

Summary
Given the pivotal roles that CD4+ T cell imbalance plays in human immune disorders, much interest centres on better understanding influences that regulate human helper T‐cell subset dominance in vivo. Here, using primary CD4+ T cells and short‐term T helper type 1 (Th1) and Th2‐like lines, we investigated roles and mechanisms by which neurotransmitter receptors may influence human type 1 versus type 2 immunity. We hypothesized that N‐methyl‐d‐aspartate receptors (NMDA‐R), which play key roles in memory and learning, can also regulate human CD4+ T cell function through induction of excitotoxicity. Fresh primary CD4+ T cells from healthy donors express functional NMDA‐R that are strongly up‐regulated upon T cell receptor (TCR) mediated activation. Synthetic and physiological NMDA‐R agonists elicited Ca2+ flux and led to marked inhibition of type 1 but not type 2 or interleukin‐10 cytokine responses. Among CD4+ lines, NMDA and quinolinic acid preferentially reduced cytokine production, Ca2+ flux, proliferation and survival of Th1‐like cells through increased induction of cell death whereas Th2‐like cells were largely spared. Collectively, the findings demonstrate that (i) NMDA‐R is rapidly up‐regulated upon CD4+ T cell activation in humans and (ii) Th1 versus Th2 cell functions such as proliferation, cytokine production and cell survival are differentially affected by NMDA‐R agonists. Differential cytokine production and proliferative capacity of Th1 versus Th2 cells is attributable in part to increased physiological cell death among fully committed Th1 versus Th2 cells, leading to increased Th2‐like dominance. Hence, excitotoxicity, beyond its roles in neuronal plasticity, may contribute to ongoing modulation of human T cell responses.
Keywords: CD4+ T cells; glutamic acid; glutamate; human; immune regulation; NMDA‐R; NMDA; Th1, Th2
Abbreviations
- IDO
indoleamine 2,3‐dioxygenase
- NMDA
N‐methyl‐d‐aspartate
- NMDA‐R
N‐methyl‐d‐aspartate receptors
- QA
quinolinic acid
Introduction
Imbalanced helper T cell subset function is a major contributor to many chronic immune diseases. Beyond their protective roles, excessive or misdirected T helper type 1 (Th1) cell activity is functionally important in various autoimmune diseases, whereas Th2‐like cells play pivotal roles in allergies, asthma and many cancers.1, 2 Although many CD4+ T cell subtypes have been described in recent years, and there is now an appreciation of their plasticity,1, 3, 4, 5, 6, 7 the broad model used of Th1‐ and Th2‐like functional phenotypes provides a ready shorthand and useful paradigm to investigate contributions of CD4+ T cells to both protective and maladaptive immunity. The continuous rebalancing known to occur between different helper cell subtypes is controlled by many influences, including regulatory T cell activity, cytokine cross‐regulation and production of anti‐inflammatory cytokines by various tissues.8, 9 One other mechanism that plays an important role in such homeostasis is activation‐induced cell death. The microenvironment for Th2‐type diseases can actively promote activation‐induced cell death of mostly Th1 but not Th2 cells,10, 11 whereas the reverse can be seen in some diseases associated with overly exuberant Th1‐type dominance. Hence, in any given disease condition, a varying proportion of cell death within distinct T‐cell subsets can make an important contribution to immune polarization12, 13, 14 and altered immune regulation. The mechanisms that contribute to such skewing are a focus of active research.
N‐Methyl‐d‐aspartate receptors (NMDA‐R) play fundamental roles in memory and learning.15, 16, 17 Our understanding of their expression and function in immunology remains limited. Excitotoxicity is a Ca2+‐dependent cell death pathway occurring in neurons that is triggered by excessive stimulation of NMDA‐R by neurotransmitters such as glutamate.18, 19 This type of cell death is clearly associated with Alzheimer's disease, Parkinson's disease, HIV‐associated neurocognitive disorders, and several other neurodegenerative conditions.20, 21, 22 Although cell biology and clinical studies to date have focused heavily on neuronal cells, interestingly, excitotoxicity can also be induced in extra‐neuronal cells.23, 24
Indoleamine 2,3‐dioxygenase (IDO) contributes to T‐cell imbalance;25, 26, 27 IDO is the first and rate‐limiting enzyme in tryptophan catabolism via the kynurenine pathway (Fig. 1a) that ultimately generates various breakdown products, collectively termed kynurenines.28, 29, 30 Acting via NMDA‐R, kynurenines exhibit diverse neurological activities. Quinolinic acid (QA) acts as an agonist, whereas kynurenic acid acts as an antagonist.31, 32 In addition to our previous study,26 data from murine models indicate that tryptophan breakdown products are crucial in Th2 cell function and survival,33 tolerance in allergic airways34 and airway smooth muscle function.35, 36
Figure 1.
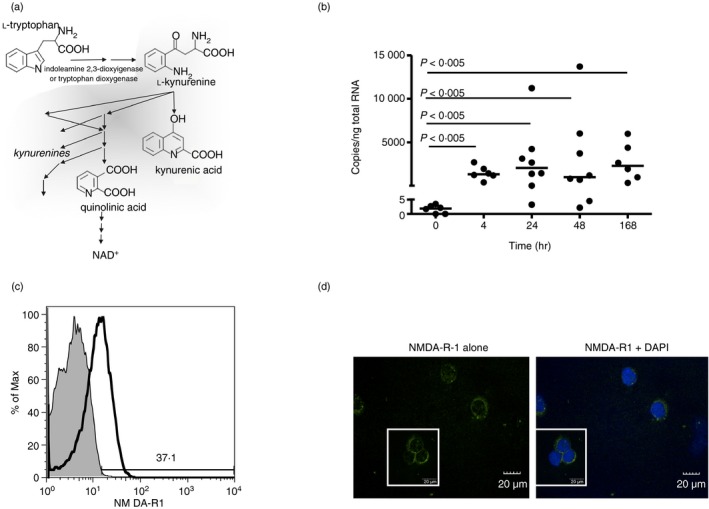
Anti‐CD3/CD28 activation induces elevated N‐methyl‐d‐aspartate receptor 1 (NMDA‐R1) expression by peripheral CD4+ T cells. (a) Scheme of kynurenine pathway in tryptophan catabolism. (b) Freshly isolated CD4+ T cells, activated with anti‐CD3/CD28 antibody for the indicated times, were examined for NMDA‐R1 expression by quantitative PCR (n = 9 for detailed kinetics) with medians (Mann–Whitney P‐values) shown. (c) Flow cytometric analysis of NMDA‐R1 protein expression on fresh CD3+/CD4+ primary T‐cell populations that were resting or stimulated with anti‐CD3/CD28 for 24 hr. Data are from one individual, representative of eight individuals examined. (d) Confocal laser‐scanning immunofluorescence microscopy of NMDA‐R1 expression at 24 hr on anti‐CD3/CD28 activated primary CD4+ T cells. NMDA‐R1 (green); DAPI (blue, right). Data are from one individual, representative of three examined.
Here, we sought to determine what role, if any, NMDA‐R play in regulation of human CD4+ T‐cell function. Specifically, we hypothesized that peripheral CD4+ T cells express functional NMDA‐R in vivo and that they are up‐regulated upon T cell receptor (TCR) ligation. We speculated that activation via this receptor acts on multiple key aspects of helper T cell function including proliferation, cytokine production and cell survival, and further, that NMDA‐R agonists exhibit differential effects on T cell subsets that can lead to Th1 cell depletion and thereby indirectly enhance Th2‐like dominance.
