

Abstract
Understanding the function of cellular systems requires describing how proteins assemble with each other into transient and stable complexes and to determine their spatial relationships. Among the tools available to perform these analyses on a large scale is Protein-fragment Complementation Assay based on the dihydrofolate reductase (DHFR PCA). Here we test how longer linkers between the fusion proteins and the reporter fragments affect the performance of this assay. We investigate the architecture of the RNA polymerases, the proteasome and the conserved oligomeric Golgi (COG) complexes in living cells and performed large-scale screens with these extended linkers. We show that longer linkers significantly improve the detection of protein-protein interactions and allow to measure interactions further in space than the standard ones. We identify new interactions, for instance between the retromer complex and proteins related to autophagy and endocytosis. Longer linkers thus contribute an enhanced additional tool to the existing toolsets for the detection and measurements of protein-protein interactions and protein proximity in living cells.
Protein-protein interactions (PPIs)1 are central to all cellular functions and are largely responsible for translating genotypes into phenotypes (1). Investigations into the organization of PPI networks have revealed important insights into the evolution of cellular functions (2–7), the robustness of protein complexes to mutations (2, 8–11) and have shown how the regulation of protein expression at the transcriptional, translational and posttranslational level contributes to the diversity of protein complex assemblies (12–16).
Methods used to investigate the organization of PPIs can be grouped into two main categories based on whether they infer co-complex memberships (or association) or detect more direct physical interactions (17). The first category includes methods based on protein affinity purification followed by mass-spectrometry (AP-MS). In this case, protein assignment to a specific complex is dependent on stable association among proteins that survive cell lysis and fractionation or affinity purification (18, 19). Unfortunately, very little is known about the structural and context dependences of PPIs inferred from co-complex membership because detecting an association does not provide information on the spatial organization of the complex (20–22). The second category of methods reports binary or pairwise interactions between proteins and reveals direct or nearly direct interactions. Such methods include the commonly used yeast-two-hybrid (Y2H) (23), protein-fragment complementation assays (PCAs) (24) and technologies based on similar principles (25).
Most PPIs derived from large-scale screening that populate interactome databases come from methods that cannot infer direct or proximate contacts, for instance AP-MS, because a single purification leads to the inference of many interactions among the co-purified proteins. For the budding yeast data stored in BioGRID (2017) (26), AP-MS has contributed more than twice as much (n = 58455) in terms of PPIs than PCA and Y2H combined (n = 6577 and 15973 respectively). Overall, these methods are complementary because on the one hand, they tell us which proteins assemble into complexes in the cell and on the other hand, how proteins may be physically located relative to one another (20, 27).
Because most of the PPI data derive from a method that does not provide information on the spatial organization of complex subunits in vivo, there is still a need for tools that can detect proximate relationships to further enhance our ability to infer the relationships among proteins within and between complexes or subcomplexes. Being able to do so at different levels of resolution in living cells is key to future development in cell and systems biology because high-resolution methods such as NMR or X-ray crystallography are not yet amenable to high-throughput analysis, cannot be applied to all protein types nor provide information on the dynamics of protein complexes in living cells. The modification of current PCAs (24, 28) may offer the technological advantages required for such an approach by complementing methods detecting co-complex membership.
PCA relies on the fusion of two proteins of interest with fragments of a reporter protein, usually at their C terminus. When a PPI takes place, the two fragments assemble into a functional protein whose activity can be detected by various means (7, 28–33). Proteins are usually connected to the reporter fragments with a linker of ten amino acids. In principle, the length of the linker limits the maximum distance among the proteins for a PPI to be detectable. In the first large-scale study performed using the dihydrofolate reductase (DHFR) PCA in yeast, it was shown that distance constraint determined by linker length could affect the ability to detect PPIs (7). For the RNA polymerase (RNApol) II complex and several other protein complexes, PPIs were 3.5 times more likely to be detected if the C-termini were within less than 82 Å of each other. In addition, an earlier study in mammalian cells showed that increasing linker length of the PCA reporter allows to detect configuration changes in a dimeric membrane receptor (34). Together, these results suggest that linkers of variable sizes could improve the detection of PPIs and even be used as a ruler to infer, albeit roughly, distances between proteins and this, in living cells. However, a recent study on luminescence PCA showed that the relationship between the linker size and the signal-to-noise ratio of PCA assays is not trivial (35). Although reducing linker size from 15 to 10 amino acid long reduces signal-to-noise ratio by 40%, further reduction has little effect until the linker is completely eliminated. Therefore, the effect of linker size requires to be tested in a quantitative manner if it is to be implemented in large-scale screen and this, possibly for each specific assay. Here we test these effects on the ability to detect PPIs by PCA in vivo using the yeast DHFR PCA. We examine how linker size influences our ability to detect PPIs in large-scale screens and in small-scale experiments by testing PPIs within complexes whose structures are known. We show that extended linkers improve the signal-to-noise ratio in these experiments and thus, helps to detect previously unreported PPIs. They also allow detecting PPIs among proteins within the same complex but that are more distant based on the location of their C-termini.
MATERIALS AND METHODS
Resources and Reagents
All resources and reagents used in this study are described in details in supplemental Table S1.
Yeast
Yeast strains used in this study were constructed (as described below) or are from the Yeast Protein Interactome Collection (7) (Table S2A). They all derive from the BY4741 (MATa his3Δ leu2Δ met15Δ ura3Δ) and BY4742 (MATα his3Δ leu2Δ lys2Δ ura3Δ) backgrounds. Cells were grown on YPD medium (1% yeast extract, 2% tryptone, 2% glucose, and 2% agar (for solid medium)) containing 100 μg/ml nourseothricin (clonNAT) and/or 250 μg/ml hygromycin B (HygB) for transformations and diploid selection. For the DHFR PCA experiment, cells were grown on MTX medium (0.67% yeast nitrogen base without amino acids and without ammonium sulfate, 2% glucose, 2.5% noble agar, drop-out without adenine, methionine and lysine, and 200 μg/ml methotrexate (MTX) diluted in DMSO).
Bacteria
Escherichia coli MC1061 was used for all DNA cloning and propagation steps. Cells were grown on 2YT medium (1% yeast extract, 1.6% tryptone, 0.2% glucose, 0.5% NaCl, and 2% agar (for solid medium)) supplemented with 100 μg/ml ampicillin (Amp).
Plasmid Construction
Plasmids pAG25-linker-DHFR-F[1,2]-ADHterm and pAG32-linker-DHFR-F[3]-ADHterm were used as templates to create new plasmids containing DHFR fragments fused to a linker of varying size. Both original plasmids contained the sequence coding for two repetitions of the motif GGGGS (2xL). Additional repetitions of the motif (one for the 3xL and two for the 4xL) were introduced between the linker present and the DHFR fragments, resulting in plasmids pAG25-3x-linker-DHFR-F[1,2]-ADHterm, pAG32-3x-linker-DHFR-F[3]-ADHterm, pAG25-4x-linker-DHFR-F[1,2]-ADHterm and pAG32-4x-linker-DHFR-F[3]-ADHterm. The new repetitions were composed of synonymous codons leading to the same peptide sequence.
