

Summary
Macrophage activation is characterized by pronounced metabolic adaptation. Classically activated macrophages show decreased rates of mitochondrial fatty acid oxidation and oxidative phosphorylation and acquire a glycolytic state together with their pro‐inflammatory phenotype. In contrast, alternatively activated macrophages require oxidative phosphorylation and mitochondrial fatty acid oxidation for their anti‐inflammatory function. Although it is evident that mitochondrial metabolism is regulated during macrophage polarization and essential for macrophage function, little is known on the regulation and role of peroxisomal β‐oxidation during macrophage activation. In this study, we show that peroxisomal β‐oxidation is strongly decreased in classically activated bone‐marrow‐derived macrophages (BMDM) and mildly induced in alternatively activated BMDM. To examine the role of peroxisomal β‐oxidation in macrophages, we used Mfp2 −/− BMDM lacking the key enzyme of this pathway. Impairment of peroxisomal β‐oxidation in Mfp2 −/− BMDM did not cause lipid accumulation but rather an altered distribution of lipid species with very‐long‐chain fatty acids accumulating in the triglyceride and phospholipid fraction. These lipid alterations in Mfp2 −/− macrophages led to decreased inflammatory activation of Mfp2 −/− BMDM and peritoneal macrophages evidenced by impaired production of several inflammatory cytokines and chemokines, but did not affect anti‐inflammatory polarization. The disturbed inflammatory responses of Mfp2 −/− macrophages did not affect immune cell infiltration, as mice with selective elimination of MFP2 from myeloid cells showed normal monocyte and neutrophil influx upon challenge with zymosan. Together, these data demonstrate that peroxisomal β‐oxidation is involved in fine‐tuning the phenotype of macrophages, probably by influencing the dynamic lipid profile during macrophage polarization.
Keywords: macrophages, metabolism, multifunctional protein 2, peroxisomal β‐oxidation
Abbreviations
- ABCD
ATP‐binding cassette transporter
- ALD
adrenoleucodystrophy
- BMDM
bone‐marrow‐derived macrophages
- CD
cluster of differentiation
- CXCL
chemokine (C–X–C motif) ligand
- DMEM
Dulbecco's modified Eagle's medium
- FAO
fatty acid oxidation
- FBS
fetal bovine serum
- GC
gas chromatography
- HPLC
high‐performance liquid chromatography
- IFN‐γ
interferon‐γ
- IL
interleukin
- LC
liquid chromatography
- LPS
lipopolysaccharide
- MFP2
multifunctional protein 2
- MS
mass spectrometry
- PE
phosphatidylethanolamine
- PGC1α
peroxisome proliferator‐activated receptor γ co‐activator 1‐α
- TAG
triacylglycerides
- TNF
tumour necrosis factor
- VLCFA
very‐long‐chain fatty acids
Introduction
Macrophages possess a high level of plasticity enabling them to respond to a variety of triggers such as inflammation, infection or tissue damage.1 Although it is becoming increasingly recognized that macrophage polarization encompasses a spectrum of activation states,2, 3 activated macrophages are often categorized according to two extremes of the spectrum, namely classically [lipopolysaccharide/interferon‐γ (LPS/IFN‐γ) induced] and alternatively [interleukin‐4 (IL‐4)/IL‐13‐induced] activated macrophages.1 After classical activation by T helper type 1‐derived IFN‐γ or Toll‐like receptor stimulation, macrophages acquire a pro‐inflammatory activation state and produce inflammatory cytokines, chemokines and reactive oxygen/nitrogen species. In contrast, T helper type 2‐derived IL‐4 induces alternative or anti‐inflammatory macrophage activation involved in parasite infections and wound healing. During polarization, macrophages adopt a specific gene expression signature that enables them to execute their highly specialized functions. A crucial aspect of this adaptation is that macrophage skewing is accompanied by metabolic reprogramming.4 Classical activation of macrophages induces a metabolic switch to aerobic glycolysis, similar to the Warburg metabolism observed in cancer cells.5, 6 In contrast, alternative activation promotes oxidative metabolism, shifting macrophages towards oxidative phosphorylation and fatty acid oxidation (FAO) as main sources of energy production.7 Importantly, the metabolic adaptation is determinant to acquire a specific phenotype as inhibition of mitochondrial FAO prevents the execution/deployment of anti‐inflammatory functions.7
In contrast to the established pivotal role of mitochondrial metabolism in macrophage polarization, it is not known how macrophages rely on other metabolic organelles, in particular peroxisomes, in different activation states. Peroxisomes exclusively perform several metabolic tasks including β‐oxidation of very‐long‐fatty acids (VLCFA), branched chain fatty acids and some polyunsaturated fatty acids.8, 9, 10 Key proteins in this pathway are the ATP‐binding cassette transporters 1 to 3 (ABCD1–3) transporters that transfer substrates across the peroxisomal membrane and multifunctional protein 2 [multifunctional protein 2 (MFP2), also known as d‐bifunctional protein and hydroxysteroid 17β‐dehydrogenase 4), a central enzyme that handles most of the substrates.8 Peroxisomal β‐oxidation is indispensable for central nervous system integrity both during development and later in life.11, 12 In addition, it is currently also hypothesized that peroxisomal metabolism might influence immunological functions in myeloid cells. This is mainly based on the therapeutic efficacy of haematopoietic stem cell transplantation in X‐linked adrenoleucodystrophy (ALD), a neurometabolic disorder caused by mutations in ABCD1 involved in the import of VLCFA into peroxisomes.13, 14, 15 Furthermore, macrophages of patients with peroxisome biogenesis disorders, MFP2 or ABCD1 deficiency were shown to accumulate characteristic inclusions.16, 17, 18 These inclusions are bi‐refringent in polarized light, can be detected with neutral lipid stains and appear as tri‐lamellar structures by ultrastructural analysis. They were found in macrophages of liver, brain, adrenals and eye and were thought to consist of VLCFA primarily esterified to cholesterol as they could be dissolved in hexane but not in acetone.19 These findings suggest that peroxisomal β‐oxidation is essential for macrophage lipid homeostasis. Given the emerging importance of lipid metabolism for macrophage function, it seems likely that peroxisomal β‐oxidation deficiency can affect macrophage activation.
To investigate the relationship between peroxisomal β‐oxidation and macrophage function, we first assessed the expression of peroxisomal β‐oxidation enzymes and the activity of this pathway in macrophages in different activation states (basal versus LPS/IFN‐γ versus IL‐4). To evaluate lipid alterations in macrophages lacking peroxisomal β‐oxidation, lipidomics was applied to control and Mfp2 −/− macrophages in non‐polarized and polarized states. Finally, we examined the consequences of peroxisomal β‐oxidation deficiency on macrophage function by using in vitro and in vivo approaches. The data indicate a mutual interaction between peroxisomal β‐oxidation and macrophage polarization.
Materials and methods
Mice
Mfp2 −/− mice and Mfp2 +/+ mice on a Swiss Webster background20 were used for isolation of bone‐marrow‐derived macrophages (BMDM) and peritoneal macrophages. Macrophage‐specific MFP2 knockout mice (LysM.Cre*MFP2 L/L) were generated by breeding Mfp2 loxP/loxP 21 with LysM‐Cre 22 mice on a C57BL/6J background. Mice were bred in the animal housing facility of the KU Leuven, had ad libitum access to water and standard rodent food, and were kept on a 12 hr : 12 hr light : dark cycle. All animal experiments were performed in accordance with the ‘Guidelines for Care and Use of Experimental Animals’ and fully approved by the Research Advisory Committee (Research Ethical committee) of the KU Leuven (#190/2012).
Bone‐marrow‐derived macrophages
Macrophages were derived from bone marrow precursors as described by Meerpohl et al.23 Briefly, femurs from control or Mfp2 −/− mice were dissected and flushed with ice‐cold PBS + 10% fetal bovine serum (FBS). Bone marrow cells were plated at a density of 9 × 106 cells in a 10‐cm Petri dish (non‐tissue‐culture‐treated, bacterial grade) in 7 ml Dulbecco's modified Eagle's medium with GlutaMAX (DMEM) (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 20% FBS, 30% L929 conditioned medium and 1% penicillin/streptomycin. After 3 days of culture, 3 ml of medium was added. At day 7, BMDM were collected by forced pipetting. Thereafter, cells were plated in DMEM complete medium (DMEM + 10% FBS + 1% penicillin/streptomycin) and stimulated for 24 hr with 100 ng/ml LPS + 20 ng/ml IFN‐γ for classical activation or 10 ng/ml IL‐4 for alternative activation. CD86 surface expression was measured by flow cytometry. Briefly, BMDM were incubated for 12 min with CD16/CD32 Fc block (1 : 500; BD Biosciences) and stained with the following conjugated antibodies all from BD Biosciences: CD11b‐phycoerythrin‐Cy7 (1 : 400), F4/80‐allophycocyanin (1 : 200) and CD86‐fluoroscein isothyocyanate (1 : 500) for 40 min. Cells were acquired with a FACSCanto (BD Biosciences, San Diego, CA, USA) and analysed with flowjo software (Treestar, Ashland, OR). To evaluate cytotoxicity, an MTT assay was performed. After polarization, 3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5,diphenyl‐2H‐tetrazolium bromide (0·5 mg/ml; AppliChem, Darmstadt, Germany) was added to the cell culture medium and incubated for 4 hr. After the incubation step, 150 μl DMSO was added to the cells and optical density of dissolved formazan was determined (570 nm) after subtraction of the background (690 nm).
