

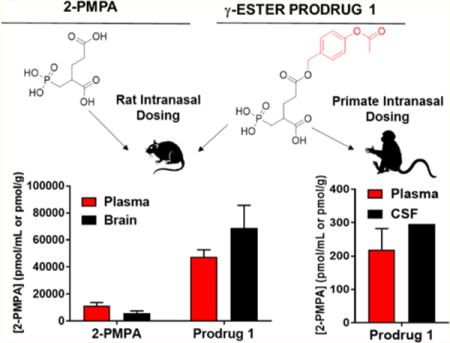
Abstract
2-(Phosphonomethyl)pentanedioic acid (2-PMPA) is a potent and selective inhibitor of glutamate carboxypeptidase-II (GCPII) with efficacy in multiple neurological and psychiatric disease models, but its clinical utility is hampered by low brain penetration due to the inclusion of multiple acidic functionalities. We recently reported an improvement in the brain-to-plasma ratio of 2-PMPA after intranasal (IN) dosing in both rodents and primates. Herein, we describe the synthesis of several 2-PMPA prodrugs with further improved brain delivery of 2-PMPA after IN administration by masking of the γ-carboxylate. When compared to IN 2-PMPA in rats at 1 h post dose, γ-(4-acetoxybenzyl)-2-PMPA (compound 1) resulted in significantly higher 2-PMPA delivery to both plasma (4.1-fold) and brain (11-fold). Subsequent time-dependent evaluation of 1 also showed high brain as well as plasma 2-PMPA exposures with brain-to-plasma ratios of 2.2, 0.48, and 0.26 for olfactory bulb, cortex, and cerebellum, respectively, as well as an improved sciatic nerve to plasma ratio of 0.84. In contrast, IV administration of compound 1 resulted in similar plasma exposure of 2-PMPA versus the IN route (AUCIV: 76 ± 9 h·nmol/mL versus AUCIN: 99 ± 24 h·nmol/mL); but significantly lower nerve and brain tissue exposures with tissue-to-plasma ratios of 0.21, 0.03, 0.04, and 0.04 in nerve, olfactory bulb, cortex, and cerebellum, respectively. In primates, IN administration of 1 more than doubled 2-PMPA concentrations in the cerebrospinal fluid relative to previously reported levels following IN 2-PMPA. The results of these experiments provide a promising strategy for testing GCPII inhibition in neurological and psychiatric disorders.
Keywords: 2-PMPA, glutamate carboxypeptidase II, neurological disease, intranasal, pharmacokinetics, prodrugs
Graphical abstract
INTRODUCTION
Glutamate carboxypeptidase II (GCPII) is an extracellular membrane-bound zinc metalloprotease that catalyzes the hydrolysis of the neuropeptide N-acetyl-aspartyl-glutamate (NAAG) to N-acetylaspartate (NAA) and glutamate.1 As a major regulator of extracellular glutamate availability in the brain,2 GCPII inhibition represents a promising drug target for multiple diseases associated with disrupted glutamate homeostasis including neuropathic pain,3–7 stroke,2 diabetic neuropathy,8–10 schizophrenia,11 addiction,12–14 multiple sclerosis,15–17 and traumatic brain injury.18,19 Selective inhibitors of GCPII have been shown to attenuate glutamate-mediated excitotoxicity, and ameliorate these disease phenotypes in preclinical models. These data were generated with structurally distinct GCPII inhibitors including thiol-based, phosphinic acid based, and urea-based compounds, providing convincing evidence that GCPII mediates their effect. In addition, several studies have demonstrated that administration of these inhibitors in vivo is accompanied by the biochemical consequences of GCPII inhibition, namely, a decrease in brain glutamate and a concomitant increase in brain NAAG. GCPII inhibition is thought to confer neuroprotection by blocking the liberation of glutamate from NAAG as well as by increasing NAAG itself which acts as an agonist at metabotropic glutamate receptor subtype 3 (mGlu3), further diminishing glutamate release.20 The mechanism of efficacy has also been confirmed by GCPII knockout mice that have been found to be resistant to ischemic and inflammatory damage in the central and peripheral nervous systems and less susceptible to traumatic brain injury.19,21
Despite promising therapeutic potential, currently available GCPII inhibitors, such as the phosphonate-based 2-(phosphonomethyl)pentanedioic acid (2-PMPA), exhibit poor pharmacokinetics due to the inclusion of highly hydrophilic moieties, which are important for maintaining potency at GCPII but impede membrane permeability and brain penetration.22 Indeed, low oral bioavailability (F < 2%) and brain penetration (brain/plasma ratio < 2%) of 2-PMPA23,24 necessitate very high systemic doses or direct brain injection for efficacy. Previous structure–activity relationship (SAR) efforts by our group and others have demonstrated that the potency of 2-PMPA is dependent upon the zinc-chelating properties of the phosphonate side chain combined with the core glutarate structure that mimics glutamate in the active site and promotes high affinity GCPII binding.25,26 Attempts to substitute these functionalities in order to increase lipophilicity and improve the physicochemical properties of the molecule resulted in substantial loss of potency,25 suggesting that SAR alone may not be sufficient to achieve significant improvement.