To replace the 2xL from pAG25-linker-DHFR-F[1,2]-ADHterm with the 3xL and 4xL, DNA fragments were synthesized and inserted in the plasmid pUC57 containing flanking BamHI and XbaI restriction sites. The 3x/4xL-DHFR F[1,2] fragments were then amplified by PCR (Table S2B-cloning), digested with DpnI and purified. The plasmid pAG25-linker-DHFR-F[1,2]-ADHterm was digested with XbaI and BamHI. The fragment corresponding to the plasmid without the 2xL-DHFR F[1,2] region was isolated by gel extraction. The fragments and plasmids were assembled by Gibson cloning (36) with an insert:vector ratio of 5:1. Cloning reactions were transformed in E. coli and clones were selected on 2YT+Amp. Finally, positive clones were verified and confirmed by double digestion with XbaI and BamHI and Sanger sequencing.
The pAG25-3x/4xL-DHFR-F[1,2]-ADHterm plasmids were used as a template to construct the pAG32-3x/4xL-DHFR-F[3]-ADHterm plasmids. 3xL and 4xL fragments were PCR amplified from pAG25-3xL-DHFR-F[1,2]-ADHterm and pAG25-4xL-DHFR-F[1,2]-ADHterm respectively (supplemental Table S2B-cloning). The DHFR F[3] fragment was amplified from pAG32-linker-DHFR-F[3]-ADHterm (supplemental Table S2B-cloning). All PCR reactions were digested with DpnI and purified. Plasmid pAG32-linker-DHFR-F[3]-ADHterm was digested with XbaI and BamHI. The fragment corresponding to the plasmid without the 2xL-DHFR F[3] region was purified on gel. The remaining steps were performed as described above for the pAG25-3x/4xL-DHFR-F[1,2]-ADHterm with an insert (linker):insert (DHFR F[3]):vector ratio of 4:4:1.
Strain Construction
Strains were constructed in BY4741 and BY4742 for the DHFR F[1,2] and DHFR F[3] fusions respectively (Table S2A). All fusions were performed at the 3′ end of genes. 2x/3x/4xL-DHFR F[1,2]/F[3] fragments along with the natMX4 (for DHFR F[1,2]) or hphMX4 (for DHFR F[3]) resistance modules (respectively for resistance to clonNAT and HygB) were amplified by PCR from their respective plasmid with oligonucleotides specific to the gene to be fused with the DHFR fragments (supplemental Table S2B-strain construction). BY4741 and BY4742 competent cells were transformed with the amplified modules following standard procedures and selection was performed on YPD+clonNAT (DHFR F[1,2]-tagged strains) or YPD+HygB (DHFR F[3]-tagged strains). PCR and Sanger sequencing for all strains confirmed proper DHFR fragment fusions.
Estimation of Protein Abundance
Protein quantification was performed for several strains with proteins fused with the 2xL and 4xL by Western blot, focusing on proteins for which antibodies directed against the proteins were available. Twenty OD600 of exponentially growing cells were resuspended in 200 μl of water containing peptidase inhibitors (1 mm PMSF, 0.7 μg/ml Pepstatin A, 0.5 μg/ml Leupeptin and 2 μg/ml Aprotinin). 425–600 μm glass beads (Sigma, Darmstadt, Germany) were added (0.1 g) and cells were vortexed using a TurboMix attachment (Scientific Industries, Bohemia, NY) for 5 min. After addition of 1% SDS, samples were boiled and supernatants were transferred in a new tube. Protein extracts equivalent to 0.1 OD600 of cells were separated on 8% (Vps35p) or 10% (Vps5p, Vps17p, Pep8p, Vps29p and Bcy1p) SDS-PAGE gel and transferred on a nitrocellulose membrane using a TE 77 PWR semi-dry device (Amersham Biosciences, Mississauga, ON, Canada). After saturation in Odyssey® Blocking Buffer (PBS) overnight at 4 °C, membranes were probed with Rabbit anti-Vps5p, anti-Vps17p, anti-Vps26p, anti-Vps29p, anti-Vps35p (kindly provided by M. N. J. Seaman) (1:2000), Goat anti-Bcy1p (1:1000) or Mouse anti-Actin (as a loading control; 1:5000) in Blocking Buffer + 0.2% Tween 20 during 2 h at room temperature. After three 10 min washes in PBS + 0.2% Tween 20, membranes were secondly probed with IRDye®680RD Goat anti-Rabbit IgG (1:10000), IRDye®680RD Donkey anti-Goat IgG (1:5000) or IRDye®800CW Goat anti-Mouse IgG (1:10000) in Blocking Buffer + 0.02% SDS + 0.2% Tween 20. Three washes of 10 min in PBS + 0.2% Tween 20 were performed and signal on membranes was detected using Odyssey® Fc Imaging System (LI-COR®, Lincoln, Nebraska). Quantifications were performed with Image StudioTM Lite software.
Protein-fragment Complementation Assays
For the global PCA experiment, baits consisted of 15 proteins fused to 2x/3x/4xL-DHFR F[1,2] that are part of seven complexes. Prey proteins fused to the 2xL-DHFR F[3] (495 strains) were selected according to the criteria that they were belonging to the same complexes as the baits (20) or that they were PPI partners of one of them based on data reported in BioGRID in October 2014 (26). A random set of 97 strains corresponding to proteins found in the cytoplasm or the nucleus was also included in the set of preys as controls. Each prey was present in four replicates, two on each prey plate, so each bait-prey pair was assayed four times. Preys were randomly positioned to avoid location biases.
To assess the specificity of the PCA experiment, we selected 13 baits that are members of large complexes (Nuclear Pore Complex (Nup100), Proteasome (Rpt6) and RNApol complexes (Rpo26)), of small complexes (Ccr4-Not (Pop2, Not3), PKA (Bcy1), COG complex (Cog6, Cog4, Cog1, Cog7) and retromer (Vps35, Vps5)) or that are not members of a known stable complex (Sla2). These baits were fused to the 2xL and 4xL-DHFR F[1,2] and were screened against the entire DHFR F[3] library of preys (n = 3600 after filtering for missing data and preys that interact non-specifically with baits). Each bait-prey pair was assayed once but replicated in an independent experiment for a subset of the PPIs observed (see below).
For the intracomplex experiment, we performed a review of the literature and considered the consensus protein complexes published by (20) to select 95 central and associated proteins members of the following complexes: the RNApol I, II and III, the proteasome, and the COG complex. These complexes were selected because they vary in size (RNApol I (n = 14), II (n = 12), III (n = 17), and associated proteins (n = 9, 7 tested), proteasome (n = 47, 44 tested), and COG complex (n = 8)) and PPIs among protein members of these complexes have been shown to be detectable at least partially by DHFR PCA (7) based on data available in (26). In addition, there are published structures available for the RNApol and proteasome complexes, making it possible to compare our results with known protein complex organization. We successfully constructed 80.0% and 76.6% of the strains in MATa and 65.0% and 70.2% in MATα for the RNApol and proteasome respectively and 100% for the COG complex. In total, 286 strains harboring proteins fused to 2xL/4xL-DHFR F[1,2] and/or 2xL/4xL-DHFR F[3] were used, a representation of 89.5% (85 out of the 95 proteins selected at first are tagged with 2xL and 4xL in at least one mating type) of the proteins. MATα 2xL/4xL-DHFR F[3] cells were used as baits. Two different prey plates of MATa cells were generated including all strains mentioned above. Baits and preys were positioned so that in a block of four strains all combinations of linker sizes could be tested for a specific interaction: 2xL−2xL4xL−2xL 2xL−4xL4xL−4xL. Each block of bait-prey pair was present in 14 replicates for the RNApol and COG complexes, and in 16 replicates for the proteasome complex. The blocks were randomly positioned on the colony arrays. Each 1536-array was finally designed to contain a double border of a strain showing a weak interaction (Pop2-2xL-DHFR F[1,2]-Arc35-2xL-DHFR F[3]) to avoid any border effects on the growth of the colonies.