Peritoneal macrophages
Peritoneal macrophages were isolated from both control and Mfp2 −/− mice as described by Ray and Dittel.24 Mice were anaesthetized and injected intraperitoneally with 5 ml ice‐cold RPMI‐1640 + 10% FBS. The peritoneal exudate was collected after softly rubbing the abdomen for 5 min and plated in 24‐well plates at 5 × 105 cells/well in RPMI complete medium (RPMI + 10% FBS + 1% penicillin/streptomycin). After 4 hr, cells were washed twice with PBS (37°) to remove contaminating cells and stimulated for 24 hr with 100 ng/ml LPS + 20 ng/ml IFN‐γ for classical activation or 10 ng/ml IL‐4 for alternative activation.
RNA isolation and real‐time PCR
RNA isolation was performed by Trizol extraction for both macrophages and tissue samples according to the manufacturer's protocol (ThermoFisher Scientific, Waltham, MA, USA). Two microgrammes of total RNA was retro‐transcribed into cDNA by qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD, USA) according to the manufacturer's instructions. Quantitative real‐time transcription PCR was performed on samples in triplicate with the LightCycler 480 SYBR Green I Master (Roche Applied Science, Basel, Switzerland) using the Light Cycler 480 (Roche, Vilvoorde, Belgium). Results were quantified using the 2−ΔΔCT method.25 The expression levels of the genes of interest were normalized to the expression levels of the reference gene Rpl32. Primer sequences used are listed in the Supplementary material (Table S1).
Cytokine measurements
Cytokine levels of tumour necrosis factor‐α (TNF‐α), IL‐6 and CXCL1 were quantified by cytometric bead array (BD Bioscience). Samples were acquired on FACSCanto flow cytometer (BD Bioscience) and analysed by fcap v3.0 analysis software (Soft Flow, Inc., Pecs, Hungary).
Inflammasome activation
Inflammasome activity was determined by measuring the production of IL‐1β (as described above) after priming the BMDM for 4 hr with LPS (500 ng/ml) followed by 45 min treatment with ATP (2·5 mm or 5 mm) (ThermoFisher Scientific).
Quantification of peroxisomes and lipid droplets
The BMDM were seeded in 24‐well plates at density of 100 000 cells/well and exposed to LPS/IFN‐γ or IL‐4 as described above. After the stimulation, cells were fixed for 20 min in 4% formaldehyde and washed with PBS. For peroxisome staining, cells were permeabilized with 1% Triton X‐100 and blocking was performed with 5% bovine serum albumin. Thereafter, cells were incubated for 1 hr with anti‐peroxisomal membrane protein 70 antibody (1 : 500; Sigma‐Aldrich, St Louis, MO, USA), followed by 1 hr incubation with Alexa‐488‐conjugated secondary antibody (1 : 1000; Molecular Probes, Eugene, OR, USA). For staining of lipid droplets, cells were incubated for 1 hr with boron‐dipyrromethene 493/503 (1 μg/ml; Sigma‐Aldrich) and washed with PBS. All images were acquired with an inverted IX‐81 microscope connected to a CCD‐FV2T digital camera (Olympus, Aartselaar, Belgium) and processed with LSM image browser software (Zeiss, Oberkochen, Germany). For each mouse 20 cells were imaged and further, during statistical analysis, considered as independent samples. The number of peroxisomes as well as the number and size of lipid droplets were determined using imagej Software (NIH, Rockvill, MD, USA).
Immunohistochemistry
Mice were transcardially perfused with PBS and 4% formaldehyde and processed for cryosections. Brain sections were incubated with rabbit anti‐mouse Iba1 (1 : 500; Wako, Richmond, VA, USA) and liver and spleen sections with rat anti‐mouse F4/80 (1 : 500; BioRad, Hercules, CA, USA). Horseradish peroxidase‐labelled secondary antibodies (1 : 200) were applied for 1 hr and fluorescently labelled with a cyanine 2 (FITC) TSA kit (Perkin Elmer, Wahtham, MA, USA). Images were acquired with an Olympus BX41 light microscope equipped with cellf software (Olympus).
Lipid analysis
The BMDM of Mfp2 +/+ and Mfp2 −/− mice were seeded in 10‐cm tissue‐culture dishes at a density of 4 million cells/dish and exposed to LPS/IFN‐γ or IL‐4 as described above. Cells were collected by scraping and cell pellets were kept at −80° until further analysis.
Total triacylglycerides (TAG) were determined on lipid extracts with a peroxidase‐coupled enzymatic assay26 and normalized to protein.27
Total fatty acids were quantified by gas chromatography mass spectrometry (GC‐MS) in negative chemical ionization mode as described by Baarine et al.28 Cell pellets, suspended in 100 μl of saline, were spiked with a mix of deuterated fatty acids [myristic acid‐d3 (14:0‐d3), palmitic acid‐d3 (16:0‐d3), stearic acid‐d3 (18:0‐d3), linoleic acid‐d3 (18:2‐d3), arachidic acid‐d3 (20:0‐d3), arachidonic acid‐d8 (20:4‐d8), behenic acid‐d3 (22:0‐d3), lignoceric acid‐d4 (24:0‐d3) and cerotic acid‐d4 (26:0‐d4)], followed by lipid extraction, and alkaline hydrolysis. Fatty acids were derivatized to pentafluorobenzyl esters. Each fatty acid concentration was determined by the relative area ratio method using the closest deuterated internal standard.
For determining the composition of TAG and phospholipids total lipids were extracted from cells by the Folch procedure29 and analysed by a Dionex Ultimate 3000 HPLC coupled on‐line to a hybrid quadrupole‐orbitrap mass spectrometer Q‐Exactive (ThermoFisher Scientific). The liquid chromatography (LC)/ESI‐MS analysis of TAG was performed using a C18 reverse phase column (Luna, 3 μm, 100 Å, 150 × 2 mm; Phenomenex, Torrance, CA, USA) and gradient solvents as previously described.30 MS spectra were acquired in positive ion mode, using range zoom (500–1000 m/z). TAG cations were formed through molecular ammonium adduction (TAG + NH4). Phospholipids (PL) were analysed using a silica column (Luna 3 μm, 100 Å, 150 × 2 mm; Phenomenex) as previously described.31 The spectra of PL were acquired in negative ion mode, using range zoom (400–1600 m/z). All scans were acquired in data‐dependent mode with isolation width of 1·0 Da. Analysis of LC/MS data was performed using software package sieve™ (ThermoFisher Scientific).
Metabolic assays
To evaluate fatty acid β‐oxidation of mitochondrial and peroxisomal substrates during macrophage polarization, BMDM were seeded in T25‐flasks and exposed to LPS/IFN‐γ or IL‐4 as described above. Subsequently, cultures were incubated with 5 μm 1–14C‐labelled fatty acid, bound to defatted albumin as described before32 for 20 hr, followed by collection of 14CO2. The medium was analysed for acid‐soluble oxidation products, and the adherent cells were collected in lysis medium (5 mm 3‐(N‐morpholino)propanesulphonic acid pH 7·2 – 1 mm EDTA – 0·1% SDS). Aliquots of the lysates were analysed for protein and subjected to lipid extraction. The extracts were analysed for phospholipid content33 and separated by thin‐layer chromatography to determine the incorporation of label in lipids.
To evaluate glycolysis and mitochondrial fatty acid β‐oxidation in Mfp2 −/− compared with control BMDM in basal and activated state, BMDM were seeded in 48‐well plates at a density of 150 000 cells/well and exposed to LPS/IFN‐γ as described above. FAO and glycolysis were measured on the basis of 3H2O production as described previously.34 Briefly, cells were incubated with DMEM complete medium containing 2 μCi/ml [9,10‐3H]palmitate (for FAO) or 2 μCi/ml [5‐3H]‐d‐glucose (for glycolysis) (Perkin Elmer). After 2 hr of incubation, cell supernatant was transferred into glass vials. The glass vials were sealed with rubber stoppers containing hanging wells with H2O‐soaked Whatman paper. After saturation was reached (48 hr at 37°), the radioactivity was determined by liquid scintillation counting. All the measurements were made in triplicate and were normalized to total protein content. For the determination of protein content, after the supernatant was collected, the cells where lysed in 30 μl radioimmunoprecipitation assay buffer (25 mm Tris–HCl pH 7·6 – 150 mm NaCl – 1% nonidet P‐40 – 1% sodium deoxycholate – 0·1% SDS) of which 5 μl was used for the assay. Total protein content was determined using a PierceTM BCA Protein Assay Kit (Thermo Scientific) following manufacturer's instructions.