Materials and methods
Healthy volunteers and isolation of CD4+ T cells
Healthy non‐atopic volunteers were recruited following a protocol approved by the Bannatyne Campus Research Ethics Board at the University of Manitoba, and the Human Research Ethics Board at the University of Alberta. Peripheral blood samples were collected from consenting healthy adults then fresh primary CD4+ T cells were immediately isolated via density gradient centrifugation on either Ficoll‐Paque (GE Healthcare, Baie‐d'Urfé, QC) or Lymphocyte Separation Medium (Lonza, Walkersville, MD) and negatively selected for using immunomagnetic bead technology either with the CD4+ T Cell Isolation Kit II (Miltenyi Biotec, Auburn, CA) or CD4+ cell Enrichment Cocktail (StemCell Technologies, Vancouver, BC).
Generation of Th1/Th2‐driven cells and cell lines
Th1 and Th2 cells were generated from naive CD4+ T cells using standard methods.26, 37 Briefly, naive T cells were obtained using negative selection with a combination of CD4+ T Cell Isolation Kit II and CD45RO microbeads (Miltenyi Biotec). anti‐CD3/CD28 activated CD4+ CD45RO– T cells were skewed into Th1 cell phenotypes for 2 weeks in the presence of interleukin‐2 (IL‐2; 8 ng/ml), IL‐12 (5 ng/ml), interferon‐γ (IFN‐γ; 10 ng/ml) (PeproTech, Rocky Hill, NJ), and anti‐IL‐4 (10 μg/ml) (R&D Systems, Minneapolis, MN); or to Th2‐like cells in the presence of IL‐2 (8 ng/ml), IL‐4 (20 ng/ml) (PeproTech), anti‐IFN‐γ (8 μg/ml) and anti‐IL‐12 (5 μg/ml) (R&D Systems). To further enrich the purity of Th1‐ and Th2‐driven dominance, short‐term cultures were enriched for IFN‐γ‐secreting cells or IL‐4‐secreting cells, using IFN‐γ or IL‐4 Secretion Assay‐Cell Enrichment and Detection Kits, respectively (Miltenyi Biotec). These cell lines were maintained in IL‐2 only (Th1‐skewed cells) or IL‐2 + IL‐4 (Th2 cells), and re‐stimulated with anti‐CD2/CD3/CD28 for further expansion every 4–6 weeks under conditioned medium. Confirmation of enriched phenotype was achieved through a combination of flow cytometry (IFN‐γ and IL‐4) and ELISA or Mesoscale assay (IFN‐γ and IL‐5)
Helper T‐cell activation and assessment of NMDA‐R agonist function
All cultures were carried out with glutamate‐deficient medium. After 24 hr activation with anti‐CD3/CD28 antibodies, primary peripheral blood mononuclear cells (PBMC) (2·5 × 106/ml), enriched primary CD4+ T cells (500 000/ml) or Th1‐ and Th2‐driven cells (1·0 × 106 cells/ml) were treated with NMDA‐R agonists (100 μm NMDA or 40 μm QA (Sigma‐Aldrich, Oakville, ON), alone or in the presence of NMDA‐R‐specific inhibitor MK‐801 (10 μm, Sigma‐Aldrich). In selected experiments, α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA), an AMPA receptor agonist, or 1‐amino‐1,3‐dicarboxycyclopentane (ACPD), a group I/II metabotropic glutamate receptor agonist, neither of which bind NMDA‐R, were used at 100 μm (R&D Systems). Cells and supernatants were harvested at the times indicated.
Flow cytometry: Ca2+ flux and NMDA‐R expression
Intracellular release of Ca2+ following stimulation of NMDA‐R was measured in T cells loaded with Ca2+‐sensitive dyes, Fluo‐3 AM and Fura Red AM according to previously described protocols.38 Briefly, cells were washed in dye loading buffer (Hanks’ balanced salt solution with 1 mm Ca2+, 1 mm Mg2+, 0·5% weight/volume bovine serum albumin, 4 mm probenecid), distributed into flow cytometry tubes (2 × 106 cells/ml) and placed in a water bath at 37° until activation and immediate acquisition on the BD Canto II flow cytometer. Background intracellular Ca2+ events were monitored by acquiring cells for 204 seconds without agonist stimulation then for the initial 50 seconds following agonist stimulation and further acquired up to 204 seconds. Positive controls consisted of cells activated with Ca2+ ionophore A23187, also used to evaluate dye partitioning to optimize the concentration of Ca2+‐sensitive dyes. Changes in intracellular Ca2+ were measured using the kinetics function in flowjo software (Tree Star Inc., Ashland, OR).
NMDA‐R1 expression on CD4+ T cells was detected by flow cytometry. Cells were treated with FcR Blocking Reagent then stained with rat monoclonal anti‐NMDA‐R1 (clone 5‐1‐G10) (Sigma‐Aldrich) or isotype control, followed by Alexa Fluor 488 goat anti‐rabbit antibody (Invitrogen, Carlsbad, CA), and anti‐CD3/CD4 monoclonal antibodies (BD Biosciences, Mississauga, ON). Samples were analysed on a BD FACS Canto II flow cytometer using diva software (BD Biosciences). Data were analysed using flowjo software, double gating on CD3+ and CD4+ cell populations.
Confocal laser scanning immunofluorescence
After 24 hr activation, cytospin samples of enriched CD4+ T cells were prepared on glass slides using 20% fetal bovine serum in PBS, dried and frozen. Cells were then fixed/permeabilized in 3% paraformaldehyde–0·5% Triton X‐100, blocked (0·5% gelatine in PBS) and stained with polyclonal rabbit anti‐NMDA‐R1 (clone D65B7, Cell Signaling Technology, Danvers, MA) followed by Alexa Fluor 488 goat anti‐rabbit immunoglobulin. Slides were analysed using an FV500 confocal microscope and fluoview 1·3 software (Olympus, Richmond Hill, ON).
Cell viability
The MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) assay was used to quantify cell viability (mitochondrial oxidation). Briefly, cells were activated with anti‐CD3/28 for the period indicated, then treated with NMDA at the indicated concentrations. A 5 mg/ml MTT (yellow tetrazolium salt; Sigma‐Aldrich) solution in PBS was added to cells at a ratio of 1 : 10 for the final 5 hr at 37°. Plates were centrifuged at 300×g, DMSO was added to solubilize the formazan crystals and optical density was read at 560 nm, with 670 nm background subtracted. The per cent of viable cells was calculated by setting the mean optical density of control groups as 100%.
Cell proliferation
Vybrant CFSE Cell Tracer Kit (Invitrogen) was used to label 1 × 106 cells/ml CD4+ T cells as well as Th1 and Th2 cells. Cells were then washed and resuspended in growth medium and cultured in the presence of various concentrations of NMDA or QA, followed by activation with anti‐CD3/CD28 antibodies. Control samples consisted of mitomycin‐C‐treated T cells and parallel experiments of similarly treated unlabelled cells were performed. Cells were harvested after 5 days in culture, washed and examined using flow cytometry. Using the proliferation platform on the flowjo, a statistical model was used to track proliferation and determine the Proliferation Index for each treatment group.
Apoptosis
Apoptosis assays were performed after 2, 4 and 24 hr in culture by detecting Alexa Fluor 488 Annexin V and membrane permeability levels using the Vybrant Apoptosis Assay Kit along with Mitotracker Red CMXRos (Invitrogen) or TO‐PRO‐3 iodide (Invitrogen) and acquired on flow cytometry. Total percentage of physiological cell death; total of Annexin V+ TO‐PRO‐3‐ cells (early state of apoptosis) and Annexin V+ TO‐PRO‐3+ cells (late stage of apoptosis or necrosis) were calculated.