Bait plates were first prepared from 10 ml saturated cultures in YPD+clonNAT (for MATa cells) or YPD+HygB (for MATα cells) that were plated on YPD Omnitray plates and incubated at 30 °C for 24 h. Cells were then printed on a 1536-array with a 1536-pin (or a 384-pin) replicating tool manipulated by a BM3-BC automated colony processing robot (S&P Robotics, North York, ON, Canada) and incubated for another 24 h at 30 °C. In parallel, two prey plates for each the global and intra complex experiments were assembled by arraying strains onto specific positions in a 96-format with a re-arraying tool. Colonies were further condensed in 384-format arrays and finally, in 1536-format arrays using a 96-pin and 384-pin replicating tool respectively. For the specificity experiment, four prey plates were obtained by printing and condensing the entire DHFR F[3] strains collection with 96-pin and 384-pin replicating tools. All 1536-format prey plates were replicated a few times with a 1536-pin replicating tool to have enough cells to perform crosses with all of the individual baits. Once having bait and prey plates, each 1536-bait plate was crossed with the two or four 1536-prey plates with a 1536-pin replicating tool on YPD and incubated for 2 days at 30 °C. Two rounds of diploid selection were performed on YPD+clonNAT+HygB with an incubation time of 2 days at 30 °C per round. Finally, diploid strains were replicated on MTX medium and incubated at 30 °C for 4 days, after which a second round of MTX selection was performed. Plates were incubated at 30 °C for another 4 days. Images were taken with an EOS Rebel T3i camera (Canon, Tokyo, Japan) each day from the second round of diploid selection to the end of the experiment.
For the global and specificity PCA experiments, we confirmed respectively by standard DHFR PCA 25 PPIs with increased signal, that did not change or that decreased (supplemental Fig. S2A) and 18 newly detected PPIs with the 4xL (supplemental Fig. S2B). The same procedure as described above was used to assess the growth on MTX medium of selected diploid cells resulting from a new cross between bait and prey strains. For the intra-complexes experiment, we confirmed results for 10 PPIs by measuring cell growth in a spot-dilution assay (supplemental Fig. S2C). Briefly, precultures of diploid cells expressing 2xL/4xL-DHFR fragments fusions to proteins of interest were adjusted to an OD600/ml of 1 in water. 5-fold serial dilutions were performed and 6 μl of each dilution were spotted on MTX and DMSO DHFR PCA media. Plates were incubated for 7 days at 30 °C and subsequently imaged with an EOS Rebel T3i camera (Canon, Tokyo, Japan).
PCA Images and Statistical Analyses
For the initial screen, colony size was estimated by measuring number of pixels using the integrated intensity function as implemented in a custom script in ImageJ64 1.44o. We applied an image correction where the intensity of each pixel was extracted and the pixel intensity matrix was smoothened using a two-way median polish and averaged with the raw image. We then converted the images to binary files and a manual threshold was applied across plates. We selected colonies for measurement with a circular selection using particle detection with the built-in function Analyze particle in ImageJ64. We excluded particles touching the edge of the selection and those that had an area inferior to 20 pixels and circularity inferior to 0.5 using the particle that is closest to the center. We considered the particle as being a colony if the mass center was within the mid-distance among two colonies.
Colony intensity values from day 4 of growth of the second MTX selection were log2 transformed after adding 1 to each value to avoid null values. All colonies with a size smaller than 16 on the diploid selection plate were eliminated.
For the global PCA experiment, bait-prey pairs with at least two replicates for all linker combinations (2xL-2xL, 3xL-2xL, 4xL-2xL) were conserved and the median of colony size was used as the interaction score (Is). Distribution of Is was modeled as a mixture of two normal distributions using the R package mixtools (function NormalmixEM) (supplemental Fig. S1B). The estimated mean (μb) and standard deviation (sdb) of the background distribution were used to convert Is into z-scores (Zs = (Is - μb)/sdb)). Bait-prey pairs with a Zs greater than 2.5 were considered as significant, detected PPIs. These Zs were used to compare the same bait-prey pairs with different linker size combinations. We considered significant changes when Zs differed by more than 2.
In the specificity PCA experiment, the set of true positive (TP) and negative (TN) PPIs for benchmarking were identified as follows. The TP reference set was derived from the BioGRID (26). Interactions were considered positive if they were detected either by (1) PCA and AP-MS, (2) Y2H and AP-MS, (3) PCA and Y2H, (4) from crystal structure, or from (5) any other method and the homologous proteins were known to interact in H. sapiens or (6) S. pombe or (7) that show physical interactions with any method in addition to genetic interactions in S. cerevisiae. Negative interactions to identify a TN set are more challenging to derive because it is impossible to completely reject that proteins might be associating. We identified candidate negative interactions as protein pairs that were not reported to interact in BioGRID physically nor genetically, that have homologs that do not interact in H. sapiens and S. pombe and that do not share GOslim go terms for molecular functions, biological process or cellular component. This led to a set of 118 positive interactions and 9283 negative interactions that were tested in this experiment, a 79-fold enrichment for negative interactions. The data was analyzed as described before by computing Zs for the 2xL and 4xL and measuring the fraction of true positives, true negatives, false positive, false negatives and total number of interactions as a function of Zs for the two linker sizes. We considered bait-prey pairs with a Zs ≥ 3 as a conservative threshold for calling a detected PPI. All strains located on the plate borders were filtered out along with preys that interact non-specifically with the DHFR fragments (3, 7).
For the intracomplex experiments, extreme outlier replicates on the MTX selection plates that were more distant from the median than Q1–3*(Q3-Q1) or Q3 + 3*(Q3-Q1) were excluded (Q1 and Q3 represent first and third quartiles). Colonies corresponding to the control PPI and positioned on the array edges were removed from downstream analyses as well as strains for which sequencing results revealed mutations in the DHFR fusion proteins. After these final filtering steps, bait-prey pairs with at least four replicates for every linker combinations were conserved and the median of colony size was used as the Is. Significant PPIs were identified as described above (supplemental Fig. S1D). For the RNApol and the proteasome, the estimated mean (μb) and standard deviation (sdb) of the background distribution were calculated for each linker combination and each complex separately. For the COG complex, because the number of bait-prey pairs is limited to 64, all the results were combined to calculate these parameters. A PPI was considered as being detected when the Zs was greater than 2.5. From the 236 bait-prey pairs presenting detected PPIs with at least one linker combination, some pairs were filtered out, mainly because they did not pass all the thresholds or because the fusion strains (Taf14 and Spt5 fused to DHFR F[3]) presented incoherent results for all tested interactions, leaving us with a total of 228 detected PPIs (197 unique) (supplemental Table S2E-detected PPIs only). At this step, bait-prey pairs presenting a new PPI (i.e. the PPI was not detected with the reference linker size (2xL-2xL) but was detected with a longer linker combination) were separated from others and classified as new PPIs. For the remaining pairs, Is values for the different linker size combinations could be compared directly. The difference with the reference 2xL-2xL Is was calculated for each linker combination: 2xL-4xL, 4xL-2xL and 4xL-4xL. A paired t-test was used to discriminate significant difference in colony size (with FDR corrected p-values). These PPIs were separated in two additional categories: unchanged interactions, in cases where the PPI was detected with the reference linker size (2xL-2xL) and also with the longer linker combinations but without any significant change (t-test FDR p-value above 0.05), and PPIs that changed quantitatively, in cases where the PPI was detected with the reference linker size (2xL-2xL) and presented significant changes for at least one longer linker combination (difference greater than 1 (positively changed) or smaller than -1 (negatively changed) with t-test FDR p-value < 0.05) (Table S2E-detected PPIs only).