Zymosan‐induced peritonitis
Peritonitis was induced in 6‐ to 8‐week‐old male and female LysM.Cre*MFP2L/L and MFP2L/L mice by injecting 1 ml of 1 mg/ml zymosan (Sigma‐Aldrich) intraperitoneally.35 Mice were killed and peritoneal exudate was harvested 6 hr post‐injection. Then, 2 × 105 cells were used for FACS analysis. First, cells were incubated for 12 min with CD16/CD32 Fc block (1 : 500; BD Biosciences). Cells were stained with the following conjugated antibodies all from BD Biosciences: CD45.2‐fluoroscein isothyocyanate (1 : 400), CD11b‐ phycoerythrin‐Cy7 (1 : 400), Ly6C‐phycoerythrin (1 : 500) and Ly6G‐allophycocyanin (1 : 200) for 40 min. Cells were acquired with a FACSCanto (BD Biosciences) and analysed with flowjo software (Treestar).
Statistical analysis
For statistical analysis graphpad prism software (version 5.0 and 6.0; GraphPad Software, Inc., San Diego, CA, USA) was used and the following tests were performed: unpaired two‐sided Student's t‐test, one‐way analysis of variance repeated measures or two‐way analysis of variance multiple comparison followed by Tukey post‐test. Data are shown as mean ± SEM and statistical significance was set at P ˂ 0·05.
Results
Peroxisomal β‐oxidation is strongly reduced in classically activated macrophages
To investigate the regulation of peroxisomal FAO during macrophage polarization, we first measured the relative expression of genes related to peroxisomal β‐oxidation in both classically (LPS/IFN‐γ) and alternatively (IL‐4) activated macrophages. Expression of acyl‐CoA oxidase 1 (Acox1), multifunctional protein‐2 (Hsd17b4) and enoyl‐CoA, hydratase/3‐hydroxyacyl CoA dehydrogenase (Ehhadh), encoding multifunctional protein 1, were strongly reduced after induction with LPS/IFN‐γ (Fig. 1a–c). In IL‐4‐induced macrophages, only Acox1 gene expression was significantly increased compared with macrophages in basal condition whereas no differences in gene expression of peroxisomal Ehhadh and Hsd17b4 were observed (Fig. 1a–c). We also assessed the expression of the mitochondrial β‐oxidation enzyme medium‐chain acyl‐CoA dehydrogenase (Acadm) and found that it was strongly reduced in classically activated macrophages, confirming previous reports5 (Fig. 1d). To assess whether the transcript changes had a functional impact, we measured mitochondrial and peroxisomal fatty acid oxidation in resting and activated macrophages. Classical activation of macrophages with LPS/IFN‐γ reduced both mitochondrial β‐oxidation of palmitate (C16:0) and peroxisomal fatty acid β‐oxidation of lignoceric acid (C24:0) and 2‐methylhexadecanoic acid (2‐Me‐C16:0) by ~ 50–60% (Fig. 1e). After alternative activation with IL‐4, we did not find changes in mitochondrial β‐oxidation in line with unaltered Acadm expression (Fig. 1e). In contrast, peroxisomal β‐oxidation (2‐Me‐C16:0 and C24:0) was clearly induced during alternative activation, which was most pronounced for 2‐Me‐C16:0 (Fig. 1e). To test whether other peroxisomal metabolic pathways were deregulated in polarized macrophages, we measured peroxisomal α‐oxidation activity by using 3‐Me‐C16:0, a substrate for α‐oxidation, but no adaptations were seen. Next, to examine whether the observed changes in peroxisomal metabolic activity were due to altered abundance of peroxisomes, we counted peroxisome numbers in polarized macrophages. Even though we found decreased peroxisomal β‐oxidation during LPS/IFN‐γ stimulation, the number of peroxisomes was up‐regulated by ~65% (Fig. 1f). Together, these data clearly show that classical macrophage polarization induces dynamic changes in peroxisomal FAO similar to mitochondrial FAO.
Figure 1.
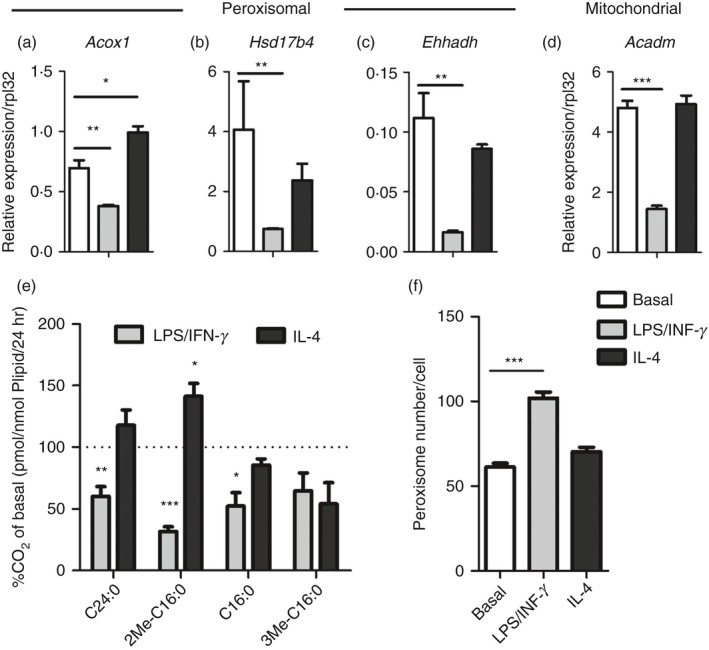
Both peroxisomal and mitochondrial β‐oxidation are strongly reduced in classically activated macrophages. (a–d) Gene expression of peroxisomal acyl‐CoA oxidase 1 (Acox1) (a), multifunctional protein‐2 (Hsd17b4) (b) and enoyl‐CoA, hydratase/3‐hydroxyacyl CoA dehydrogenase (Ehhadh) (c) and mitochondrial medium chain acyl‐CoA dehydrogenase (Acadm) (d) in classically (LPS/IFN‐γ) and alternatively (IL‐4) activated bone‐marrow‐derived macrophages (BMDM) compared with basal conditions (n = 4 versus 4 versus 4); (e) Degradation of substrates by peroxisomal β‐oxidation (2‐Me‐C16:0 and C24:0), mitochondrial β‐oxidation (C16:0) and peroxisomal α‐oxidation (3Me C16:0) in classically and alternatively activated BMDM (n = 4 versus 4 versus 4) expressed as percentage of CO 2 release in basal conditions; (f) Number of peroxisomes per cell in classically and alternatively activated BMDM compared with basal conditions (n > 15). Bars represent mean ± SEM. Statistical differences based on one‐way analysis of variance test: *P < 0·05, **P < 0·01, ***P < 0·001.
