

Abstract
Lipopolysaccharide (LPS) is a component of the outer membrane of Gram-negative bacteria that induces strong proinflammatory reactions of mammals. These processes are triggered upon sequential binding of LPS to CD14, a GPI-linked plasma membrane raft protein, and to the TLR4/MD2 receptor complex. We have found earlier that upon LPS binding, CD14 triggers generation of phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2], a lipid controlling subsequent proinflammatory cytokine production. Here we show that stimulation of RAW264 macrophage-like cells with LPS induces global changes of the level of fatty-acylated, most likely palmitoylated, proteins. Among the acylated proteins that were up-regulated in those conditions were several enzymes of the phosphatidylinositol cycle. Global profiling of acylated proteins was performed by metabolic labeling of RAW264 cells with 17ODYA, an analogue of palmitic acid functionalized with an alkyne group, followed by detection and enrichment of labeled proteins using biotin-azide/streptavidin and their identification with mass spectrometry. This proteomic approach revealed that 154 fatty-acylated proteins were up-regulated, 186 downregulated, and 306 not affected in cells stimulated with 100 ng/ml LPS for 60 min. The acylated proteins affected by LPS were involved in diverse biological functions, as found by Ingenuity Pathway Analysis. Detailed studies of 17ODYA-labeled and immunoprecipitated proteins revealed that LPS induces S-palmitoylation, hence activation, of type II phosphatidylinositol 4-kinase (PI4KII) β, which phosphorylates phosphatidylinositol to phosphatidylinositol 4-monophosphate, a PI(4,5)P2 precursor. Silencing of PI4KIIβ and PI4KIIα inhibited LPS-induced expression and production of proinflammatory cytokines, especially in the TRIF-dependent signaling pathway of TLR4. Reciprocally, this LPS-induced signaling pathway was significantly enhanced after overexpression of PI4KIIβ or PI4KIIα; this was dependent on palmitoylation of the kinases. However, the S-palmitoylation of PI4KIIα, hence its activity, was constitutive in RAW264 cells. Taken together the data indicate that LPS triggers S-palmitoylation and activation of PI4KIIβ, which generates PI(4)P involved in signaling pathways controlling production of proinflammatory cytokines.
Lipopolysaccharide (LPS) is a component of the outer membrane of Gram-negative bacteria. Upon infection, LPS induces strong proinflammatory responses that facilitate eradication of bacteria. These proinflammatory reactions are triggered upon recognition of LPS by Toll-like receptor 4 (TLR4) (1), which is expressed in cells of myeloid lineage, such as monocytes, macrophages, and dendritic cells, and certain nonimmune cells like endothelial and epithelial cells. TLR4 belongs to so-called pattern recognizing receptors specialized in identification of evolutionarily conserved constituents of cell wall/membranes of microbes, their RNA, and unmethylated CpG DNA motifs. These receptors trigger innate immune responses constituting the first line of defense against pathogens and initiate appropriate adaptive immune responses (2). The LPS-induced proinflammatory reaction, although initially beneficial, when exaggerated can lead to a potentially fatal systemic inflammation called sepsis, severe sepsis, and septic shock (3). In addition, a prolonged low-grade inflammation caused by LPS derived from intestinal bacteria has been recognized as a factor in the development of several chronic diseases (4), which additionally drives interest in TLR4-induced signaling.
TLR4 is a transmembrane protein that associates with an extracellular protein MD2 crucial for LPS binding. Upon the binding, up to five of the six acyl chains of LPS are buried within a hydrophobic pocket of one MD2 protein while the remaining acyl chain interacts with TLR4 of a neighboring TLR4/MD2 complex, inducing dimerization of the TLR4/MD2 complexes (5). This facilitates recruitment of two sets of adaptor proteins to the signaling Toll/interleukin-1 receptor (TIR) homology domain of TLR4. First, a pair of adaptor proteins consisting of TIR domain-containing adaptor protein (TIRAP) and MyD88 binds to the receptor, and this interaction is guided by simultaneous binding of TIRAP to both the TIR domain of TLR4 and to a plasma membrane lipid, phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] (6). MyD88, in turn, recruits interleukin-1 receptor-associated kinase (IRAK)4 and IRAK1/2, forming a multimolecular complex called the myddosome (7). The myddosome assembly initiates a signaling cascade, eventually leading to activation of NFκB and activator protein-1 transcription factors, the latter acting downstream of MAP kinases. Expression of genes encoding proinflammatory cytokines, like tumor necrosis factor α (TNFα), follows (8). Subsequently, the TLR4/MD2 complexes are internalized, and at the endosomal membrane, the first set of adaptor proteins is replaced by another one consisting of TIR domain-containing adaptor inducing interferon-β (TRIF) and TRIF-related adaptor molecule (TRAM) proteins (9–11). The signaling pathway triggered thereby leads to activation of IRF3/7 transcription factors and expression of type I interferons and some other cytokines exemplified by C-C motif chemokine ligand 5/regulated upon activation, normal T cell expressed and secreted (CCL5/RANTES) (8).
In cells of the myeloid lineage, the recognition of LPS by TLR4 and LPS-induced signaling are assisted by CD14. The protein contains the glycosylphosphatidylinositol (GPI) moiety attached to its C terminus, which anchors CD14 in the outer leaflet of the plasma membrane within nanodomains of the membrane rich in cholesterol and lipids with saturated fatty acids (mainly sphingolipids), called rafts (12, 13). Once in the plasma membrane, CD14 binds an LPS molecule in a large hydrophobic pocket located in the N-terminal part of the protein and facilities transfer of the LPS onto the MD2 protein of the TLR4/MD2 complex (14, 15). A growing body of evidence indicates that the role of CD14 in LPS-induced signaling goes beyond the binding of LPS. CD14 controls the internalization of LPS-activated TLR4/MD2, and with some exceptions, this CD14-controlled uptake of TLR4 is required for the initiation of the endosomal TRIF-dependent signaling pathway of TLR4 (16–19). Our recent studies have shown a direct link between CD14 and production of PI(4,5)P2 in LPS-stimulated cells. Upon binding of LPS, CD14 induces biphasic production and accumulation of PI(4,5)P2 in macrophages, whereas TLR4 can fine tune the process (20, 21). The PI(4,5)P2 is generated by phosphorylation of phosphatidylinositol 4-monophosphate [PI(4)P] by type I phosphatidylinositol 4-phosphate 5-kinase (PIP5KI), isoforms Iα and Iγ (20, 22). In LPS-stimulated cells, PI(4,5)P2 serves as a binding site for TIRAP in the MyD88-dependent pathway and undergoes hydrolysis to diacylglycerol and inositol 1,4,5-trisphosphate (IP3) or, alternatively, phosphorylation to phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3]. Both the hydrolysis and phosphorylation of PI(4,5)P2 are required for internalization of LPS-activated TLR4/MD2 and initiation of the TRIF-dependent signaling (18, 23, 24). Additionally, PI(4,5)P2 can also control TLR4-independent stimulation of cells by LPS that has entered the cytosol (25, 26). These data point to phosphatidylinositols as crucial factors affecting all LPS-induced responses of cells. During the CD14- and TLR4/MD2-mediated stimulation of cells, the newly formed PI(4,5)P2 accumulates in the Triton X-100-resistant fraction of cells that is enriched in membrane raft components, including CD14 itself (20). These data corroborate the thesis on the location of the machinery engaged in LPS-induced signaling in plasma membrane rafts (13).
One of the key factors controlling the association of proteins with rafts is their S-acylation, frequently referred to as S-palmitoylation and defined as the attachment of a palmitic acid (C16:0) residue to a cysteine residue of the protein through a thioester bond (27). In general, S-palmitoylation or, more broadly, S-acylation, affects a diversity of protein functions, including their stability, cellular trafficking, membrane localization, and protein–protein interactions (28, 29). These functions are likely to be disturbed when, instead of the saturated palmitic acid, dietary unsaturated fatty acids are attached via the thioester linkage to proteins; however, biological consequences of this type of S-acylation remain mostly unknown (30, 31). Beside S-palmitoylation, several other modifications consist in the attachment of fatty acid residues to proteins. These are myristoylation, lysine side chain acylation, and rare O- and N-palmitoylation (32). Among all these acylations, S-palmitoylation is the best characterized reversible modification, and 24 palmitoyl acyltransferases (zinc finger DHHC (zDHHC) domain containing enzyme family) and several depalmitoylating enzymes have been identified in mammals (33–35). It has been shown that S-palmitoylation controls the engagement of proteins in signaling pathways of receptors other than TLR4 (36–38). We have found earlier that LPS induces accumulation of S-palmitoylated Lyn kinase in the raft-enriched fraction of RAW264 macrophage-like cells, which determines negative regulation of TLR4 signaling by the kinase (39). Taking into account the redistribution and activation of proteins involved in the MyD88- and TRIF-dependent signaling pathways of TLR4, we assumed that changes of the level of S-palmitoylated proteins could contribute substantially to those complex cascades of events. In order to identify acylated proteins affected by LPS, in this study we metabolically labeled RAW264 cells with a palmitic acid analogue, 17-octadecynoic acid (17ODYA)1 and then tagged the 17ODYA-labeled proteins with biotin-azide in a click chemistry reaction and analyzed them by mass spectrometry. Proteomic analysis has recently been used to identify global protein acylation in diverse cells, including RAW264 macrophage-like cells and the dendritic cell line DC2.4 (31, 40, 41). It should be emphasized, however, that none of the earlier studies examined the influence of cells stimulation with LPS on protein palmitoylation, and thus the contribution of palmitoylated proteins to LPS-induced signaling remains largely unknown.
We identified 154 fatty-acylated proteins up-regulated and 186 downregulated ones in cells stimulated with 100 ng/ml LPS for 60 min, which is a time window where the signaling events related to both MyD88- and TRIF-dependent pathways of TLR4 occur. Ingenuity Pathway Analysis (IPA) revealed several functional networks involving the fatty-acylated proteins affected by LPS. By adding an immunoprecipitation step to the above procedure, we confirmed the proteomic data pointing to LPS-induced S-palmitoylation, hence activation, of type II phosphatidylinositol 4-kinase (PI4KII) β, which phosphorylates phosphatidylinositol to PI(4)P. S-palmitoylation of the related isoform PI4KIIα was constitutive in RAW264 cells. Our data indicate that LPS induces production of PI(4,5)P2 and its PI(4)P precursor controlling downstream proinflammatory signaling and that S-palmitoylation of PI4KIIβ is a crucial step in this cascade of events.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatment
RAW264.7 and J774A.1 macrophage-like cells, and HEK293 cells were cultured in DMEM containing 10% fetal bovine serum (FBS), 2 mm l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Metabolic labeling of RAW264 cells with 17ODYA (Sigma-Aldrich, Poznan, Poland) was performed in DMEM containing 2% charcoal-stripped FBS (ThermoFisher Scientific, Waltham, MA, USA), l-glutamine, and antibiotics as above and 30 mm Hepes, pH 7.4. For stimulation, cells were exposed to 100 ng/ml LPS (ultrapure smooth LPS from Escherichia. coli O111:B4, List Biological Laboratories, Campbell, CA, USA). In some experiments, prior to labeling with 17ODYA, cells were incubated with 125–250 μm bromohexadecanoic acid (BPA) for 1 h (37 °C) in DMEM/2% charcoal-stripped FBS, and the drug was present during subsequent labeling of cells with 17ODYA, stimulation with LPS, and cytokine production. BPA was precomplexed to fatty-acid-free bovine serum albumin (BSA; Sigma-Aldrich) at a 4:1 molar ratio, essentially as described earlier (42). When indicated, cells cultured overnight in DMEM/2% FBS were incubated with 150–500 μm palmitic acid, prepared according to (43), for 30 min (37 °C), and labeled with 17ODYA in its presence.
Plasmids
pCMV5-Myc plasmid encoding rat PI4KIIα or its deletion mutant lacking the 173CCPCC177 motif, as well as pCMV5-Myc plasmid encoding human PI4KIIβ were kindly provided by Professor Helen L. Yin (University of Texas Southwestern Medical Center, Dallas, TX). Deletion mutant of PI4KIIβ lacking the S-palmitoylation motif 170CCPCC174 was generated by site directed mutagenesis using primers 5′-GGACCAAATATGTCCATAAGGTCTTTGGCCGAGGCTGCCTGATTCCTAATC-3′ and 5′-GATTAGGAATCAGGCAGCCTCGGCCAAA GACCTTATGGACATATTTGGTCC-3′ as described earlier (44).
Plasmid encoding murine CD14, pUNO-mCD14, was from InvivoGen (Toulouse, France). The CD14-VSVG fusion protein was generated by replacing the C-terminal 21 amino acids of CD14, which contain the GPI-anchor signal (45), with the transmembrane and cytoplasmic domains of the G-protein of vesicular stomatitis virus (VSVG) encompassing 49 C-terminal amino acids of the protein (46). For this, CD14 cDNA was amplified by PCR using primers 5′-GGTGAAGCTTACCATGGAGCGTGTGCTTGG-3′ and 5′-GAATATGGATCCTGGAGCTCCGGCGGTG-3′ that contained HindIII and BamHI restriction sites (italicized), respectively, and pUNO1-mCD14 plasmid as template. In a separate reaction, VSVG cDNA was amplified using primers 5′-GCGCCGGATCCAGCTCTATTGCCTCTTTTTTC-3′ and 5′-GTAAATTGCGGCCGCCTTTCCAAGTCGGTTCATC-3′ that contained BamHI and NotI restriction sites (italicized), respectively, and pCMV-VSVG plasmid (a gift from Dr. Robert D. Weinberg, Whitehead Institute for Biomedical Research, Cambridge, MA; Addgene plasmid #8454 described in (47)) as a template. In order to obtain cDNA encoding CD14-VSVG with 3xHA tag, the two PCR products isolated and digested with respective enzymes were ligated consecutively with pcDNA3.1/Hygro(+)-3xHA plasmid (prepared in our laboratory by addition of three repeats of a sequence encoding HA tag to pcDNA3.1/Hygro(+) from Invitrogen) in two separate reactions. The obtained CD14-VSVG-3xHA fusion cDNA was amplified using above template and primers 5′-GATTATACCGGTCACCATGGAGCGTGTGC-3′ and 5′-GGTCGCTAGCTTAGGCATAGTCTGGCACATC-3′ containing AgeI and NheI restriction sites (italicized), respectively. The amplified and purified DNA fragment was digested with respective enzymes and eventually ligated with the pUNO1 vector yielding pUNO-CD14-VSVG used in experiments. The plasmid encoding CD14-VSVG fusion protein mutated in the S-palmitoylation site (Cys489Ala substitution, numbered as in full length VSVG), pUNO-CD14-VSVGmut, was generated by site-directed mutagenesis using pUNO-CD14-VSVG as template and primers 5′-CGAGTTGGTATCCATCTTGCCATTAAATTAAAGCACACC-3′ and 5′-GGTGTGCTTTAATTTAATGGCAAGATGGATACCAACTCG-3′. Plasmid pDUO-3xFLAG-mTLR4/MD2 encoding murine FLAG-tagged TLR4 and MD2 was obtained as described earlier (21). All constructs were verified be sequencing. NFκB-firefly luciferase reporter plasmid pNFκB-Luc was from Invitrogen, interferon-stimulated response element (ISRE)-firefly luciferase reporter plasmid pISRE-Luc from Stratagene, Renilla luciferase pRL-TK plasmid from Promega (Warsaw, Poland). Plasmids were introduced into E. coli DH5α, purified using GenElute Endotoxin-free Plasmid HP Midiprep (Sigma-Aldrich) and used for cell transfection.