All PPIs data related to the global PCA, specificity PCA and intra-complex experiments can be found in supplemental Table S2C–S2E.
Analysis of Protein Distances Within Complexes
Yeast protein sequences of the RNApol I, II and III were obtained from SGD (http://www.yeastgenome.org) and queried through the RNApol I, II and III protein complexes present in the RCSB protein data bank (http://www.rcsb.org) using USEARCH. PDB files 4C3I, 4V1N and 5FJA were selected as representative monomeric complexes for the RNApol I, II and III respectively as they included the largest number of proteins from the experimental set with the highest sequence identities. Similarly, structure 4C2M was selected as the representative RNApol I dimeric complex. Table S3A presents the identity between each RNApol structures and the experimental sequences.
The proteasome is composed of three sections, the barrel-shaped core particle, the base and the lid (supplemental Fig. S3A, top). There was no complete structure of the yeast proteasome complex in the RCSB protein data bank at the time of the analyses. Sequence alignment of the experimental protein sequences of the individual sections of the proteasome complex with the sequences of the RCSB protein data bank identified PDB IDs 5A5B and 5CZ4. Structure PDB ID 5A5B is composed of the base, the lid and half of the core. Structure PDB ID 5CZ4 is composed of a full core. A complete proteasome structure was built by superposing two PDB 5A5B structures onto the structure of 5CZ4, one on each side of the CP using the super command in PyMOL. Visual inspection of the resulting superposed 5A5B structures showed an incorrect overlap in the central core (supplemental Fig. S3B). This overlap is well solved in 5CZ4. Thus, the final proteasome structure was composed of 5A5B for the base, the lid and the outer rings of the core. The inner rings of the core were from structure 5CZ4. Supplemental Fig. S3A summarizes the methodology used to build the final proteasome structure. Supplemental Table S3B presents the identity between the built structure and the experimental sequences.
The distances between the different proteins within a complex were calculated between C-terminal residues (supplemental Table S3C). In several cases, the structure of the protein is not complete in the C-terminal section. In these cases, the last available residue was used instead to calculate the distance (a list is provided in supplemental Table S3D). The distances were calculated from the weighted shortest path using the dijkstra algorithm as implemented in NetworkX (example of shortest path between Scl1p and Rpn5p is presented in supplemental Fig. S3C). Surface residues Cα were used as nodes to build the graph. The edges of the graph were placed between each pair of nodes using a distance cutoff of 15 Å for the RNApol II and of 30 Å for the proteasome. The weight of the edges was equal to the distance between node pairs. Surface residues were identified as follows. First, the structure of the protein complex was represented using the show dots and set dots_solvent commands in PyMOL, using a solvent radius of 10 Å for the RNApol II complex and of 20 Å for the proteasome, respectively. These dots were exported in the “.wrl” graphic file format. From this file, each dot coordinates were extracted. Residues within 15 Å of any dot of the RNApol II structure, and within 20 Å of the proteasome structure, were considered as surface residues (see supplemental Fig. S3D for a representation of the method for the proteasome). In cases where multiple copies of the proteins were present within the complexes, the mean of the minimal distances possible was used for the analyses.
RESULTS AND DISCUSSION
Longer Linkers Increase Signal-To-Noise Ratio in Large-scale Screens
The standard linker used in DHFR PCA consists of two repetitions of the peptide GGGGS (7), which we refer to as the 2x-linker (2xL). We modified existing plasmids to include three and four repetitions of this sequence (referred to as 3xL and 4xL) and used them as PCR template for both complementary DHFR fragments (DHFR F[1,2] and DHFR F[3]) to be introduced in yeast (supplemental Table S2A for strains used in this study). We assessed whether longer linkers destabilize proteins and therefore interfere with the detection of PPIs. We could test this effect only for proteins for which we had antibodies, but because we found no evidence of protein degradation for the six proteins tested (supplemental Fig. S1A), we conclude that if linker length affects protein stability, it has a minor effect that is not generalized.
To verify the effect of longer linker length on the detection of PPIs by DHFR PCA (7), we constructed reporter strains for 15 proteins that are part of seven complexes with the 2xL, 3xL, and 4xL fused to the DHFR F[1,2] fragment. Using high-density yeast colony arrays (37), we queried these baits (n = 45) against 592 prey proteins fused to DHFR F[3] (with regular 2xL). These include proteins known to interact with the baits, that are within the same complexes as the baits or that are random proteins used as controls, for a total of 26,640 bait-prey pairs (Table S2C).
We detected 99, 110 and 126 PPIs (Zs greater than 2.5) with the 2xL, 3xL, and 4xL respectively (supplemental Fig. S1B), revealing a significant increase in signal-to-noise ratio with longer linkers, particularly for the 4xL. Four and seven PPIs showed greater than 2-fold Zs differences with the 3xL (two decreases, two increases) and the 4xL (seven increases) as compared with the 2xL assay (Fig. 1A). The decreased signals may represent steric effects because of the fusion of the DHFR fragments or reduce protein stability in some specific cases. Four out of nine cases of increased PPI signal were reported by AP-MS (38) but not by PCA with standard linkers, showing that longer linkers may allow for the detection of PPIs that have C-termini that are further in space, among proteins that do not have a binding interface, or simply increase signal-to-noise ratio for PPIs that are not detectable with the standard linker. Moreover, the four PPIs with the strongest PCA signal represent cases between baits and preys within the same complexes, suggesting that there is no decrease in specificity with the elongated linkers. Finally, the cases where proteins were not members of the same complex or were not previously shown to interact may represent false positives. However, they likely reflect actual PPIs not detected before in living cells because they are supported by independent observations. For example, many genetic interactions and PPIs (in vitro and in vivo) (39, 40) have been described between the actin cytoskeleton and the proteasome. Here, we detect some PPIs in living cells among subunits of these complexes such as between Arc18 and Pup1 (supplemental Table S2C). All of these results thus show that the DHFR PCA with increased linker length reveals new PPIs and could be an improved tool to study PPIs among complexes, although these specific cases may require further investigation. Because the 4xL has a better ability to detect PPIs compared with the 3xL, we focused on this linker size for the remainder of the experiments.
Fig. 1.