Lipid changes in polarized control and MFP2‐deficient murine macrophages
As indicated above, we show that peroxisomal β‐oxidation is dynamically regulated during macrophage polarization, similar to mitochondrial β‐oxidation. It has been reported that macrophage activation induces pronounced changes in lipid metabolism including increased triacylglyceride storage and lipid droplet accumulation.36 Moreover, blocking mitochondrial FAO significantly affects the polarization of macrophages.7 It is however unknown whether peroxisomal β‐oxidation activity plays a modulatory role in activated macrophages. Given the pronounced decrease in peroxisomal β‐oxidation in classically activated macrophages, we investigated whether peroxisomal β‐oxidation deficiency affects macrophage activation. To this end, we characterized the metabolic and inflammatory response of classically activated BMDM lacking MFP2, a central enzyme of the peroxisomal β‐oxidation pathway (Mfp2 −/−). First, we confirmed that the oxidation of C24:0 and 2M‐C16:0 was reduced in Mfp2 −/− BMDM (see Supplementary material, Fig. S1a). As expected mitochondrial β‐oxidation and peroxisomal α‐oxidation were unaffected (see Supplementary material, Fig. S1 b,c). Next, we investigated the impact of deficient peroxisomal β‐oxidation on the lipid profile of BMDM derived from control and Mfp2 −/− mice. In line with previous reports, we confirmed that classical macrophage activation had a pronounced effect on lipid droplet dynamics. Although lipid droplet number was mildly reduced after LPS/IFN‐γ treatment, the size of lipid droplets was significantly increased in classically activated macrophages (Fig. 2b). Both lipid droplet number and lipid droplet size were unaltered in alternatively activated macrophages compared with the basal state. In Mfp2 −/− macrophages, similar changes were observed but no differences were detected between control and Mfp2 −/− macrophages with regard to lipid droplet content both in non‐polarized and polarized conditions. The only exception was a slight increase of lipid droplet number in alternatively activated Mfp2 −/− macrophages compared with controls. Similarly, quantification of triglyceride content showed a tendency for increased neutral lipid storage in classically activated macrophages of both genotypes versus the respective controls, but no differences between control and Mfp2 −/− macrophages were observed (Fig. 2c). Because we did not detect differences in general lipid droplet content between control and Mfp2 −/− macrophages, we next performed general and detailed lipidomic analysis using GC‐MS‐NCI and LC‐MS/MS to obtain better insight into the fatty acid profile, as well as TAG and PL composition of control and Mfp2 −/− BMDM mainly focusing on peroxisomal β‐oxidation substrates (data summarized in the Supplementary material, Table [Link], [Link], [Link]). We observed that peroxisomal β‐oxidation‐deficient BMDM show pronounced accumulation of VLCFA, including C24:0, C24:1, C26:0 and C26:1 in basal, LPS/IFN‐γ polarized and IL‐4 polarized states, whereas these fatty acids were present at extremely low levels in control BMDM (Fig. 3a–d). On the other hand, no differences were observed in saturated long‐chain fatty acids between control and Mfp2 −/− macrophages, which is in line with the unaltered mitochondrial β‐oxidation (C16:0 oxidation) in Mfp2 −/− macrophages (Fig. 3e and see Supplementary material, Table S2). We also observed accumulation of several polyunsaturated fatty acids including γ‐linoleic acid (C18:3 n‐6), mead acid (C20:3 n‐9) and eicosapentaenoic acid (20:5 n‐3) in Mfp2 −/− macrophages (Fig. 3f–h). In contrast, arachidonic acid (C20:4 n‐6) and docosahexaenoic acid (C22:6 n‐3), which are known as fatty acids with pro‐ and anti‐inflammatory functions, respectively, showed no changes between activation states or BMDM phenotype (Fig. 2i,j). Furthermore, we checked whether stored TAG contained higher levels of VLCFA. Detailed lipidomic analysis showed that peroxisomal β‐oxidation deficiency resulted in increased levels of TAG species containing a single saturated or mono‐unsaturated fatty acid with chain lengths ≥ C24 (2·6–3·1 fold compared with control) (Fig. 4a,b). Notably, these TAG species also accumulated when control macrophages were converted from the basal to LPS/IFN‐γ polarized phenotype, in accordance with the suppressed peroxisomal β‐oxidation activity. It was also remarkable that TAG species containing VLCFA at two or all three positions (e.g. 68:4 and 70:4) were detectable in Mfp2 −/− but not in control macrophages, albeit at extremely low levels (see Supplementary material, Table S3). We further examined whether the fatty acid profile in phospholipids was altered as a consequence of peroxisomal β‐oxidation deficiency by analysing phosphatidylethanolamine (PE) species (see Supplementary material, Table S4 and selected species shown in Fig. 4c). We found that minor PE species containing VLCFA showed dynamic changes during macrophage polarization, as evidenced by a two‐ to threefold increase after stimulation with LPS/IFN‐γ. Although similar polarization‐dependent dynamics were seen in Mfp2 −/− macrophages, these species were strongly enriched in all states (three‐ to tenfold) compared with control (Fig. 4c). Taken together, we show that macrophage polarization with LPS/IFN‐γ induces dynamic changes in TAG and PE lipid species containing peroxisomal β‐oxidation substrates. In addition, an increased accumulation of VLCFA is observed in Mfp2 −/− macrophages in all activation states, but always in very low levels compared to the total lipid fraction.
Figure 2.
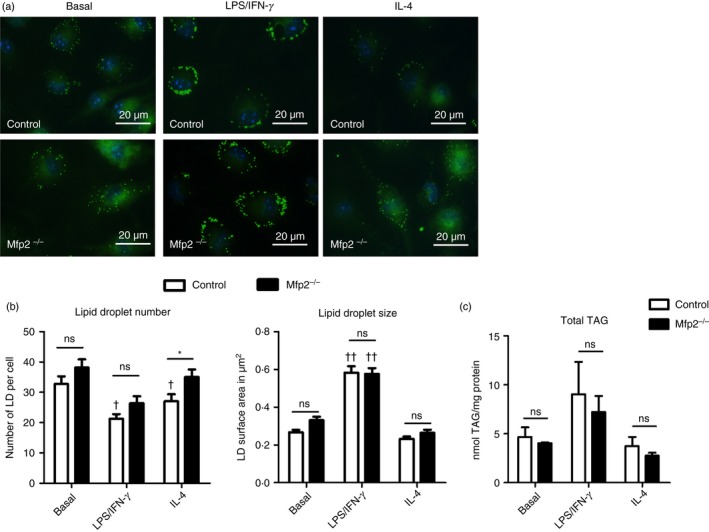
Neutral lipid storage is similar in control and Mfp2 −/− bone‐marrow‐derived macrophages (BMDM). (a) Boron‐dipyrromethene 493/503 staining (b) number and size of lipid droplets (LD) (n > 15) and (c) total content of triacylglycerides (TAG) (n = 4 versus 4) in BMDM. Bars represent mean ± SEM. Statistical differences based on two‐way analysis of variance test: ns P > 0·05, *P < 0·05 (Mfp2 −/− compared to control BMDM); †P < 0·01, ††P < 0·0001 (activated Mfp2 −/− and control BMDM compared to respective basal condition). [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 3.
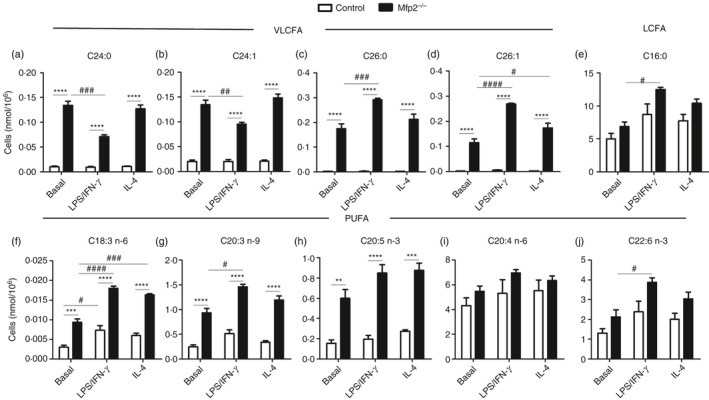
Fatty acid composition of control and Mfp2 −/− bone‐marrow‐derived macrophages (BMDM) in different polarization states. (a–j) Representative examples (taken from the Supplementary material, Table S2) of fatty acid. (a–d) VLCFA – C24:0 (a), C24:1 (b), C26:0 (c) and C26:1 (d); (e) LCFA – palmitic acid (C16:0); (f–j) polyunsaturated fatty acids (PUFA) – γ‐linoleic acid (C18:3 n‐6) (f), mead acid (C20:3 n‐9) (g), eicosapentaenoic acid (20:5 n‐3) (h), arachidonic acid (C20:4 n‐6) (i) and docosahexaenoic acid (C22:6 n‐3) (j) in basal, classically or alternatively activated control and Mfp2 −/− BMDM (n = 3 versus 3). Bars represent mean ± SEM. Statistical differences based on two‐way analysis of variance test: **P < 0·01, ***P < 0·001, ****P < 0·0001 (Mfp2 −/− compared with control BMDM); #P < 0·05, ##P < 0·01, ###P < 0·001, ####P < 0·0001 (activated Mfp2 −/− and wild‐type BMDM compared with respective basal condition).
Figure 4.
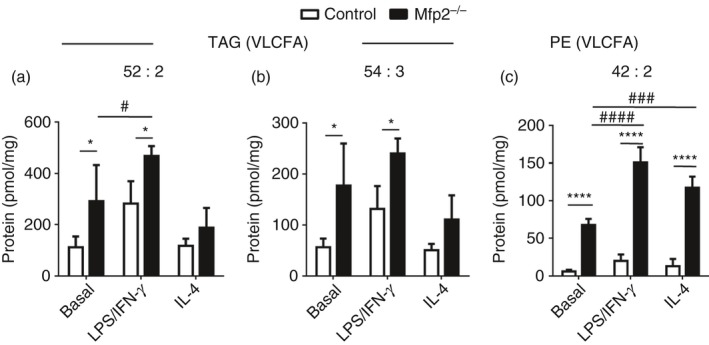
Triacylglycerides (TAG) and phospholipid species containing very‐long‐chain fatty acids (VLCFA) are enriched in Mfp2 −/− bone‐marrow‐derived macrophages (BMDM). (a, b) Representative examples (taken from the Supplementary material, Table S3) of TAG species containing VLCFA; (c) Representative examples (taken from the Supplementary material, Table S4) of phosphatidylethanolamine containing VLCFA in basal, classically or alternatively activated control and Mfp2 −/− BMDM (n = 4 versus 4). Bars represent mean ± SEM. Statistical differences based on two‐way analysis of variance test: *P < 0·05, ****P < 0·0001 (Mfp2 −/− compared to control BMDM); #P < 0·05, ###P < 0·001, ####P < 0·0001 (activated Mfp2 −/− and wild type BMDM compared to respective basal condition).
