receptors have not been deleted during development and thus these T‐cells can be efficiently expanded. High mutation index has been indicated to be the underlying mechanism behind successful immune checkpoint inhibitor therapy.133 Although it is as yet unclear whether the mutation index of CSCs is different from that of primary cancer cells, there are data available that neoantigens are present in colorectal cancer both in CSCs and non‐CSCs in a manner that is targetable with T‐cells.134 Identifying CSC‐specific neoantigens may lead to further breakthroughs in cancer immunotherapy.
Immune targeting CSCs/TICs – clinical applications
Antigen‐specific immunological targeting of CSCs/TICs requires the generation of primary T‐cell responses, reactivation of memory responses or the adoptive transfer of engineered antigen‐specific T‐cells into the host.
Generation of T‐cell responses
Most clinical trials, listed on the http://clinicaltrials.gov website that use immunotherapy and target cancer stem cells, are based on isolating CSCs/TICs from solid tumours and loading them onto DC, which are then used as a cancer vaccine. The approach is based on preclinical data showing the success of this approach in immunocompetent mice.66 Trials are listed in pancreatic, nasopharyngeal, colorectal, ovarian, lung, liver and brain tumours. One trial studied the outcome of vaccination with DC, transfected with hTert and survivin as amplified ovarian CSC mRNA (NCT01334047). The stem‐like cell‐associated antigen(s), serving as a vaccine in these trials, are undetermined and likely to be individual patient‐specific. The outcome of these trials is not yet publicly available; however, as with non‐CSCs‐DC vaccines, it is unlikely that any monotherapy alone will be hugely effective. The explosion of approved immunotherapies with checkpoint inhibitors opens up the way for designing combination therapies. However, a shift in treatment design must be considered: vaccines or redirected T‐cells should be targeting multiple antigens, including CSC‐specific ones; this treatment should then be combined either with appropriate chemotherapy based on patient stratification data or high‐dose radiation and/or immune checkpoint inhibitor treatments in order to provide the best chance for generating robust and long‐lasting T‐cell responses leading to tumour rejection.
T‐cell transfer
The success of chimeric antigen receptor (CAR) T‐cells in treating haematological malignancies (https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm574058.htm) has generated interest in using the approach against solid tumours and also CSCs. CAR T‐cells express a CAR which consists of an extracellular binding domain of a single‐chain fragment of the antibody variable region (scFv), providing antigen‐specificity and the intracellular signalling domains of CD3‐zeta chain. The receptor can be coupled with co‐stimulatory molecules, such as CD28 and CD137. Cell surface antigens, expressed on CSCs, such as CD44, CD133, aldehyde dehydrogenases (ALDH) and EpCAM can be targeted in this way. Preclinical work using these CAR T‐cells in glioblastoma, prostate cancer and gynecological tumours have been encouraging (reviewed by Guo135). Clinical trials are currently ongoing against EGFR and CD133 (NCT01869166 and NCT02541370).
Disclosures
The authors have no competing interests to declare.