Mass Spectrometry Analysis: Experimental Design and Statistical Rationale
A) Cell Labeling with 17ODYA
RAW264 cells (5 × 106/sample) were transferred into DMEM containing 2% charcoal-stripped FBS and 30 mm Hepes, pH 7.4, and supplemented with 50 μm 17ODYA in DMSO or 0.05% DMSO carrier in control samples. After 4 h (37 °C), cells were either left unstimulated or were supplemented with 100 ng/ml LPS for 60 min. Three independent experiments were performed according to this protocol, each comprising four above-mentioned samples. Cells were collected and washed with ice-cold phosphate buffered saline (PBS) by centrifugation (5 min, 400 × g, 4 °C) and lysed (15 min, 25 °C) in 1.5 ml of SDS buffer (4% SDS, 150 mm NaCl, 50 mm triethanolamine, pH 7.4, 250 units Benzonase (Sigma-Aldrich), EDTA-free protease inhibitor mixture (Roche, Warsaw, Poland), phosphatase inhibitors (10 mm p-nitrophenyl phosphate, 1 mm Na3VO4, and 50 mm phenylarsine phosphate)). Then the lysates were subjected to Cu(I)-catalyzed click reaction with biotin-azide and enriched on streptavidin beads (48). For this purpose, cell lysates (about 2.2 mg of total protein) were supplemented with 100 μm biotin-azide (PEG4 carboxamide-6-azidohexanyl biotin, Life Technologies, Warsaw, Poland), 1 mm Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Sigma-Aldrich), 100 μm Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, Sigma-Aldrich) and 1 mm CuSO4, vortexed, and incubated for 1.5 h at 25 °C in the darkness. To remove unreacted biotin, protein was precipitated by adding eight volumes of methanol (−20 °C, 18 h). Protein pellets were centrifuged (30 min, 9000 × g, 4 °C), washed two times with methanol (−20 °C), and air dried for 5–10 min. Subsequently, the pellet was dissolved in 300 μl of the SDS buffer containing 10 mm EDTA; for further steps, 2 mg of total protein were taken. The solutions were diluted to 0.5% SDS with 2.1 ml of Brij buffer (1% Brij 97 (Sigam-Aldrich), 150 mm NaCl, 50 mm triethanolamine, pH 7.4, protease inhibitors (10 μg/ml aprotinin, leupeptin, and pepstatin each and 1 mm PMSF) and phosphatase inhibitors as above) and supplemented with 60 μl of high-capacity streptavidin-agarose beads (ThermoFisher Scientific) prewashed three times with the Brij buffer. Samples were incubated for 1.5 h at 25 °C with end-over-end rotation. The beads were washed extensively with 3 ml of 8 m urea in 10 mm Hepes, pH 7.4 (five times for 1 min), 4 ml of 10% acetonitrile (three times for 1 min), 4 ml of 1% SDS in PBS (five times for 1 min), 4 ml of PBS (five times for 1 min), and 4 ml of 100 mm ammonium bicarbonate (once for 1 min) and subjected to mass spectrometry analysis.
B) Sample Preparation for Mass Spectrometry
Proteins bound to streptavidin beads were subjected to a standard procedure of trypsin digestion, during which proteins were reduced with TCEP (5 mm final concentration from 0.5 m stock in 100 mm ammonium bicarbonate buffer, pH 8.0) for 1 h at 60 °C, blocked with methyl methanethiosulfonate (10 mm final concentration from 200 mm stock in 100 mm ammonium bicarbonate buffer, pH 8.0) for 10 min at 25 °C, and digested overnight with 10 ng/μl trypsin. The resulting peptide mixtures were applied on an RP-18 precolumn (Waters, Milford, MA, USA) using water containing 0.1% formic acid as a mobile phase and then transferred to a nano-HPLC RP-18 column (internal diameter 75 μm, Waters) using acetonitrile gradient (0–35% acetonitrile in 160 min) in the presence of 0.1% formic acid at a flow rate of 250 nl/min. The column outlet was coupled directly to the ion source of an Orbitrap Velos mass spectrometer (Thermo Electron Corp., San Jose, CA, USA) working in the regime of data-dependent MS to MS/MS switch. A blank run ensuring absence of cross-contamination from previous samples preceded each analysis.
C) Analysis of Mass Spectrometry Data
The acquired MS/MS data were preprocessed with Mascot Distiller software (v. 2.5.1, MatrixScience, London, U.K.) and a search was performed with the Mascot Search Engine (MatrixScience, Mascot Server 2.4.1) against Mus sp. proteins from the SwissProt protein database (Swissprot 2017_02; 16,905 sequences). To reduce mass errors, the peptide and fragment mass tolerance settings were established separately for individual LC-MS/MS runs after a measured mass recalibration, as described previously (49). After the recalibration, the mass tolerance for proteins was in the range 5–10 ppm and for peptides 0.01–0.05 Da. The Mascot search parameters were as follows: enzyme, Trypsin; missed cleavages, 1; fixed modifications, Methylthio (C); variable modifications, Oxidation (M); instrument, HCD; Decoy option, active. False discovery rate (FDR) was estimated with Mascot Decoy search, and score threshold was adjusted for each sample to keep the FDR below 1%. Detailed protein/peptide identification data are supplemented to this text (supplemental Table 1). Only proteins represented by at least two unique peptides in at least two 17ODYA-labeled samples are shown and were further considered. Subsequently, probable contaminants (keratin, albumin) were removed from the list, and redundantly identified proteins were curated manually. For evaluation of the relative protein abundance in each sample, spectral count values determined using exponentially modified protein abundance index (emPAI) scores (50) were used. Only proteins that met the acceptance criteria: FDR<1%, at least two unique peptides, Mascot score over 25, nonredundant proteins, were taken for further analysis. Spectral count approach was used to reveal which acylated proteins were significantly enriched in 17ODYA-treated samples in comparison to DMSO-treated ones (48, 51). For this, missing values were imputed as 0, and the spectral counts of significantly identified peptides of a given protein were summed up over three samples of unstimulated cells labeled with 17ODYA (data set A), three samples of LPS-stimulated cells labeled with 17ODYA (data set C), three samples of unstimulated control cells exposed to DMSO without 17ODYA (data set B), and three samples of stimulated control cells exposed to DMSO without 17ODYA (data set D). At this point, all zeros were replaced by 1. Subsequently, for each protein, a ratio of the sum of spectral counts of A versus B and C versus D, and next, natural logarithms (ln) of the ratios were calculated. Proteins with a negative ln value, i.e. those with more spectral counts in DMSO-treated than in 17ODYA-treated samples (a total of 84), were removed from further considerations. It was assumed that an ln A/B and/or ln C/D ≥ 2.5 reflected tentative identification of a given 17ODYA-labeled acylated protein. Then, statistical significance of the difference between spectral counts summed in the A and B or C and D data sets for each protein was estimated with the unequal variance one-tailed, heteroschedastic t test. It was assumed that a fatty-acylated protein was identified with high confidence when the p value of the difference between 17ODYA- and DMSO-treated samples was ≤ 0.05 and with medium confidence when 0.05 < p ≤ 0.275 (51). All proteins identified with an ln of the ratio of 17ODYA- to DMSO-treated samples (A/B and/or C/D) ≥ 2.5 had to meet at least the medium confidence criterion both in unstimulated and LPS-stimulated cells to be considered further. The obtained list of fatty-acylated proteins served as a basis to reveal proteins up-regulated or downregulated in LPS-treated cells. For each protein on this list, the ratio of spectral counts, named RL, was obtained by dividing the C/D ratio by the A/B ratio. Proteins with RL value ≥ 1.5 were considered as up-regulated while those with an RL ≤ 0.5 as downregulated in LPS-stimulated cells. The statistical significance of the difference between the A/B and C/D ratios in a given group of proteins (up-regulated, downregulated, and not affected ones) was calculated using the unequal variance two-tailed heteroschedastic t test. Eventually from the list of up-regulated proteins extracted were those which were found in two or three experiments in LPS-stimulated cells and no more than once in unstimulated cells to form a final short list of LPS-up-regulated proteins. Analogous approach yielded a short list of downregulated proteins.
Pathway Analysis of Proteomics Data
IPA (Qiagen, Hilden, Germany), version from June 2017, was used to identify the molecular interaction networks involving the acylated proteins detected with mass spectrometry. The IPA library contains experimentally confirmed interactions, and its content is curated manually. Information on the up- or downregulation of the acylated proteins was incorporated into the uploaded data set. The molecular networks were created automatically using default settings of the IPA Network algorithm, taking into account direct and indirect interactions in all cells and tissues. The networks were visualized using IPA Path Designer.
Immunoprecipitation and Analysis of S-palmitoylation of PI4KIIα and PI4KIIβ
RAW264 cells (8 × 105 in 60-mm dish) were transfected with 4 μg of pCMV5-Myc plasmid encoding wild-type PI4KIIα or its mutant form lacking the S-palmitoylation site, and 8 μl of TurboFect (ThermoFisher Scientific) according to manufacturer's instructions. For expression of PI4KIIβ, the cells were transfected with 6 μg of pCMV5-Myc plasmid encoding wild-type PI4KIIβ or its non-palmitoylatable mutant, and 12 μl TurboFect. After 24 h, cells were treated or not with 250 μm BPA (1 h, 37 °C), labeled with 50 μm 17ODYA or exposed to 0.05% DMSO in DMEM/2% charcoal-stripped FBS (4 h, 37 °C), and were either left unstimulated or were stimulated with 100 ng/ml LPS for 15–60 min. Subsequently, the cells were collected, washed with ice-cold PBS by centrifugation (3 min, 400 × g, 4 °C), frozen in liquid nitrogen, and stored at −80 °C. The cell pellets were resuspended at 4 °C in 200 μl of lysis buffer containing 0.5% Nonidet P-40, 100 mm NaCl, 50 mm phosphate buffer, pH 7.4, protease and phosphatase inhibitors described above, and protein thioesterase inhibitors (10 μm palmostatin; Merck, Warsaw, Poland, and 0.2 mm 1-hexadecanesulfonyl fluoride, Cayman Chemicals, Tallinn, Estonia). After 30 min, lysates were clarified by centrifugation (10 min, 16,000 × g, 4 °C), and supernatants were diluted with two volumes of the lysis buffer without the detergent, supplemented with 20 μl of EZView Red agarose affinity gel bearing rabbit anti-c-Myc IgG (Sigma-Aldrich), and incubated for 3 h at 4 °C with end-over-end rotation. Subsequently, samples were washed three times with ice-cold wash buffer containing 0.05% Nonidet P-40, 100 mm NaCl, 50 mm phosphate buffer, pH 7.4, protease, phosphatase, and protein thioesterase inhibitors as above, and once with the wash buffer devoid of the detergent. Finally, the agarose beads were pelleted (400 × g, 4 °C) and suspended in 45 μl of ice-cold PBS containing EDTA-free protease inhibitor mixture and 1 mm PMSF. For the click reaction, the mixture was supplemented with 1 mm TCEP, 100 μm TBTA, 1 mm CuSO4, and 10 μm IRDye 800CW-azide (LI-COR, Lincoln, NE). The final volume of the reaction mixture was 50 μl. The reaction was carried out in the darkness with gentle rotation. Subsequently, the samples were washed as after immunoprecipitation then washed once in PBS, suspended in 30 μl of SDS-sample buffer, and heated for 5 min at 95 °C. Some samples were treated with 2.5% hydroxylamine, pH 7.5 (30 min, 25 °C) before heating. Proteins were separated by 10% SDS-PAGE, transferred onto nitrocellulose (1 h, 400 mA), and analyzed in an Odyssey CLx Imager (LI-COR) or subjected to immunoblotting with mouse anti-Myc IgG, as described below.
Analysis of Fatty Acylation of Proteins Isolated by Affinity Enrichment
To analyze lipidation of CD14, HEK293 cells (5 × 105 in 60-mm dish) were transfected with 6 μg of total DNA containing 1 μg pUNO-mCD14 and 5 μg of empty pcDNA3.1/Hygro(+) plasmid or 6 μg of pUNO-CD14-VSVG or pUNO-CD14-VSVGmut using FuGENE (Promega) according to manufacturer's instruction. After 24 h, cells were labeled with 50 μm 17ODYA or exposed to 0.05% DMSO in DMEM/2% charcoal-stripped FBS (4 h, 37 °C), lysed, treated with biotin-azide (500 μm biotin-PEG3-azide; Sigma-Aldrich), and biotin-tagged proteins were affinity enriched on streptavidin-coupled beads, essentially as described for sample preparation for mass spectrometry. To elute proteins, the beads were incubated with the following solutions: twice with 150 μl H2O with heating to 70 °C at ∼1 °C per 10 s followed by 5 min at 70 °C with shaking (52), once with 100 μl 2X SDS-sample buffer (5 min at 95 °C), once with 100 μl 2X SDS-sample buffer supplemented according to (53) with 14% β-mercaptoethanol and 5 mm EDTA (5 min at 95 °C), and finally once with 100 μl H2O (5 min at 95 °C). All eluted fractions were combined, and proteins were precipitated overnight with 20% TCA (4 °C), pelleted (30 min, 16,000 × g, 25 °C), washed twice with acetone, air dried and dissolved in 33 μl of 2X SDS-sample buffer (5 min, 95 °C), resolved by 10% SDS-PAGE, transferred to nitrocellulose as above, and analyzed for the presence of CD14 or flotillin-2.
In a series of experiments, samples were subjected to delipidation (54, 55). For this purpose, proteins precipitated with methanol after the click reaction were resuspended in 200 μl of 0.73% NaCl, sonicated, supplemented with 0.8 ml of chloroform:methanol (2:1, v:v), and vortexed for 5 min. Then 9 ml of cold methanol were added to precipitate proteins (1 h, −20 °C). Proteins were pelleted (50 min, 4000 × g, 4 °C), pellets air dried, dissolved in 140 μl of the SDS buffer/10 mm EDTA, and processed for affinity enrichment on streptavidin beads as above.