As an alternative means, intranasal (IN) administration has been shown to improve central nervous system (CNS) penetration of 2-PMPA24 and many other small molecules, peptides, and biologics.27 Compounds administered IN reach the CNS by bypassing the blood–brain barrier (BBB) through initial absorption into the olfactory epithelium of the nasal cavity prior to intracellular and/or interstitial diffusion through the olfactory bulb (OB) to other brain regions and cerebrospinal fluid (CSF).27 IN delivery also results in systemic exposure by absorption through the nasal respiratory epithelium.27 Recently, we demonstrated that IN administration of 2-PMPA significantly improved CNS exposure in rodents as well as nonhuman primates.24 Although the brain penetration of 2-PMPA was improved through the IN versus systemic route, its diffusion across the olfactory epithelium and throughout the brain was limited, perhaps by its extreme hydrophilicity (cLogP = −1.54). Thus, we hypothesized that lipophilic prodrugs of 2-PMPA could afford enhanced permeability and tissue exposure, and further optimize delivery of this compound to the CNS.
We recently employed a 2-PMPA prodrug strategy to decrease its polarity and showed that we could improve its oral availability by >20-fold in both rodents and dogs.23 Oral administration of 2-PMPA prodrugs did not significantly enhance brain exposure, however, likely due to metabolism of 2-PMPA prodrugs while traversing the gastrointestinal tract, and in plasma, prior to BBB penetration. Thus, we hypothesized that if we combined this prodrug strategy with IN dosing, we could obtain high 2-PMPA CNS exposure. In the present work, we synthesized five prodrugs of 2-PMPA containing a single promoiety at the γ-carboxylic acid. We subsequently tested the prodrugs for in vitro GCPII inhibition and in vivo distribution at 1 h following IN dosing in rats. The compound with the best brain penetration, γ-(4-acetoxybenzyl)-2-PMPA (compound 1), was then further characterized for time-dependent delivery of 2-PMPA to plasma, various brain regions, and peripheral nerve via both IV and IN routes. To more closely reflect human nasal anatomy, nonhuman primates were used to confirm the rodent findings using a brain targeted IN device.24
EXPERIMENTAL SECTION
2-PMPA was synthesized internally by our laboratory as reported previously.25 Reagents, such as LC/MS grade acetonitrile and water (LC/MS grade) with 0.1% formic acid, were obtained from Fisher Scientific (Hanover Park, IL). Drug-free (blank) heparinized rat plasma was obtained from Innovative Research Inc. (Plymouth, MN). All other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Synthesis of 2-PMPA Prodrugs
The acetoxybenzyl (1) and n-butyl (3) derivatives were prepared as shown in Scheme 1. Starting from common intermediate 623 the benzyl group was removed using palladium catalyzed hydrogenation to yield the carboxylate 7. The carboxylate 7 was coupled to n-butanol using DCC and DMAP to yield ester 8a, or Mitsunobu coupling conditions were used with 4-(hydroxymethyl)phenyl acetate to yield intermediate 8b. Ester 8a was deprotected using bromotrimethylsilane (TMSBr) to yield the phosphonic acid 9a in a quantitative yield, followed by acidic hydrolysis of the t-butyl ester using trifluoroacetic acid (TFA) to yield the PMPA prodrug 3. In a similar manner, ester 8a was deprotected using TFA to yield the carboxylate 9b, which was then subsequently converted to the phosphonic acid 1 using TMSBr. Prodrug 3 was prepared in total 69% yield over 4 steps; in the case of prodrug 1, the overall yield was 31%.
Scheme 1. Synthesis of Compounds 1 and 3a.
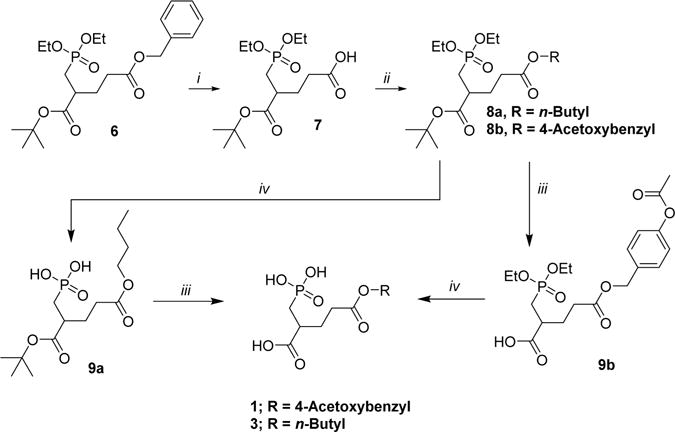
aReagents and conditions: (i) Pd/C, H2 (balloon), THF, rt, 22 h. (ii) For 8a: n-butanol, DCC, DMAP, DCM, rt, 20 h. For 8b: 4-(hydroxymethyl)phenyl acetate, PPh3, DIAD, 1 h, rt. (iii) TFA/DCM 1:1, 0 °C to rt, 3 h. (iv) TMS-Br, DCM, 0 °C to rt, 18 h.