Quantitative PCR
Total RNA was isolated from fresh PBMC and human CD4+ T cells directly ex vivo as well as Th1 and Th2 lines using an RNeasy Plus Mini Kit (Qiagen, Toronto, ON). RNA integrity ranged from 9·6 to 10. Complemtary DNA was synthesized using iScript Reverse Transcription Supermix (Bio‐Rad, Mississauga, ON). Quantitative PCR was performed using intron‐spanning primers (see Supplementary material, Table S1) and Thunderbird SYBR qPCR Mix (Toyobo, Japan) in a CFX96 real‐time PCR Detection System (Bio‐Rad). For improved precision in quantifying changes in mRNA levels, standards were made for each primer set using Human Universal Reference Total RNA (Clontech, Mountain View, CA). Data are expressed as copy number/ng cDNA rather than fold induction. All data were normalized using 18s rRNA as housekeeping gene. Replicate assays of the same sample on independent days typically resulted in 5–10% CV in copy number values obtained.
Determination of cytokine protein levels
Cytokine levels in cryopreserved supernatants were determined using MesoScale Discovery (MSD, Gaithersburg, MD) electrochemiluminescence assays to quantify binding events on patterned arrays. To provide uniformity in comparing data between different plate lots, constant internal laboratory standards (Peprotech, Rocky Hill, NJ and R&D Systems, Minneapolis, MN) were used throughout the study. Samples and standards were incubated for 3 hr (instead of 2 hr) and plates were incubated with detection antibody for 3 hr (instead of 2 hr) before washing, with all other steps as per the manufacturers’ recommendations. Analysis was on a SECTOR™ 2400 instrument (MSD). The operator was blind to the nature of all samples during processing, with subsequent statistical analysis also performed independently. Interassay variation was generally 5–10%.
Statistical analysis
Analyses were carried out with prism 5 software (GraphPad, San Diego, CA). Results were analysed for statistical significance using Wilcoxon or Mann–Whitney tests when non‐parametric distributions occurred, and paired t‐tests when parametric distributions were present. Friedman's test, a non‐parametric test similar to the parametric repeated measures analysis of variance, was used to assess differences across multiple treatment concentrations. Two‐tailed P‐values were reported with differences considered significant if P < 0·05. Throughout the study, each n value represents the number of independent donors examined.
Results
CD4+ T cell activation induces strong NMDA‐R1 up‐regulation
Although NMDA‐R expression by resting lymphocytes is well established, the impact of TCR‐mediated activation on receptor expression is poorly understood. We initially focused on NMDA‐R1, an essential subunit for functional NMDA‐R expression39 in primary CD4+ T cells obtained from peripheral blood following acute TCR‐mediated activation with anti‐CD3/CD28 antibodies. Figure 1(b) demonstrates that NMDA‐R1 expression was present at low levels in resting cells in vivo. Upon activation, it was rapidly enhanced with median 600‐fold increases by 4 hr, peak expression at 24 hr (median 1500‐fold enhanced) and was sustained for at least a week. Subsequent experiments with 18 additional individuals demonstrated that in total 23 of the 27 healthy adults examined up‐regulated NMDA‐R1 at the 24 hr time‐point following TCR stimulation. NMDA‐R1 protein expression was subsequently confirmed by flow cytometry of TCR‐activated CD4+ T cells (Fig. 1c) and confocal laser scanning microscopy (Fig. 1d).
Functional activity of activated primary CD4+ T cells is altered upon NMDA‐R stimulation
To determine if NMDA‐R1 expression was functional, intracellular Ca2+ flux was assessed. NMDA is a synthesized ligand that does not typically occur in nature. It is used here to complement glutamate or QA (two natural NMDA‐R ligands) and because of its specificity for NMDA‐R, whereas glutamate can bind to receptors other than the NMDA‐R. TCR‐activated primary human CD4+ T cells were cultured with NMDA or glutamate. Dose‐dependent increases in intracellular Ca2+ (Fig. 2) were evident, with specific, non‐competitive, NMDA‐R inhibitor MK‐801 largely abolishing such responses. NMDA‐R specificity was also confirmed using independent glutamate receptor subtype‐specific agonists; AMPA (an AMPA receptor agonist) (Fig. 2c) and ACPD (a group I/II metabotropic glutamate receptor agonist) (Fig. 2d). Neither of these stimulated Ca2+ flux.
Figure 2.
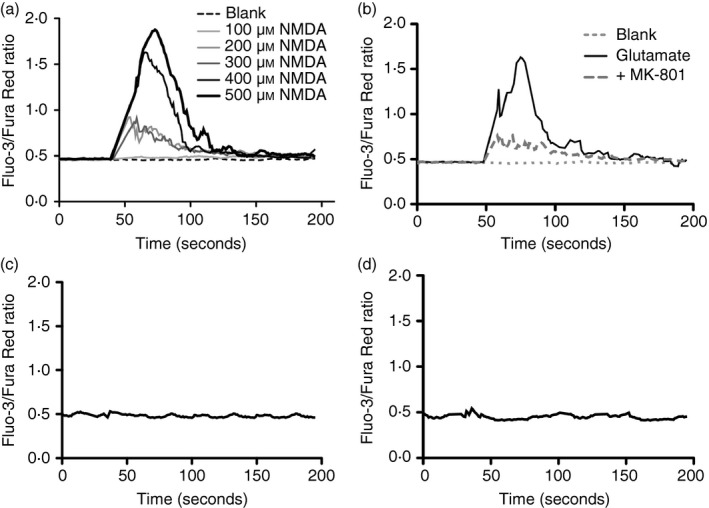
Specificity and functional activity of N‐methyl‐d‐aspartate receptor (NMDA‐R) expression on CD4+ T cells. Dose‐dependent intracellular Ca2+ influx by enriched primary CD4+ T cells following (a) NMDA stimulation or (b) glutamate stimulation (200 μm) alone and in the presence of NMDA‐R antagonist MK‐801 (10 μm) or (c, d) stimulation with AMPA (100 μm) or ACPD, NMDA‐R‐independent glutamate receptor subtype‐specific agonists. Data are from one individual, representative of three individuals examined.
NMDA‐R stimulation has a differential impact on Type 1 versus Type 2 cytokine responses
To determine if there was an impact of NMDA‐R agonist exposure on cytokine production by fresh human T cells directly ex vivo, we stimulated PBMC from 12 asymptomatic donors with anti‐CD3/CD28 alone and in the presence of NMDA or QA. QA, a physiological tryptophan breakdown product that acts as an NMDA‐R agonist in the nervous system,31, 32 is also found in blood and local tissues. Type 1 cytokine production was substantially decreased in the presence of either NMDA‐R ligand [Fig. 3a, i.e. median decreases 65% (P < 0·005) and 75% (P = 0·002) for IFN‐γ; 40% (P = 0·015) and 50% (P = 0·002) for tumour necrosis factor‐α (TNF‐α)]. Conversely, type 2 cytokine production stimulated by anti‐CD3/CD28 monoclonal antibodies (Fig. 3b left columns) was not hindered. Recognizing that anti‐CD3/CD28 is a strong type 1 stimulus but relatively weak stimulus of type 2 cytokine production40 and that the positive controls in these experiments exhibited low levels of cytokine production, phytohaemagglutinin (PHA) was used as a recognized stimulus of type 2 recall responses. PHA elicited ~ 10‐fold to 100‐fold increased type 2 responses compared with those elicited with anti‐CD3/CD28 (Fig. 3b, right columns). None of these robust type 2 responses were inhibited in the presence of QA or NMDA.
Figure 3.