References
- 1. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres‐Cortes J et al A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994; 367:645–8. [DOI] [PubMed] [Google Scholar]
- 2. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100:3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maccalli C, Volonte A, Cimminiello C, Parmiani G. Immunology of cancer stem cells in solid tumours. A review. Eur J Cancer 2014; 50:649–55. [DOI] [PubMed] [Google Scholar]
- 4. Qureshi‐Baig K, Ullmann P, Haan S, Letellier E. Tumor‐initiating cells: a criTICal review of isolation approaches and new challenges in targeting strategies. Mol Cancer 2017; 16:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 2008; 8:755–68. [DOI] [PubMed] [Google Scholar]
- 6. Matsui WH. Cancer stem cell signaling pathways. Medicine (Balt) 2016; 95:S8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer 2003; 3:903–11. [DOI] [PubMed] [Google Scholar]
- 8. Ingham PW, Placzek M. Orchestrating ontogenesis: variations on a theme by sonic hedgehog. Nat Rev Genet 2006; 7:841–50. [DOI] [PubMed] [Google Scholar]
- 9. Clevers H. Wnt/beta‐catenin signaling in development and disease. Cell 2006; 127:469–80. [DOI] [PubMed] [Google Scholar]
- 10. Miele L, Miao H, Nickoloff BJ. NOTCH signaling as a novel cancer therapeutic target. Curr Cancer Drug Targets 2006; 6:313–23. [DOI] [PubMed] [Google Scholar]
- 11. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M et al Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol 2015; 12:445–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang J, Sullenger BA, Rich JN. Notch signaling in cancer stem cells. Adv Exp Med Biol 2012; 727:174–85. [DOI] [PubMed] [Google Scholar]
- 13. Radtke F, Fasnacht N, Macdonald HR. Notch signaling in the immune system. Immunity 2010; 32:14–27. [DOI] [PubMed] [Google Scholar]
- 14. Palaga T, Miele L, Golde TE, Osborne BA. TCR‐mediated Notch signaling regulates proliferation and IFN‐gamma production in peripheral T cells. J Immunol 2003; 171:3019–24. [DOI] [PubMed] [Google Scholar]
- 15. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene 2017; 36:1461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Sousa EM, Vermeulen L, Richel D, Medema JP. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res 2011; 17:647–53. [DOI] [PubMed] [Google Scholar]
- 17. Takahashi‐Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res 2010; 16:3153–62. [DOI] [PubMed] [Google Scholar]
- 18. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z et al Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 2009; 15:808–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gattinoni L, Ji Y, Restifo NP. Wnt/beta‐catenin signaling in T‐cell immunity and cancer immunotherapy. Clin Cancer Res 2010; 16:4695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gonnissen A, Isebaert S, Haustermans K. Targeting the Hedgehog signaling pathway in cancer: beyond smoothened. Oncotarget 2015; 6:13899–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Michel KD, Uhmann A, Dressel R, van den Brandt J, Hahn H, Reichardt HM. The hedgehog receptor patched1 in T cells is dispensable for adaptive immunity in mice. PLOS ONE 2013; 8:e61034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rowbotham NJ, Hager‐Theodorides AL, Cebecauer M, Shah DK, Drakopoulou E, Dyson J et al Activation of the Hedgehog signaling pathway in T‐lineage cells inhibits TCR repertoire selection in the thymus and peripheral T‐cell activation. Blood 2007; 109:3757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de la Roche M, Ritter AT, Angus KL, Dinsmore C, Earnshaw CH, Reiter JF et al Hedgehog signaling controls T cell killing at the immunological synapse. Science 2013; 342:1247–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crompton T, Outram SV, Hager‐Theodorides AL. Sonic hedgehog signalling in T‐cell development and activation. Nat Rev Immunol 2007; 7:726–35. [DOI] [PubMed] [Google Scholar]