Benzyl substituted prodrug 4 (Scheme 2) was prepared from analogue 1028 in three steps. Intermediate 10 was converted to the 1-allyl-5-benzyl-2-PMPA diester 11 using Eschenmoser’s salt, and 11 was then converted to the phosphonic ester 12 using diethyl phosphite and trimethylaluminum. Ethyl groups in phosphonic ester 12 were removed by TMSBr, and subsequently the allyl moiety was removed using Pd(PPh3)4 with phenylsilane as scavenger. Prodrug 4 was obtained in an overall yield of 45%.
Scheme 2. Synthesis of Compound 4a.
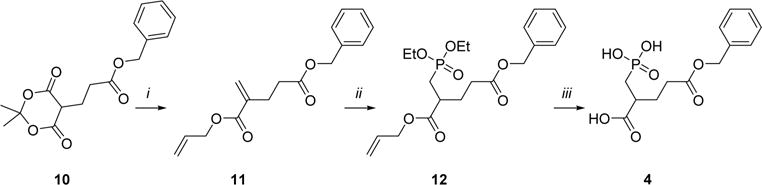
aReagents and conditions: (i) Eschenmoser’s salt, allyl alcohol, 70 °C, 21 h. (ii) Diethyl phosphite, Al(CH3)3, DCM, 0 °C to rt, 16 h. (iii) (1) TMS-Br, DCM, 0 °C to rt, 18 h; (2) phenylsilane, Pd(PPh3)4, THF, rt, 1 h.
Prodrugs 2 and 5 were prepared from the common intermediate 13, Scheme 3. Intermediate 13 was reacted with Eschenmoser’s salt in benzyl alcohol to yield the 5-allyl-1- benzyl-2-PMPA diester 14. The phosphonic ester 15 was formed from compound 14 using diethyl phosphite and trimethylaluminum. The allylic moiety of compound 15 was deprotected to form the carboxylate 16 using Pd(PPh3)4 in the presence of phenylsilane. The carboxylic acid 16 was then condensed under basic conditions with either chloromethyl isopropyl carbonate to form 17a or 1-bromooctane to form 17b. Esters 17a and 17b were subsequently deprotected with TMSBr and palladium catalyzed hydrogenation to form prodrugs 2 (35% overall yield) and 5 (25% overall yield), respectively.
Scheme 3. Synthesis of Compounds 2 and 5a.
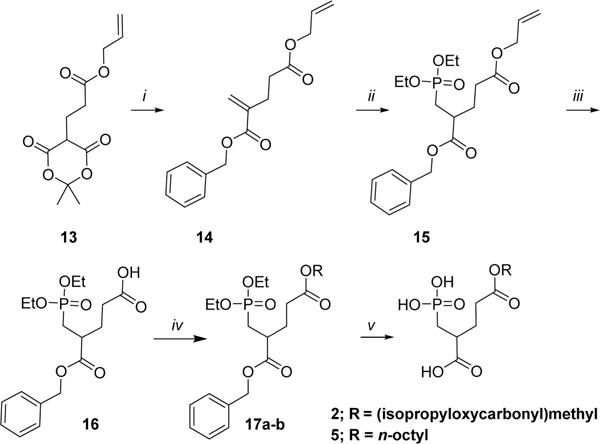
aReagents and conditions: (i) Eschenmoser’s salt, BnOH, 70 °C, 21 h. (ii) Diethyl phosphite, Al(CH3)3, DCM, 0 °C to rt, 16 h. (iii) Phenylsilane, Pd(PPh3)4, THF, rt, 1 h. (iv) For 17a: POC-Cl, Nal, K2CO3, acetonitrile, rt to 50 °C, overnight. For 17b: C8H17Br, Nal, K2CO3, acetonitrile, rt to 50 °C, overnight. (v) (1) TMS-Br, DCM, 0 °C to rt, 18 h; (2) Pb/C, H2 (balloon), THF, rt, 22 h.
Detailed synthesis and characterization for each compound and mass transitions used for LC–MS/MS analysis of prodrugs 1–5 are provided in the Supporting Information.