Fatty acylation of endogenous proteins of RAW264 cells was validated by performing an analogous procedure as for CD14 using 2 × 106 untransfected cells per sample, which were lysed in 640 μl of 4% SDS buffer. Proteins were tagged with 500 μm biotin-PEG3-azide, subjected to chloroform:methanol precipitation, and tagged proteins were bound to streptavidin-agarose beads and after elution processed for immunoblotting. Parallel nitrocellulose membranes from one experiment were blotted with a set of antibodies or subjected to stripping of antibodies followed by subsequent immunoblotting to verify changes in the amounts of palmitoylated proteins.
Gene Reporter Assay
HEK293 cells (2.5 × 104 in 48-well dish) were transfected using FuGENE (Promega), essentially as described earlier (22) with a mixture of DNA containing 50 ng pUNO-mCD14, 20 ng pDUO-3xFLAG-mMD2/TLR4, 12.5–50 ng pCMV5-Myc plasmid encoding wild-type PI4KIIα or PI4KIIβ or their non-palmitoylatable mutants, 20 ng Renilla luciferase pRL-TK plasmid, 100 ng pNFκB-Luc or 100 ng pISRE-Luc luciferase reporter plasmid, and appropriate amounts of empty pcDNA3.1/Hygro(+) plasmid to bring the amount of DNA in all samples to 345 ng. After 24 h, cells were stimulated with 100 ng/ml LPS, lysed 24 h later in passive lysis buffer (Promega), and analyzed for reporter protein luminescence using dual luciferase assay reagent (21), or the lysates were subjected to SDS-PAGE and immunoblotting analysis for the presence of indicated proteins.
Cell Fractionation
HEK293 cells were transfected with CD14, TLR4/MD2 and PI4KIIα or PI4KIIβ variants as for the gene reporter assay; to equalize the amount of overexpressed kinases, two and four times more DNA encoding wild type than mutant kinases were used for PI4KIIα and PI4KIIβ, respectively. After 24 h, 1 × 106 cells were solubilized in 220 μl of 0.5% Brij 98 in 100 mm NaCl, 2 mm EDTA, 2 mm EGTA, 30 mm Hepes, pH 7.4, with proteases and phosphatase inhibitors (10 min, 25 °C) and fractionated in a 0–40% Optiprep density gradient containing the detergent (170,000 × g, 3.5 h, 4 °C), essentially as described earlier (56). Seven fractions were collected from the top of the gradient and analyzed for the presence of selected proteins by immunoblotting.
Immunoblotting
Proteins were separated by 10% or 4–20% SDS-PAGE and transferred on nitrocellulose by electroblotting. Nitrocellulose strips were probed with the following antibodies: mouse anti-FLAG (Sigma-Aldrich), mouse anti-Myc (ThermoFisher Scientific), mouse anti-β actin, rat anti-CD14 (both BD Biosciences, Warsaw, Poland), rabbit anti-flotillin-1 or flotillin-2, anti-JAK1, anti-Lyn A, anti-TNFα (all from Cell Signaling, Leiden, The Netherlands), rabbit anti-PAG (Exbio, Prague, Czech Republic), rabbit anti-eukaryotic translation initiation factor 5A2 (eIF5A2; Sigma-Aldrich). They were followed by goat anti-mouse IgG-HRP (Jackson ImmunoResearch, West Grove, PA, USA), goat anti-rat IgG-HRP (Sigma-Aldrich), or anti-rabbit-HRP (Millipore, Burlington, MA). Immunoreactive bands were visualized with chemiluminescence using SuperSignal WestPico substrate (ThermoFisher Scientific) and analyzed densitometrically using the ImageJ program. Prestained molecular mass standards were from BioRad (Puchheim, Germany).
Gene Silencing
Silencing of Pi4k2a and Pi4k2b was performed essentially as described earlier (57). Cell suspension (8 × 105 in 1 ml RPMI containing 5% FBS) was mixed with 1 ml of RPMI containing 200 pmol of Pi4k2a or Pi4k2b siRNA or scrambled siRNA (Qiagen) and 20 μl of TrueFect Lipo (United Biosystems, Herndon, VA). Cells were seeded in 96-well (0.8 × 105/well) or 48-well (1.6 × 105/well) plates, and after 6 h the medium was exchanged for DMEM/10% FBS. Cells were cultured for 18 h and used for ELISA tests or quantitative polymarase chain reaction (qPCR) analysis.
TNFα and CCL5/RANTES Assays
Cells were plated at 0.8 × 105/well in 96-well plates and stimulated as described above. After 4 h of stimulation, levels of TNFα and CCL5/RANTES in culture supernatants were determined using murine ELISA kits (R&D Systems, Abingdon, U.K.; BioLegend, Katowice, Poland). The product absorbance was measured using a Sunrise plate reader (Tecan Group, Zurich, Switzerland).
qPCR Analysis
Total RNA was isolated from cells with TRI reagent (Sigma-Aldrich) and precipitated with ethanol. cDNA was synthetized using High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific) according to the manufacturer's protocol. qPCR analysis was performed on a StepOnePlus instrument using fast SYBR Green Master Mix (ThermoFisher Scientific). The primer sequences for Pip5k1a, Pip5k1b, Pip5k1c, and Hprt genes were described by us earlier (20); those for Tnfa were according to (58). The following primers were used for Pi4k2a: 5′-GCGAAAGTCCCGTTCTCTCA-3′ and 5′-CGAAGCCAGGATCTCTCTTGAA-3′; for Pi4k2b: 5′-CAGGAACACAGACAGGGGAAA-3′ and 5′-TATGCTCGCCATTCATCAGG-3′; and for Ccl5 (encoding CCL5/RANTES): 5′-GCTCCAATCTTGCAGTCGTGT-3′ and 5′-CCATTTTCCCAGGACCGAGT-3′. Relative mRNA levels for the investigated genes were calculated using the ΔΔCT method using Hprt gene expression as an internal reference for all determinations.
Data Analysis
The significance of observed differences, except for proteomic data analysis described above, was calculated using Student's t test or by two-way or one-way analysis of variance (ANOVA) with Tukey's post hoc test when indicated. ANOVA was performed with the aov function from the stats package in the R software environment. Significance levels are provided in figure legends.
RESULTS
LPS-Induced Changes in Protein Acylation Revealed with Click Chemistry in RAW264 Cells
Taking into account that S-acylation (S-palmitoylation) of proteins controls their involvement in the signaling cascades of several plasma membrane receptors (28), we examined whether protein acylation is affected during stimulation of RAW264 macrophage-like cells with LPS. To identify fatty-acylated proteins, cells were metabolically labeled with 17ODYA, an analogue of palmitic acid functionalized with an alkyne group (59–61), and the labeled proteins were tagged with biotin-azide, captured on streptavidin-coupled beads, and analyzed by mass spectrometry (Fig. 1A). In all experiments, unstimulated cells and cells stimulated with 100 ng/ml LPS for 60 min were analyzed. Additionally, to control the reliability of the identification of fatty-acylated proteins with 17ODYA, both unstimulated and stimulated cells were in parallel incubated with the DMSO carrier without 17ODYA. Three independent experiments were performed, eventually giving three lists of 17ODYA-labeled proteins in unstimulated cells, three lists in LPS-stimulated cells, and corresponding lists of proteins found in DMSO-treated unstimulated and LPS-stimulated cells (two sets combining three lists each). After removal of contaminants and redundant proteins, 950 proteins remained; they are presented in supplemental Table 2 together with their spectral counts in each sample.
Fig. 1.
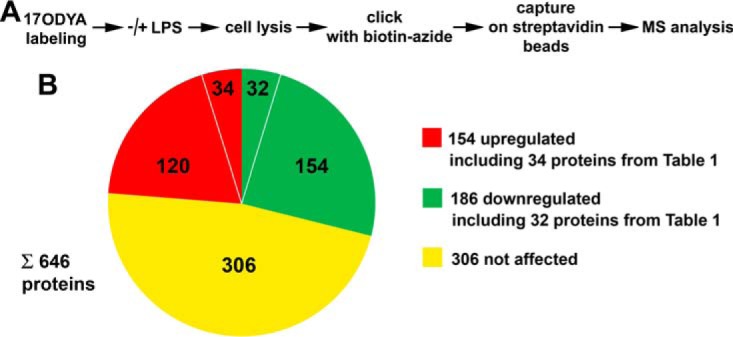
Stimulation of RAW264 cells with LPS up-regulates and downregulates fatty-acylated proteins identified by metabolic labeling with 17ODYA and proteomic analysis. (A) Scheme of experimental procedure comprising metabolic labeling of cells with 50 μm 17ODYA or exposed to 0.05% DMSO carrier for 4 h, stimulation with 100 ng/ml LPS for 60 min, cell lysis, click chemistry reaction with biotin-azide, and capture of tagged proteins on streptavidin-coupled beads followed by mass spectrometry. Three experiments were performed. The abundance of proteins was estimated using a semiquantitative spectral counting approach described under “Experimental Procedures.” A total of 646 fatty-acylated proteins that were eventually identified with high and medium confidence are listed in supplemental Table 3. (B) Acylated proteins up- or downregulated or not affected by LPS treatment of cells revealed by calculating the RL ratio and listed in supplemental Table 4. Up-regulated and downregulated proteins include those (34 and 32 proteins listed in Table I) selected basing on the number of experiments they were detected in.
Subsequent statistical analysis aimed at: (i) establishing which fatty-acylated proteins were identified with high and medium confidence by 17ODYA labeling and (ii) revealing which of those proteins were affected by the LPS stimulation. For this purpose, spectral counts were summed up over three samples of each experimental variant yielding data sets A and B for unstimulated cells labeled with 17ODYA and treated with DMSO, respectively, and corresponding data sets C and D for LPS-stimulated cells. Subsequently, the ratios of the A/B pair and the C/D pair were calculated for each protein and expressed as ln. We assumed that a given protein was tentatively identified as fatty-acylated by 17ODYA labeling if the ln value (ln(A/B) and/or ln(C/D)) for that protein was ≥ 2.5, corresponding to about 12-fold excess of the sum of spectral counts in the 17ODYA-labeled sample over that in the corresponding DMSO-treated sample. Furthermore, the statistical significance of the difference between spectral counts summed for each protein in A and B as well as in C and D had to reach p ≤ 0.275. A total of 646 fatty-acylated proteins fulfilling those criteria were identified in unstimulated and/or stimulated cells (of which 167, common to these two sets, were with high confidence, p ≤ 0.05). These proteins are listed in supplemental Table 3.
Of the 646 acylated proteins found here, 149 met the criteria of confident identification in unstimulated cells only (supplemental Table 3, green and blue rows) and 78 exclusively following LPS stimulation (supplemental Table 3, orange and red rows). Thus, these proteins can be considered to be downregulated and up-regulated, respectively, in cells exposed to LPS. We reasoned, however, that the definition of LPS-affected acylated proteins should also take into account the different intensity of the labeling of a given protein with 17ODYA in unstimulated versus stimulated cells. Therefore, for all the 646 acylated proteins, the parameter named RL was calculated. RL was equal to the ratio of the value reflecting protein labeling in LPS-stimulated cells (C/D ratio) to that in unstimulated cells (A/B ratio). Arbitrary thresholds were set to consider a protein as up-regulated (RL ≥ 1.5) or downregulated (RL ≤ 0.5) in LPS-stimulated cells. Using these criteria, 154 acylated proteins were up-regulated (Fig. 1B; supplemental Table 4, red rows) and, as could be expected, this number included the vast majority (73 of 78) of the proteins identified with high or medium confidence in stimulated cells only. On the other hand, 186 proteins were downregulated in LPS-stimulated cells (Fig. 1B; supplemental Table 4, green rows), including 138 of the 149 proteins identified with high or medium confidence exclusively in unstimulated cell. Finally, 306 acylated proteins were unaffected by LPS (Fig. 1B; supplemental Table 4, yellow rows).
To characterize the fatty acylated proteins, especially those affected by LPS, their putative cellular functions were inferred using IPA. Taking into account all 646 proteins labeled with 17ODYA, IPA generated 25 networks of proteins/molecules that were involved in various biological processes. Those included amino acid and lipid metabolism, transcription and translation, cellular assembly and organization, cell-to-cell signaling, infectious diseases, and others (supplemental Table 5). Several such networks were composed nearly exclusively of the fatty-acylated proteins identified in this study, which suggests that acylation is a modification widely utilized to regulate the biological activity of interrelated proteins. This was the case for network 2 of proteins involved in RNA posttranscriptional modification, infectious diseases, and dermatological diseases. Moreover, more than half of the fatty acylated proteins from this network were affected by stimulation of cells with LPS (supplemental Fig. 1).
To strengthen the identification of LPS-regulated fatty acylated proteins, from the 154 up-regulated and 186 downregulated proteins, we selected those found in two or three experiments in stimulated cells and absent or found only once in unstimulated cells and vice versa. In this manner, a short list of 34 proteins up-regulated and of 32 downregulated proteins in LPS-stimulated cells was obtained (Table I, Fig. 1B). Next, we validated the identification of protein fatty acylation by mass spectrometry using immunoblotting. For this purpose, 17ODYA-labeled proteins present in lysates of unstimulated or LPS-stimulated cells were tagged with biotin and captured on streptavidin-coupled beads, as for the mass spectrometry analysis. These proteins were subsequently eluted from the beads, and the eluates analyzed by immunoblotting for the presence of selected proteins from Table I (Fig. 2A). As shown in Fig. 2B, the amount of fatty acylated tyrosine kinase Jak1 recovered from LPS-stimulated cells was strongly reduced in comparison to that in unstimulated cells. In contrast, the amounts of fatty acylated flotillin-1, transmembrane TNFα precursor, and eIF5A2 were markedly higher in LPS-stimulated than unstimulated cells. For comparison, no significant differences in the level of fatty-acylated PAG adaptor protein and Lyn tyrosine kinase were detected between unstimulated and stimulated cells (Fig. 2B). These results are in full agreement with the above results of the proteomic mass spectrometry analysis (compare Fig. 2B with Table I and supplemental Table 3). No proteins were recovered from lysates of cells exposed to DMSO instead of 17ODYA, confirming the specificity of the method (Fig. 2B).
Table I. Acylated proteins up-regulated and downregulated in LPS-stimulated cell selected depending on frequency of their identification.