- 25. Xie J. The hedgehog's trick for escaping immunosurveillance: the molecular mechanisms driving myeloid‐derived suppressor cell recruitment in hedgehog signaling‐dependent tumors. Oncoimmunology 2014; 3:e29180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou J, Wang H, Cannon V, Wolcott KM, Song H, Yates C. Side population rather than CD133(+) cells distinguishes enriched tumorigenicity in hTERT‐immortalized primary prostate cancer cells. Mol Cancer 2011; 10:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T et al CD133 expression is not restricted to stem cells, and both CD133+ and CD133− metastatic colon cancer cells initiate tumors. J Clin Invest 2008; 118:2111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu Y, Wu PY. CD133 as a marker for cancer stem cells: progresses and concerns. Stem Cells Dev 2009; 18:1127–34. [DOI] [PubMed] [Google Scholar]
- 29. Jaszai J, Fargeas CA, Florek M, Huttner WB, Corbeil D. Focus on molecules: prominin‐1 (CD133). Exp Eye Res 2007; 85:585–6. [DOI] [PubMed] [Google Scholar]
- 30. Hermansen SK, Christensen KG, Jensen SS, Kristensen BW. Inconsistent immunohistochemical expression patterns of four different CD133 antibody clones in glioblastoma. J Histochem Cytochem 2011; 59:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Corbeil D, Röper K, Hellwig A, Tavian M, Miraglia S, Watt SM et al The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J Biol Chem 2000; 275:5512–20. [DOI] [PubMed] [Google Scholar]
- 32. Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M et al The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res 2010; 70:719–29. [DOI] [PubMed] [Google Scholar]
- 33. Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M et al CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014; 14:342–56. [DOI] [PubMed] [Google Scholar]
- 34. Thapa R, Wilson GD. The importance of CD44 as a stem cell biomarker and therapeutic target in cancer. Stem Cells Int 2016; 2016:2087204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mashita N, Yamada S, Nakayama G, Tanaka C, Iwata N, Kanda M et al Epithelial to mesenchymal transition might be induced via CD44 isoform switching in colorectal cancer. J Surg Oncol 2014; 110:745–51. [DOI] [PubMed] [Google Scholar]
- 36. Lee Y, Shin JH, Longmire M, Wang H, Kohrt HE, Chang HY et al CD44+ cells in head and neck squamous cell carcinoma suppress T‐cell‐mediated immunity by selective constitutive and inducible expression of PD‐L1. Clin Cancer Res 2016; 22:3571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Menke‐van der Houven CW, van Oordt CG, van Herpen C, Coveler AL, Mahalingam D, Verheul HM et al First‐in‐human phase I clinical trial of RG7356, an anti‐CD44 humanized antibody, in patients with advanced, CD44‐expressing solid tumors. Oncotarget 2016; 7:80046–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baaten BJ, Li CR, Deiro MF, Lin MM, Linton PJ, Bradley LM. CD44 regulates survival and memory development in Th1 cells. Immunity 2010; 32:104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schumann J, Stanko K, Schliesser U, Appelt C, Sawitzki B. Differences in CD44 surface expression levels and function discriminates IL‐17 and IFN‐gamma producing helper T cells. PLoS ONE 2015; 10:e0132479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ying J, Tsujii M, Kondo J, Hayashi Y, Kato M, Akasaka T et al The effectiveness of an anti‐human IL‐6 receptor monoclonal antibody combined with chemotherapy to target colon cancer stem‐like cells. Int J Oncol 2015; 46:1551–9. [DOI] [PubMed] [Google Scholar]
- 41. Krishnamurthy S, Warner KA, Dong Z, Imai A, Nör C, Ward BB et al Endothelial interleukin‐6 defines the tumorigenic potential of primary human cancer stem cells. Stem Cells 2014; 32:2845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yi H, Cho HJ, Cho SM, Jo K, Park JA, Kim NH et al Blockade of interleukin‐6 receptor suppresses the proliferation of H460 lung cancer stem cells. Int J Oncol 2012; 41:310–6. [DOI] [PubMed] [Google Scholar]
- 43. Hunter CA, Jones SA. IL‐6 as a keystone cytokine in health and disease. Nat Immunol 2015; 16:448–57. [DOI] [PubMed] [Google Scholar]