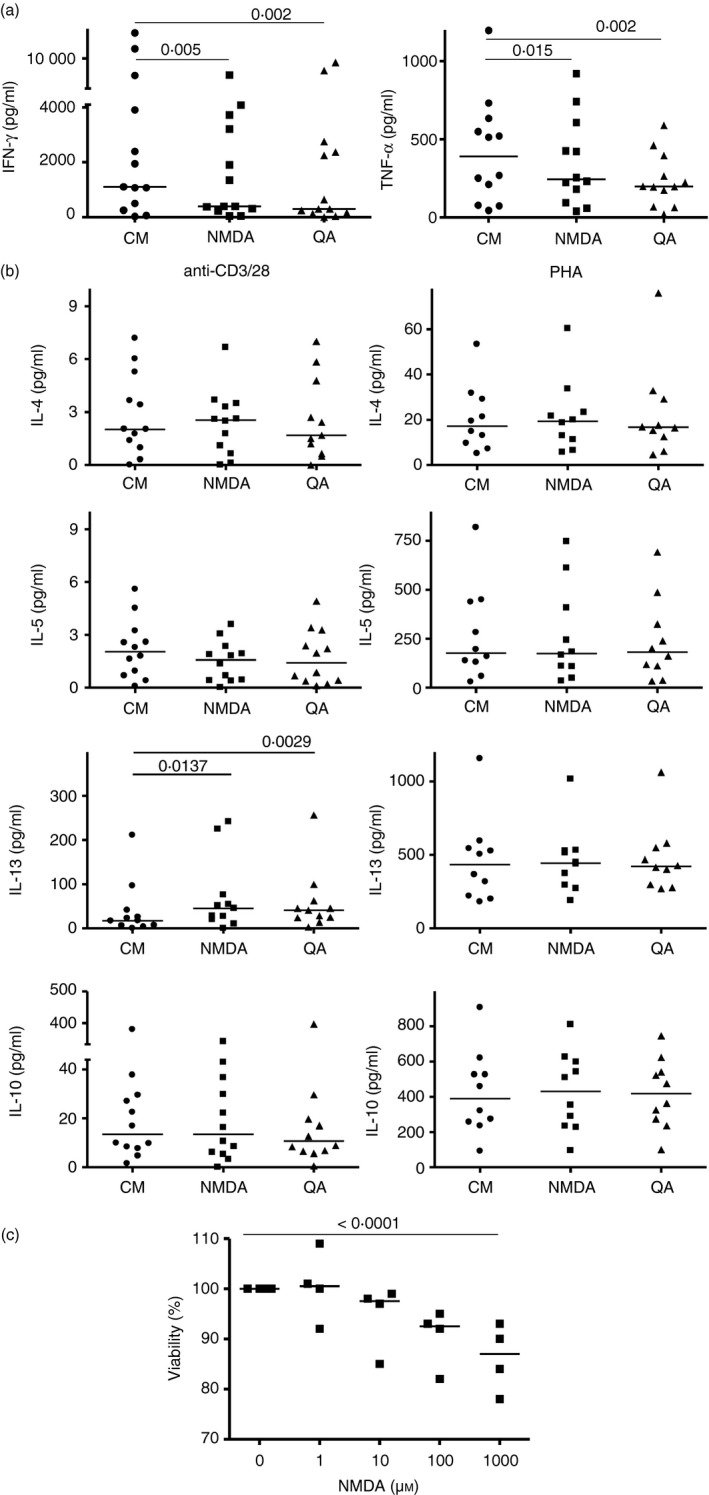
Type 1 but not type 2 cytokine responses by fresh human T cells are inhibited in the presence of N‐methyl‐d‐aspartate (NMDA) or kynurenine metabolite, quinolinic acid (QA). (a) Freshly obtained peripheral blood mononuclear cells were stimulated with anti‐CD3/CD28 for 24 hr then NMDA receptor (NMDA‐R) agonists (100 μm NMDA or 40 μm QA) were added. Cytokine levels were assessed at 48 hr. Each point represents a unique individual. Median values shown with paired non‐parametric analyses. (b) T helper type 2 (Th2) cytokine responses from anti‐CD3/CD28 cultures or, in separate wells, phytohaemagglutinin (10 μg/ml) in the presence/absence of NMDA‐R agonists were quantified. All supernatants in 12 experiments with unique donors were harvested at 48 hr. Median values and Wilcoxon analyses are shown. All other comparisons were not statistically significant. (c) CD4+ cells were anti‐CD3/CD28 activated for 24 hr then exposed to NMDA for 3 days, with MTT added for the final 5 hr. Cell viability was calculated as a ratio by setting the absorbance of cells in the absence of NMDA as 100% (medians and Friedman's test shown for n = 4 individuals).
To determine if the selective effect of NMDA or QA on type 1 responses was linked to increased IL‐10 production, the impact of NMDA‐R activation was assessed in responses elicited by either anti‐CD3/CD28 or PHA activation. Interleukin‐10 production was not affected in either case.
Collectively these data demonstrate that NMDA‐R agonists are capable of eliciting Ca2+ flux in primary human CD4+ T cells and of selectively inhibiting type 1 cytokine production, while sparing type 2 cytokine responses.
Impact of NMDA‐R triggering on cell survival
To examine the effect of NMDA‐R ligation on survival of primary CD4+ T cells, fresh CD4+ T cell‐enriched populations were stimulated with anti‐CD3/CD28 monoclonal antibodies in the presence of increasing concentrations of NMDA. NMDA‐R stimulation was found to dose‐dependently decrease cellular viability (Fig. 3c).
Differential sensitivity of Th1 versus Th2 polarized cells to NMDA‐R regulation
To assess how NMDA‐R stimulation might differentially affect more fully committed human Th1 versus Th2 cells, we induced short‐term Th1 and Th2 cell lines in vitro using CD4+ T cells from five different individuals. Real‐time PCR, ELISA and intracellular staining for IFN‐γ and IL‐4 followed by flow cytometric analysis were used to confirm typical Th1 and Th2 cell phenotypes in the 10 lines derived. Transient Ca2+ flux was evident in both Th1‐ and Th2‐driven cell lines (Fig. 4a). In the nine experiments conducted with Th1 cells, each demonstrated robust Ca2+ flux. Considerably more variability was evident with Th2 lines, with 8 of 11 experiments exhibiting weak or undetectable Ca2+ flux. Hence, although both cell types are capable of exhibiting NMDA‐R‐dependent activation, it was most intensely and consistently evident in Th1‐driven cells. Use of QA to elicit activation revealed similar strong responses by Th1 clones whereas Th2 clones were usually weakly responsive or unresponsive to this physiological NMDA‐R ligand (Fig. 4b). As seen above for fresh (largely uncommitted) primary T cell populations directly ex vivo, IFN‐γ and TNF‐α production driven by TCR engagement was reduced in the presence of NMDA or its physiological counterpart, QA. Cytokine production by Th2 cells was again, unaffected (Fig. 5).
Figure 4.
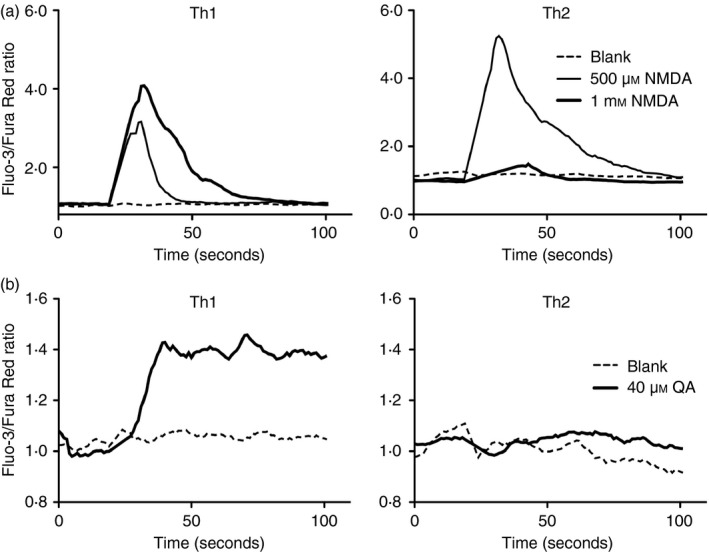
Differential Ca2+ flux responses occur in activated T helper type 1 (Th1) versus Th2 cells upon N‐methyl‐d‐aspartate receptor (NMDA‐R) stimulation. Intracellular Ca2+ responses in anti‐CD3/CD28‐activated Th1 and Th2 short‐term lines pre‐loaded with Ca2+‐sensitive dyes then stimulated with (a) 500 μm and 1 mm NMDA or (b) 40 μm quinolinic acid (QA). Date are representative of 8 and 11 experiments for Th1 and Th2 respectively in (a) for NMDA and of five Th1 and Th2 lines in (b) for QA.