Cells were labeled with 50 μm 17ODYA or exposed to 0.05% DMSO carrier for 4 h then stimulated with 100 ng/ml LPS for 60 min or left unstimulated and labeled proteins were captured on streptavidin-coupled beads. Subsequently, mass spectrometry was performed, as described in Fig. 1. Three sets of experiments were performed, followed by statistical analysis of the data based on spectral counts. Fatty-acylated proteins identified with high and medium confidence are listed in supplemental Table 3, and those considered to be up-regulated or downregulated in LPS-stimulated cells based on the RL value are listed in supplemental Table 4. The proteins shown have been selected from those in supplemental Table 4, taking into account the number of experiments in which they were identified. Up-regulated proteins: acylated proteins identified in two or three experiments in stimulated cells and absent or found only once in unstimulated cells. Downregulated proteins: acylated proteins identified in two or three experiments in unstimulated cells and absent or found only once in stimulated cells. Numbers in columns A, B, C, D indicate in how many experiments represented by those symbols (see text) a given protein was identified.* Protein with GPI anchor or an enzyme involved in GPI anchor attachment that can incorporate 17ODYA into the GPI anchor.
| UP-REGULATED |
DOWNREGULATED |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UniProt accession number | UniProt protein name | No. in A | No. in B | No. in C | No. in D | RL | UniProt accession number | UniProt protein name | No. in A | No. in B | No. in C | No. in D | RL |
| P06804 | Tumor necrosis factor | 0 | 0 | 3 | 0 | 119.000 | O35381 | Acidic leucine-rich nuclear phosphoprotein 32 family member A | 2 | 0 | 1 | 0 | 0.500 |
| Q8BGY2 | Eukaryotic translation initiation factor 5A-2 | 0 | 0 | 2 | 0 | 84.000 | Q9EST5 | Acidic leucine-rich nuclear phosphoprotein 32 family member B | 2 | 0 | 1 | 0 | 0.500 |
| *O08603 | Retinoic acid early-inducible protein 1-beta | 0 | 0 | 2 | 0 | 46.000 | Q80YP0, P30285 | Cyclin-dependent kinase | 2 | 0 | 1 | 0 | 0.500 |
| *Q9CXY9 | GPI-anchor transamidase | 0 | 0 | 3 | 0 | 37.000 | P97377 | Cyclin-dependent kinase 2 | 2 | 0 | 1 | 0 | 0.500 |
| Q9R1J0 | Sterol-4-alpha-carboxylate 3-dehydrogenase, decarboxylating | 0 | 0 | 2 | 0 | 33.000 | Q69ZN6 | N-acetylglucosamine-1-phosphotransferase subunits alpha/beta | 3 | 0 | 1 | 0 | 0.500 |
| O88693 | Ceramide glucosyltransferase | 0 | 0 | 2 | 0 | 28.000 | Q8VC90 | Probable palmitoyltransferase ZDHHC12 | 2 | 0 | 1 | 0 | 0.500 |
| Q8R480 | Nuclear pore complex protein Nup85 | 0 | 0 | 3 | 0 | 23.000 | Q8CIG8 | Protein arginine N-methyltransferase 5 | 2 | 0 | 1 | 0 | 0.500 |
| Q8K4Z5 | Splicing factor 3A subunit 1 | 0 | 0 | 2 | 0 | 15.000 | Q8CGC6 | RNA-binding protein 28 | 2 | 0 | 1 | 0 | 0.500 |
| Q8BHN3 | Neutral alpha-glucosidase AB | 1 | 1 | 3 | 0 | 8.941 | Q571J5 | Zinc finger protein 354C | 2 | 0 | 1 | 0 | 0.500 |
| Q91W98 | Solute carrier family 15 member 4 | 1 | 0 | 3 | 0 | 8.428 | Q8C827 | Zinc finger protein 62 | 2 | 0 | 1 | 0 | 0.500 |
| P15920 | V-type proton ATPase 116 kDa subunit a isoform 2 | 1 | 0 | 3 | 0 | 6.500 | E9Q9A9 | 2′-5′-oligoadenylate synthase 2 | 2 | 0 | 1 | 0 | 0.400 |
| Q91WU6 | Palmitoyltransferase ZDHHC7 | 1 | 0 | 3 | 0 | 6.333 | P49710 | Hematopoietic lineage cell-specific protein | 2 | 0 | 1 | 0 | 0.372 |
| P0DOV2 | Interferon-activable protein 204 | 1 | 0 | 3 | 0 | 6.000 | Q6ZQL4 | WD repeat-containing protein 43 | 2 | 0 | 1 | 0 | 0.353 |
| Q8K1M6 | Dynamin-1-like protein | 1 | 0 | 2 | 0 | 5.400 | Q9QUJ7 | Long-chain-fatty-acid–CoA ligase 4 | 3 | 0 | 1 | 0 | 0.333 |
| P59268 | Palmitoyltransferase ZDHHC9 | 1 | 0 | 3 | 0 | 5.200 | O55234 | Proteasome subunit beta type-5 | 2 | 0 | 1 | 0 | 0.326 |
| P48962 | ADP/ATP translocase 1 | 1 | 0 | 3 | 0 | 5.154 | Q8R016 | Bleomycin hydrolase | 2 | 0 | 1 | 0 | 0.320 |
| P00493, Q64531 | Hypoxanthine-guanine phosphoribosyltransferase | 1 | 0 | 3 | 0 | 3.846 | P60122 | RuvB-like 1 | 2 | 0 | 1 | 0 | 0.320 |
| Q9R1C6 | Diacylglycerol kinase epsilon | 1 | 0 | 2 | 0 | 3.167 | Q3UEB3 | Poly(U)-binding-splicing factor PUF60 | 2 | 0 | 1 | 0 | 0.318 |
| Q8VEE4 | Replication protein A 70 kDa DNA-binding subunit | 1 | 0 | 2 | 0 | 3.167 | A2AVZ9 | Solute carrier family 43 member 3 | 2 | 0 | 1 | 0 | 0.318 |
| Q9CPY7 | Cytosol aminopeptidase | 1 | 0 | 2 | 0 | 3.143 | Q8BJS4 | SUN domain-containing protein 2 | 2 | 0 | 1 | 0 | 0.313 |
| O08917 | Flotillin-1 | 1 | 0 | 2 | 0 | 3.111 | Q921H8, Q8VCH0 | 3-ketoacyl-CoA thiolase | 2 | 0 | 1 | 0 | 0.250 |
| Q9Z0H4 | CUGBP Elav-like family member2 | 1 | 0 | 2 | 0 | 3.000 | Q8VCM8 | Nicalin | 3 | 0 | 1 | 0 | 0.250 |
| Q8BH24 | Transmembrane 9 superfamily member 4 | 1 | 0 | 2 | 0 | 3.000 | Q5SSI6 | U3 small nucleolar RNA-associated protein 18 homolog | 2 | 0 | 1 | 0 | 0.250 |
| Q8VEC4 | Calcium homeostasis modulator protein 2 | 1 | 0 | 3 | 0 | 2.520 | Q9JJ00 | Phospholipid scramblase 1 | 3 | 0 | 1 | 0 | 0.245 |
| P68033, P68134 | Actin | 1 | 0 | 3 | 0 | 2.364 | Q8CIF6 | SID1 transmembrane family member 2 | 3 | 0 | 1 | 0 | 0.235 |
| P22892 | AP-1 complex subunit gamma-1 | 1 | 0 | 2 | 0 | 2.111 | P62317 | Small nuclear ribonucleoprotein Sm D2 | 2 | 0 | 1 | 0 | 0.194 |
| Q9JL26 | Formin-like protein 1 | 1 | 0 | 2 | 0 | 2.000 | Q99LG1 | Transmembrane protein 51 | 3 | 0 | 1 | 0 | 0.185 |
| D3Z7P3 | Glutaminase kidney isoform, mitochondrial | 1 | 0 | 2 | 0 | 2.000 | Q5SYD0 | Unconventional myosin-Id | 2 | 0 | 1 | 0 | 0.182 |
| Q9CPP6 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 5 | 1 | 0 | 2 | 0 | 2.000 | P52332 | Tyrosine-protein kinase JAK1 | 2 | 0 | 1 | 0 | 0.150 |
| Q8VEK2 | Rhomboid domain-containing protein 2 | 1 | 0 | 3 | 0 | 2.000 | Q6T707 | Acyl-CoA desaturase 4 | 2 | 0 | 1 | 1 | 0.071 |
| Q924S7 | Sprouty-related, EVH1 domain-containing protein 2 | 1 | 0 | 2 | 0 | 2.000 | Q922F4 | Tubulin beta-6 chain | 2 | 0 | 0 | 0 | 0.040 |
| P12382 | ATP-dependent 6-phosphofructokinase, liver type | 1 | 0 | 2 | 0 | 1.500 | O35153 | BET1-like protein | 2 | 0 | 0 | 0 | 0.009 |
| Q8CBQ5 | Phosphatidylinositol 4-kinase type 2-beta | 1 | 0 | 2 | 0 | 1.500 | |||||||
| P54823 | Probable ATP-dependent RNA helicase DDX6 | 1 | 0 | 2 | 0 | 1.500 | |||||||
Fig. 2.

Immunoblotting analysis confirms changes of fatty acylation of proteins induced by stimulation of RAW264 cells with LPS. BPA affects LPS-induced signaling. (A) Scheme of experimental procedure mirroring that in Fig. 1 but finalized by elution of 17ODYA-labeled and biotin-tagged proteins from streptavidin beads and their separation by SDS-PAGE. In a series of experiments, cells were pretreated with 250 μm BPA (1h, 37 °C), labeled with 17ODYA and stimulated with LPS in the presence of the drug. (B, C) Immunoblotting analysis of indicated proteins in cells unstimulated and stimulated with 100 ng/ml LPS for 60 min (B) and cells untreated or treated with BPA and stimulated with LPS (C). For comparison, 2% or 0.5% (eIF5A2 detection) of total cell lysate was also run. Relative protein levels were quantified by densitometry and are presented as mean from three experiments. Positions of transmembrane TNFα precursor are indicated by arrows. Molecular weight markers are shown on the left. (D) Production of TNFα and CCL5/RANTES by cells exposed to 250 μm BPA. Data shown are mean ± s.d. from four experiments. ***, Significantly different at p < 0.001 from cells stimulated with LPS.
We then used immunoblotting of affinity-enriched 17ODYA-labeled proteins to examine whether the LPS-affected fatty acylation of proteins is sensitive to BPA, a drug with pleiotropic effects, including inhibition of protein palmitoylation (62). BPA variably affected the labeling of proteins with 17ODYA. It markedly reduced it for Jak1, flotillin-1, and TNFα but not for PAG or Lyn (Fig. 2C). Among the proteins tested, the amount of TNFα precursor detected in input cell lysates was also reduced by BPA (Fig. 2C). Furthermore, in the presence of BPA, the LPS-induced release of TNFα and CCL5/RANTES cytokines from cells was inhibited by about 53 and 61%, respectively (Fig. 2D). These data suggest that protein palmitoylation can control the LPS-induced proinflammatory signaling pathways leading to cytokine production and release.
From this point of view, it was of major interest that PI4KIIβ was one of the fatty acylated proteins up-regulated in LPS-stimulated cells (Table I). PI4KIIβ is one of the four kinases phosphorylating phosphatidylinositol to PI(4)P (63). In turn, PI(4)P is a substrate for the production of PI(4,5)P2 required for maximal release of proinflammatory cytokines (20, 21) and possibly also for motility of LPS-stimulated cells (64). The only other S-palmitoylated kinase involved in PI(4)P generation, PI4KIIα, was labeled relatively strongly with 17ODYA in both unstimulated and stimulated cells (supplemental Tables 2–4). S-palmitoylation is required for the activity of both PI4KIIα and PI4KIIβ (44, 65, 66). Thus, the proteomic data suggested that LPS stimulates the activity of PI4KIIβ. Because of the importance of phosphatidylinositols in LPS-induced signaling, we analyzed in detail the palmitoylation of PI4KIIβ and PI4KIIα in the course of cell stimulation with LPS and its role in triggered signaling.
Palmitoylation of PI4KIIα and PI4KIIβ in LPS-Stimulated Cells
In order to follow their palmitoylation, Myc-tagged PI4KIIα and PI4KIIβ were expressed separately in RAW264 cells that were labeled metabolically with 17ODYA and stimulated with 100 ng/ml LPS for up to 1 h. The palmitoylation status of immunoprecipitated kinases was analyzed with the use of azide-functionalized fluorescent IRDye 800CW (Fig. 3A). Relatively high labeling of PI4KIIα was detected in resting cells, and its level did not change markedly upon stimulation of the cells with LPS (Figs. 3B and 3C). The labeling was sensitive to 2.5% hydroxylamine, abrogated by deletion of the palmitoylation site of PI4KIIα, and also absent when 17ODYA was omitted (Figs. 3B and 3C).
Fig. 3.

Stimulation of RAW264 cells with LPS induces S-palmitoylation of PI4KIIβ. (A) Scheme of experimental procedure. RAW264 cells were transfected with plasmids encoding Myc-tagged PI4KIIα or PI4KIIβ kinase wild type (wt) or their deletion mutants lacking the S-palmitoylation site (mut). After 24 h, cells were treated or not with 250 μm BPA (1 h, 37 °C), subjected to metabolic labeling with 50 μm 17ODYA or exposed to 0.05% DMSO carrier as control (- 17ODYA) for 4 h, and stimulated with 100 ng/ml LPS for 15, 30 and 60 min or left unstimulated. After lysis, the kinases were immunoprecipitated with anti-Myc rabbit IgG-agarose and subjected to click chemistry reaction with IRDye 800CW-azide. (B) 17ODYA labeling of PI4KIIα and (D) PI4KIIβ. (B, D, upper panels) In-gel fluorescence showing labeling with 17ODYA followed by IRDye-azide. (B, D, lower panels) Efficiency of immunoprecipitation of kinases determined by blotting with anti-Myc antibody. HXA, samples treated with 2.5% hydroxylamine prior to SDS-PAGE. Molecular weight markers are shown on the right. (C, E) Labeling of PI4KIIα (C) and PI4KIIβ (E) normalized against content of respective Myc-tagged kinase and expressed relative to that in unstimulated cells or cells stimulated with LPS for 60 min (for BPA-treated cells). Data shown are mean ± s.d. from three experiments. *, ***, Significantly different at p < 0.05 and p < 0.001 from unstimulated cells expressing wild-type kinases. ##, Significantly different from LPS-stimulated cells at p < 0.01.
In contrast to PI4KIIα, palmitoylation of PI4KIIβ was clearly induced by LPS. Incorporation of 17ODYA into PI4KIIβ increased about twofold after 30 min of cell stimulation with LPS and remained at that level during the next 30 min of LPS action (Figs. 3D and 3E). As for PI4KIIα, the labeling of PI4KIIβ was abolished by deletion of the S-palmitoylation site of the kinase or by an exposure of the 17ODYA-labeled wild-type kinase to 2.5% hydroxylamine and was absent in DMSO-treated cells (Figs. 3D and 3E). As S-palmitoylation closely correlates with the kinase activity (44), these data indicate that PI4KIIβ is activated during stimulation of cells with LPS. We also examined whether BPA affected the S-palmitoylation of PI4KIIβ and PI4KIIα in LPS-stimulated cells. This was indeed the case: The S-palmitoylation of PI4KIIα was inhibited by nearly 50% and of PI4KIIβ by about 70% by the drug (Figs. 3B–3E). Taken together, the data confirm constitutive S-palmitoylation, hence activity, of endogenous PI4KIIα, and LPS-induced S-palmitoylation/activation of PI4KIIβ revealed in RAW264 cells by mass spectrometry.