- 44. Sellers ZP, Schneider G, Bujko K, Suszynska M, Pedziwiatr D. Do cancer cell lines have fixed or fluctuating stem cell phenotypes? Studies with the NTera2 cell line. Stem Cell Rev 2017; 13:603–10. [DOI] [PubMed] [Google Scholar]
- 45. Prasetyanti PR, Medema JP. Intra‐tumor heterogeneity from a cancer stem cell perspective. Mol Cancer 2017; 16:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tomita H, Tanaka K, Tanaka T, Hara A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016; 7:11018–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pors K, Moreb JS. Aldehyde dehydrogenases in cancer: an opportunity for biomarker and drug development? Drug Discov Today 2014; 19:1953–63. [DOI] [PubMed] [Google Scholar]
- 48. van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, Guzmán‐Ramírez N et al High aldehyde dehydrogenase activity identifies tumor‐initiating and metastasis‐initiating cells in human prostate cancer. Cancer Res 2010; 70:5163–73. [DOI] [PubMed] [Google Scholar]
- 49. Nishida S, Hirohashi Y, Torigoe T, Kitamura H, Takahashi A, Masumori N et al Gene expression profiles of prostate cancer stem cells isolated by aldehyde dehydrogenase activity assay. J Urol 2012; 188:294–9. [DOI] [PubMed] [Google Scholar]
- 50. Hellsten R, Johansson M, Dahlman A, Sterner O, Bjartell A. Galiellalactone inhibits stem cell‐like ALDH‐positive prostate cancer cells. PLOS ONE 2011; 6:e22118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen X, Li Q, Liu X, Liu C, Liu R, Rycaj K et al Defining a population of stem‐like human prostate cancer cells that can generate and propagate castration‐resistant prostate cancer. Clin Cancer Res 2016; 22:4505–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fiñones RR, Yeargin J, Lee M, Kaur AP, Cheng C, Sun P et al Early human prostate adenocarcinomas harbor androgen‐independent cancer cells. PLoS ONE 2013; 8:e74438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kryczek I, Liu S, Roh M, Vatan L, Szeliga W, Wei S et al Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int J Cancer 2012; 130:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuroda T, Hirohashi Y, Torigoe T, Yasuda K, Takahashi A, Asanuma H et al ALDH1‐high ovarian cancer stem‐like cells can be isolated from serous and clear cell adenocarcinoma cells, and ALDH1 high expression is associated with poor prognosis. PLOS ONE 2013; 8:e65158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ginestier C, Hur MH, Charafe‐Jauffret E, Monville F, Dutcher J, Brown M et al ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007; 1:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA et al High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med 2009; 13:2236–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alamgeer M, Peacock CD, Matsui W, Ganju V, Watkins DN. Cancer stem cells in lung cancer: evidence and controversies. Respirology 2013; 18:757–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ucar D, Cogle CR, Zucali JR, Ostmark B, Scott EW, Zori R et al Aldehyde dehydrogenase activity as a functional marker for lung cancer. Chem Biol Interact 2009; 178:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jiang F, Qiu Q, Khanna A, Todd NW, Deepak J, Xing L et al Aldehyde dehydrogenase 1 is a tumor stem cell‐associated marker in lung cancer. Mol Cancer Res 2009; 7:330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shao C, Sullivan JP, Girard L, Augustyn A, Yenerall P, Rodriguez‐Canales J et al Essential role of aldehyde dehydrogenase 1A3 for the maintenance of non‐small cell lung cancer stem cells is associated with the STAT3 pathway. Clin Cancer Res 2014; 20:4154–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fitzgerald TL, McCubrey JA. Pancreatic cancer stem cells: association with cell surface markers, prognosis, resistance, metastasis and treatment. Adv Biol Regul 2014; 56:45–50. [DOI] [PubMed] [Google Scholar]