MFP2‐deficient BMDM have reduced inflammatory cytokine production during classical activation
It has been proposed that accumulation of VLCFA might enhance inflammatory signalling in diverse cell types.37, 38, 39, 40 To investigate whether the accumulation of VLCFA in different lipid species affects classical and alternative polarization in BMDM Mfp2 −/− macrophages, we first measured gene expression of a panel of inflammatory genes, including cytokines (Tnf, Il1b, Il1a, Il6, Il12a, Il12b) and chemokines (Cxcl1, Cxcl2, Ccl3, Ccl5, Ccl8) after classical activation with LPS/IFN‐γ. The gene expression of inflammatory cytokines and chemokines as well as their production, was low or below the detection limit in basal state both in control and Mfp2 −/− macrophages (Fig. 5a,b). Unexpectedly, after activation with LPS/IFN‐γ, Mfp2 −/− macrophages showed blunted inflammatory responses as the transcript expression of Il6, Il1a, Il12a, Il12b, Cxcl1, Cxcl2 and Ccl3 was significantly lower compared with control macrophages (Fig. 5a,b). Next, we measured cytokine secretion by Mfp2 −/− macrophages. In the basal state all cytokines and chemokines measured were undetectable in the medium of both control and Mfp2 −/− macrophages (data not shown). After stimulation with LPS/IFN‐γ, Mfp2 −/− macrophages produced lower levels of IL‐6 and CXCL1 compared with control macrophages in line with the transcript data (Fig. 5c). Moreover, although we could not detect differences in gene expression of Tnfa, TNF‐α production was significantly reduced in Mfp2 −/− macrophages compared with control macrophages (Fig. 5c). To evaluate possible effects of peroxisomal β‐oxidation deficiency on inflammasome activation, IL‐1β production was measured after stimulation with LPS and ATP. Control and Mfp2 −/− macrophages showed similar production of IL‐1β after the challenge, indicating that peroxisomal β‐oxidation deficiency does not affect inflammasome activation (see Supplementary material, Fig. S2). To exclude that the blunted inflammatory response of Mfp2 −/− macrophages was due to reduced cell survival upon stimulation, we performed an MTT‐viability assay. But did not observe a reduction in cell survival of Mfp2 −/− compared with control macrophages upon LPS/IFN‐γ treatment (see Supplementary material, Fig. S3). To investigate whether reduced cytokine and chemokine production by classically activated Mfp2 −/− macrophages was due to defective polarization, we evaluated the expression of CD86, a co‐stimulatory molecule that is strongly expressed by classically activated macrophages, by flow cytometry. Although macrophage activation induced CD86 expression on CD11b+ F480+ BMDM, no differences were detected between control and Mfp2 −/− macrophages in basal state and during classical activation indicating adequate polarization of Mfp2 −/− macrophages (Fig. 5d).
Figure 5.
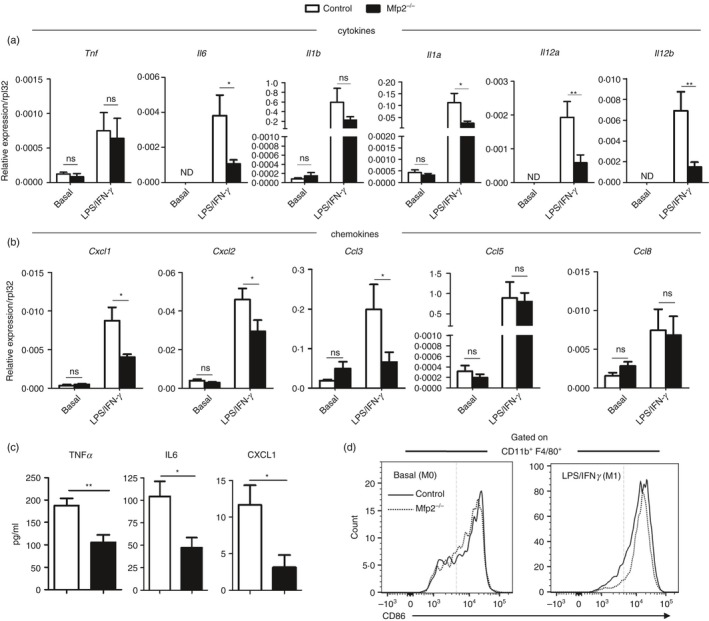
Reduced inflammatory response of Mfp2 −/− bone‐marrow‐derived macrophages (BMDM). (a, b) Gene expression of pro‐inflammatory cytokines (a) and chemokines (b) in basal and classically activated Mfp2 −/− BMDM compared with control BMDM (n = 5–8 versus 5–8); (c) Secretion of pro‐inflammatory cytokines and chemokines in classically activated Mfp2 −/− BMDM compared with control BMDM (n = 6 versus 6); (d) Expression of CD86 marker does not differ in classically activated Mfp2 −/− BMDM compared with control BMDM (n = 3 versus 3). Bars represent mean ± SEM. Statistical differences based on t‐test or two‐way analysis of variance test: ns P > 0·05, *P < 0·05, **P < 0·01.
Together, these data show that MFP2 deficiency has subtle dampening effects on the classical activation of macrophages, mainly influencing cytokine production. To determine whether Mfp2 −/− macrophages also respond differently during alternative polarization, we measured the gene expression of typical anti‐inflammatory markers in control and Mfp2 −/− BMDM. Stimulation with IL‐4 robustly induced gene expression of anti‐inflammatory genes such as Arg1, Mrc1, Retnla, Ym1 and Vegf in both control and Mfp2 −/− BMDM but no differences were observed between the two genotypes with the exception of Ym1, which was slightly more induced in Mfp2 −/− macrophages upon IL‐4 stimulation (Fig. 6).
Figure 6.
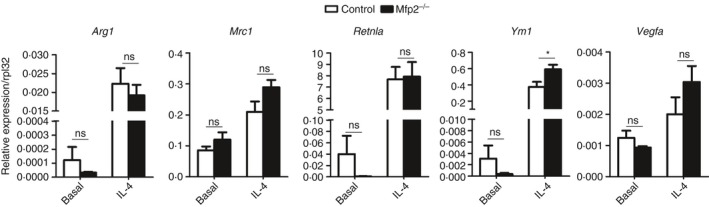
Unaltered anti‐inflammatory response of Mfp2 −/− bone‐marrow‐derived macrophages (BMDM). Gene expression of anti‐inflammatory markers in basal and alternatively activated Mfp2 −/− BMDM compared with control BMDM (n = 6 versus 6). Bars represent mean ± SEM. Statistical differences based on two‐way analysis of variance test: ns P > 0·05, *P < 0·05.
MFP2‐deficiency does not influence metabolic adaptation during macrophage activation
Recent evidence indicates that metabolic adaptation is an important determinant of the functional phenotype acquired by macrophages. During inflammatory conditions (e.g. activation with LPS and/or IFN‐γ), macrophages display a metabolic shift towards the anaerobic glycolytic pathway whereas FAO is down‐regulated. Although this metabolic adaptation is probably necessary to meet the rapid energy requirement of classically activated macrophages, it has been shown that disturbances in metabolic energy expenditure strongly influence the functional phenotype of macrophages.7, 41 To investigate whether the reduced inflammatory activation of Mfp2 −/− macrophages is due to an imbalance in this metabolic adaptation, we measured glycolysis and C16:0 FAO in basal and LPS/IFN‐γ conditions. Although we could confirm that the switch from basal to classically activated macrophages was associated with increased glycolysis and decreased C16:0 FAO, we did not detect any differences between control and Mfp2 −/− macrophages, indicating that the blunted inflammatory response during classical activation is not due to a defect in metabolic adaptation (Fig. 7).
Figure 7.
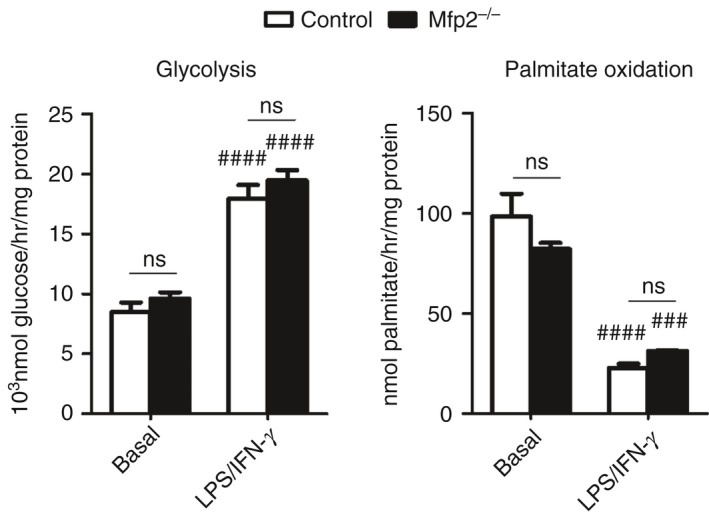
Metabolic shift during classical activation is not affected in Mfp2 −/− bone‐marrow‐derived macrophages (BMDM). Rates of glycolysis and palmitate oxidation in control and Mfp2 −/− BMDM in basal conditions and after classical activation (n = 6 versus 6). Bars represent mean ± SEM. Statistical differences based on two‐way analysis of variance test: ns P > 0·05 (Mfp2 −/− compared to control BMDM); ###P < 0·001, ####P < 0·0001 (activated Mfp2 −/− and control BMDM compared to respective basal condition).