Participation of PI4KIIα and PI4KIIβ in LPS-Induced Cytokine Expression
Having established the profile of S-palmitoylation of PI4KIIα and PI4KIIβ in LPS-stimulated RAW264 cells, we analyzed their contribution to the mechanisms controlling production of cytokines by the cells. An estimation of the relative abundance of the two kinases with qPCR revealed that the level of mRNA encoding PI4KIIα in unstimulated RAW264 cells was about seven times higher than that of PI4KIIβ (Fig. 4A). Following stimulation with LPS, the expression of PI4KIIα nearly doubled, but only after 4 h, while the expression of PI4KIIβ remained fairly constant (Figs. 4B and 4C). Silencing of PI4KIIα or PI4KIIβ significantly reduced LPS-induced production of TNFα and CCL5/RANTES, the latter produced exclusively in the TRIF-dependent pathway of TLR4 (67). The inhibition of cytokine production was correlated with a significant reduction of TNFα and CCL5/RANTES mRNA levels (Figs. 4D–4G). The production of CCL5/RANTES was more sensitive to the depletion of either kinase, and it was reduced by 47% after PI4KIIβ and by 70% after PI4KIIα silencing in comparison with a reduction by only about 27% of the TNFα release in response to the depletion of one or the other kinase (Figs. 4E and 4G).
Fig. 4.

PI4KIIα and PI4KIIβ contribute to cytokine production in LPS-stimulated RAW264 cells. (A) Relative expression of genes encoding PI4KIIα and PI4KIIβ in unstimulated cells. (B, C) Expression of PI4KIIα (B) and PI4KIIβ (C) in cells prior to and after stimulation of cells with 100 ng/ml LPS for 1–4 h. (D–G) Expression and production of TNFα (D, E) and CCL5/RANTES (F, G) in cells transfected with scrambled siRNA (scr) or siRNA targeting PI4KIIα or PI4KIIβ. NS, unstimulated cells, LPS, cells stimulated with 100 ng/ml LPS for 4 h. Expression of genes was analyzed with qPCR (D, F) while concentration of TNFα (E) and CCL5/RANTES (G) in culture supernatants was measured with ELISA. (H, I) Expression of PI4KIIα (H) and PI4KIIβ (I) in cells exposed to scrambled siRNA or siRNA targeting PI4KIIα or PI4KIIβ. (J) Expression of PIP5KIα, Iβ and Iγ in cells exposed to scrambled siRNA or siRNA targeting PI4KIIα or PI4KIIβ. Data are mean ± s.d. from three (A–D, F, J) or five (H, I) experiments run in duplicates, or of three experiments run in triplicates (E, G). ###, Significantly different from unstimulated cells at p < 0.001. ***, Significantly different at p < 0.001 from cells transfected with scrambled siRNA and left unstimulated (H, I) or stimulated with LPS (D-G).
We confirmed that transfection of cells with siRNA targeting PI4KIIα or PI4KIIβ led to a profound depletion of the respective kinase at the mRNA level (Figs. 4H and 4I). On the other hand, the level of mRNA encoding PI4KIIα was unaffected by the silencing of PI4KIIβ and vice versa (Figs. 4H and 4I). It has been reported that cells can compensate for a loss of PI4KIIIα by increasing the level of PIP5KIγ and PIP5KIβ, both kinases phosphorylating PI(4)P to PI(4,5)P2 (68). However, this was not the case for PI4KII silencing in RAW264 cells, as depletion of either PI4KIIα or PI4KIIβ did not affect the mRNA level of any PIP5KI isoform, including the most abundant PIP5KIα (Fig. 4J).
Taking into account the LPS-induced S-palmitoylation/activation of PI4KIIβ and its role in LPS-induced cytokine production observed in RAW264 cells, we examined its contribution to this event in another macrophage-like line, J774. In unstimulated J774 cells, the PI4KIIβ mRNA level was about fivefold lower than that of PI4KIIα, similarly as in RAW264 cells. Effective silencing of PI4KIIβ was achieved using RNA interference, and it did not affect expression of PI4KIIα (Figs. 5A–5C). The depletion of J774 cells of PI4KIIβ substantially (by over 55%) inhibited the expression and production of CCL5/RANTES following cell stimulation with LPS but had no significant effect on TNFα expression or production (Figs. 5D–5G). Taking into account the relatively strong inhibitory effect of PI4KIIβ depletion on CCL5/RANTES production found in RAW264 cells, it can be suggested that activity of PI4KIIβ is crucial for the TRIF-dependent production of cytokines in LPS-stimulated cells.
Fig. 5.

PI4KIIβ controls production of CCL5/RANTES in J774 cells. (A) Expression of genes encoding PI4KIIα and PI4KIIβ in unstimulated cells. (B, C) Expression of PI4KIIα (B) and PI4KIIβ (C) in cells exposed to scrambled siRNA or siRNA targeting PI4KIIβ. (D–G) Expression and production of TNFα (D, E) and CCL5/RANTES (F, G) in cells transfected with scrambled siRNA (scr) or siRNA targeting PI4KIIβ. Expression of genes was analyzed with qPCR (D, F) while concentration of TNFα (E) and CCL5/RANTES (G) in culture supernatants was measured with ELISA. NS, unstimulated cells, LPS, cells stimulated with 100 ng/ml LPS for 4 h. Data are mean ± s.d. from three experiments run in duplicates (A–D, F) or in triplicates (E, G). ***, Significantly different at p < 0.001 from cells transfected with scrambled siRNA and left unstimulated (C) or stimulated with LPS (F, G).
Effect of S-Palmitoylation on PI4KIIα and PI4KIIβ Involvement in LPS-Induced Signaling
In order to examine whether the engagement of PI4KIIα and PI4KIIβ in LPS-induced signaling depends on their S-palmitoylation, we analyzed the influence of overexpression of wild-type kinases and their mutant forms lacking the palmitoylation sites on the LPS-induced activation of NFκB and IRF(s). Among those transcription factors, IRF3/7 are activated by LPS exclusively in the TRIF-dependent signaling pathway of TLR4 (8). Thus, PI4KIIα or PI4KIIβ was co-expressed in HEK293 cells with CD14 and TLR4/MD2, which sensitize the cell to LPS, as revealed by expression of firefly luciferase controlled by promoters containing NFκB or ISRE transcription factor-binding sites (Fig. 6). Immunoblotting analysis showed that the cellular level of ectopically expressed wild-type PI4KIIα or PI4KIIβ was in these transfection conditions lower than that of respective nonpalmitoylated form, and this disproportion was stronger for PI4KIIβ (Figs. 6A and 6E). No differences in the level of CD14 or TLR4 proteins were detected between the transfectants tested (Figs. 6A and 6E). Neither wild-type nor nonpalmitoylated PI4KIIα or PI4KIIβ affected significantly the LPS-induced NFκB-luciferase reporter activity (Figs. 6B and 6F). Only in cells producing relatively high amounts of the mutant nonpalmitoylated PI4KIIβ was a moderate inhibition of the activity found (Fig. 6F). In contrast, the ISRE-dependent induction of luciferase was increased by wild-type PI4KIIα or PI4KIIβ depending on their overproduction level, reaching 2.0-fold and 1.4- to 1.6-fold, respectively, in comparison to cells nontransfected with either kinase. The nonpalmitoylated forms of PI4KIIα and PI4KIIβ did not have the ability to enhance the ISRE-driven induction of luciferase (Figs. 6C and 6G). These data indicate that S-palmitoylation, hence activity of PI4KIIα and PI4KIIβ, affects especially strongly the TRIF-dependent signaling pathway of TLR4 leading to IRF(s) activation.
Fig. 6.

PI4KIIα and PI4KIIβ up-regulate activity of IRF(s) in palmitoylation-dependent manner. Palmitoylation affects subcellular distribution of kinases. HEK293 cells were co-transfected with plasmids encoding CD14 and FLAG-tagged TLR4/MD2 and indicated amounts of plasmids encoding Myc-tagged PI4KIIα (A–D) or PI4KIIβ (E–H) kinases wild type (wt) or their deletion mutants lacking the S-palmitoylation site (mut). In control cells, PI4KII-bearing plasmids were omitted (“0”). In (A–C) and (E–G) the DNA mixture used for cell transfection also contained plasmid encoding Renilla luciferase and firefly luciferase controlled by either NFκB-dependent or ISRE-dependent promoter. Cells were either left unstimulated (NS) or were stimulated with 100 ng/ml LPS for 24 h. (A, E) Immunoblotting analysis of ectopically expressed PI4KIIα, PI4KIIβ, CD14, and TLR4 using anti-Myc, anti-CD14 or anti-FLAG antibodies. Equal protein loading was verified by probing membranes with anti-actin antibody. Numbers below upper panels indicate relative mean level of PI4KIIα or PI4KIIβ quantified by densitometry in four experiments. (B, F) NFκB activity and (C, G) ISRE activity assessed by corresponding firefly luciferase activity measured in cell lysates. The firefly luciferase activity was normalized against constitutive Renilla luciferase activity in the same sample. The relative luciferase activity (RLA) is expressed as a fold increase over the RLA value found in LPS-stimulated cells lacking the ectopically expressed PI4KIIα or PI4KIIβ (“0”). Data are mean ± s.d. from four experiments. &, &&&, Significant differences at p < 0.05 and p < 0.001 between LPS-stimulated samples established with two-way ANOVA with interactions. *, **, ***, Significantly different at p < 0.05, p < 0.01, and p < 0.001 from RLA in LPS-stimulated cells lacking the ectopically expressed PI4KIIα or PI4KIIβ (“0”), as estimated using 1-way ANOVA with Tukey's post hoc test. (D, H) Distribution of PI4KIIα (D) and PI4KIIβ (H) in density gradient fractions. HEK293 transfectants were lysed in 0.5% Brij 98 and the lysates were fractionated over 0–40% OptiPrep density gradient. Seven fractions with increasing density were collected and analyzed for the presence of indicated proteins. Molecular weight markers are shown on the right.
Taking into account the observed palmitoylation-dependent influence of both PI4KIIα and PI4KIIβ on LPS-induced signaling, we examined whether S-palmitoylation affects their subcellular distribution. For this purpose, HEK293 transfectants were solubilized in Brij 98 and subjected to OptiPrep density gradient centrifugation to separate the buoyant detergent-resistant membrane fraction expected to accommodate PI4KIIα (65). About 20.3 ± 7% of wild-type PI4KIIα and 17.6 ± 5% of PI4KIIβ (n = 3) floated to fractions 1–3 of the gradient. However, they only partially overlapped with CD14, a typical raft protein, which was recovered nearly exclusively in fraction 1 of the lowest density (Figs. 6D and 6H). The major fraction of both kinases was found in the high density fractions 5–7 of the gradient that contained detergent-soluble proteins, including TLR4 receptor (Figs. 6D and 6H). Deletion of the palmitoylation site substantially (by over 70%) reduced the amounts of PI4KIIα and PI4KIIβ found in the low-density fractions of the gradient (Figs. 6D and 6H). On the other hand, the distribution of PI4KIIα and PI4KIIβ in the gradient was not affected by stimulation of cells with LPS (not shown).
Lipidation of CD14
Among the proteins labeled with 17ODYA in RAW264 cells, we detected CD14 (supplemental Tables 2–4), which was of interest in view of its essential role in PI(4,5)P2 generation in LPS-stimulated cells (20, 21). CD14 bears a GPI moiety that incorporates palmitic acid, as described, e.g. for the GPI anchor of placental alkaline phosphatase (69). To check whether the labeling with 17ODYA reflects true acylation or whether the 17ODYA simply incorporates into GPI, we removed the C-terminal 21 amino acids of CD14 containing the GPI-anchor attachment signal, devoid of any cysteine residue (70, 71), and fused the truncated protein with a 49-amino acid long VSVG protein fragment containing its transmembrane and cytoplasmic parts. This VSVG fragment includes a juxtamembrane cysteine residue (amino acid 489 of full-length VSVG) that undergoes S-palmitoylation (72). We also created a CD14-VSVG fusion protein in which the cysteine 489 residue was mutated to alanine (Fig. 7). We overexpressed wild-type CD14 (CD14 WT), CD14-VSVG, or CD14-VSVGmut in HEK293 cells lacking endogenous expression of CD14, labeled the cells with 17ODYA, performed the click reaction with biotin-azide, and analyzed the amounts of CD14 captured on streptavidin-coupled beads by immunoblotting for CD14 (Fig. 7A), in an approach analogous to that used to analyze palmitoylation of endogenous proteins in RAW264 cells. This analysis confirmed incorporation of 17ODYA into CD14 WT; however, the labeled protein constituted a small fraction of the whole pool of CD14 (Fig. 7B, left panel). As CD14 contains a large hydrophobic pocket located in the N-terminal part that binds LPS, we reasoned that 17ODYA could not only be incorporated into the GPI anchor of the protein but could also be trapped in the pocket. To examine this possibility, samples after the click reaction were subjected to delipidation according to Folch et al. (54). Following the delipidation, the labeling of CD14 with 17ODYA was still detectable at a similar level, pointing to its covalent attachment to the protein (Fig. 7B, left panel). In contrast, no 17ODYA labeling of CD14-VSVGmut was detected (Fig. 7B, right panel) while high amounts of CD14-VSVG fusion protein were recovered via the biotin-streptavidin interaction, reflecting a high level of its fatty acylation regardless of delipidation (Fig. 7B, middle panel). In all the 17ODYA-treated samples, flotillin-2 was detected serving as a positive control for the recovery of labeled proteins (Fig. 7B). Taken together, the data indicate that during metabolic labeling of cells with 17ODYA it incorporates into the GPI anchor of CD14. By analogy, for the seven more GPI-anchored proteins and GPI-anchor transamidase found among the 17ODYA-labeled proteins (supplemental Table 3, Table I) incorporation of 17ODYA into their GPI moieties rather than direct S-palmitoylation seems plausible.
Fig. 7.

Labeling of CD14 with 17ODYA is confined to its GPI anchor. (A) Scheme of experimental procedure. CD14 wild type (WT), CD14-VSVG, or CD14-VSVGmut were expressed in HEK293 cells. Cells were labeled with 50 μm 17ODYA or exposed to 0.05% DMSO carrier for 4 h, lysed, and cell lysates were subjected to click chemistry reaction with biotin-azide. Biotin-tagged proteins were captured on streptavidin-coupled beads, and, after elution, separated by SDS-PAGE. A parallel set of samples was subjected to delipidation after the click chemistry reaction (DeLip). (B) The presence of CD14 or flotillin-2 in eluates from streptavidin beads and in lysates (10% of total cell lysate). As CD14 WT did not contain the HA tag, all CD14 variants were detected with anti-CD14 antibody. Molecular weight markers are shown on the right. (C) Structure of CD14 constructs. TM, CP, transmembrane and cytoplasmic fragments of VSVG, respectively. HA, HA tag. Results of one representative experiment of three are shown.