Longer linkers increase signal-to-noise ratio in large-scale Protein-fragment complementation assay (PCA) screens and allow the detection of new PPIs. A, PPIs Zs (representing a quantitative deviation from the background noise) obtained in a large-scale screen using baits fused to the DHFR F[1,2] fragment with a 3xL (left) and a 4xL (right) compared with a 2xL. PPIs with a significant difference are highlighted with red triangles (3xL) and squares (4xL). B, Distribution of colony size in the DHFR PCA assay with the 2xL and 4xL. 13 baits were screened against the DHFR F[3] yeast collection (n = 3600) with both the 2xL and 4xL (26 baits total). The distribution of normalized colony sizes (interaction score, Is) is shown for a set of computationally identified most likely true negative and positive interaction partners, TN and TP respectively. The two modes of the bimodal distribution of colony sizes for the TP represent PPIs that are detected (highest mode) and those that cannot be distinguished from the background growth (lowest mode). The signal-to-noise ratio, illustrated as the difference between the medians of the TP over the TN, increases with the 4xL. C, Precision and number of detected PPIs as a function of Zs thresholds. Precision, or fraction of TP, was calculated as the ratio of TP over all positives (TP + FP) at various values of Zs for the 2xL and 4xL. A conservative threshold corresponding to a Zs of 3 is indicated by a dotted vertical line on the graph. D, Retromer PPI network. Colored nodes: retromer subunits. Blue nodes: Vps35 interaction partners. Black lines: all known PPIs between Vps35 and its interaction partners and among these interaction partners. Red lines: newly detected PPIs with Vps35 in the large-scale experiment with the 4xL and confirmed in small scale experiments.
To specifically test the sensitivity and specificity and the ability to uncover new PPIs with the 4xL in standard PCA large-scale screens (37), we screened 13 baits fused to the DHFR F[1,2] with the entire DHFR F[3] libraries of preys (n = 3,600 after filtering). These baits are members of large complexes such as the Nuclear Pore Complex (Nup100), the Proteasome (Rpt6) and the RNApol complexes (Rpo26) and small complexes, namely the Ccr4-Not complex (Pop2, Not3), the PKA (Bcy1), the COG complex (Cog6, Cog4, Cog1, Cog7), the retromer (Vps35, Vps5) or not a member of a known stable complex (Sla2). For this set of baits, we identified a set of true positive (TP) and true negative (TN) PPIs (see methods) so we could estimate and compare the precision, sensitivity and specificity of the two linker sizes in a genome-wide screen. We observed that positive PPIs and negative PPIs are clearly distinguishable from background growth based on colony size alone, with a larger difference in colony size between the TP and TN for the 4xL (Fig. 1B). We also observed that the signal-to-noise ratio was higher for the 4xL than from 2xL, with larger Zs in 65% of cases for the TPs. Using this set of baits, we find that at a Zs of 3 based on colony size analysis, the two linker sizes achieve the same precision (89%) but the number of PPIs detected is larger for the 4xL (187 versus 149), confirming the significant increase in sensitivity observed above (Fig. 1C). At this threshold, the majority of the 2xL PPIs are also detected with the 4xL (137/149). Among the 50 PPIs that are specific to the 4xL at a Zs of 3, 1 is a TP, 3 were previously reported PPIs not considered as TP based on the criteria described in the method and 8 are unknown PPI partners but are known to interact genetically, confirming their functional relationships, which can be better detected here with the 4xL. The performance in terms of sensitivity and specificity is also comparable for the two-linker sizes (supplemental Fig. S1C).
The increase in sensitivity with the 4xL allowed to detect previously not reported PPIs (supplemental Table S2D). We re-rested 18 of those in a highly replicated experiment (n = 11), which also included negative and positive PPIs, to confirm that these were not false positives in the large-scale assay and that the signal was stronger with the 4xL. These 18 novel PPIs appear to be of intermediate intensity between negative and positive controls (supplemental Fig. S2B left panel), which explains why they may have been missed before. The confirmation shows the increased signal for the 4xL for these hits and a slight increase for the positive controls but not for the negative controls (supplemental Fig. S2B right panel). Note that these PPIs are also visible with the 2xL in the confirmation experiments but they are closer to the background distribution (Zs<3) in the large-scale screening because of the lower signal-to-noise ratio.
These novel PPIs include Atg20, Tms1, Ypq2, Vma7, Atg27, and YML018C as partners (supplemental Table S2D-new PPIs only) for the retromer complex subunit Vps35, many of which were previously known to interact with other retromer subunits (Fig. 1D). Atg20 is a sorting nexin family member that was not known before to interact with the retromer but like the Vps5 retromer subunit, it is a phosphatidylinositol-3-phosphate binding protein that locates to the endosome (41). Further analyses will be required to examine whether the PCA signal comes from direct association or proximate co-localization. Another set of novel PPIs includes partners of the RNase Pop2, which is a subunit of the Ccr4-Not complex (supplemental Table S2D-new PPIs only). These novel PPIs involve a partner with stress granule components Pbp1 and Scd6, a protein that contains Lsm domains, that associates with P-bodies and that forms foci during DNA replication stress (42). These results confirm that the added sensitivity of the 4xL allows for the identification of novel PPIs across various conserved eukaryotic cellular processes.
PCA Signal Reflects The Superorganization of Protein Complexes
To examine the effect of a longer linker on the detection of PPIs among members of complexes, we selected five complexes (RNApol I, II and III, proteasome and COG complexes), which differ in their number of subunits. We used four combinations of linker lengths (2xL-2xL, 2xL-4xL, 4xL-2xL, 4xL-4xL) for all proteins within a complex. As a negative control, tests for PPIs between the RNApol and the COG complex were also performed. Among the 10,192 unique pairs of baits and preys tested, 755 were considered as showing PPIs when considering all linker sizes (supplemental Fig. S1D and supplemental Table S2E), representing PPIs among 228 protein pairs (197 unique - reciprocal interactions such as X-DHFR F[1,2]-Y-DHFR F[3] and Y-DHFR F[1,2]-X-DHFR F[3] accounting for only one PPI) after filtering (supplemental Table S2E-detected PPIs only).
As expected, no significant signal was detected between the RNApol and COG proteins. Reciprocal PPI signals were correlated, as previously noted (7) (supplemental Fig. S1E - 4xL-4xL PPIs). Also, for almost 60% of PPIs (135/228 or 114/197 unique), no significant change in the PCA signal was observed when using the 4xL compared with the 2xL, reinforcing the fact that no overall decrease in specificity is seen with the elongated linkers. However, the increased linker length had an obvious impact for 93 (83 unique) PPIs (Fig. 2). PCA signal was indeed quantitatively changed for 19 (18 unique) PPIs and 74 (65 unique) new PPIs were detected using at least one 4xL. Thus, doubling the linker length can substantially widen the repertoire of detected PPIs within complexes in living cells.
Fig. 2.