Macrophage‐specific deletion of MFP2 does not affect immune cell influx in zymosan‐induced peritonitis
As a next step, we wanted to evaluate the functional relevance of the decreased inflammatory activation of MFP2‐deficient macrophages in vivo. To this end, we generated mice with myeloid‐cell‐specific deletion of MFP2 by crossing LysM.Cre mice with Mfp2 LoxP/LoxP mice (LysM‐Mfp2 −/−). These mice developed normally and showed no overt phenotype. Gene expression analysis and immunohistochemistry showed normal expression of inflammatory markers (Tnf, Il6 and Cxcl1) and normal macrophage morphology/abundance in spleen, liver and brain under basal conditions (see Supplementary material, Fig. S4). In addition, flow cytometric analysis of the peritoneal exudate showed normal abundance of CD11b+/F4/80+ peritoneal macrophages in LysM‐Mfp2 −/− compared with Mfp2 L/L mice (Fig. 8a). To characterize the classical activation of MFP2‐deficient macrophages in vivo, LysM‐Mfp2 −/− mice were subjected to zymosan‐induced peritonitis. In this model, Toll‐like receptor 2 activation by zymosan activates resident peritoneal macrophages that in turn produce chemokines to attract other innate effector cells (mainly neutrophils and monocytes) to the peritoneal cavity. In the acute phase of the induced peritonitis (i.e. 6 hr post‐injection), we observed no differences in number of monocytes (Ly6Chi, Ly6Gneg) and neutrophils (Ly6Cmid/low, Ly6Gpos) in peritoneal exudate between control and LysM‐Mfp2 −/− mice suggesting that MFP2‐deficiency does not affect acute activation of resident macrophages in vivo (Fig. 8b–d). To remove doubt of possible differences between in vitro and in vivo data due to different responses of Mfp2 −/− peritoneal macrophages compared with Mfp2 −/− BMDM upon LPS/IFN‐γ stimulation, we evaluated the response of Mfp2 −/− peritoneal macrophages to classical activation. Gene expression analysis of inflammatory cytokines and chemokines confirmed the blunted activation of peritoneal Mfp2 −/− macrophages upon stimulation with LPS/IFN‐γ compared with controls (Fig. 9a). Upon IL‐4 stimulation, there were no changes in gene expression of anti‐inflammatory markers between Mfp2 −/− and control peritoneal macrophages with the exception of Retnla as observed in BMDM (Fig. 9b). Altogether, these data indicate that even though Mfp2 −/− macrophages show decreased production of inflammatory chemokines in vitro, they are able to mount a normal immune response after an acute inflammatory trigger in vivo.
Figure 8.
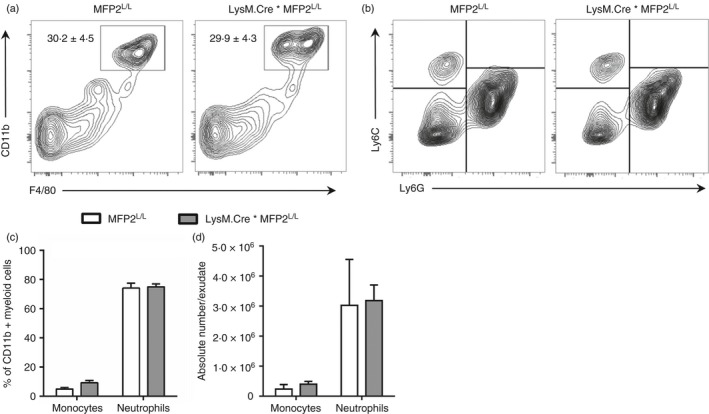
Reduced pro‐inflammatory response of Mfp2 −/− macrophages does not affect their function in vivo. (a) Percentage of macrophages defined as CD11b+/F480+ cells in peritoneal exudate of LysM.Cre*Mfp2 L/L mice compared with Mfp2 L/L mice (n = 4 versus 4); (b–d) Number of monocytes and neutrophils in peritoneal exudate after zymosan treatment expressed as % of CD11b+ myeloid cells (c) or as absolute cell number (d) in LysM.Cre*Mfp2 L/L mice compared with Mfp2 L/L mice (n = 4 versus 6). Bars represent mean ± SEM.
Figure 9.
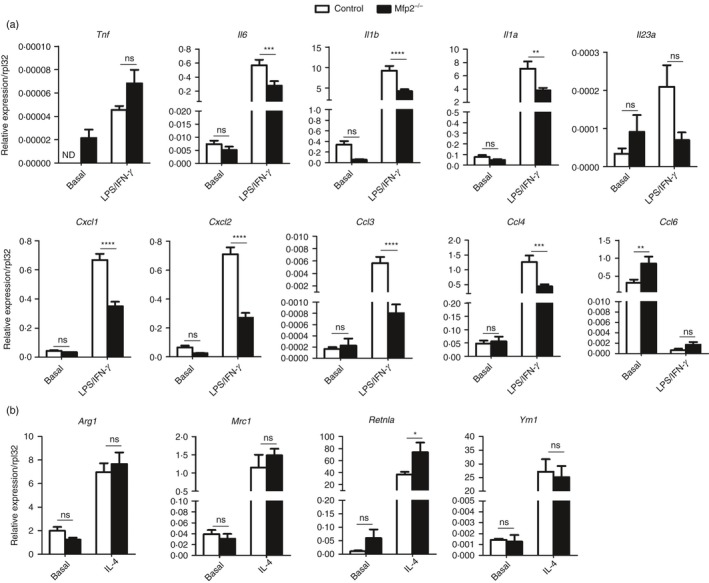
Reduced pro‐inflammatory and unaltered anti‐inflammatory response of Mfp2 −/− peritoneal macrophages. (a, b) Gene expression of pro‐inflammatory (a) and anti‐inflammatory (b) markers in classically and alternatively activated Mfp2 −/− compared with control peritoneal macrophages (n = 6 versus 6). Bars represent mean ± SEM. Statistical differences based on t‐test: ns P > 0·05, *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001.
Discussion
In this study we report that, similar to mitochondrial fatty acid oxidation, peroxisomal fatty acid oxidation is strongly impaired during classical macrophage activation and mildly induced during alternative activation. The suppression of peroxisomal β‐oxidation in classical macrophages leads to increased incorporation of peroxisomal substrates in triacylglycerides and phospholipids. We further demonstrated that BMDM lacking peroxisomal β‐oxidation (Mfp2 −/− macrophages) showed a mildly reduced inflammatory response upon stimulation with LPS/IFN‐γ in vitro, but mice lacking peroxisomal β‐oxidation in the myeloid cell compartment mounted a normal immune response in the peritoneal cavity. Our data shed new light on the dynamic regulation of lipid metabolic pathways during macrophage polarization and provide unanticipated evidence that peroxisomal β‐oxidation deficiency does not lead to increased inflammatory activation of macrophages.
Recent advances in immunometabolism underscore the essential role of lipid metabolism in regulating macrophage function and phenotype. Depending on the activation state, macrophages employ different metabolic pathways to meet their energy requirements. Metabolic adaptation of polarized macrophages is not only required for proper energy expenditure, but also drives specific effector functions in macrophages.42, 43 It has indeed been shown that a shift toward glycolysis and fatty acid synthesis, and away from Krebs cycle and FAO, skews macrophages into a pro‐inflammatory state.44, 45, 46 This metabolic shift, which can be induced by Toll‐like receptor agonists such as LPS, seems to be essential for their functional phenotype as inhibition of glycolysis reduces the production of certain pro‐inflammatory cytokines by classically activated macrophages.41 Alternatively activated macrophages on the other hand rely completely on fatty acid oxidation and oxidative metabolism to meet their energy demand, and inhibition of mitochondrial FAO affects alternative polarization.7, 47 The reprogramming to oxidative metabolism was shown to be mediated by peroxisome proliferator‐activated receptor γ co‐activator 1‐β (PGC1β).7 In contrast, little attention has been paid to the molecular determinants driving the strong suppression of mitochondrial β‐oxidation in classically activated macrophages. Our findings that this is accompanied by profoundly reduced expression of key enzymes of peroxisomal β‐oxidation, leading to reduced activity of this pathway, indicate that common transcriptional regulators of both mitochondrial and peroxisomal fatty acid oxidation are involved. Potential candidates are the nuclear receptors, peroxisome proliferator‐activated receptors, oestrogen‐related receptors and hepatocyte nuclear factor 4 that are known to regulate mitochondrial fatty acid catabolism in concert with the PGC1 co‐activators. To date, for peroxisomal β‐oxidation only peroxisome proliferator‐activated receptor‐α 48 and PGC1α 49 were shown to control expression of the enzymes but it cannot be excluded that additional factors are involved. The activity of these transcriptional regulators not only depends on their expression levels but also on ligand concentration and post‐translational modifications, calling for an extensive investigation of the underlying mechanisms.