GCPII Enzymatic Activity
The potency of each 2-PMPA γ-ester prodrug 1–5 in inhibiting GCPII was determined using a radioenzymatic activity assay as previously described.29 Briefly, recombinant human GCPII protein was purified and incubated with 3H-NAAG (radiolabeled on the terminal glutamate) prior to separation of cleaved glutamate by ion exchange chromatography, which was subsequently measured by liquid scintillation. Increasing concentrations of each 2-PMPA prodrug were dissolved in 10% DMSO and coincubated with the radioenzymatic assay mixture to determine their IC50 values.
Plasma Stability of 2-PMPA Prodrugs
For stability studies, naive plasma from rat and humans was used. Prodrugs (10 μM) were spiked in each plasma matrix and incubated in an orbital shaker at 37 °C. At predetermined times (0 and 60 min), 100 μL aliquots of the mixture in duplicate were removed and the reaction was quenched by addition of three times the volume of ice cold acetonitrile spiked with the internal standard (losartan 0.5 μM). The samples were vortex mixed for 30 s and centrifuged at 12000g for 10 min. Fifty microliters of the supernatant was diluted with 50 μL of water and transferred to a 250 μL polypropylene vial sealed with a Teflon cap. Prodrug disappearance was monitored over time using liquid chromatography and tandem mass spectrometry (LC–MS/MS). Briefly, prodrugs were analyzed on a Thermo Scientific Accela UPLC system coupled to an Accela open autosampler on an Agilent C18 (100 × 2.1 mm i.d.) UPLC column. The autosampler was temperature controlled and operated at 10 °C. The mobile phase used for the chromatographic separation was composed of acetonitrile/water containing 0.1% formic acid and had a flow rate of 0.5 mL/min for 4.5 min using gradient elution. The column effluent was monitored using a TSQ Vantage triple-quadrupole mass spectrometric detector, equipped with an electrospray probe set in the positive ionization mode. Samples were introduced into the ionization source through a heated nebulized probe (350 °C). Disappearance of prodrugs was measured from ratio of peak areas of analyte to IS.
Rodent Studies
All rodent studies were reviewed and approved by the Johns Hopkins Institutional Animal Care and Use Committee in compliance with the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) and the Public Health Service Policy on the Humane Care and Use of Laboratory Animals (PHS Policy). Studies were conducted using 6–8 week old, 200–250 g, male, outbred Wistar rats (Envigo, Indianapolis, IN). Animals were housed in individually ventilated cages (Allentown Caging Equipment, Allentown, PA) with ad libitum access to autoclaved rodent chow (Envigo, Teklad, Indianapolis, IN) and filtered reverse-osmosis-treated water.
Pharmacokinetic Evaluation of 2-PMPA Prodrugs Following IN Administration in Rats
An initial pharmacokinetic screen (n = 3 rats/group) was conducted by dosing 2-PMPA or prodrugs (10 mg/kg or molar equivalent, IN) prior to collecting plasma and OB for bioanalysis 1 h later. IN administrations were performed per previously described methods with minor modifications.24 Briefly, rats were anesthetized with 1–1.5 mL (IP) of 10% chloral hydrate and kept under anesthesia throughout the entire experiment. To prevent drainage, the nasal cavity was isolated by transecting the trachea; the lower part of the trachea was cannulated to allow air breathing. Rats were then given 10 μL of the experimental compound solution per nostril using a micro-syringe connected to a 1.5 cm PE-10 tube, over a period of 5– 10 s. The rats were maintained under anesthesia for the entire duration of the experiment until sacrificed. Based on the initial screening data, compound 1 was selected for full pharmacokinetic evaluation (n = 3 rats/time point) by collecting plasma and tissues (OB, cortex, cerebellum, and sciatic nerve) at 0.25, 0.5, 1, 2, 3, 5 h following IN administration. For collection of tissues, animals were euthanized with CO2, and blood samples were collected in heparinized microtubes by cardiac puncture. Tissues were dissected and immediately flash frozen (−80 °C). Plasma was prepared by centrifugation immediately after collection of blood samples. All samples were stored at −80 °C until bioanalysis of 2-PMPA and intact prodrug as described below.
Evaluation of Compound 1 Following IV Administration in Rats
For IV studies, compound 1 was administered as a single IV dose. The dosing solution was prepared on the day of the experiment in 50 mM HEPES buffered saline, and pH was adjusted to 7.4 before injection. At various time points following drug administration (0.25, 0.5, 1, 2, 3, 5 h post dose), animals (n = 3 per time point) were euthanized with CO2, blood samples were collected in heparinized microtubes by cardiac puncture, and tissues (OB, cortex, cerebellum, and sciatic nerve) were dissected and immediately flash frozen (−80 °C). Plasma was prepared by centrifugation immediately after collection of blood samples. All samples were stored in −80 °C until bioanalysis.