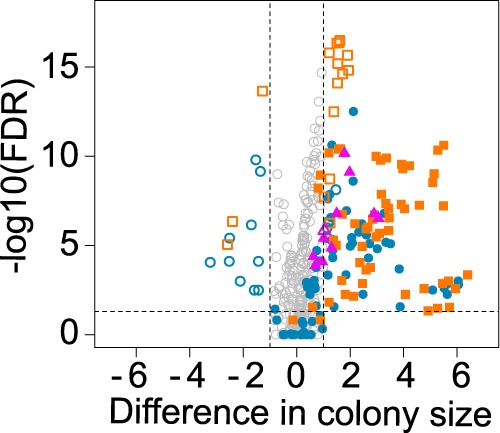
PCA signal increases using a longer linker. Detected PPIs after data filtering for the intra complex PCA experiment. Blue circles: RNApol I, II, and III. Orange squares: proteasome. Purple triangles: COG complex. Empty shapes: quantitatively changed PPIs (significantly decreased or increased when compared with 2xL-2xL reference interactions). Solid shapes: new PPIs (PPI not detected with the 2xL-2xL reference linker but detected with a longer linker combination). The p-values are from FDR-corrected t-tests on colony sizes.
In general, having only one longer linker (mainly 4xL-DHFR F[1,2]) was sufficient for the detection of new PPIs or to increase the PCA signal of a previously detected one (2xL-4xL compared with 2xL-2xL). However, the signal was often improved with the 4xL-4xL combination. In rare cases, increasing linker length had an opposite effect, leading to PPI loss or signal reduction. Rpo21 was particularly affected. This protein, one of the two largest components of the RNApol II, contributes to five out of the nine quantitatively decreased PPIs. PPIs involving Rpo21–4xL with its main partners are preserved (Rpb2 and Rpb3 (43)) but all the others are lost. If these losses were because of complete protein destabilization, one would expect all PPIs involving Rpo21 to be lost but this is not the case. Other mechanisms could therefore be involved, for instance partial destabilization that reduces signal below the detection threshold for some PPIs and not others, or steric effects of the 4xL that affects the PPIs directly.
Quantitative changes were observed for about 5–10% of the detected PPIs across complexes. However, a larger proportion (about 30–40%) of new PPIs were detected for the RNApol complexes compared with the proteasome and the COG complex (Fig. 3A). Within the RNApol complexes, more than half of the new PPIs were found among proteins common to the three polymerases (Rpb5, Rpb10 and Rpo26) and proteins specific to each of the individual polymerase (Fig. 3B left panel). In the proteasome, five new PPIs involved Nas6, an assembly chaperone for the proteasome, and proteins from the base subunit (Fig. 3B center panel). In the COG complex, new PPIs were seen between Cog1 from the core subunit and proteins from the lobe a or lobe b (Fig. 3B right panel). All these results show that doubling the linker length of central proteins in complexes expands the range of PPIs detected by DHFR PCA and helps to better describe the organization of protein complexes in living cells.
Fig. 3.

The use of a longer linker proves to be useful to infer the super-organization of protein complexes. A, Proportions of quantitatively changed PPIs and new PPIs versus unchanged PPIs for all complexes considering every reciprocal interactions such as X-DHFR F[1,2]-Y-DHFR F[3] and Y-DHFR F[1,2]-X-DHFR F[3] as a single PPI. B, Circle plots of all detected PPIs for each complex. Line thickness is proportional to the difference between the 4xL-4xL and 2xL-2xL PCA signal for each PPI. Gray lines: unchanged PPIs. Green lines: negatively changed PPIs. Pink lines: positively changed and new PPIs. Stripe patterns inside the colored boxes represent proteins that were absent from the experiment. C, Proportion of PPIs detected for each combination of subcomplexes within complexes.
In addition to uncovering new PPIs, PCA signal using longer linkers allowed a better discrimination among the different subunits of large complexes. This is particularly well illustrated with the proteasome (Fig. 3B and 3C center panel). More PPIs are detected when the two proteins are in the same subcomplex (such as base-base, core-core and lid-lid) regardless of the linker length though the fraction is systematically higher with longer linkers. The same trend is observed for the RNApol and COG complexes (Fig. 3B and 3C left and right panels). Structural biology in living cells could thus gain from PPI data obtained with several linker lengths.
Longer Linkers Allow the Detection of PPIs Among More Distant Proteins Within Complexes
Because structural data for the RNApol and proteasome complexes were available, we could test whether PCA signal with longer linkers reflects, at least partly, the proximity of proteins within complexes, as suggested by the analysis on subcomplexes (Fig. 3C). As a proxy for distance, we measured the shortest path among C-termini of the proteins of interest at the surface of the complexes (supplemental Table S3C). We illustrate some of the PPIs and the distances among the C-termini of the proteins involved in (Fig. 4A). For the proteasome, the complex for which we have the most distance values, a negative correlation is observed between the pairwise distance and Zs of PPIs across the range of distances and all linkers (Fig. 4B left panel). The enhanced ability to detect PPIs at larger distance values with the 4xL is clearly visible from the cumulative distribution of Zs as a function of pairwise distances, where positive Zs accumulate to a longer distance for the 4xL-4xL combination than the other combinations (Fig. 4B right panel). The density distribution of distances within complexes is also slightly shifted toward larger distances for 4xL, showing that greater distances are better detectable with these linkers sizes (supplemental Fig. S1F). Finally, we find that distance among the C-termini of proteins is significantly longer for cases in which the 4xL increases signal or leads to the detection of new PPIs (Fig. 4C). This demonstrates once again that the 4xL enhances the ability to detect PPIs, especially for proteins that are more distant in space. These result are consistent with the observation made with a luminescent PCA in which the relationship among the signal-to-noise ratio was maximized with the longest linker sizes examined, in this case a 15 amino acid peptide (35).
Fig. 4.

Longer linkers allow for the detection of PPIs among more distant proteins within complexes. A, Structures of the RNApol I, II, and III and of the proteasome. Green: proteins shared by at least two out of the three RNApol. Dark red: proteasome catalytic subunit. Red: proteasome base. Orange: proteasome lid. Proteins located at different distances or in different subunits are highlighted on each structure. Examples of distances between C-termini of these proteins and their associated PPI Zs. PPIs detected with longer linkers but not with the 2xL are indicated in the tables. DHFR fragments have also been modeled and are presented at the same scale as the proteasome structure. B, (Left) Correlation between all of the detected PPIs in the proteasome (Zs) and the distance between the C-termini (2xL-2xL: Spearman r = -0.34, p-value = 2.249e-15; 2xL-4xL: r = -0.36, p-value < 2.2e-16; 4xL-2xL: r = -0.36, p-value < 2.2e-16; 4xL-4xL: r = -0.40, p-value < 2.2e-16). Data were binned into 10 classes for representation only, the correlation tests were performed on raw data. (Right) Distribution of cumulative Zs for the proteasome PPIs as a function of protein pairwise distances. C, Pairwise C-termini distances for the PPI that are not affected by linker length, those that have enhanced interaction score (Is) and those that were not detected with the 2xL but are detectable with the 4xL. Data from the RNApol and proteasome complexes are combined in these analyses. p-values of Wilcoxon tests are shown.
CONCLUSION
Understanding the molecular organization of the cell at the scale of protein complexes (27) remains challenging largely because it is difficult to study how proteins interact directly and indirectly in vivo. Making progress requires that we adapt or develop tools to detect and measure protein proximity in living cells and among endogenously expressed proteins. Much of our knowledge of cell biology, including on protein interaction networks and protein localization, derives from reporter proteins that require the use of short peptides to link proteins and protein fragments together (44). The effect of the length of these linkers on assay performance is rarely established. This could have important consequences on large-scale assays, whose performance is often affected by high rates of false negatives.