- 62. Ruscito I, Castillo‐Tong DC, Vergote I, Ignat I, Stanske M, Vanderstichele A et al Exploring the clonal evolution of CD133/aldehyde‐dehydrogenase‐1 (ALDH1)‐positive cancer stem‐like cells from primary to recurrent high‐grade serous ovarian cancer (HGSOC). A study of the ovarian cancer therapy‐innovative models prolong survival (OCTIPS) consortium. Eur J Cancer 2017; 79:214–25. [DOI] [PubMed] [Google Scholar]
- 63. Le Magnen C, Bubendorf L, Rentsch CA, Mengus C, Gsponer J, Zellweger T et al Characterization and clinical relevance of ALDHbright populations in prostate cancer. Clin Cancer Res 2013; 19:5361–71. [DOI] [PubMed] [Google Scholar]
- 64. Prasmickaite L, Engesæter BØ, Skrbo N, Hellenes T, Kristian A, Oliver NK et al Aldehyde dehydrogenase (ALDH) activity does not select for cells with enhanced aggressive properties in malignant melanoma. PLoS ONE 2010; 5:e10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lu L, Tao H, Chang AE, Hu Y, Shu G, Chen Q et al Cancer stem cell vaccine inhibits metastases of primary tumors and induces humoral immune responses against cancer stem cells. Oncoimmunology 2015; 4:e990767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ning N, Pan Q, Zheng F, Teitz‐Tennenbaum S, Egenti M, Yet J et al Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res 2012; 72:1853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kanakry CG, Ganguly S, Zahurak M, Bolaños‐Meade J, Thoburn C, Perkins B et al Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci Transl Med 2013; 5:211ra157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bettum IJ, Gorad SS, Barkovskaya A, Pettersen S, Moestue SA, Vasiliauskaite K et al Metabolic reprogramming supports the invasive phenotype in malignant melanoma. Cancer Lett 2015; 366:71–83. [DOI] [PubMed] [Google Scholar]
- 69. Xie H, Hanai JI, Ren JG, Kats L, Burgess K, Bhargava P et al Targeting lactate dehydrogenase‐a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor‐initiating cells. Cell Metab 2014; 19:795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Feng W, Gentles A, Nair RV, Huang M, Lin Y, Lee CY et al Targeting unique metabolic properties of breast tumor initiating cells. Stem Cells 2014; 32:1734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Song K, Kwon H, Han C, Zhang J, Dash S, Lim K et al Active glycolytic metabolism in CD133(+) hepatocellular cancer stem cells: regulation by MIR‐122. Oncotarget 2015; 6:40822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ye XQ, Li Q, Wang GH, Sun FF, Huang GJ, Bian XW et al Mitochondrial and energy metabolism‐related properties as novel indicators of lung cancer stem cells. Int J Cancer 2011; 129:820–31. [DOI] [PubMed] [Google Scholar]
- 73. Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H et al Evidence for label‐retaining tumour‐initiating cells in human glioblastoma. Brain 2011; 134:1331–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Morrison SJ, Kimble J. Asymmetric and symmetric stem‐cell divisions in development and cancer. Nature 2006; 441:1068–74. [DOI] [PubMed] [Google Scholar]
- 75. Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S et al Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 2010; 140:62–73. [DOI] [PubMed] [Google Scholar]
- 76. Roesch A, Fukunaga‐Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A et al A temporarily distinct subpopulation of slow‐cycling melanoma cells is required for continuous tumor growth. Cell 2010; 141:583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell‐like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis 2009; 26:611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hirschmann‐Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U et al A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA 2004; 101:14228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Davidson AL, Dassa E, Orelle C, Chen J. Structure, function, and evolution of bacterial ATP‐binding cassette systems. Microbiol Mol Biol Rev 2008; 72:317–64, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Scharenberg CW, Harkey MA, Torok‐Storb B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 2002; 99:507–12. [DOI] [PubMed] [Google Scholar]