Another remarkable finding of this study is the blunted inflammatory response of Mfp2 −/− macrophages. This is surprising given the accumulation of VLCFA in Mfp2 −/− macrophages, which further increased upon stimulation with LPS/IFN‐γ, together with the proposed role of VLCFA as a potential trigger of inflammatory activation. It has indeed been suggested that VLCFA accumulation, a typical hallmark of peroxisomal β‐oxidation disorders such as X‐linked ALD, might trigger an inflammatory cascade in immune cells that can subsequently lead to the neuroinflammatory disease seen in disorders such as cerebellar ALD. Several studies have indeed reported that abnormal VLCFA accumulation could increase inflammatory activation of murine astrocytes, peritoneal macrophages, human fibroblasts and mononuclear cells.37, 38, 39, 40 In murine astrocytes, silencing of Abcd1 and Abcd2, two peroxisomal transporters involved in shuttling substrates into peroxisomes, leads to a threefold increase of C26:0 together with activation of nuclear factor‐κB, activator protein‐1 and CCAAT‐enhancer‐binding protein and increased production of inflammatory cytokines such as TNF‐α and IL‐1β.38, 50, 51 Treatment of Abcd1/2 silenced astrocytes with a mixture of oleic acid/erucic acid (Lorenzo's Oil) not only normalizes VLCFA levels but also significantly reduces the production of inflammatory cytokines, suggesting that VLCFA accumulation is indeed underlying the inflammatory activation of peroxisomal β‐oxidation deficient astrocytes. Also in Abcd1 −/− peritoneal macrophages, accumulation of VLCFA coincides with enhanced production of inflammatory cytokines such as TNF‐α, IL‐6 and IL‐12p70.37 In this study, we wanted to further validate these observations by investigating the inflammatory activation of bone‐marrow‐derived and peritoneal macrophages lacking the central enzyme of peroxisomal β‐oxidation, MFP2. Although we observed a pronounced accumulation of different VLCFA in both the neutral lipid and PE fraction, Mfp2 −/− macrophages show a decreased rather than increased inflammatory activation upon stimulation with LPS/IFN‐γ. In addition, our in vivo studies also provide new insight that macrophages lacking peroxisomal β‐oxidation are able to mount a normal immune response upon stimulation, as myeloid‐cell‐specific MFP2 knockout mice attract similar amounts of monocytes and neutrophils upon classical activation. These findings do not support the paradigm that VLCFA accumulation potentiate inflammatory activation of macrophages, thereby initiating an inflammatory cascade. Our data nevertheless indicate that peroxisomal β‐oxidation deficiency can affect macrophage homeostasis and macrophages can serve as relevant cellular tools to study altered innate immune cell function in peroxisomal β‐oxidation deficiencies. This is particularly relevant given the proposed role of macrophages as pathogenic drivers of the neuroinflammatory cascade in cerebellar ALD. Of interest, a recent study provided new evidence that mainly monocytes of ALD patients are metabolically affected whereas lymphocytes are not.52 In this respect, it is interesting to further follow up on how peroxisomal β‐oxidation could affect different signalling pathways involved in macrophage homeostasis. Recent integrated genomics, metabolomics and lipidomics studies on mononuclear cells isolated from ALD patients indeed identified novel pathway alterations, including insulin signalling, glycerophospholipid metabolism and bile acid metabolism.53, 54 It was recently shown that induced pluripotent stem cells derived from ALD patients strongly induce Ch25h, the gene encoding cholesterol 25‐hydroxylase.55 Importantly, it was shown that 25‐hydroxycholesterol, but not VLCFA, acts as an endogenous regulator of NLRP3‐induced inflammasome activation in BMDM, possibly underlying the neuroinflammatory cascade associated with cerebellar ALD. In line with this study, we did not find potentiation of inflammasome activation in Mfp2 −/− macrophages, supporting the idea that the observed alterations in Abcd1‐deficient macrophages are not merely due to VLCFA accumulation. These results nevertheless underscore the importance of identifying novel mediators associated with disturbed immune cell function in peroxisomal β‐oxidation deficiency. In view of the fact that the brain is the prime site where inflammation develops in peroxisomal β‐oxidation deficiency, it will be of interest to also assess the inflammatory reactivity of microglia specifically lacking peroxisomal β‐oxidation enzymes. Although these resident macrophages of the brain bear many similarities with peripheral monocytes/macrophages they also display distinct characteristics.
Taken together, we provide novel insights on the regulation of peroxisomal β‐oxidation during macrophage polarization and provide unanticipated novel information on the impact of peroxisomal β‐oxidation deficiency on inflammatory activation of macrophages.
Authors contribution
SV and IG designed the study, performed experiments, analysed data and wrote the manuscript. MB designed the study and wrote the manuscript. PPVV, YYT and VEK designed, performed and analysed experiments regarding lipidomics and commented on the manuscript. EDS, OK and DVK assisted in experiments and commented on the manuscript. The authors thank Benny Das for excellent technical assistance and group of Jean‐Paul Pais De Barros in Dijon for performing fatty acid analysis.
Disclosure
The authors declare no conflict of interest.
Supporting information
Figure S1. Reduced peroxisomal β‐oxidation in Mfp2 −/− macrophages.
Figure S2. No differences in inflammasome activation between control and Mfp2 −/− bone‐marrow‐derived macrophages.
Figure S3. No difference in cell viability between control and Mfp2 −/− BMDM.
Figure S4. Mice with selective deletion of multifunctional protein 2 in macrophages show no overt phenotype.
Table S1. Sequences of primers used in quantitative RT‐PCR.
Table S2. Quantitative assessment of fatty acids in control and multifunctional protein 2‐deficient macrophages in basal state or after classical and alternative activation (n = 3).
Table S3. Quantitative assessment of triacylglycerides1 in control and multifunctional protein 2‐deficient macrophages in basal state or after classical and alternative activation (n = 4).
Table S4. Quantitative assessment of phosphatidylethanolamine1 molecular species in control and multifunctional protein 2‐deficient macrophages in basal or after classical and alternative activation (n = 4).
Acknowledgements
SV is supported by a postdoctoral fellowship of the Flemish Research Foundation (FWO) and IG by an ERAWEB fellowship. Research was supported by grants from UGhent (BOF14.GOA/019, to DK and OK), from FWO (G.0675.12 and G.0A15.13, to MB, DK and OK), and from KU Leuven (OT12/78 to MB).
Note
The gene encoding multifunctional protein 2 (MFP2) is Hsd17b4, a name related to the initial description of this enzyme as a steroid dehydrogenase. In this text, the knockout will be referred to as Mfp2 −/−.