Here we show that DHFR PCA, with a modest increase in linker size from 41 Å to 82 Å, can be used to detect PPIs in these specific conditions with an increased signal-to-noise ratio and with an enhanced ability to detect PPIs among distant proteins, including PPIs among complexes and subcomplexes within large complexes. It would be difficult to assess the relative contribution of the enhanced ability to detect PPIs between distant proteins and increased signal-to-noise for the 4xL with the data in hand but both are affected. Because only a single longer linker is generally sufficient to detect new PPIs, the current strains from the DHFR PCA collection could directly be used as preys while requiring only the construction of baits with different linker sizes. The DHFR PCA is therefore an additional method that allows to measure low resolution structural information among subunits of complexes, and complements other methods that are amenable to analysis performed in living cells, for instance chemical cross-linking of protein complexes (45), FRET-based analyses (46) and Bio-ID proximity-dependent biotinylation in mammalian cells (47). Despite major advances in these other technologies in the recent years, PCA still remains the simplest assay because it requires minimal infrastructure investment and can be adapted for high-throughput screening, which is still difficult to achieve with other approaches.
Supplementary Material
Acknowledgments
We thank the members of the Landry laboratory, Yves Bourbonnais and Nick Bisson for feedback on the manuscript and Marie Filteau for guidance on the statistical analyses.
Footnotes
* This work was supported by the Canadian Institute of Health Research Grants 299432 and 324265 to CRL. CRL holds the Canadian Research Chair in Evolutionary Cell and Systems Biology. AEC was supported by fellowships from CIHR and FRSQ. CL was supported by a NSERC NRSA Scholarship and PCD by a CIHR/ICS-IG summer studentship award.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PPIs
- Protein-protein interactions.
REFERENCES
- 1. Vidal M., Cusick M. E., and Barabasi A. L. (2011) Interactome networks and human disease. Cell 144, 986–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Diss G., Dube A. K., Boutin J., Gagnon-Arsenault I., and Landry C. R. (2013) A systematic approach for the genetic dissection of protein complexes in living cells. Cell Rep. 3, 2155–2167 [DOI] [PubMed] [Google Scholar]
- 3. Gagnon-Arsenault I., Marois Blanchet F. C., Rochette S., Diss G., Dube A. K., and Landry C. R. (2013) Transcriptional divergence plays a role in the rewiring of protein interaction networks after gene duplication. J. Proteomics 81, 112–125 [DOI] [PubMed] [Google Scholar]
- 4. Vo T. V., Das J., Meyer M. J., Cordero N. A., Akturk N., Wei X., Fair B. J., Degatano A. G., Fragoza R., Liu L. G., Matsuyama A., Trickey M., Horibata S., Grimson A., Yamano H., Yoshida M., Roth F. P., Pleiss J. A., Xia Y., and Yu H. (2016) A Proteome-wide Fission Yeast Interactome Reveals Network Evolution Principles from Yeasts to Human. Cell 164, 310–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Obado S. O., Brillantes M., Uryu K., Zhang W., Ketaren N. E., Chait B. T., Field M. C., and Rout M. P. (2016) Interactome Mapping Reveals the Evolutionary History of the Nuclear Pore Complex. PLos Biol. 14, e1002365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arabidopsis Interactome Mapping, C. (2011) Evidence for network evolution in an Arabidopsis interactome map. Science 333, 601–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tarassov K., Messier V., Landry C. R., Radinovic S., Serna Molina M. M., Shames I., Malitskaya Y., Vogel J., Bussey H., and Michnick S. W. (2008) An in vivo map of the yeast protein interactome. Science 320, 1465–1470 [DOI] [PubMed] [Google Scholar]
- 8. Filteau M., Vignaud H., Rochette S., Diss G., Chretien A. E., Berger C. M., and Landry C. R. (2015) Multi-scale perturbations of protein interactomes reveal their mechanisms of regulation, robustness and insights into genotype-phenotype maps. Brief Funct. Genomics 15, 130–137 [DOI] [PubMed] [Google Scholar]
- 9. Sahni N., Yi S., Taipale M., Fuxman Bass J. I., Coulombe-Huntington J., Yang F., Peng J., Weile J., Karras G. I., Wang Y., Kovacs I. A., Kamburov A., Krykbaeva I., Lam M. H., Tucker G., Khurana V., Sharma A., Liu Y. Y., Yachie N., Zhong Q., Shen Y., Palagi A., San-Miguel A., Fan C., Balcha D., Dricot A., Jordan D. M., Walsh J. M., Shah A. A., Yang X., Stoyanova A. K., Leighton A., Calderwood M. A., Jacob Y., Cusick M. E., Salehi-Ashtiani K., Whitesell L. J., Sunyaev S., Berger B., Barabasi A. L., Charloteaux B., Hill D. E., Hao T., Roth F. P., Xia Y., Walhout A. J., Lindquist S., and Vidal M. (2015) Widespread macromolecular interaction perturbations in human genetic disorders. Cell 161, 647–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sahni N., Yi S., Zhong Q., Jailkhani N., Charloteaux B., Cusick M. E., and Vidal M. (2013) Edgotype: a fundamental link between genotype and phenotype. Curr. Opin. Genet. Dev. 23, 649–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diss G., Gagnon-Arsenault I., Dion-Cote A. M., Vignaud H., Ascencio D. I., Berger C. M., and Landry C. R. (2017) Gene duplication can impart fragility, not robustness, in the yeast protein interaction network. Science 355, 630–634 [DOI] [PubMed] [Google Scholar]
- 12. Yang X., Coulombe-Huntington J., Kang S., Sheynkman G. M., Hao T., Richardson A., Sun S., Yang F., Shen Y. A., Murray R. R., Spirohn K., Begg B. E., Duran-Frigola M., MacWilliams A., Pevzner S. J., Zhong Q., Trigg S. A., Tam S., Ghamsari L., Sahni N., Yi S., Rodriguez M. D., Balcha D., Tan G., Costanzo M., Andrews B., Boone C., Zhou X. J., Salehi-Ashtiani K., Charloteaux B., Chen A. A., Calderwood M. A., Aloy P., Roth F. P., Hill D. E., Iakoucheva L. M., Xia Y., and Vidal M. (2016) Widespread expansion of protein interaction capabilities by alternative splicing. Cell 164, 805–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bisson N., James D. A., Ivosev G., Tate S. A., Bonner R., Taylor L., and Pawson T. (2011) Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat. Biotechnol. 29, 653–658 [DOI] [PubMed] [Google Scholar]
- 14. Ori A., Iskar M., Buczak K., Kastritis P., Parca L., Andres-Pons A., Singer S., Bork P., and Beck M. (2016) Spatiotemporal variation of mammalian protein complex stoichiometries. Genome Biol. 17, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rochette S., Gagnon-Arsenault I., Diss G., and Landry C. R. (2014) Modulation of the yeast protein interactome in response to DNA damage. J. Proteomics 100, 25–36 [DOI] [PubMed] [Google Scholar]
- 16. Grossmann A., Benlasfer N., Birth P., Hegele A., Wachsmuth F., Apelt L., and Stelzl U. (2015) Phospho-tyrosine dependent protein-protein interaction network. Mol. Syst. Biol. 11, 794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Landry C. R., Levy E. D., Abd Rabbo D., Tarassov K., and Michnick S. W. (2013) Extracting insight from noisy cellular networks. Cell 155, 983–989 [DOI] [PubMed] [Google Scholar]