Figure 5.
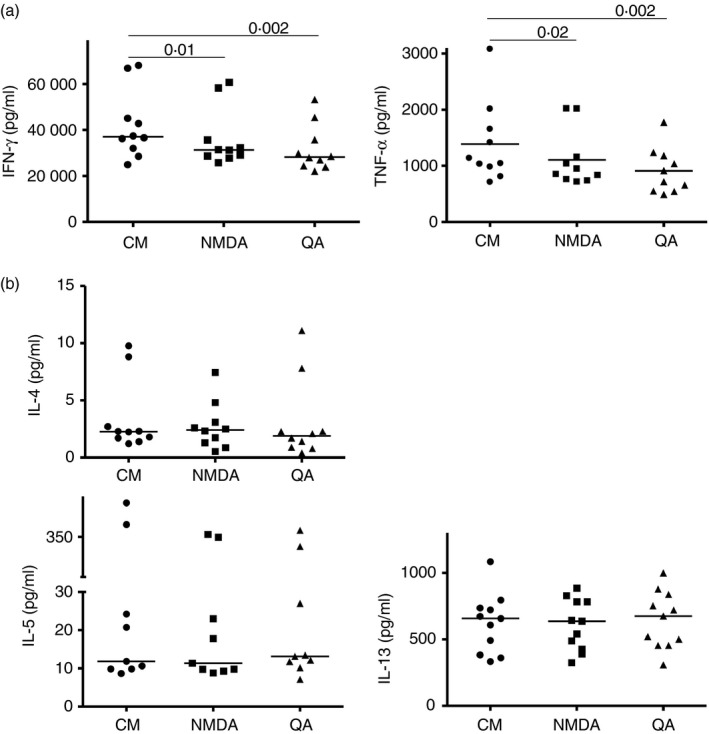
Differential responses of T helper type 1 (Th1) versus Th2 cell lines to N‐methyl‐d‐aspartate receptor (NMDA‐R) stimulation. (a) Th1 and (b) Th2 cells were anti‐CD3/CD28 stimulated for 24 hr then NMDA (100 μm) or quinolinic acid (QA) (40 μm) was added for 24 hr after which cytokine production was determined. Median values (Wilcoxon matched pairs tests) from replicate experiments, using Th1 or Th2 cell lines derived from five independent donors for each Th phenotype are shown.
We therefore directly tested the hypothesis that exposure to NMDA‐R agonists differentially affects proliferation and/or survival of committed Th1 versus Th2 cells. CFSE‐loaded Th1 and Th2 cultures were activated with anti‐CD3/CD28 mAbs for 24 hr then cultured in the presence of different concentrations of NMDA. Figure 6(a,b) demonstrates that NMDA had no impact on the proliferation of TCR‐induced Th2‐driven proliferation over 11 independent experiments, even at concentrations up to 1600 μm. Conversely, Th1‐driven cell proliferation was markedly inhibited by NMDA, even at 100 μm (P < 0·001). Figure 6(b) provides sample CFSE data. Similarly, NMDA resulted in sharp decreases in Th1 cell numbers whereas Th2 cultures exposed to 200 μm NMDA were unchanged (Fig. 6c,d). Collectively these data indicate that differential cytokine production and proliferative capacity of Th1 versus Th2 cell lines were attributable to an increased frequency of physiological cell death among fully committed Th1 versus Th2 cells upon NMDA‐R‐dependent triggering.
Figure 6.
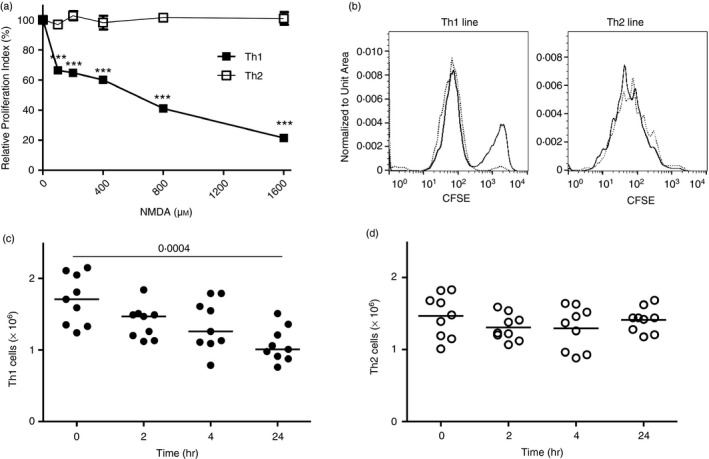
(a) Reduced proliferation of T‐cell receptor (TCR) ‐activated T helper type 1 (Th1) versus Th2 cell lines in the presence of N‐methyl‐d‐aspartate (NMDA). CFSE‐labelled Th1 and Th2 cells stimulated with anti‐CD3/CD28 for 24 hr then incubated with NMDA at the concentrations shown. Proliferation was determined at day 4 by flow cytometry. Relative proliferation indices were calculated in comparison to the proliferation index for groups lacking NMDA exposure as 100%. n = 6 experiments in total, ***P < 0·001. (b) CFSE FACS plots for representative Th1 and Th2 lines stimulated with anti‐CD3/CD28 in the presence/absence of 200 μm NMDA treatment. (c) Live cell number transition of Th1 and Th2 cell lines. Th1‐ or Th2‐driven cells obtained from five unique donors for each Th phenotype were anti‐CD3/CD28 stimulated as in (a), then incubated with 200 μm NMDA. Live cell numbers were determined by trypan blue counting at the indicated times. Paired t‐test is shown.
TCR‐stimulated Th1 and Th2 lines cultured with NMDA (200 μm) were stained with TO‐PRO‐3 and Annexin V then analysed by flow cytometry (Fig. 7). Th1 cells exhibited strong sensitivity to NMDA (mean 33% death at 24 hr), whereas only a small proportion of Th2 cells died. These effects of NMDA were largely blocked in the presence of MK‐801 (10 μm). This pattern of differential Th1 versus Th2 cell sensitivity with respect to cell death following NMDA‐R ligation was also evident in independent experiments using physiological ligand QA (40 μm) (Fig. 7c).
Figure 7.