Evaluation of 2-PMPA Prodrugs Following IN Administration in Nonhuman Primates
All nonhuman primate studies were reviewed and approved by the Johns Hopkins Institutional Animal Care and Use Committee in compliance with AAALAC, the PHS Policy, and the Animal Welfare Act, Regulations and Care Policies. Studies were conducted using three healthy nine-year-old male rhesus macaques (Macaca mulatta) weighing an average of 8.9 kg (range of 8.3–9 kg). Animals were maintained on a 14:10 h light:dark cycle and provided a commercial macaque diet (Envigo, Teklad, Indianapolis, IN) with ad libitum access to filtered reverse-osmosis-treated water and fresh enrichment (fruit/vegetables) supplemented daily. Animals were singly housed while on study to monitor behavior and clinical health following drug administration.
Animals were anesthetized with ketamine 20 mg/kg intramuscularly, intubated, and maintained under isoflurane inhalant anesthesia for intranasal drug administration and subsequent blood and CSF collection. Compound 1 was administered as an aqueous solution (87.5 mg/mL and pH adjusted to 7.4) via IN delivery employing a drug delivery device (Kurve Technology, Bothell, WA), designed to deliver drugs to the olfactory region to maximize transport to the CNS. The device was actuated for a period of 4 min in one nostril (depositing 600 μL). The nosepiece was cleaned with a mist of air, and then the same procedure was performed in the second nostril (600 μL). Total dose delivered was 2.5 mg/kg 2-PMPA equivalent. CSF (target of 100 μL) was obtained at 0.5 and 1 h post dose using a 1.5 in. spinal needle placed in the intrathecal space of the cisterna magna. Whole blood was obtained at 30 min post dose using a peripheral saphenous vein catheter. Plasma separated using low speed centrifugation at 1500g for 10 min was flash-frozen on dry ice. Plasma and CSF samples were stored at −80 °C until bioanalysis.
Bioanalysis of 2-PMPA
Quantification of analytes in plasma and brain tissues was performed after protein precipitation by LC–MS/MS analysis as previously described.24 Briefly, standard curves were prepared in naive plasma and brain tissues ranging from 10 to 50,000 nM. For extraction of 2-PMPA, plasma samples were thawed on ice, and then 50 μL of the calibration standard or sample was transferred into silanized vials. Three hundred microliters of methanol with internal standard was added to each plasma sample for protein extraction, followed by mixing and centrifugation at 12000g for 10 min. The supernatant was then transferred and evaporated to dryness at 40 °C under a gentle stream of nitrogen. Brain samples were diluted 1:5 w/v with methanol, stored at −20 °C for 1 h, and then homogenized. Homogenized brain samples were vortex mixed and centrifuged as above; 100 μL of the supernatant was mixed with 100 μL of internal standard in methanol, and then evaporated to dryness.
2-PMPA samples and standards were derivatized using N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide with 1% tert-butyldimethylchlorosilane (MTBSTFA + 1% TBDMSCl), to enable reverse phase chromatography, as described previously.24 Briefly, samples and standards were reconstituted in 100 μL of acetonitrile with vortex mixing followed by 50 μL of MTBSTFA and were set to react at 60 °C in a water bath for 30 min. Upon completion of the reaction the samples were then centrifuged again at 10,000 rpm at 5 °C for 2 min. An aliquot of 100 μL of supernatant was transferred to 250 μL of polypropylene autosampler vials sealed with Teflon caps and analyzed via LC–MS/MS. Separation of the analyte was achieved using a Waters X-terra, RP18, 3.5 μm, and 2.1 × 50 mm. The mobile phase was composed of 0.1% formic acid in acetonitrile and 0.1% formic acid in H2O with gradient elution. Chromatographic analysis was performed on an Accela UPLC and a TSQ vantage mass spectrometer. The [M + H]+ ion transitions of derivatized 2-PMPA at m/z 683.0 → 551.4 and that of the internal standard at m/z 669.0 → 537.2 were monitored.
Bioanalysis of Intact Prodrugs
For quantification on intact prodrugs in plasma and brain, extraction of the prodrugs from the tissues was done by protein precipitation with methanol containing losartan as internal standard. Following extraction and evaporation of organic, the residue was reconstituted in acetonitrile/water (70:30 v/v) and subjected to liquid chromatography/tandem mass spectrometry (LC–MS/MS).
Briefly, quantification was performed in multiple reaction monitoring (MRM) mode using mass transitions in Table S1. Chromatographic analysis was performed using an Accela ultrahigh-performance system consisting of an analytical pump, and an autosampler coupled with a TSQ Vantage mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). Separation of the analyte was achieved at ambient temperature using an Agilent Eclipse Plus column (100 × 2.1 mm i.d.) packed with a 1.8 μm C18 stationary phase. The mobile phase used was composed of 0.1% formic acid in acetonitrile and 0.1% formic acid in water with gradient elution, starting with 10% organic linearly increasing to 99% at 2 min, maintaining at 99% (2–2.5 min), re-equilibrating to 10% by 2.7 min, and maintaining 10% organic until the end of the run. The total run time for each analyte was 5 min.