- 18. Wan C., Borgeson B., Phanse S., Tu F., Drew K., Clark G., Xiong X., Kagan O., Kwan J., Bezginov A., Chessman K., Pal S., Cromar G., Papoulas O., Ni Z., Boutz D. R., Stoilova S., Havugimana P. C., Guo X., Malty R. H., Sarov M., Greenblatt J., Babu M., Derry W. B., Tillier E. R., Wallingford J. B., Parkinson J., Marcotte E. M., and Emili A. (2015) Panorama of ancient metazoan macromolecular complexes. Nature 525, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kristensen A. R., Gsponer J., and Foster L. J. (2012) A high-throughput approach for measuring temporal changes in the interactome. Nat. Methods 9, 907–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benschop J. J., Brabers N., van Leenen D., Bakker L. V., van Deutekom H. W., van Berkum N. L., Apweiler E., Lijnzaad P., Holstege F. C., and Kemmeren P. (2010) A consensus of core protein complex compositions for Saccharomyces cerevisiae. Mol. Cell 38, 916–928 [DOI] [PubMed] [Google Scholar]
- 21. Ideker T., and Krogan N. J. (2012) Differential network biology. Mol. Syst. Biol. 8, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baker M. (2012) Proteomics: The interaction map. Nature 484, 271–275 [DOI] [PubMed] [Google Scholar]
- 23. Vidal M., and Fields S. (2014) The yeast two-hybrid assay: still finding connections after 25 years. Nat. Methods 11, 1203–1206 [DOI] [PubMed] [Google Scholar]
- 24. Michnick S. W., Ear P. H., Manderson E. N., Remy I., and Stefan E. (2007) Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat. Rev. Drug Discov. 6, 569–582 [DOI] [PubMed] [Google Scholar]
- 25. Johnsson N., and Varshavsky A. (1994) Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. U.S.A. 91, 10340–10344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N. K., O'Donnell L., Oster S., Theesfeld C., Sellam A., Stark C., Breitkreutz B. J., Dolinski K., and Tyers M. (2017) The BioGRID interaction database: 2017 update. Nucleic Acids Res. 45, D369–D379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robinson C. V., Sali A., and Baumeister W. (2007) The molecular sociology of the cell. Nature 450, 973–982 [DOI] [PubMed] [Google Scholar]
- 28. Michnick S. W., Ear P. H., Landry C., Malleshaiah M. K., and Messier V. (2010) A toolkit of protein-fragment complementation assays for studying and dissecting large-scale and dynamic protein-protein interactions in living cells. Methods Enzymol. 470, 335–368 [DOI] [PubMed] [Google Scholar]
- 29. Ear P. H., and Michnick S. W. (2009) A general life-death selection strategy for dissecting protein functions. Nat. Methods 6, 813–816 [DOI] [PubMed] [Google Scholar]
- 30. Remy I., and Michnick S. W. (2015) Mapping biochemical networks with protein fragment complementation assays. Methods Mol. Biol. 1278, 467–481 [DOI] [PubMed] [Google Scholar]
- 31. Stefan E., Aquin S., Berger N., Landry C. R., Nyfeler B., Bouvier M., and Michnick S. W. (2007) Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 16916–16921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tchekanda E., Sivanesan D., and Michnick S. W. (2014) An infrared reporter to detect spatiotemporal dynamics of protein-protein interactions. Nat. Methods 11, 641–644 [DOI] [PubMed] [Google Scholar]
- 33. Kerppola T. K. (2009) Visualization of molecular interactions using bimolecular fluorescence complementation analysis: characteristics of protein fragment complementation. Chem. Soc. Rev. 38, 2876–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Remy I., Wilson I. A., and Michnick S. W. (1999) Erythropoietin receptor activation by a ligand-induced conformation change. Science 283, 990–993 [DOI] [PubMed] [Google Scholar]
- 35. Dixon A. S., Schwinn M. K., Hall M. P., Zimmerman K., Otto P., Lubben T. H., Butler B. L., Binkowski B. F., Machleidt T., Kirkland T. A., Wood M. G., Eggers C. T., Encell L. P., and Wood K. V. (2016) NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 11, 400–408 [DOI] [PubMed] [Google Scholar]
- 36. Gibson D. G., Young L., Chuang R.-Y., Venter J. C., Hutchison C. A., and Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods 6, 343–345 [DOI] [PubMed] [Google Scholar]
- 37. Rochette S., Diss G., Filteau M., Leducq J. B., Dube A. K., and Landry C. R. (2015) Genome-wide protein-protein interaction screening by protein-fragment complementation assay (PCA) in living cells. J. Vis. Exp. 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krogan N. J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A. P., Punna T., Peregrin-Alvarez J. M., Shales M., Zhang X., Davey M., Robinson M. D., Paccanaro A., Bray J. E., Sheung A., Beattie B., Richards D. P., Canadien V., Lalev A., Mena F., Wong P., Starostine A., Canete M. M., Vlasblom J., Wu S., Orsi C., Collins S. R., Chandran S., Haw R., Rilstone J. J., Gandi K., Thompson N. J., Musso G., St Onge P., Ghanny S., Lam M. H., Butland G., Altaf-Ul A. M., Kanaya S., Shilatifard A., O'Shea E., Weissman J. S., Ingles C. J., Hughes T. R., Parkinson J., Gerstein M., Wodak S. J., Emili A., and Greenblatt J. F. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 [DOI] [PubMed] [Google Scholar]
- 39. Haarer B., Aggeli D., Viggiano S., Burke D. J., and Amberg D. C. (2011) Novel interactions between actin and the proteasome revealed by complex haploinsufficiency. PLoS Genet. 7, e1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guerrero C., Milenkovic T., Przulj N., Kaiser P., and Huang L. (2008) Characterization of the proteasome interaction network using a QTAX-based tag-team strategy and protein interaction network analysis. Proc. Natl. Acad. Sci. U.S.A. 105, 13333–13338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hettema E. H., Lewis M. J., Black M. W., and Pelham H. R. (2003) Retromer and the sorting nexins Snx4/41/42 mediate distinct retrieval pathways from yeast endosomes. EMBO J. 22, 548–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tkach J. M., Yimit A., Lee A. Y., Riffle M., Costanzo M., Jaschob D., Hendry J. A., Ou J., Moffat J., Boone C., Davis T. N., Nislow C., and Brown G. W. (2012) Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat. Cell Biol. 14, 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Archambault J., and Friesen J. D. (1993) Genetics of eukaryotic RNA polymerases I, II, and III. Microbiol. Rev. 57, 703–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen X., Zaro J. L., and Shen W. C. (2013) Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 65, 1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leitner A., Walzthoeni T., and Aebersold R. (2014) Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat. Protoc. 9, 120–137 [DOI] [PubMed] [Google Scholar]
- 46. Vogel S. S., van der Meer B. W., and Blank P. S. (2014) Estimating the distance separating fluorescent protein FRET pairs. Methods 66, 131–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.