- 81. Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer 2010; 10:147–56. [DOI] [PubMed] [Google Scholar]
- 82. van de Ven R, Scheffer GL, Scheper RJ, de Gruijl TD. The ABC of dendritic cell development and function. Trends Immunol 2009; 30:421–9. [DOI] [PubMed] [Google Scholar]
- 83. Krause M, Dubrovska A, Linge A, Baumann M. Cancer stem cells: radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv Drug Deliv Rev 2017; 109:63–73. [DOI] [PubMed] [Google Scholar]
- 84. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN et al Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009; 458:780–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. de Jong MC, Pramana J, van der Wal JE, Lacko M, Peutz‐Kootstra CJ, de Jong JM et al CD44 expression predicts local recurrence after radiotherapy in larynx cancer. Clin Cancer Res 2010; 16:5329–38. [DOI] [PubMed] [Google Scholar]
- 86. Pearce EJ, Everts B. Dendritic cell metabolism. Nat Rev Immunol 2015; 15:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E et al Hypoxia‐inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol 2013; 14:1173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Spary LK, Al‐Taei S, Salimu J, Cook AD, Ager A, Watson HA et al Enhancement of T cell responses as a result of synergy between lower doses of radiation and T cell stimulation. J Immunol 2014; 192:3101–10. [DOI] [PubMed] [Google Scholar]
- 89. Salimu J, Spary LK, Al‐Taei S, Clayton A, Mason MD, Staffurth J et al Cross‐presentation of the oncofetal tumor antigen 5T4 from irradiated prostate cancer cells–a key role for heat‐shock protein 70 and receptor CD91. Cancer Immunol Res 2015; 3:678–88. [DOI] [PubMed] [Google Scholar]
- 90. Di Tomaso T, Mazzoleni S, Wang E, Sovena G, Clavenna D, Franzin A et al Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin Cancer Res 2010; 16:800–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Schatton T, Schütte U, Frank NY, Zhan Q, Hoerning A, Robles SC et al Modulation of T‐cell activation by malignant melanoma initiating cells. Cancer Res 2010; 70:697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F et al Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin‐4. Cell Stem Cell 2007; 1:389–402. [DOI] [PubMed] [Google Scholar]
- 93. Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W et al Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro‐Oncol 2010; 12:1113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kawasaki BT, Farrar WL. Cancer stem cells, CD200 and immunoevasion. Trends Immunol 2008; 29:464–8. [DOI] [PubMed] [Google Scholar]
- 95. Erin N, Podnos A, Tanriover G, Duymuş Ö, Cote E, Khatri I et al Bidirectional effect of CD200 on breast cancer development and metastasis, with ultimate outcome determined by tumor aggressiveness and a cancer‐induced inflammatory response. Oncogene 2015; 34:3860–70. [DOI] [PubMed] [Google Scholar]
- 96. Messai Y, Gad S, Noman MZ, Le Teuff G, Couve S, Janji B et al Renal cell carcinoma programmed death‐ligand 1, a new direct target of hypoxia‐inducible factor‐2 alpha, is regulated by von Hippel‐Lindau gene mutation status. Eur Urol 2016; 70:623–32. [DOI] [PubMed] [Google Scholar]
- 97. Zhi Y, Mou Z, Chen J, He Y, Dong H, Fu X et al B7H1 expression and epithelial‐to‐mesenchymal transition phenotypes on colorectal cancer stem‐like cells. PLOS ONE 2015; 10:e0135528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yang Y, Wu KE, Zhao E, Li W, Shi L, Xie G et al B7‐H1 enhances proliferation ability of gastric cancer stem‐like cells as a receptor. Oncol Lett 2015; 9:1833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Noh KH, Lee YH, Jeon JH, Kang TH, Mao CP, Wu TC et al Cancer vaccination drives Nanog‐dependent evolution of tumor cells toward an immune‐resistant and stem‐like phenotype. Cancer Res 2012; 72:1717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Brown CE, Starr R, Martinez C, Aguilar B, D'Apuzzo M, Todorov I et al Recognition and killing of brain tumor stem‐like initiating cells by CD8+ cytolytic T cells. Cancer Res 2009; 69:8886–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wu A, Wiesner S, Xiao J, Ericson K, Chen W, Hall WA et al Expression of MHC I and NK ligands on human CD133+ glioma cells: possible targets of immunotherapy. J Neurooncol 2007; 83:121–31. [DOI] [PubMed] [Google Scholar]