Analysis of 2-PMPA in Nonhuman Primate CSF Samples
Analysis of 2-PMPA in CSF samples was done as described previously, with minor modifications. Briefly, standard curves were generated in blank nonhuman primate CSF. Calibration curves were constructed in the range of 50–10000 nM. Samples were analyzed by LC–MS/MS. Samples (20 μL) were injected and separated on an Agilent 1290 LC equipped with an Agilent Eclipse Plus C18 column (2.1 × 100 mm) packed with 1.8 μm stationary phase. The mobile phase consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile with an isocratic elution at 2.5% organic. Analytes were detected with an Agilent 6520 QTOF mass spectrometer in negative mode using [M – H]−ions for 2-PMPA (225.0163) and internal standard (325.1043). Calibration curves were generated in an analogous manner as described above with a correlation coefficient >0.99.
Pharmacokinetic Analysis of 2-PMPA
Mean plasma and tissue concentrations of 2-PMPA were analyzed using non-compartmental method as implemented in the computer software program Phoenix WinNonlin version 6.4 (Certara USA, Inc., Princeton, NJ). The maximum plasma concentration (Cmax) and time to Cmax (Tmax) were the observed values. The area under the plasma concentration time curve (AUC) value was calculated to the last quantifiable sample (AUClast) by use of the log–linear trapezoidal rule. The tissue to plasma partition coefficients were calculated as a ratio of mean AUCs (AUC0−t,tissue/AUC0−t,plasma).
Statistical Analysis
For single time point studies, compound concentrations were statistically compared by one-way ANOVA with Dunnett’s post hoc test for 2-PMPA and Tukey’s post hoc test for intact prodrugs. Comparison of Cmax values between IV and IN routes for each corresponding tissue was performed by t test. The method of Bailer was used to estimate the variance of AUClast based on the mean concentration at each time point.30,31 To determine whether there was a significant difference between exposure as expressed by AUCs of IV versus IN plasma, brain, and nerve tissue, a pairwise comparison was performed using a Z test.32 The a priori level of significance for all analyses was defined as p < 0.05.
RESULTS
Relative to 2-PMPA, Compounds 1–5 Showed Enhanced Lipophilicity, Nanomolar GCPII Inhibitory Potency, and Differential Plasma Metabolic Lability
Five different prodrugs using 4-acetoxybenzyl (PAB), isopropoxycarbonyloxymethyl (POC), n-butyl, benzyl, and n-octyl promoieties added at the γ-carboxylate of 2-PMPA were synthesized (Schemes 1–3) to yield compounds 1, 2, 3, 4, and 5, respectively. All prodrugs exhibited an increase in lipophilicity (cLogP = from −0.39 to 2.6) compared to 2-PMPA (cLogP = −1.54). Moreover, all 5 prodrugs displayed nanomolar potency as GCPII inhibitors (1–20 nM) (Table 1). When incubated in rat and human plasma, compounds 1 and 2 were metabolically labile (<40% remaining at 60 min), and compound 4 was moderately stable, while compounds 3 and 5 were stable with >50% remaining following 1 h incubation. (Table 1).
Table 1.
GCPII Activity, Lipophilicity, and Plasma Stability of γ-Protected 2-PMPA Prodrugs
| Compound | Structure | IC50(±SEM) (nM) | cLogP | Plasma stability % remaining @60 min | |
|---|---|---|---|---|---|
| Rat | Human | ||||
| 2-PMPA |
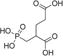
|
0.2 ± 0.02 | −1.5 | – | – |
| 1 |
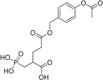
|
3 ± 0.2 | 0.024 | 7% | 42% |
| 2 |
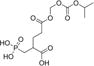
|
1 ±0.1 | −0.39 | 32% | 39% |
| 3 |
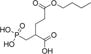
|
1±0.1 | 0.53 | 82% | 87% |
| 4 |
|
20 ± 1.0 | 0.67 | 40% | 50% |
| 5 |
|
2 ± 0.2 | 2.6 | 67% | 75% |
Relative to 2-PMPA, IN Delivery of Compound 1 in Rats Showed Enhanced 2-PMPA Plasma and Olfactory Bulb Delivery
In an initial pharmacokinetic screening study, compounds 1–5 were delivered IN to rats, and plasma and OB were collected 1 h post dose. Relative to 10 mg/kg IN 2-PMPA, a 10 mg/kg molar equivalent dose of compound 1 led to substantially higher concentrations of 2-PMPA in both the plasma (4.1-fold; 47 ± 7.5 nmol/mL versus 11.5 ± 2.2 nmol/mL) [F5,12 = 40.05, p < 0.0001] (Figure 1A) and OB (11-fold, 68 ± 23 nmol/g versus 6.1 ± 1.5 nmol/g) [F5,12 = 14.69, p < 0.0001] (Figure 1B). Compound 2 also resulted in an approximate 2-fold increase in 2-PMPA concentrations in the plasma (22.7 ± 2.9 nmol/mL), but similar OB levels (4.3 ± 2.1 nmol/g). Compounds 3, 4, and 5 exhibited similar or lower 2-PMPA concentrations in the plasma and OB.