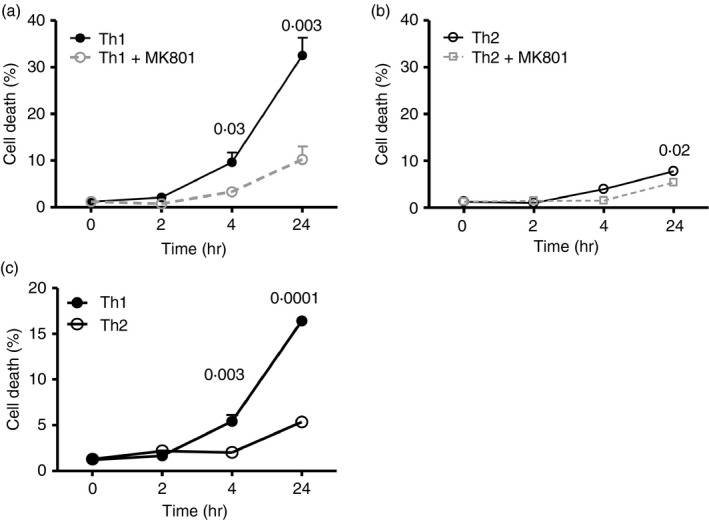
T helper type 1 (Th1) cells differ from Th2 cells in their sensitivity to cell death upon N‐methyl‐d‐aspartate receptor (NMDA‐R) ligation. Kinetic analysis of cell death among Th1 versus Th2 cells stimulated with anti‐CD3/CD28 for 24 hr then cultured as indicated with (a, b) NMDA for the period indicated plus/minus the inhibitor MK‐801 (10 mm) or (c) quinolinic acid (QA) (40 μm). The total percentage of physiological cell death was determined by adding Annexin V+ TO‐PRO‐3‐ cells (early stage of apoptosis) and Annexin V+ TO‐PRO‐3+ cells (late stage of apoptosis or necrosis) at each time‐point. n = 4 independent experiments, each with unique Th1 and Th2.
Discussion
There is increasing recognition of the importance of neurotransmitter signalling within the human immune system.41, 42 In this study, we found that anti‐CD3/CD28 activation induces mature human CD4+ T cells to markedly up‐regulate functional NMDA‐R on their surface. Specific stimulation with NMDA, or with the physiological tryptophan metabolite QA, resulted in reduced expression of Th1‐like cytokine production concomitant with unaltered Th2‐like or IL‐10 responses. Th1, but not Th2, cells exhibited reduced proliferation upon TCR‐dependent activation. The mechanism underlying these differential effects on Th1‐ versus Th2‐like function was linked to a pronounced susceptibility of Th1 cells, and relative resistance of Th2 cells, to NMDA‐R‐dependent physiological cell death. This differential sensitivity to cell death in Th1 versus Th2‐like cells is the first report of which we are aware that demonstrates differential responses of CD4+ T cell subsets to NMDA‐R stimulation. The data suggest that NMDA‐R‐mediated cytotoxicity of Th1 cells could contribute to the CD4+ T cell imbalance that is characteristic of a range of human chronic immune disorders, including allergies and asthma.
This observation, that NMDA‐R stimulation selectively modulates TCR‐dependent Th1 versus Th2 function and survival has analogies to NMDA‐R‐driven effects on neurons, where both neuroprotective and neurotoxic outcomes are observed. In the nervous system, NMDA‐R stimulation can lead cells to death when they are excited beyond homeostatic levels.43 An overabundance of cytosolic Ca2+ results in over‐activation of Ca2+‐dependent degradative enzymes leading to cell death.44 Microglia can release glutamate, promoting neurotoxicity in neurons expressing NMDA‐R.45 In the immune system, glutamate from antigen‐presenting cells contributes to regulate glutamate levels in the microenvironment of lymph nodes that may modulate T cell cytokine secretion.46 Similarly, Banday et al. recently published findings from the NOD model of type 2 diabetes that glutamic acid is a potent inducer of apoptosis among healthy murine β cells.47 Physical interaction of glutamate and T cells has been demonstrated using radio‐labelled glutamate.48 In addition to elevated glutamate levels in white brain matter of patients with multiple sclerosis,49 glutamate levels were found to be elevated in plasma of patients with several immune diseases, including HIV infection,50, 51 cancer,52, 53 and rheumatoid arthritis.54, 55
Here, glutamate, NMDA and QA were all used to examine NMDA‐R function in peripheral human CD4+ T cells. Importantly, although glutamate is an agonist for NMDA‐R, it can bind to several different subtypes of receptors.56 Using the synthetic but highly specific NMDA‐R ligand NMDA allowed for highly targeted stimulation. In contrast, QA, a tryptophan metabolite, exists in vivo and has a higher excitatory activity than other kynurenines.57 QA promotes apoptosis in oligodendrocytes,58 neurons59, 60 and astrocytes,61 as well as thymocytes62 at pathophysiological concentrations. In the immune system, dendritic cells63 as well as eosinophils26 are known to express IDO, which catalyses kynurenine synthesis. Hence, in addition to glutamate, QA via IDO activity can be an efficient source of NMDA‐R stimulation in vivo.
Hence, as found here, NMDA‐R ligands are implicated as important modulators of immune capacity. In contrast, unlike the mature peripheral T cells or Th1/Th2‐driven lines described above, thymocytes in isolation were found to exhibit neither NMDA‐R nor NMDA‐R‐dependent functional activity (i.e. Ca2+ flux). In 2011, Affaticati et al.64 proposed, based on experiments examining murine thymocyte development, that in T cell–dendritic cell co‐cultures an un‐identified dendritic cell‐derived glutamate agonist induces Ca2+ flux and caspase‐3 activation in immature T cells. Hence, as found here, NMDA‐R ligands were implicated as important modulators of immune capacity.
In 2014, Beurel et al.65 reported that during culture of murine thymocytes with astrocytes, glutamate was an effective promoter of naive CD4+ T cell commitment to Th1 differentiation. In a collagen‐induced arthritis model, in vivo use of an NMDA‐R antagonist enhanced the expression of Foxp3 in splenic T cells, resulted in increased CD4+ CD25+ regulatory T cells and attenuated the development of arthritis.66 If reproduced in humans, these data suggest that, as for many other biological response modifiers, the same molecule (or its metabolites) may exert distinct effects on the physiology of naive versus fully committed cells. Notwithstanding the fact that different experimental designs and species were used, these data reinforce the concept of a multifaceted role for NMDA‐R in both generation and function of mature immune systems.67
This study has several limitations. Unbalanced Th1‐like and Th2‐like activities play key roles in the pathogenesis and maintenance of many human immune disorders. We recognize that multiple other helper T cell subset phenotypes and regulatory T cell phenotypes have been described over the last decade. The roles that NMDA‐R expression may play in the initial induction, or in the protracted maintenance of other subsets remains to be examined. Second, although the data demonstrate differential sensitivity of a broad array of Th1 versus Th2 function among fresh CD4+ T cells ex vivo and their in vitro derived T cell line counterparts, we did not explicitly quantify NMDA‐R subunit density on Th1 and Th2 lines. In addition, the experiments on helper T cell physiology in this report were performed with PBMC, enriched CD4+ T cells, and Th1‐ or Th2‐driven cell lines of healthy adults. Future studies will need to examine CD4+ T cell function, as well as putatively differential in vivo expression of physiological NMDA‐R ligands, in volunteers with well‐established type 1‐ or type 2‐biased chronic inflammatory diseases.
In summary, this study demonstrates that upon TCR‐mediated activation, human CD4+ T cells up‐regulate functional NMDA‐R and that the triggering of such receptors by biologically relevant ligands causes a differential impact on cytokine production, proliferation and cell viability, both ex vivo in fresh helper T cells, and in Th1‐ versus Th2‐driven cell lines. The selective and pronounced cytotoxicity displayed by Th1 cells provides insight into a mechanism underlying T cell imbalance‐mediated disorders that share aspects analogous to excitotoxicity in the nervous system. Taken with recent advances in neurology, the findings suggest opportunities19 for better understanding of, and perhaps intervention into, immune processes that underlie the persistence of well‐established human maladaptive immune responses and clinical disease.
Disclosures
The authors declare no competing financial interests.
Supporting information
Table S1. Primer sequences used for quantitative real‐time PCR.