- 102. Wang B, Wang Q, Wang Z, Jiang J, Yu SC, Ping YF et al Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res 2014; 74:5746–57. [DOI] [PubMed] [Google Scholar]
- 103. Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A et al NK cells recognize and kill human glioblastoma cells with stem cell‐like properties. J Immunol 2009; 182:3530–9. [DOI] [PubMed] [Google Scholar]
- 104. Tseng HC, Arasteh A, Paranjpe A, Teruel A, Yang W, Behel A et al Increased lysis of stem cells but not their differentiated cells by natural killer cells; de‐differentiation or reprogramming activates NK cells. PLOS ONE 2010; 5:e11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tallerico R, Todaro M, Di Franco S, Maccalli C, Garofalo C, Sottile R et al Human NK cells selective targeting of colon cancer‐initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol 2013; 190:2381–90. [DOI] [PubMed] [Google Scholar]
- 106. Todaro M, Orlando V, Cicero G, Caccamo N, Meraviglia S, Stassi G et al Chemotherapy sensitizes colon cancer initiating cells to Vgamma9Vdelta2 T cell‐mediated cytotoxicity. PLOS ONE 2013; 8:e65145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Lai D, Wang F, Chen Y, Wang C, Liu S, Lu B et al Human ovarian cancer stem‐like cells can be efficiently killed by gammadelta T lymphocytes. Cancer Immunol Immunother 2012; 61:979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chen HC, Joalland N, Bridgeman JS, Alchami FS, Jarry U, Khan MW et al Synergistic targeting of breast cancer stem‐like cells by human gammadelta T cells and CD8+ T cells. Immunol Cell Biol 2017; 95:620–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Engelmann K, Shen H, Finn OJ. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res 2008; 68:2419–26. [DOI] [PubMed] [Google Scholar]
- 110. Inoda S, Hirohashi Y, Torigoe T, Morita R, Takahashi A, Asanuma H et al Cytotoxic T lymphocytes efficiently recognize human colon cancer stem‐like cells. Am J Pathol 2011; 178:1805–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Morita R, Hirohashi Y, Torigoe T, Ito‐Inoda S, Takahashi A, Mariya T et al Olfactory receptor family 7 subfamily C member 1 is a novel marker of colon cancer‐initiating cells and is a potent target of immunotherapy. Clin Cancer Res 2016; 22:3298–309. [DOI] [PubMed] [Google Scholar]
- 112. Verdegaal EM, De Miranda NF, Visser M, Harryvan T, Van Buuren MM, Andersen RS et al Neoantigen landscape dynamics during human melanoma‐T cell interactions. Nature 2016; 536:91–5. [DOI] [PubMed] [Google Scholar]
- 113. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT et al The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res 2009; 15:5323–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget 2015; 6:15772–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hirohashi Y, Torigoe T, Inoda S, Morita R, Kochin V, Sato N. Cytotoxic T lymphocytes: sniping cancer stem cells. Oncoimmunology 2012; 1:123–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Yamada R, Takahashi A, Torigoe T, Morita R, Tamura Y, Tsukahara T et al Preferential expression of cancer/testis genes in cancer stem‐like cells: proposal of a novel sub‐category, cancer/testis/stem gene. Tissue Antigens 2013; 81:428–34. [DOI] [PubMed] [Google Scholar]
- 117. Nishizawa S, Hirohashi Y, Torigoe T, Takahashi A, Tamura Y, Mori T et al HSP DNAJB8 controls tumor‐initiating ability in renal cancer stem‐like cells. Cancer Res 2012; 72:2844–54. [DOI] [PubMed] [Google Scholar]