Figure 1.
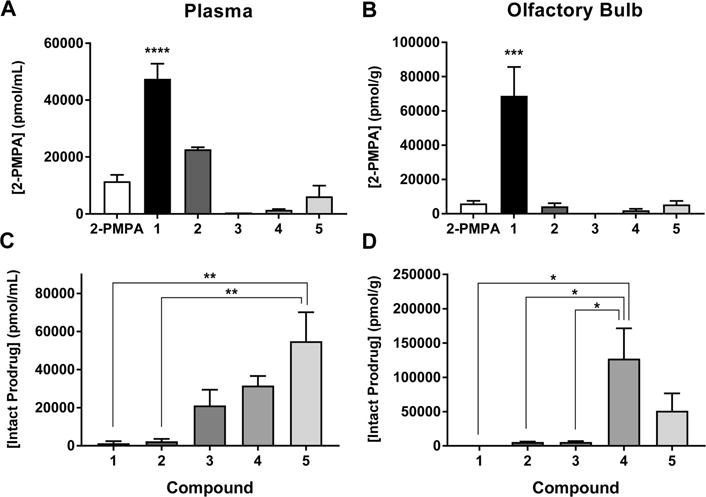
Relative to 2-PMPA, IN delivery of compound 1 in rats showed enhanced 2-PMPA plasma and olfactory bulb delivery at 1 h post dose. 10 mg/kg (equivalent 2-PMPA) doses of compounds 1–5 were administered IN to rats. Plasma and olfactory bulb were extracted 1 h later, and 2-PMPA was quantified via LC–MS/MS. Compound 1 showed the best improvement, delivering 2-PMPA at concentrations greater than 4-fold and 11-fold higher in (A) plasma and (B) olfactory bulb (OB), respectively, versus administration of equivalent IN doses of 2-PMPA.Compounds 3–5 liberated lower concentrations of 2-PMPA in both plasma and OB, and compound 2 liberated lower concentrations of 2-PMPA in OB. Concentrations of intact prodrug were highest for compounds 4 and 5 in (C) plasma and (D) OB. By contrast, intact compound 1 was undetectable in plasma and OB. Data are presented as mean ± SEM (n = 3/group). Compound concentrations compared by one-way ANOVA with Dunnett’s post hoc test for 2-PMPA: ***p < 0.001, ****p < 0.0001 versus 2-PMPA. Tukey’s post hoc test for intact prodrug: *p < 0.05, **p < 0.01.
Intact prodrug quantification showed that compounds 1 and 2 were very low/undetectable in both plasma (0.27 ± 0.18 and 3.34 ± 1.34 nmol/mL, respectively) [F4,10 = 7.58, p = 0.0045] (Figure 1C) and OB (<0.1 nmol/g and 5.69 ± 1.24 nmol/g, respectively) [F4,10 = 5.62, p = 0.0123] (Figure 1D). Intact compound 3 was detectable at significant levels in plasma (29.25 ± 3.06 nmol/mL) and low levels in OB (5.10 ± 2.18 nmol/g). Compounds 4 and 5 were both detectable at elevated levels in plasma (31.64 ± 4.98 and 60.59 ± 24.47 nmol/mL, respectively) and OB (73.37 ± 21.50 and 127.18 ± 44.08 nmol/g, respectively).