17ODYA as an Indicator of Protein Palmitoylation
In view of the ability of 17ODYA to incorporate into the GPI anchor of proteins, its utility as an indicator of protein palmiotylation in our system had to be verified. For this, unstimulated RAW264 cells were labeled with 17ODYA alone, with 17ODYA in the presence of palmitic acid as a competitor, or with 17ODYA and BPA. Alternatively, after labeling, samples were treated with hydroxylamine cleaving thioester linkages. Global profiles of 17ODYA-labeled proteins tagged with IRDye 800CW-azide proteins were then analyzed by in-gel fluorescence (Fig. 8A). Compared with the 17ODYA-alone labeling all the additional treatments reduced the fluorescence to various extents. Thus, the labeling was most strongly reduced by the hydroxylamine treatment, diminished by 50.4 ± 14% (n = 3) in the presence of 500 μm palmitic acid and by 32.1 ± 5% (n = 3) in the presence of 250 μm BPA (Fig. 8B and 8C). Taken together, these data show that the metabolic labeling of proteins by 17ODYA reflects preferentially their S-palmitoylation.
Fig. 8.

In-gel profiling reveals acylated proteins in RAW264 cells. (A) Scheme of experimental procedure comprising preincubation of cells with 150 μm or 500 μm palmitic acid (30 min, 37 °C; PA) or 125 μm or 250 μm BPA (1 h, 37 °C) or with 30 μm BSA in controls followed by metabolic labeling of cells with 50 μm 17ODYA or exposed to 0.05% DMSO carrier (- 17ODYA) for 4 h. After lysis, click chemistry reaction was performed with IRDye 800CW-azide and proteins were separated by SDS-PAGE. A set of samples was exposed to 2.5% hydroxylamine (HXA, 30 min, 25 °C) before SDS-PAGE. (B, C) Profile of 17ODYA-labeled proteins in cells exposed to PA (B) or BPA (C) revealed by in-gel fluorescence (left panels) and stained with Coomassie Blue to verify equal loading (right panels). Molecular weight markers are shown on the right.
DISCUSSION
Stimulation of cells with LPS triggers redistribution and activation of numerous proteins involved in the formation of MyD88- and TRIF-dependent signaling complexes of TLR4 (7, 10). As S-palmitoylation is known to affect localization and activity of proteins (28), we asked whether stimulation of cells with LPS affects protein palmitoylation in RAW264 macrophage-like cells. To identify fatty-acylated proteins, we treated living cells with the labeling agent 17ODYA, which mimics palmitic acid (59–61), and stimulated them with 100 ng/ml LPS for 60 min. Our earlier studies indicated that in such conditions both signaling pathways of TLR4 are triggered, judging from the activation of NFκB and IRF3 transcription factors (20, 39). In addition, full development of the events related to the phosphatidylinositol cycle, crucial to TLR4 signaling, could be expected at this duration of treatment since 100 ng/ml LPS induces biphasic accumulation of PI(4,5)P2 in cells with peaks at 5–10 and 60 min of stimulation (20, 21). A proteomic analysis of the 17ODYA-labeled proteins revealed that the stimulation of cells with LPS induced profound global changes of the level of palmitoylated proteins. As many as 154 acylated proteins were markedly up-regulated and even more—186 proteins—were downregulated in comparison with 306 proteins not affected by LPS. One should note here that the approach used to quantify palmitoylated (i.e. labeled with 17ODYA) proteins did not allow distinguishing between a changed amount of a protein (due to its de novo synthesis and/or degradation) and a changed degree of its palmitoylation. A combination of both factors likely affected individual proteins to varying extent, as exemplified by the following two cases. One of the acylated proteins up-regulated by LPS was TNFα, whose rapid synthesis is a hallmark of LPS-induced inflammation. TNFα is synthesized as a transmembrane protein S-palmitoylated at cysteine 30 adjacent to the plasma membrane, which modification facilitates incorporation of TNFα into membrane rafts (73, 74). The other end of the spectrum is represented by PI4KIIβ, whose expression was unaffected by LPS treatment, as shown by a constant mRNA level for up to 4 h of cell stimulation, but the level of S-palmitoylation doubled after 30 min of LPS action. An even faster S-palmitoylation was recently found with application of 17ODYA for the Lck tyrosine kinase in Fas-exposed T cells (38).
We aimed at revealing changes of the level of S-acylated proteins related to LPS-induced signaling. According to the SwissPalm database (http://swisspalm.epfl.ch), 422 of the proteins from our list of 646 have been reported previously as palmitoylated in murine cells and as many as 570 (88%) proteins as palmitoylated in the mouse or other species. Thus, these data provided a good basis for an analysis of the regulation of protein palmitoylation in LPS-stimulated cells. To our knowledge, no earlier studies have addressed this subject. S-palmitoylated proteins were identified with application of the acyl-biotin exchange technique in unstimulated RAW264 cells only (40). That analysis yielded 101 S-acylated proteins, 72 of which were also found in our study (supplemental Table 3). The same group showed LPS-induced accumulation of proteins in Triton X-100-insoluble (raft) fraction of these cells, but contribution of protein S-palmitoylation to their redistribution has not been analyzed in detail (75). In yet another study RAW64 cells were used to examine heterogeneous S-acylation of proteins with application of click chemistry. The cells were first stimulated with the high dose of 0.5 μg/ml LPS together with 100 U/ml INFγ for as long as 12 h and subsequently were labeled for 8 h with alkyne-containing analogues of saturated and unsaturated fatty acids, alk-16, alk-16:1, alk-17:1, or alk-18:1, in concentrations reaching 200 μm (31). The observed protein acylation was related to formation of lipid droplets in the lipid-loaded cells, and no comparison of protein acylation in LPS/INFγ-treated cells with that in resting cells was made. After the prolonged lipid loading, over 680 proteins acylated with various lipids were revealed, 361 of which are also found in our list (supplemental Table 3). Due to the substantially different conditions of cell treatment, labeling, and analysis between those and our studies, such discrepancies could be expected.
We confirmed our mass spectrometry results showing changes of the level of palmitoylated protein in LPS-stimulated RAW264 cells by performing immunoblotting for selected 17ODYA-labeled proteins recovered from cell lysates by affinity enrichment on streptavidin beads and also by immunoprecipitation of PI4KIIα and PI4KIIβ. Interestingly, the 17ODYA labeling of those proteins was to various extents reduced by BPA, which is often considered an inhibitor of protein S-palmitoylation. However, BPA is highly reactive toward thiols, and for this reason, it is a promiscuous inhibitor that, besides inhibiting zDHHC palmitoyl acyltransferases, also affects several other enzymes (62). Among others, BPA has been found to inhibit acyl protein thioesterases (APT1 and APT2), most likely by their direct alkylation (76). The nonuniform influence of BPA on the labeling of proteins with 17ODYA found in our study can account for its moderate inhibition of the overall extent of protein labeling demonstrated by in-gel fluorescence. On the other hand, the strong reduction of the 17ODYA labeling by an excess of palmitic acid and its high sensitivity to hydroxylamine confirm that the labeling mostly represents S-palmitoylation. Notably, during metabolic labeling, 17ODYA can also be incorporated at sites of O- and N-acylation of proteins, but these modification are rare (32). We did find that 17ODYA also incorporates into the GPI anchor of proteins. In addition to CD14, for which we showed such incorporation experimentally, only a few (eight) other proteins from among the 646 labeled ones were predicted to be labeled in the GPI moiety instead of being S-palmitoylated. Hence, this type of labeling does not affect substantially the final list of acylated proteins revealed by the click chemistry reaction.
Prediction of the functions of the 17ODYA-labeled proteins using the IPA algorithm showed their involvement in a variety of cellular processes, with a strong representation of proteins engaged in transcription and translation. In addition, a manual comparison of the protein palmitoylation patterns in resting and LPS-stimulated cells has revealed that the stimulation affects several enzymes involved in phosphatidylinositol synthesis and turnover (Fig. 9). This is of interest in view of earlier studies that have revealed that LPS induces accumulation of PI(4,5)P2 in cells, and that PI(4,5)P2 and its derivatives control several aspects of LPS-induced signaling (6, 20, 21, 23–26). Here we established that LPS triggers S-palmitoylation of PI4KIIβ, one of the four mammalian enzymes catalyzing phosphorylation of phosphatidylinositol to PI(4)P. PI4Ks are classified as kinases of type II or III based on their structure and sensitivity to inhibitors such as wortmannin (63). Among them, PI4KIIα and PI4KIIβ contain a conserved catalytic domain comprising a common CCPCC motif that is S-palmitoylated. Once S-palmitoylated, these enzymes behave as integral membrane proteins, and the S-palmitoylation is also required for their enzymatic activity (44, 65, 66). Both PI4KIIα and PI4KIIβ contribute to the signaling pathways triggered by LPS since depletion of either kinase inhibited the LPS-induced production of cytokines, especially the TRIF-dependent CCL5/RANTES chemokine. In agreement, overexpression of PI4KIIα and PI4KIIβ enhanced the TRIF-dependent signaling of TLR4. This enhancement was dependent on the S-palmitoylation of either kinase. We have also found earlier that overexpressed PI4KIIα enhances production of PI(4)P and PI(4,5)P2 in LPS-stimulated cells in a palmitoylation-dependent manner (21). However, PI4KIIα is S-palmitoylated, hence active, constitutively, as found here and in previous studies (77), while the S-palmitoylation of PI4KIIβ is induced by LPS. Thus, LPS triggers PI(4)P synthesis by inducing S-palmitoylation and thereby activation of PI4KIIβ.
Fig. 9.
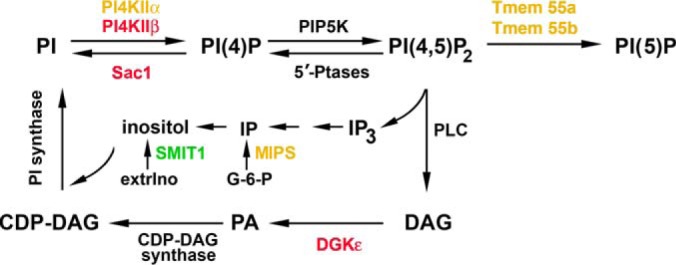
Essential steps of phosphatidylinositol synthesis and turnover are mediated by palmitoylated proteins. Scheme shows major steps of the phosphatidylinositol cycle. Red, acylated proteins up-regulated in LPS-stimulated cells; green, acylated protein downregulated in LPS-stimulated cells. Yellow, proteins whose acylation is not affected by LPS. Nonacylated proteins are shown in black. CDP-DAG, cytidine diphosphate-diacylglycerol; extrIno, extracellular myo- inositol; G-6-P, d-glucose 6-phosphate; IP, myo-inositol 3-phosphate; IP3, inositol 1,4,5-trisphosphate; MIPS, inositol-3-phosphate synthase 1; PA, phosphatidic acid; PI, phosphatidylinositol; PLC, phospholipase C; SMIT1, acylated sodium/myo-inositol transporter 1; Tmem55a and Tmem55b, type 2 and type 1 phosphatidylinositol 4,5-bisphosphate 4-phosphatase, respectively; 5′-Ptases, inositol polyphosphate 5-phosphatases; PIP5KI, type I phosphatidylinositol 4-phosphate 5-kinase.
In unstimulated cells, PI4KIIβ is distributed almost evenly between the cytosol where it is sequestered by Hsp90, and membranes of the trans-Golgi apparatus, a subset of endosomes and the plasma membrane. In the membrane-bound fraction, only half of the kinase seems to be S-palmitoylated, hence in unstimulated cells only about 25–30% of PI4KIIβ is palmitoylated/active (78–80). This estimate agrees with the relatively weak labeling of PI4KIIβ with 17ODYA in unstimulated RAW264 cells that, however, increased markedly in cells exposed to LPS. Upon stimulation with LPS, the cytosolic pool of the enzyme can shift toward the membranes, where it is S-palmitoylated and activated. Thus far, PDGF and a downstream effector of PDGF receptor, Rac GTPase, have been identified as activators of PI4KIIβ inducing its association with the plasma membrane (78). Also cross-linking of T cell receptor CD3ζ chains induces PI4KIIβ activation (81). No further details of the mechanisms governing the S-palmitoylation/activation of PI4KIIβ are known yet, but those deciphered for PI4KIIα can provide a clue. PI4KIIα is associated with trans-Golgi membranes and endosomes (78, 82, 83). At the Golgi, PI4KIIα can be docked in cholesterol-enriched domains (rafts?) by palmitoyl acyltransferases zDHHC3 and zDHHC7 that subsequently palmitoylate the enzyme, endowing it with an anchor for firm “integral” membrane association (65, 77, 84). It is tempting to speculate that zDHHC(s) could serve as docking molecules also for PI4KIIβ at its target membranes. The list of acylated proteins found in our analysis includes as many as ten zDHHCs, in agreement with the earlier data showing that autopalmitoylation is the first catalytic step in the reaction carried out by zDHHC (85). Among the zDHHCs found in our analysis, zDHHC5, 6, 7, 9, and 17 were up-regulated in LPS-stimulated cells, suggesting that they can be involved in the palmitoylation of the proteins of the TLR4 signaling pathways, possibly including PI4KIIβ. Also, tetraspanins, transmembrane S-palmitoylated adaptor proteins that form domains separated from rafts containing GPI-anchored proteins, could serve as anchors of PI4KIIα and PI4KIIβ. The tetraspanins CD9, CD63, and CD81 have been found to associate with PI4KII, although the isoform was not determined (86, 87). During density gradient centrifugation of Brij 99 lysates of cells, a fraction of the CD63-PI4KII complexes has been found in low-density fractions, but the majority has shifted to high-density fractions of the gradients (86). In our hands, the distribution of PI4KIIα and PI4KIIβ in Brij 98 gradient factions resembled that of PI4KII/IIα and CD63 from earlier studies (65, 77, 86), suggesting an interaction of PI4KIIα and PI4KIIβ with tetraspanin(s) that indeed were identified among the fatty acylated proteins in RAW264 cells (supplemental Tables 1–4). This type of interaction could explain the lack of redistribution of PI4KIIβ in the gradient after stimulation of cells with LPS despite its induced S-palmitoylation and thus membrane binding.