References
- 1. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature 2013; 496:445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S et al Gene‐expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 2012; 13:1118–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I et al Transcriptome‐based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014; 40:274–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity 2013; 38:633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ et al Toll‐like receptor‐induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010; 115:4742–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodriguez‐Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin‐Sanz P et al Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 2010; 185:605–14. [DOI] [PubMed] [Google Scholar]
- 7. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR et al Oxidative metabolism and PGC‐1β attenuate macrophage‐mediated inflammation. Cell Metab 2006; 4:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Veldhoven PP. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J Lipid Res 2010; 51:2863–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Islinger M, Grille S, Fahimi HD, Schrader M. The peroxisome: an update on mysteries. Histochem Cell Biol 2012; 137:547–74. [DOI] [PubMed] [Google Scholar]
- 10. Moore SA, Hurt E, Yoder E, Sprecher H, Spector AA. Docosahexaenoic acid synthesis in human skin fibroblasts involves peroxisomal retroconversion of tetracosahexaenoic acid. J Lipid Res 1995; 36:2433–43. [PubMed] [Google Scholar]
- 11. Baes M, Aubourg P. Peroxisomes, myelination, and axonal integrity in the CNS. Neuroscientist 2009; 15:367–79. [DOI] [PubMed] [Google Scholar]
- 12. Berger J, Dorninger F, Forss‐Petter S, Kunze M. Peroxisomes in brain development and function. Biochim Biophys Acta 2016; 1863:934–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cartier N, Hacein‐Bey‐Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I et al Hematopoietic stem cell gene therapy with a lentiviral vector in X‐linked adrenoleukodystrophy. Science 2009; 326:818–23. [DOI] [PubMed] [Google Scholar]
- 14. Cartier N, Aubourg P. Hematopoietic stem cell transplantation and hematopoietic stem cell gene therapy in X‐linked adrenoleukodystrophy. Brain Pathol 2010; 20:857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kemp S, Berger J, Aubourg P. X‐linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta 2012; 1822:1465–74. [DOI] [PubMed] [Google Scholar]
- 16. Depreter M, Espeel M, Roels F. Human peroxisomal disorders. Microsc Res Tech 2003; 61:203–23. [DOI] [PubMed] [Google Scholar]
- 17. Glasgow BJ, Brown HH, Hannah JB, Foos RY. Ocular pathologic findings in neonatal adrenoleukodystrophy. Ophthalmology 1987; 94:1054–60. [DOI] [PubMed] [Google Scholar]
- 18. Van Maldergem L, Espeel M, Wanders RJ, Roels F, Gerard P, Scalais E et al Neonatal seizures and severe hypotonia in a male infant suffering from a defect in peroxisomal beta‐oxidation. Neuromuscul Disord 1992; 2:217–24. [DOI] [PubMed] [Google Scholar]
- 19. Johnson AB, Schaumburg HH, Powers JM. Histochemical characteristics of the striated inclusions of adrenoleukodystrophy. J Histochem Cytochem 1976; 24:725–30. [DOI] [PubMed] [Google Scholar]
- 20. Baes M, Huyghe S, Carmeliet P, Declercq PE, Collen D, Mannaerts GP et al Inactivation of the peroxisomal multifunctional protein‐2 in mice impedes the degradation of not only 2‐methyl‐branched fatty acids and bile acid intermediates but also of very long chain fatty acids. J Biol Chem 2000; 275:16329–36. [DOI] [PubMed] [Google Scholar]
- 21. Verheijden S, Bottelbergs A, Krysko O, Krysko DV, Beckers L, De Munter S et al Peroxisomal multifunctional protein‐2 deficiency causes neuroinflammation and degeneration of Purkinje cells independent of very long chain fatty acid accumulation. Neurobiol Dis 2013; 58:258–69. [DOI] [PubMed] [Google Scholar]
- 22. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 1999; 8:265–77. [DOI] [PubMed] [Google Scholar]
- 23. Meerpohl HG, Lohmann‐Matthes ML, Fischer H. Studies on the activation of mouse bone marrow‐derived macrophages by the macrophage cytotoxicity factor (MCF). Eur J Immunol 1976; 6:213–7. [DOI] [PubMed] [Google Scholar]
- 24. Ray A, Dittel BN. Isolation of mouse peritoneal cavity cells. J Vis Exp 2010; 35:1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐ΔΔ C(T)) Method. Methods 2001; 25:402–8. [DOI] [PubMed] [Google Scholar]
- 26. Van Veldhoven PP, Swinnen JV, Esquenet M, Verhoeven G. Lipase‐based quantitation of triacylglycerols in cellular lipid extracts: requirement for presence of detergent and prior separation by thin‐layer chromatography. Lipids 1997; 32:1297–300. [DOI] [PubMed] [Google Scholar]
- 27. Peterson GL. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem 1977; 83:346–56. [DOI] [PubMed] [Google Scholar]
- 28. Baarine M, Andreoletti P, Athias A, Nury T, Zarrouk A, Ragot K et al Evidence of oxidative stress in very long chain fatty acid–treated oligodendrocytes and potentialization of ROS production using RNA interference‐directed knockdown of ABCD1 and ACOX1 peroxisomal proteins. Neuroscience 2012; 213:1–18. [DOI] [PubMed] [Google Scholar]
- 29. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957; 226:497–509. [PubMed] [Google Scholar]
- 30. Tyurin VA, Cao W, Tyurina YY, Gabrilovich DI, Kagan VE. Mass‐spectrometric characterization of peroxidized and hydrolyzed lipids in plasma and dendritic cells of tumor‐bearing animals. Biochem Biophys Res Commun 2011; 413:149–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthu TS et al A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem 2014; 6:542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van Veldhoven PP, Huang S, Eyssen HJ, Mannaerts GP. The deficient degradation of synthetic 2‐ and 3‐methyl‐branched fatty acids in fibroblasts from patients with peroxisomal disorders. J Inherit Metab Dis 1993; 16:381–91. [DOI] [PubMed] [Google Scholar]
- 33. Van Veldhoven PP, Mannaerts GP. Inorganic and organic phosphate measurements in the nanomolar range. Anal Biochem 1987; 161:45–8. [DOI] [PubMed] [Google Scholar]
- 34. Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I et al Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015; 520:192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cash JL, White GE, Greaves DR. Chapter 17. Zymosan‐induced peritonitis as a simple experimental system for the study of inflammation. Methods Enzymol 2009; 461:379–96. [DOI] [PubMed] [Google Scholar]
- 36. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS et al Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol 2012; 92:829–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yanagisawa N, Shimada K, Miyazaki T, Kume A, Kitamura Y, Sumiyoshi K et al Enhanced production of nitric oxide, reactive oxygen species, and pro‐inflammatory cytokines in very long chain saturated fatty acid‐accumulated macrophages. Lipids Health Dis 2008; 7:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh J, Khan M, Singh I. Silencing of Abcd1 and Abcd2 genes sensitizes astrocytes for inflammation: implication for X‐adrenoleukodystrophy. J Lipid Res 2009; 50:135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. El Hajj HI, Vluggens A, Andreoletti P, Ragot K, Mandard S, Kersten S et al The inflammatory response in acyl‐CoA oxidase 1 deficiency (pseudoneonatal adrenoleukodystrophy). Endocrinology 2012; 153:2568–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Di Biase A, Merendino N, Avellino C, Cappa M, Salvati S. Th 1 cytokine production by peripheral blood mononuclear cells in X‐linked adrenoleukodystrophy. J Neurol Sci 2001; 182:161–5. [DOI] [PubMed] [Google Scholar]
- 41. Tannahill GM, Curtis AM, Adamik J, Palsson‐McDermott EM, McGettrick AF, Goel G et al Succinate is an inflammatory signal that induces IL‐1β through HIF‐1α . Nature 2013; 496:238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mills E, O'Neill LA. Succinate: a metabolic signal in inflammation. Trends Cell Biol 2014; 24:313–20. [DOI] [PubMed] [Google Scholar]
- 43. Mills EL, O'Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti‐inflammatory signal. Eur J Immunol 2016; 46:13–21. [DOI] [PubMed] [Google Scholar]
- 44. Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long‐chain fatty acids and ketone bodies by murine macrophages. Biochem J 1986; 239:121–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo‐starvation. Nature 2013; 493:346–55. [DOI] [PubMed] [Google Scholar]
- 46. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E et al Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015; 42:419–30. [DOI] [PubMed] [Google Scholar]
- 47. Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM et al Cell‐intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 2014; 15:846–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rakhshandehroo M, Knoch B, Muller M, Kersten S. Peroxisome proliferator‐activated receptor α target genes. PPAR Res 2010; 2010:612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bagattin A, Hugendubler L, Mueller E. Transcriptional coactivator PGC‐1α promotes peroxisomal remodeling and biogenesis. Proc Natl Acad Sci USA 2010; 107:20376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singh J, Khan M, Singh I. HDAC inhibitor SAHA normalizes the levels of VLCFAs in human skin fibroblasts from X‐ALD patients and downregulates the expression of proinflammatory cytokines in Abcd1/2‐silenced mouse astrocytes. J Lipid Res 2011; 52:2056–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Singh J, Khan M, Singh I. Caffeic acid phenethyl ester induces adrenoleukodystrophy (Abcd2) gene in human X‐ALD fibroblasts and inhibits the proinflammatory response in Abcd1/2 silenced mouse primary astrocytes. Biochim Biophys Acta 2013; 1831:747–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weber FD, Wiesinger C, Forss‐Petter S, Regelsberger G, Einwich A, Weber WH et al X‐linked adrenoleukodystrophy: very long‐chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. Hum Mol Genet 2014; 23:2542–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schluter A, Espinosa L, Fourcade S, Galino J, Lopez E, Ilieva E et al Functional genomic analysis unravels a metabolic‐inflammatory interplay in adrenoleukodystrophy. Hum Mol Genet 2012; 21:1062–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ruiz M, Jove M, Schluter A, Casasnovas C, Villarroya F, Guilera C et al Altered glycolipid and glycerophospholipid signaling drive inflammatory cascades in adrenomyeloneuropathy. Hum Mol Genet 2015; 24:6861–76. [DOI] [PubMed] [Google Scholar]
- 55. Jang J, Park S, Jin Hur H, Cho HJ, Hwang I, Pyo Kang Y et al 25‐hydroxycholesterol contributes to cerebral inflammation of X‐linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat Commun 2016; 7:13129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Reduced peroxisomal β‐oxidation in Mfp2 −/− macrophages.
Figure S2. No differences in inflammasome activation between control and Mfp2 −/− bone‐marrow‐derived macrophages.
Figure S3. No difference in cell viability between control and Mfp2 −/− BMDM.
Figure S4. Mice with selective deletion of multifunctional protein 2 in macrophages show no overt phenotype.
Table S1. Sequences of primers used in quantitative RT‐PCR.
Table S2. Quantitative assessment of fatty acids in control and multifunctional protein 2‐deficient macrophages in basal state or after classical and alternative activation (n = 3).
Table S3. Quantitative assessment of triacylglycerides1 in control and multifunctional protein 2‐deficient macrophages in basal state or after classical and alternative activation (n = 4).
Table S4. Quantitative assessment of phosphatidylethanolamine1 molecular species in control and multifunctional protein 2‐deficient macrophages in basal or after classical and alternative activation (n = 4).