- 118. Asano T, Hirohashi Y, Torigoe T, Mariya T, Horibe R, Kuroda T et al Brother of the regulator of the imprinted site (BORIS) variant subfamily 6 is involved in cervical cancer stemness and can be a target of immunotherapy. Oncotarget 2016; 7:11223–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Chang WW, Lee CH, Lee P, Lin J, Hsu CW, Hung JT et al Expression of Globo H and SSEA3 in breast cancer stem cells and the involvement of fucosyl transferases 1 and 2 in Globo H synthesis. Proc Natl Acad Sci USA 2008; 105:11667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Damelin M, Geles KG, Follettie MT, Yuan P, Baxter M, Golas J et al Delineation of a cellular hierarchy in lung cancer reveals an oncofetal antigen expressed on tumor‐initiating cells. Cancer Res 2011; 71:4236–46. [DOI] [PubMed] [Google Scholar]
- 121. Kerk SA, Finkel KA, Pearson AT, Warner KA, Zhang Z, Nör F et al 5T4‐targeted therapy ablates cancer stem cells and prevents recurrence of head and neck squamous cell carcinoma. Clin Cancer Res 2017; 23:2516–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Hirohashi Y, Torigoe T, Tsukahara T, Kanaseki T, Kochin V, Sato N. Immune responses to human cancer stem‐like cells/cancer‐initiating cells. Cancer Sci 2016; 107:12–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM et al HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012; 150:549–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Flores AN, McDermott N, Meunier A, Marignol L. NUMB inhibition of NOTCH signalling as a therapeutic target in prostate cancer. Nat Rev Urol 2014; 11:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Garcia‐Heredia JM, Verdugo Sivianes EM, Lucena‐Cacace A, Molina‐Pinelo S, Carnero A. Numb‐like (NumbL) downregulation increases tumorigenicity, cancer stem cell‐like properties and resistance to chemotherapy. Oncotarget 2016; 7:63611–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Mine T, Matsueda S, Li Y, Tokumitsu H, Gao H, Danes C et al Breast cancer cells expressing stem cell markers CD44+ CD24 lo are eliminated by Numb‐1 peptide‐activated T cells. Cancer Immunol Immunother 2009; 58:1185–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Yasuda K, Hirohashi Y, Mariya T, Murai A, Tabuchi Y, Kuroda T et al Phosphorylation of HSF1 at serine 326 residue is related to the maintenance of gynecologic cancer stem cells through expression of HSP27. Oncotarget 2017; 8:31540–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Yasuda K, Hirohashi Y, Kuroda T, Takaya A, Kubo T, Kanaseki T et al MAPK13 is preferentially expressed in gynecological cancer stem cells and has a role in the tumor‐initiation. Biochem Biophys Res Commun 2016; 472:643–7. [DOI] [PubMed] [Google Scholar]
- 129. Weina K, Utikal J. SOX2 and cancer: current research and its implications in the clinic. Clin Transl Med 2014; 3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Nakatsugawa M, Takahashi A, Hirohashi Y, Torigoe T, Inoda S, Murase M et al SOX2 is overexpressed in stem‐like cells of human lung adenocarcinoma and augments the tumorigenicity. Lab Invest 2011; 91:1796–804. [DOI] [PubMed] [Google Scholar]
- 131. Saijo H, Hirohashi Y, Torigoe T, Horibe R, Takaya A, Murai A et al Plasticity of lung cancer stem‐like cells is regulated by the transcription factor HOXA5 that is induced by oxidative stress. Oncotarget 2016; 7:50043–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Vonderheide RH, Anderson KS, Hahn WC, Butler MO, Schultze JL, Nadler LM. Characterization of HLA‐A3‐restricted cytotoxic T lymphocytes reactive against the widely expressed tumor antigen telomerase. Clin Cancer Res 2001; 7:3343–8. [PubMed] [Google Scholar]
- 133. Alexandrov LB, Nik‐Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV et al Signatures of mutational processes in human cancer. Nature 2013; 500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Mennonna D, Maccalli C, Romano MC, Garavaglia C, Capocefalo F, Bordoni R et al T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut 2017; 66:454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Guo Y, Feng K, Wang Y, Han W. Targeting cancer stem cells by using chimeric antigen receptor‐modified T cells: a potential and curable approach for cancer treatment. Protein Cell 2017; 9:604. [DOI] [PMC free article] [PubMed] [Google Scholar]