PI4KIIα and PI4KIIβ control various aspects of vesicular trafficking between trans-Golgi and endosomes and between sorting and recycling endosomes. The PI(4)P produced by these enzymes is either an intermediate in the chain of phosphoinositide conversions required for that trafficking or itself facilitates the binding of adaptor proteins involved in these processes (80, 82, 83). Cumulative evidence indicates that PI4KIIα and PI4KIIβ have nonredundant functions, suggesting that also in LPS-treated cells the enzymes produce different pools of PI(4)P. For instance, PIKIIβ produces PI(4)P that is required for the functioning of adaptor protein-1 (AP-1), a key adaptor protein complex controlling the assembly of clathrin-coated vesicles involved in bidirectional trans-Golgi–endosomes trafficking (80). AP-1 binds to the trans-Golgi and endosomal membranes via cooperative interactions with PI(4)P, Arf1, and cargo proteins. It is of interest that the γ subunit of AP-1, the same that binds PI(4)P (88), was among the acylated proteins up-regulated in LPS-stimulated cells. Although the role of palmitoylation in the regulation of AP-1 activity has not been studied, it is possible that the increased palmitoylation of PIK4IIβ and AP-1γ can facilitate their interaction with Golgi/endosome membranes and the assembly of the clathrin coat at the membrane. By controlling AP-1, PI4KIIβ contributes to the recycling of Fz receptors in canonical Wnt signaling, thereby preventing degradation of the receptor (80). In a similar manner, PI4KIIβ can support TLR4 proinflammatory signaling. Proper trafficking of TLR4 and CD14 can be of special importance for the endosomal TRIF-dependent signaling of TLR4, which we found to be particularly sensitive to PI4KIIβ or PI4KIIα activity. Of these two enzymes, PI4KIIβ is also likely to produce PI(4)P at the plasma membrane, as it has been found to be recruited there in response to some stimuli (78, 81). Thus, its recruitment to the plasma membrane in response to CD14 and/or TLR4 ligation with LPS and local production of PI(4)P cannot be ruled out even though PIKIIIα is considered as the main kinase producing the plasma membrane PI(4)P (89).
In addition to PI4KIIα and PI4KIIβ, the palmitoylated proteins of the phosphatidylinositol cycle also include phosphatidylinositide phosphatase Sac1 (Fig. 9), which dephosphorylates PI(4)P (90). Our proteomic analysis indicates that palmitoylated Sac1 is up-regulated in LPS-stimulated cells similarly to PI4KIIβ, suggesting that PI(4)P level is strictly regulated in these conditions. Diacylglycerol kinase-ε (DGKε) also deserves attention (Fig. 9) because it has a strict substrate specificity for one type of diacylglycerol only, 1-stearoyl-2-arachidonoyl glycerol, which is released from PI(4,5)P2 upon hydrolysis. The phosphatidic acid produced by DGKε is in turn an intermediate in the synthesis of phosphatidylinositol (91, 92). DGKε simultaneously exhibits properties of an integral and a peripheral membrane protein (91). Our data suggest that palmitoylation could contribute to the membrane binding of DGKε, similarly as for PI4KIIβ. In contrast to PI4KIIβ, type 1 and 2 phosphatidylinositol 4,5-bisphosphate 4-phosphatases (Tmem55b and Tmem55a, respectively), which dephosphorylate PI(4,5)P2 to PI(5)P, and also inositol-3-phosphate synthase 1, which is the rate-limiting enzyme in de novo synthesis of myo-inositol, were found to be acylated constitutively (Fig. 9). Finally, acylated sodium/myo-inositol transporter 1, was downregulated under the influence of LPS. This transporter mediates the uptake of extracellular myo-inositol, which also originates from the de novo synthesis and recycling of phosphatidylinositol (Fig. 9).
Taken together, our studies have revealed that stimulation of cells with LPS induces profound changes in the levels of numerous fatty-acylated, most likely S-palmitoylated, proteins paving the way for further studies on the role of this protein modification in LPS-triggered signaling cascades. In particular, S-palmitoylation of enzymes catalyzing phosphatidylinositol synthesis and turnover deserves attention as a likely factor regulating the LPS-induced pro-inflammatory responses.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (93) partner repository (http://www.ebi.ac.uk/pride/archive/) with the dataset identifier PXD007918.
Supplementary Material
Acknowledgments
We thank Prof. Helen L. Yin from the Department of Physiology, University of Texas Southwestern Medical Center (Dallas, TX, USA), for providing pCMV5-Myc plasmids encoding PI4KIIα and PI4KIIβ. We also thank Prof. Andrzej Sobota and Dr. Anna Ciesielska from the Laboratory of Molecular Membrane Biology of the Nencki Institute (Warsaw, Poland) and Prof. Jan Fronk from the Faculty of Biology, University of Warsaw for critical discussion, and Dr. Michał Dąbrowski from the Laboratory of Bioinformatics of the Nencki Institute for preliminary analysis of proteomic data and for ANOVA analysis. The mass spectrometry analysis was performed in the Laboratory of Mass Spectrometry (IBB PAS).
Footnotes
* The studies were supported by the National Science Centre, Poland, grant number DEC-2013/08/A/NZ3/00850 to K.K. This project also received funding from the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant No. 665735.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- 17ODYA
- 17-octadecynoic acid
- AP-1
- adaptor protein-1
- BPA
- bromohexadecanoic acid
- BSA
- bovine serum albumin
- CCL5/RANTES
- C-C motif chemokine ligand 5/regulated upon activation, normal T cell expressed and secreted
- DGKε
- diacylglycerol kinase-ε
- eIF5A2
- eukaryotic translation initiation factor 5A2
- FBS
- fetal bovine serum
- GPI
- glycosylphosphatidylinositol
- IP3
- inositol 1,4,5-trisphosphate
- IPA
- Ingenuity Pathway Analysis
- ISRE
- interferon-stimulated response element
- LPS
- lipopolysaccharide
- PBS
- phosphate buffered saline
- PI
- phosphatidylinositol
- PI(4)P
- phosphatidylinositol 4-monophosphate
- PI
- phosphatidylinositol
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PI(3,4,5)P3
- phosphatidylinositol 3,4,5-trisphosphate
- PI4KII
- type II phosphatidylinositol 4-kinase
- PIP5KI
- type I phosphatidylinositol 4-phosphate 5-kinase
- TBTA
- Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine
- TCEP
- Tris(2-carboxyethyl)phosphine hydrochloride
- TIR
- Toll/interleukin-1 receptor
- TLR4
- Toll-like receptor 4
- VSVG
- vesicular stomatitis virus G-protein
- zDHHC
- zinc finger DHHC
- emPAI
- exponentially modified protein abundance index
- FDR
- false discovery rate
- IRAK
- interleukin-1 receptor-associated kinase
- IRF
- interferon regulatory factor
- TIRAP
- TIR domain-containing adaptor protein
- TNFα
- tumor necrosis factor α
- TRAM
- TRIF-related adaptor molecule
- TRIF
- TIR domain-containing adapter-inducing interferon-β
- qPCR
- quantitative polymerase chain reaction.
REFERENCES
- 1. Poltorak A., He X., Smirnova I., Liu M. Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., and Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 282, 2085–2088 [DOI] [PubMed] [Google Scholar]
- 2. Brubaker S. W., Bonham K. S., Zanoni I., and Kagan J. C. (2015) Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 33, 257–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Angus D. C., and van der Poll T. (2013) Severe sepsis and septic shock. N. Engl. J. Med. 369, 840–851 [DOI] [PubMed] [Google Scholar]
- 4. Kaliannan K., Wang B., Li X. Y., Kim K. J., and Kang J. X. (2015) A host-microbiome interaction mediates the opposing effects of omega-6 and omega-3 fatty acids on metabolic endotoxemia. Sci. Rep. 5, 11276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park B. S., Song D. H., Kim H. M., Choi B. S., Lee H., and Lee J. O. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 [DOI] [PubMed] [Google Scholar]
- 6. Kagan J. C., and Medzhitov R. (2006) Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 125, 943–955 [DOI] [PubMed] [Google Scholar]
- 7. Motshwene P. G., Moncrieffe M. C., Grossmann J. G., Kao C., Ayaluru M., Sandercock A. M., Carol V., Robinson C. V., Latz E., and Gay N. J. (2009) An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 284, 25404–25411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawai T., and Akira S. (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 [DOI] [PubMed] [Google Scholar]
- 9. Husebye H., Halaas Ø., Stenmark H., Tunheim G., Sandanger Ø., Bogen B., Brech A., Latz E., and Espevik T. (2006) Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 25, 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kagan J. C., Su T., Horng T., Chow A., Akira S., and Medzhitov R. (2008) TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 9, 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Enokizono Y., Kumeta H., Funami K., Horiuchi M., Sarmiento J., Yamashita K., Standley D. M., Matsumoto M., Seya T., and Inagaki F. (2013) Structures and interface mapping of the TIR domain-containing adaptor molecules involved in interferon signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 19908–19913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simmons D. L., Tan S., Tenen D. G., Nicholson-Weller A., and Seed B. (1989) Monocyte antigen CD14 is a phospholipid anchored membrane protein. Blood 73, 284–289 [PubMed] [Google Scholar]
- 13. Płóciennikowska A., Hromada-Judycka A., Borzęcka K., and Kwiatkowska K. (2015) Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 72, 557–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim J.-I., Lee C. J., Jin M. S., Lee C. H., Paik S. G., Lee H., and Lee J. O. (2005) Crystal structure of CD14 and its implications for lipopolysaccharide signaling. J. Biol. Chem. 280, 11347–11351 [DOI] [PubMed] [Google Scholar]
- 15. Gioannini T. L., Teghanemt A., Zhang D., Levis E. N., and Weiss J. P. (2005) Monomeric endotoxin:protein complexes are essential for TLR4-dependent cell activation. J. Endotoxin Res. 11, 117–123 [DOI] [PubMed] [Google Scholar]
- 16. Jiang Z., Georgel P., Du X., Shamel L., Sovath S., Mudd S., Huber M., Kalis C., Keck S., Galanos C., Freudenberg M., and Beutler B. (2005) CD14 is required for MyD88-independent LPS signaling. Nat. Immunol. 6, 565–570 [DOI] [PubMed] [Google Scholar]
- 17. Husebye H., Aune M. H., Stenvik J., Samstad E., Skjeldal F., Halaas O., Nilsen N. J., Stenmark H., Latz E., Lein E., Mollnes T. E., Bakke O., and Espevik T. (2010) The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity 33, 583–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zanoni I., Ostuni R., Marek L. R., Barresi S., Barbalat R., Barton G. M., Granucci F., and Kagan J. C. (2011) CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147, 868–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tan Y., Zanoni I., Cullen T. W., Goodman A. L., and Kagan J. C. (2015) Mechanisms of Toll-like receptor 4 endocytosis reveal a common immune-evasion strategy used by pathogenic and commensal bacteria. Immunity 43, 909–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Płóciennikowska A., Zdioruk M. I., Traczyk G., Świątkowska A, Kwiatkowska K. (2015) LPS-induced clustering of CD14 triggers generation of PI(4,5)P2. J. Cell Sci. 128, 4096–4111 [DOI] [PubMed] [Google Scholar]
- 21. Płóciennikowska A., Hromada-Judycka A., Dembińska J., Roszczenko P., Ciesielska A., and Kwiatkowska K. (2016) Contribution of CD14 and TLR4 to changes of the PI(4,5)P2 level in LPS-stimulated cells. J. Leukoc. Biol. 100, 1363–1373 [DOI] [PubMed] [Google Scholar]
- 22. Nguyen T. T., Kim Y. M., Kim T. D., Le O. T., Kim J. J., Kang H. C., Hasegawa H., Kanaho Y., Jou I., and Lee S. Y. (2013) Phosphatidylinositol 4-phosphate 5-kinase α facilitates Toll-like receptor 4-mediated microglial inflammation through regulation of the Toll/interleukin-1 receptor domain-containing adaptor protein (TIRAP) location. J. Biol. Chem. 288, 5645–5659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chiang C.-Y., Veckman V., Limmer K., and David M. (2012) Phospholipase Cγ-2 and intracellular calcium are required for lipopolysaccharide-induced Toll-like receptor 4 (TLR4) endocytosis and interferon regulatory factor 3 (IRF3) activation. J. Biol. Chem. 287, 3704–3709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aksoy E., Taboubi S., Torres D., Delbauve S., Hachani A., Whitehead M. A., Pearce W. P., Berenjeno I. M., Nock G., Filloux A., Beyaert R., Flamand V., and Vanhaesebroeck B. (2012) The p110δ isoform of the kinase PI(3)K controls the subcellular compartmentalization of TLR4 signaling and protects from endotoxic shock. Nat. Immunol. 13, 1045–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ding J., Wang K., Liu W., She Y., Sun Q., Shi J., Sun H., Wang D. C., and Shao F. (2016) Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 [DOI] [PubMed] [Google Scholar]
- 26. Liu X., Zhang Z., Ruan J., Pan Y., Magupalli V. G., Wu H., and Lieberman J. (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Levental I., Lingwood D., Grzybek M., Coskun U., and Simons K. (2010) Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc. Natl. Acad. Sci. U.S.A. 107, 22050–22054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chamberlain L. H., and Shipston M. J. (2015) The physiology of protein S-acylation. Physiol. Rev. 95, 341–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sobocińska J., Roszczenko-Jasińska P., Ciesielska A., and Kwiatkowska K. (2018) Protein palmitoylation and its role in bacteria and virus pathogenicity. Frontiers in Immunology 10.3389/fimmu.2017.02003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liang X., Nazarian A., Erdjument-Bromage H., Bornmann W., Tempst P., and Resh M. D. (2001) Heterogeneous fatty acylation of Src family kinases with polyunsaturated fatty acids regulates raft localization and signal transduction. J. Biol. Chem. 276, 30987–30994 [DOI] [PubMed] [Google Scholar]
- 31. Thinon E., Percher A., and Hang H. C. (2016) Bioorthogonal chemical reporters for monitoring unsaturated fatty-acylated proteins. ChemBioChem 17, 1800–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Resh M. D. (2016) Fatty acylation of proteins: The long and the short of it. Prog. Lipid Res. 63, 120–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohno Y., Kihara A., Sano T., and Igarashi Y. (2006) Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim. Biophys. Acta 1761, 474–483 [DOI] [PubMed] [Google Scholar]
- 34. Blaskovic S., Adibekian A., Blanc M., and van der Goot G. F. (2014) Mechanistic effects of protein palmitoylation and the cellular consequences thereof. Chem. Phys. Lipids 180, 44–52 [DOI] [PubMed] [Google Scholar]