Acknowledgements
The authors thank Drs Darryl Adamko, Francis Davoine (University of Alberta), Kenji Matsumoto and Hirohisa Saito (National Research Institute for Child Health and Development, Tokyo) for valuable scientific discussions. We thank Caroline Graham for extensive assistance. We also thank Kelsey Falk and Rishma Chooniedass for expert assistance. This work was supported by Canadian Institutes of Health Research grants to KTH and RM, Canada Research Chairs funding for KTH, the Alberta Heritage Foundation for Medical Research for RM, Research Manitoba and the University of Manitoba.
KO and SOO are joint first authors.
Dr Moqbel is deceased.
References
- 1. Yamane H, Paul WE. Early signaling events that underlie fate decisions of naive CD4+ T cells toward distinct T‐helper cell subsets. Immunol Rev 2013; 252:12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akdis M, Aab A, Altunbulakli C, Azkur K, Costa RA, Crameri R et al Interleukins (from IL‐1 to IL‐38), interferons, transforming growth factor β, and TNF‐α: receptors, functions, and roles in diseases. J Allergy Clin Immunol 2016; 138:984–1010. [DOI] [PubMed] [Google Scholar]
- 3. Akdis M, Palomares O, van de Veen W, van Splunter M, Akdis CA. TH17 and TH22 cells: a confusion of antimicrobial response with tissue inflammation versus protection. J Allergy Clin Immunol 2012; 129:1438–49. quiz50‐1. [DOI] [PubMed] [Google Scholar]
- 4. Vahedi G, C Poholek A, Hand TW, Laurence A, Kanno Y, O'Shea JJ et al Helper T‐cell identity and evolution of differential transcriptomes and epigenomes. Immunol Rev 2013; 252:24–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hirahara K, Nakayama T. CD4+ T‐cell subsets in inflammatory diseases: beyond the Th1/Th2 paradigm. Int Immunol 2016; 28:163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who's who of T‐cell differentiation: human memory T‐cell subsets. Eur J Immunol 2013; 43:2797–809. [DOI] [PubMed] [Google Scholar]
- 7. Cheng Y, Newell EW. Deep profiling human T cell heterogeneity by mass cytometry. Adv Immunol 2016; 131:101–34. [DOI] [PubMed] [Google Scholar]
- 8. Banchereau J, Pascual V, O'Garra A. From IL‐2 to IL‐37: the expanding spectrum of anti‐inflammatory cytokines. Nat Immunol 2012; 13:925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sawant DV, Vignali DA. Once a Treg, always a Treg? Immunol Rev 2014; 259:173–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akkoc T, de Koning PJ, Ruckert B, Barlan I, Akdis M, Akdis CA. Increased activation‐induced cell death of high IFN‐γ‐producing TH1 cells as a mechanism of TH2 predominance in atopic diseases. J Allergy Clin Immunol 2008; 121:652–8. e1. [DOI] [PubMed] [Google Scholar]
- 11. Akdis CA, Akdis M. Mechanisms of immune tolerance to allergens: role of IL‐10 and Tregs. J Clin Invest 2014; 124:4678–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Akdis CA, Blaser K, Akdis M. Apoptosis in tissue inflammation and allergic disease. Curr Opin Immunol 2004; 16:717–23. [DOI] [PubMed] [Google Scholar]
- 13. Jeevanandam M, Ramias L, Schiller WR. Altered plasma free amino acid levels in obese traumatized man. Metabolism 1991; 40:385–90. [DOI] [PubMed] [Google Scholar]
- 14. Potapinska O, Demkow U. T lymphocyte apoptosis in asthma. Eur J Med Res 2009; 14(Suppl. 4):192–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 2013; 14:383–400. [DOI] [PubMed] [Google Scholar]
- 16. Morisot N, Ron D. Alcohol‐dependent molecular adaptations of the NMDA receptor system. Genes Brain Behav 2017; 16:139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bick SK, Eskandar EN. Neuromodulation for restoring memory. Neurosurg Focus 2016; 40:E5. [DOI] [PubMed] [Google Scholar]
- 18. Spalloni A, Nutini M, Longone P. Role of the N‐methyl‐d‐aspartate receptors complex in amyotrophic lateral sclerosis. Biochim Biophys Acta 2013; 1832:312–22. [DOI] [PubMed] [Google Scholar]
- 19. Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 2014; 115:157–88. [DOI] [PubMed] [Google Scholar]
- 20. Danysz W, Parsons CG. Alzheimer's disease, β‐amyloid, glutamate, NMDA receptors and memantine – searching for the connections. Br J Pharmacol 2012; 167:324–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sgambato‐Faure V, Cenci MA. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson's disease. Prog Neurobiol 2012; 96:69–86. [DOI] [PubMed] [Google Scholar]
- 22. Williams CJ, Dexter DT. Neuroprotective and symptomatic effects of targeting group III mGlu receptors in neurodegenerative disease. J Neurochem 2014; 129:4–20. [DOI] [PubMed] [Google Scholar]
- 23. Anegawa NJ, Lynch DR, Verdoorn TA, Pritchett DB. Transfection of N‐methyl‐d‐aspartate receptors in a nonneuronal cell line leads to cell death. J Neurochem 1995; 64:2004–12. [DOI] [PubMed] [Google Scholar]
- 24. Sturgill JL, Mathews J, Scherle P, Conrad DH. Glutamate signaling through the kainate receptor enhances human immunoglobulin production. J Neuroimmunol 2011; 233:80–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Odemuyiwa SO, Ebeling C, Duta V, Abel M, Puttagunta L, Cravetchi O et al Tryptophan catabolites regulate mucosal sensitization to ovalbumin in respiratory airways. Allergy 2009; 64:488–92. [DOI] [PubMed] [Google Scholar]
- 26. Odemuyiwa SO, Ghahary A, Li Y, Puttagunta L, Lee JE, Musat‐Marcu S et al Cutting edge: human eosinophils regulate T cell subset selection through indoleamine 2,3‐dioxygenase. J Immunol 2004; 173:5909–13. [DOI] [PubMed] [Google Scholar]
- 27. Xu H, Oriss TB, Fei M, Henry AC, Melgert BN, Chen L et al Indoleamine 2,3‐dioxygenase in lung dendritic cells promotes Th2 responses and allergic inflammation. Proc Natl Acad Sci U S A 2008; 105:6690–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barth H, Raghuraman S. Persistent infectious diseases say – IDO. Role of indoleamine‐2,3‐dioxygenase in disease pathogenesis and implications for therapy. Crit Rev Microbiol 2014; 40:360–8. [DOI] [PubMed] [Google Scholar]
- 29. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol 2013; 34:137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adams S, Braidy N, Bessede A, Brew BJ, Grant R, Teo C et al The kynurenine pathway in brain tumor pathogenesis. Cancer Res 2012; 72:5649–57. [DOI] [PubMed] [Google Scholar]
- 31. Stone TW. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev 1993; 45:309–79. [PubMed] [Google Scholar]
- 32. Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 2012; 13:465–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hayashi T, Mo JH, Gong X, Rossetto C, Jang A, Beck L et al 3‐Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc Natl Acad Sci U S A 2007; 104:18619–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taher YA, Piavaux BJ, Gras R, van Esch BC, Hofman GA, Bloksma N et al Indoleamine 2,3‐dioxygenase‐dependent tryptophan metabolites contribute to tolerance induction during allergen immunotherapy in a mouse model. J Allergy Clin Immunol 2008; 121:983–91. e2. [DOI] [PubMed] [Google Scholar]
- 35. Anaparti V, Ilarraza R, Orihara K, Stelmack GL, Ojo OO, Mahood TH et al NMDA receptors mediate contractile responses in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2015; 308:L1253–64. [DOI] [PubMed] [Google Scholar]