- 35. Yokoi N., Fukata Y., Sekiya A., Murakami T., Kobayashi K., and Fukata M. (2016) Identification of PSD-95 depalmitoylating enzymes. J. Neurosci. 36, 6431–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang M. M., Tsou L. K., Charron G., Raghavan A. S., and Hang H. C. (2010) Tandem fluorescence imaging of dynamic S-acylation and protein turnover. Proc. Natl. Acad. Sci. U.S.A. 107, 8627–8632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jia L., Chisari M., Maktabi M. H., Sobieski C., Zhou H., Konopko A. M., Martin B. R., Mennerick S. J., and Blumer K. J. (2014) A mechanism regulating G protein-coupled receptor signaling that requires cycles of protein palmitoylation and depalmitoylation. J. Biol. Chem. 289, 6249–6257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Akimzhanov A. M., and Boehning D. (2015) Rapid and transient palmitoylation of the tyrosine kinase Lck mediates Fas signaling. Proc. Natl. Acad. Sci. U.S.A. 112, 11876–11880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Borzęcka-Solarz K., Dembińska J., Hromada-Judycka A., Traczyk G., Ciesielska A., Ziemlińska E., Świątkowska A, Kwiatkowska K. (2017) Association of Lyn kinase with membrane rafts determines its negative influence on LPS-induced signaling. Mol. Biol. Cell 28, 1147–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Merrick B. A., Dhungana S., Williams J. G., Aloor J. J., Peddada S., Tomer K. B., and Fessler M. B. (2011) Proteomic profiling of S-acylated macrophage proteins identifies a role for palmitoylation in mitochondrial targeting of phospholipid scramblase 3. Mol. Cell. Proteomics 10, M110.006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chesarino N. M., Hach J. C., Chen J. L., Zaro B. W., Rajaram M. V., Turner J., Schlesinger L. S., Pratt M. R., Hang H. C., and Yount J. S. (2014) Chemoproteomics reveals Toll-like receptor fatty acylation. BMC Biol. 12, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kwiatkowska K., Frey J., and Sobota A. (2003) Phosphorylation of FcγRIIA is required for the receptor-induced actin rearrangement and capping: the role of membrane rafts. J. Cell Sci. 116, 537–550 [DOI] [PubMed] [Google Scholar]
- 43. Jin J., Zhang X., Lu Z., Perry D. M., Li Y., Russo S. B., Cowart L. A., Hannun Y. A., and Huang Y. (2013) Acid sphingomyelinase plays a key role in palmitic acid-amplified inflammatory signaling triggered by lipopolysaccharide at low concentrations in macrophages. Am. J. Physiol. Endocrinol. Metab. 305, E853–E867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jung G., Wang J., Wlodarski P., Barylko B., Binns D. D., Shu H., Yin H. L., and Albanesi J. P. (2008) Molecular determinants of activation and membrane targeting of phosphoinositol 4-kinase IIβ. Biochem. J. 409, 501–509 [DOI] [PubMed] [Google Scholar]
- 45. Kitchens R. L., Wang P., and Munford R. S. (1998) Bacterial lipopolysaccharide can enter monocytes via two CD14-dependent pathways. J. Immunol. 161, 5534–5545 [PubMed] [Google Scholar]
- 46. Berger J., Micanovic R., Greenspan R. J., and Udenfriend S. (1989) Conversion of placental alkaline phosphatase from a phosphatidylinositol-glycan-anchored protein to an integral transmembrane protein. Proc. Natl. Acad. Sci. U.S.A. 86, 1457–1460, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stewart S. A., Dykxhoorn D. M., Palliser D., Mizuno H., Yu E. Y., An D. S., Sabatini D. M., Chen I. S., Hahn W. C., Sharp P. A., Weinberg R. A., and Novina C. D. (2003) Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilson J. P., Raghavan A. S., Yang Y. Y., Charron G., and Hang H. C. (2011) Proteomic analysis of fatty-acylated proteins in mammalian cells with chemical reporters reveals S-acylation of histone H3 variants. Mol. Cell. Proteomics 10, M110.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Malinowska A., Kistowski M., Bakun M., Rubel T., Tkaczyk M., Mierzejewska J., and Dadlez M. (2012) Diffprot—Software for non-parametric statistical analysis of differential proteomics data. J. Proteomics 75, 4062–4073 [DOI] [PubMed] [Google Scholar]
- 50. Arike L., and Peil L. (2014) Spectral counting label-free proteomics. Methods Mol. Biol. 1156, 213–222 [DOI] [PubMed] [Google Scholar]
- 51. Yang W., Di Vizio D., Kirchner M., Steen H., and Freeman M. R. (2010) Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol. Cell. Proteomics 9, 54–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Holmberg A., Blomstergren A., Nord O., Lukacs M., Lundeberg J., and Uhlén M. (2005) The biotin-streptavidin interaction can be reversibly broken using water at elevated temperatures. Electrophoresis 26, 501–510 [DOI] [PubMed] [Google Scholar]
- 53. Jenne A., and Famulok M. (1999) Disruption of the streptavidin interaction with biotinylated nucleic acid probes by 2-mercaptoethanol. BioTechniques 26, 249–252, 254 [DOI] [PubMed] [Google Scholar]
- 54. Folch J., Lees M., and Sloane S. G. H. (1957) A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 55. Axelsson M., and Gentili F. (2014) A single-step method for rapid extraction of total lipids from green microalgae. PLoS ONE 9, e89643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Szymańska E., Korzeniowski M., Raynal P., Sobota A., and Kwiatkowska K. (2009) Contribution of PIP-5 kinase Iα to raft-based FcγRIIA signaling. Exp. Cell Res. 315, 981–995 [DOI] [PubMed] [Google Scholar]
- 57. Borzęcka K., Płóciennikowska A., Bjorkelund H., Sobota A., and Kwiatkowska K. (2013) CD14 mediates binding of high doses of LPS but is dispensable for TNF-α production. Mediators Inflam. 2013, 824919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kobayashi E., Kobayashi M., Tsuneyama K., Fukami T., Nakajima M., and Yokoi T. (2009) Halothane-induced liver injury is mediated by interleukin-17 in mice. Toxicol. Sci. 111, 302–310 [DOI] [PubMed] [Google Scholar]
- 59. Martin B. R., and Cravatt B. F. (2009) Large-scale profiling of protein palmitoylation in mammalian cells. Nat. Methods 6, 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Charron G., Zhang M. M., Yount J. S., Wilson J., Raghavan A. S., Shamir E., and Hang H. C. (2009) Robust fluorescent detection of protein fatty-acylation with chemical reporters. J. Am. Chem. Soc. 131, 4967–4975 [DOI] [PubMed] [Google Scholar]
- 61. Martin B. R., Wang C., Adibekian A., Tully S. E., and Cravatt B. F. (2011) Global profiling of dynamic protein palmitoylation. Nat Methods 9, 84–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Davda D., El Azzouny M. A., Tom C. T., Hernandez J. L., Majmudar J. D., Kennedy R. T., and Martin B. R. (2013) Profiling targets of the irreversible palmitoylation inhibitor 2-bromopalmitate. ACS Chem. Biol. 8, 1912–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Balla T. (2013) Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 93, 1019–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kleveta G., Borzęcka K., Zdioruk M., Czerkies M., Kuberczyk H., Sybirna N., Sobota A., and Kwiatkowska K. (2012) LPS induces phosphorylation of actin-regulatory proteins leading to actin reassembly and macrophage motility. J. Cell. Biochem. 113, 80–92 [DOI] [PubMed] [Google Scholar]
- 65. Barylko B., Mao Y. S., Wlodarski P., Jung G., Binns D. D., Sun H. Q., Yin H. L., and Albanesi J. P. (2009) Palmitoylation controls the catalytic activity and subcellular distribution of phosphatidylinositol 4-kinase IIα. J. Biol. Chem. 284, 9994–10003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhou Q., Li J., Yu H., Zhai Y., Gao Z., Liu Y., Pang X., Zhang L., Schulten K., Sun F., and Chen C. (2014) Molecular insights into the membrane-associated phosphatidylinositol 4-kinase IIα. Nat Commun. 5, 3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Björkbacka H., Fitzgerald K. A., Huet F., Li X., Gregory J. A., Lee M. A., Ordija C. M., Dowley N. E., Golenbock D. T., and Freeman M. W. (2004) The induction of macrophage gene expression by LPS predominantly utilizes Myd88-independent signaling cascades. Physiol. Genomics. 19, 319–330 [DOI] [PubMed] [Google Scholar]
- 68. Nakatsu F., Baskin J. M., Chung J., Tanner L. B., Shui G., Lee S. Y., Pirruccello M., Hao M., Ingolia N. T., Wenk M. R., and De Camilli P. (2012) PtdIns4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. J. Cell Biol. 199, 1003–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Berger J., Howard A. D., Brink L., Gerber L., Hauber J., Cullen B. R., and Udenfriend S. (1988) COOH-terminal requirements for the correct processing of a phosphatidylinositol-glycan anchored membrane protein. J. Biol. Chem. 263, 10016–10021 [PubMed] [Google Scholar]
- 70. Orlean P., and Menon A. K. (2007) Thematic review series: lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: How we learned to stop worrying and love glycophospholipids. J. Lipid Res. 48, 993–1011 [DOI] [PubMed] [Google Scholar]
- 71. Galian C., Björkholm P., Bulleid N., and von Heijne G. (2012) Efficient glycosylphosphatidylinositol (GPI) modification of membrane proteins requires a C-terminal anchoring signal of marginal hydrophobicity. J. Biol. Chem. 287, 16399–16409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rose J. K., Adams G. A., and Gallione C. J (1984) The presence of cysteine in the cytoplasmic domain of the vesicular stomatitis virus glycoprotein is required for palmitate addition. Proc. Natl. Acad. Sci. U.S.A. 81, 2050–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Utsumi T., Takeshige T., Tanaka K., Takami K., Kira Y., Klostergaard J., and Ishisaka R. (2001) Transmembrane TNF (pro-TNF) is palmitoylated. FEBS Lett. 500, 1–6 [DOI] [PubMed] [Google Scholar]
- 74. Poggi M., Kara I., Brunel J. M., Landrier J. F., Govers R., Bonardo B., Fluhrer R., Haass C., Alessi M. C., and Peiretti F. (2013) Palmitoylation of TNFα is involved in the regulation of TNF receptor 1 signalling. Biochim. Biophys. Acta 1833, 602–612 [DOI] [PubMed] [Google Scholar]
- 75. Dhungana S., Merrick B. A., Tomer K. B., and Fessler M. B. (2009) Quantitative proteomics analysis of macrophage rafts reveals compartmentalized activation of the proteasome and of proteasome-mediated ERK activation in response to lipopolysaccharide. Mol. Cell. Proteomics 8, 201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pedro M. P., Vilcaes A. A., Tomatis V. M., Oliveira R. G., Gomez G. A., and Daniotti J. L. (2013) 2-Bromopalmitate reduces protein deacylation by inhibition of acyl-protein thioesterase enzymatic activities. PLoS ONE 8, e75232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lu D., Sun H. Q., Wang H., Barylko B., Fukata Y., Fukata M., Albanesi J. P., and Yin H. L. (2012) Phosphatidylinositol 4-kinase IIα is palmitoylated by Golgi-localized palmitoyltransferases in cholesterol-dependent manner. J. Biol. Chem. 287, 21856–21865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wei Y. J., Sun H. Q., Yamamoto M., Wlodarski P., Kunii K., Martinez M., Barylko B., Albanesi J. P., and Yin H. L. (2002) Type II phosphatidylinositol 4-kinase β is a cytosolic and peripheral membrane protein that is recruited to the plasma membrane and activated by Rac-GTP. J. Biol. Chem. 277, 46586–46593 [DOI] [PubMed] [Google Scholar]
- 79. Jung G., Barylko B., Lu D., Shu H., Yin H., and Albanesi J. P. (2011) Stabilization of phosphatidylinositol 4-kinase type IIβ by interaction with Hsp90. J. Biol. Chem. 286, 12775–12784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wieffer M., Cibrián Uhalte E., Posor Y., Otten C., Branz K., Schütz I., Mössinger J., Schu P., Abdelilah-Seyfried S., Krauβ M., and Haucke V. (2013) PI4K2β/AP-1-based TGN-endosomal sorting regulates Wnt signaling. Curr. Biol. 23, 2185–2190 [DOI] [PubMed] [Google Scholar]
- 81. Srivastava R., Sinha R. K., and Subrahmanyam G. (2006) Type II phosphatidylinositol 4-kinase β associates with TCR-CD3 ζ chain in Jurkat cells. Mol. Immunol. 43, 454–463 [DOI] [PubMed] [Google Scholar]
- 82. Henmi Y., Morikawa Y., Oe N., Ikeda N., Fujita A., Takei K., Minogue S., and Tanabe K. (2016) PtdIns4KIIα generates endosomal PtdIns(4)P and is required for receptor sorting at early endosomes. Mol. Biol. Cell. 27, 990–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ketel K., Krauss M., Nicot A. S., Puchkov D., Wieffer M., Müller R., Subramanian D., Schultz C., Laporte J., and Haucke V. (2016) A phosphoinositide conversion mechanism for exit from endosomes. Nature 529, 408–412 [DOI] [PubMed] [Google Scholar]
- 84. Minogue S., Chu K. M., Westover E. J., Covey D. F., Hsuan J. J., and Waugh M. G. (2010) Relationship between phosphatidylinositol 4-phosphate synthesis, membrane organization, and lateral diffusion of PI4KIIα at the trans-Golgi network. J. Lipid Res. 51, 2314–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jennings B. C., and Linder M. E. (2012) DHHC protein S-acyltransferases use similar ping-pong kinetic mechanisms but display different acyl-CoA specificities. J. Biol. Chem. 287, 7236–7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Claas C., Stipp C. S., and Hemler M. E. (2001) Evaluation of prototype transmembrane 4 superfamily protein complexes and their relation to lipid rafts. J. Biol. Chem. 276, 7974–7984 [DOI] [PubMed] [Google Scholar]
- 87. Yauch R. L., and Hemler M. E. (2000) Specific interactions among transmembrane superfamily (TM4SF) proteins and phosphoinositide 4-kinase. Biochem. J. 351, 629–637 [PMC free article] [PubMed] [Google Scholar]
- 88. Ren X., Farías G. G., Canagarajah B. J., Bonifacino J. S., and Hurley J. H. (2013) Structural basis for recruitment and activation of the AP-1 clathrin adaptor complex by Arf1. Cell 152, 755–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chung J., Nakatsu F., Baskin J. M., and De Camilli P. (2015) Plasticity of PI4KIIIα interactions at the plasma membrane. EMBO Rep. 16, 312–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hsu F., and Mao Y. (2015) The structure of phosphoinositide phosphatases: Insights into substrate specificity and catalysis. Biochim. Biophys. Acta 1851, 698–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Epand R. M. (2016) Features of the phosphatidylinositol cycle and its role in signal transduction. J. Membr. Biol., in press [DOI] [PubMed] [Google Scholar]
- 92. Epand R. M., So V., Jennings W., Khadka B., Gupta R. S., and Lemaire M. (2016) Diacylglycerol kinase-ε: properties and biological roles. Front. Cell. Dev. Biol. 4, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Resiniger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (93) partner repository (http://www.ebi.ac.uk/pride/archive/) with the dataset identifier PXD007918.