- 36. Anaparti V, Pascoe CD, Jha A, Mahood TH, Ilarraza R, Unruh H et al Tumor necrosis factor regulates NMDA receptor‐mediated airway smooth muscle contractile function and airway responsiveness. Am J Physiol Lung Cell Mol Physiol 2016; 311:L467–80. [DOI] [PubMed] [Google Scholar]
- 37. Fitch FW, Gajewski TF, Hu‐Li J. Production of TH1 and TH2 cell lines and clones. Curr Protoc Immunol 2006; doi: 10.1002/0471142735.im0313s72. Chapter 3:Unit 3.13. [DOI] [PubMed] [Google Scholar]
- 38. June CH, Moore JS. Measurement of intracellular ions by flow cytometry. Curr Protoc Immunol 2004; doi: 10.1002/0471142735.im0505s64. Chapter 5:Unit 5.5. [DOI] [PubMed] [Google Scholar]
- 39. Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Loron G et al Activation of microglial N‐methyl‐d‐aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol 2012; 72:536–49. [DOI] [PubMed] [Google Scholar]
- 40. Su RC, Becker AB, Kozyrskyj AL, Hayglass KT. Epigenetic regulation of established human type 1 versus type 2 cytokine responses. J Allergy Clin Immunol 2008; 121:57–63. e3. [DOI] [PubMed] [Google Scholar]
- 41. Estrada LD, Agac D, Farrar JD. Sympathetic neural signaling via the β2‐adrenergic receptor suppresses T‐cell receptor‐mediated human and mouse CD8+ T‐cell effector function. Eur J Immunol 2016; 46:1948–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Braun S, Gaza N, Werdehausen R, Hermanns H, Bauer I, Durieux ME et al Ketamine induces apoptosis via the mitochondrial pathway in human lymphocytes and neuronal cells. Br J Anaesth 2010; 105:347–54. [DOI] [PubMed] [Google Scholar]
- 43. Dickman KG, Youssef JG, Mathew SM, Said SI. Ionotropic glutamate receptors in lungs and airways: molecular basis for glutamate toxicity. Am J Respir Cell Mol Biol 2004; 30:139–44. [DOI] [PubMed] [Google Scholar]
- 44. Slemmer JE, De Zeeuw CI, Weber JT. Don't get too excited: mechanisms of glutamate‐mediated Purkinje cell death. Prog Brain Res 2005; 148:367–90. [DOI] [PubMed] [Google Scholar]
- 45. Centonze D, Muzio L, Rossi S, Furlan R, Bernardi G, Martino G. The link between inflammation, synaptic transmission and neurodegeneration in multiple sclerosis. Cell Death Differ 2010; 17:1083–91. [DOI] [PubMed] [Google Scholar]
- 46. Pacheco R, Gallart T, Lluis C, Franco R. Role of glutamate on T‐cell mediated immunity. J Neuroimmunol 2007; 185:9–19. [DOI] [PubMed] [Google Scholar]
- 47. Banday VS, Lejon K. Elevated systemic glutamic acid level in the non‐obese diabetic mouse is Idd linked and induces β cell apoptosis. Immunology 2017; 150:162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kostanyan IA, Merkulova MI, Navolotskaya EV, Nurieva RI. Study of interaction between l‐glutamate and human blood lymphocytes. Immunol Lett 1997; 58:177–80. [DOI] [PubMed] [Google Scholar]
- 49. Tisell A, Leinhard OD, Warntjes JB, Aalto A, Smedby O, Landtblom AM et al Increased concentrations of glutamate and glutamine in normal‐appearing white matter of patients with multiple sclerosis and normal MR imaging brain scans. PLoS ONE 2013; 8:e61817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou Y, Liu J, Xiong H. HIV‐1 glycoprotein 120 enhancement of N‐methyl‐d‐aspartate NMDA receptor‐mediated excitatory postsynaptic currents: implications for HIV‐1‐associated neural injury. J Neuroimmune Pharmacol 2017; 12:314–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ferrarese C, Aliprandi A, Tremolizzo L, Stanzani L, De Micheli A, Dolara A et al Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology 2001; 57:671–5. [DOI] [PubMed] [Google Scholar]
- 52. Castro‐Bello F, Ramos F, Vivanco F, Marina‐Fiol C. High serum glutamic acid levels in patients with carcinoma of the pancreas. Digestion 1976; 14:360–3. [DOI] [PubMed] [Google Scholar]
- 53. Michalak S, Rybacka‐Mossakowska J, Ambrosius W, Gazdulska J, Golda‐Gocka I, Kozubski W et al The markers of glutamate metabolism in peripheral blood mononuclear cells and neurological complications in lung cancer patients. Dis Markers 2016; 2016:2895972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hajati AK, Alstergren P, Nasstrom K, Bratt J, Kopp S. Endogenous glutamate in association with inflammatory and hormonal factors modulates bone tissue resorption of the temporomandibular joint in patients with early rheumatoid arthritis. J Oral Maxillofac Surg 2009; 67:1895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lowin T, Straub RH. Synovial fibroblasts integrate inflammatory and neuroendocrine stimuli to drive rheumatoid arthritis. Expert Rev Clin Immunol 2015; 11:1069–71. [DOI] [PubMed] [Google Scholar]
- 56. Rojas A, Dingledine R. Ionotropic glutamate receptors: regulation by G‐protein‐coupled receptors. Mol Pharmacol 2013; 83:746–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Erreger K, Geballe MT, Dravid SM, Snyder JP, Wyllie DJ, Traynelis SF. Mechanism of partial agonism at NMDA receptors for a conformationally restricted glutamate analog. J Neurosci 2005; 25:7858–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cammer W. Apoptosis of oligodendrocytes in secondary cultures from neonatal rat brains. Neurosci Lett 2002; 327:123–7. [DOI] [PubMed] [Google Scholar]
- 59. Kelly WJ, Burke RE. Apoptotic neuron death in rat substantia nigra induced by striatal excitotoxic injury is developmentally dependent. Neurosci Lett 1996; 220:85–8. [DOI] [PubMed] [Google Scholar]
- 60. Jang S, Jeong HS, Park JS, Kim YS, Jin CY, Seol MB et al Neuroprotective effects of (‐)‐epigallocatechin‐3‐gallate against quinolinic acid‐induced excitotoxicity via PI3K pathway and NO inhibition. Brain Res 2010; 1313:25–33. [DOI] [PubMed] [Google Scholar]
- 61. Guillemin GJ, Wang L, Brew BJ. Quinolinic acid selectively induces apoptosis of human astrocytes: potential role in AIDS dementia complex. J Neuroinflammation 2005; 2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A et al T cell apoptosis by tryptophan catabolism. Cell Death Differ 2002; 9:1069–77. [DOI] [PubMed] [Google Scholar]
- 63. Hwu P, Du MX, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3‐dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol 2000; 164:3596–9. [DOI] [PubMed] [Google Scholar]
- 64. Affaticati P, Mignen O, Jambou F, Potier MC, Klingel‐Schmitt I, Degrouard J et al Sustained calcium signalling and caspase‐3 activation involve NMDA receptors in thymocytes in contact with dendritic cells. Cell Death Differ 2011; 18:99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Beurel E, Harrington LE, Buchser W, Lemmon V, Jope RS. Astrocytes modulate the polarization of CD4+ T cells to Th1 cells. PLoS ONE 2014; 9:e86257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lindblad SS, Mydel P, Hellvard A, Jonsson IM, Bokarewa MI. The N‐methyl‐d‐aspartic acid receptor antagonist memantine ameliorates and delays the development of arthritis by enhancing regulatory T cells. Neurosignals 2012; 20:61–71. [DOI] [PubMed] [Google Scholar]
- 67. Pacheco R, Riquelme E, Kalergis AM. Emerging evidence for the role of neurotransmitters in the modulation of T cell responses to cognate ligands. Cent Nerv Syst Agents Med Chem 2010; 10:65–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences used for quantitative real‐time PCR.