Relative to IV Administration, IN Delivery of Compound 1 to Rats Resulted in High Brain Exposure of 2-PMPA
Given that compound 1 looked most promising in the screening study, it was chosen for a full pharmacokinetic evaluation. Compound 1 (10 mg/kg 2-PMPA equivalent dose) was delivered IN or IV in rats, and plasma and tissues (OB, cortex, cerebellum, and sciatic nerve) were collected at predetermined time points. Concentration time profiles for intact prodrug and liberated 2-PMPA are shown in Figure 2 for IN (panels A and B) and IV (panels C and D) administration, respectively. Pharmacokinetic parameters are summarized in Table 2. Following IN administration, compound 1 delivered peak 2-PMPA concentrations (Cmax) of 90 ± 13 nmol/g, 34.4 ± 23 nmol/g, and 22.4 ± 6.6 nmol/g to the OB, cortex, and cerebellum, respectively (Figure 2A) at 30 min post IN administration (Tmax). Compound 1 also afforded excellent plasma levels of 2-PMPA with a Cmax of 152 ± 42 nmol/mL (Figure 2A). AUC0−t in plasma, OB, cortex, and cerebellum were 99 ± 24 h·nmol/mL, 217 ± 23 h·nmol/g, 30 ± 10 h·nmol/g, and 26 ± 5 h·nmol/g, respectively, resulting in brain-to-plasma ratios (AUC0−t,brain/AUC0−t,plasma) of 2.2, 0.48, and 0.26 for OB, cortex, and cerebellum, respectively (Table 2). In the sciatic nerve, compound 1 delivered 2-PMPA at a Cmax of 28.2 ± 8.4 nmol/g and AUC0−t of 84 ± 25 h·nmol/g (Figure 2A, Table 2) resulting in a high peripheral nerve to plasma ratio of 0.84.
Figure 2.
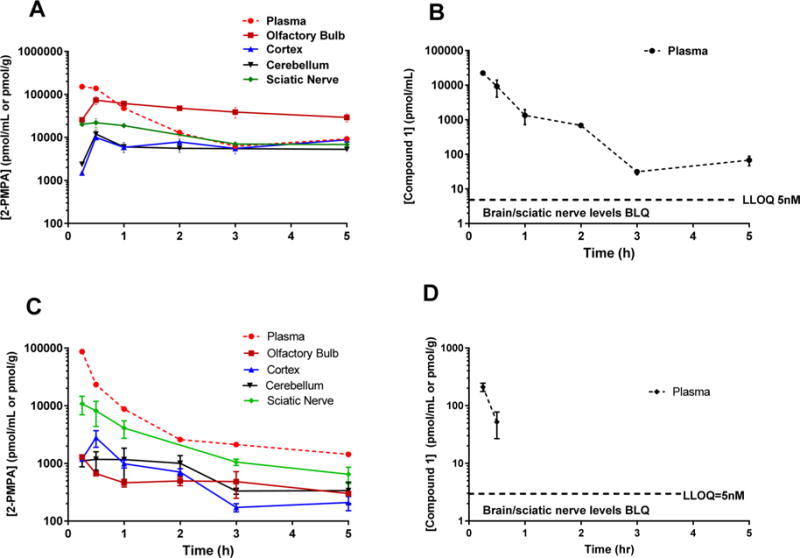
IN delivery of compound 1 to rats resulted in high brain and peripheral 2-PMPA exposures versus the IV route, which delivered high peripheral exposures but low brain exposures. (A) Compound 1 administered IN in rats delivered significant concentrations of 2-PMPA to CNS and sciatic nerve, achieving high tissue-to-plasma ratios. (C) Compound 1 administered IV (10 mg/kg equivalent dose) showed high plasma and moderate sciatic nerve 2-PMPA levels but low brain exposures achieving low brain-to-plasma ratios in all brain regions. Intact prodrug concentrations in the plasma following IN (B) and IV (D) were low and variable. No intact prodrug of compound 1 was measured in CNS or sciatic nerve. Data are presented as mean ± SEM (n = 3/group).
Table 2.
Mean ± SEM Pharmacokinetic Parameters of 2-PMPA in Plasma, Brain, and Sciatic Nerve Following 10 mg/kg Equivalent IN or IV Administration of Compound 1 to Ratsa
| route | tissue | Tmax (h) | 2-PMPA Cmax (nmol/mL) | 2-PMPA AUC0−t (h·nmol/mL) | tissue/plasma ratio |
|---|---|---|---|---|---|
| IN | plasma | 0.25 | 152 ± 42 | 99 ± 24 | 1 |
| olfactory | 0.5 | 90 ± 13* | 217 ± 23* | 2.2 | |
| cortex | 0.5 | 34.4 ± 23 | 30 ± 10* | 0.48 | |
| cerebellum | 0.5 | 22.4 ± 6.6** | 26 ± 5* | 0.26 | |
| sciatic nerve | 2 | 28.2 ± 8.4* | 84 ± 25* | 0.84 | |
| IV | plasma | 0.25 | 87 ± 12 | 76 ± 9.0 | 1 |
| olfactory | 0.25 | 1.2 ± 0.0 | 2.7 ± 0.3 | 0.03 | |
| cortex | 0.25 | 2.8 ± 0.9 | 3.3 ± 0.5 | 0.04 | |
| cerebellum | 0.25 | 2.0 ± 0.3 | 3.2 ± 0.3 | 0.04 | |
| sciatic nerve | 0.25 | 12 ± 4 | 15.8 ± 16.0 | 0.21 |
Tissue-to-plasma ratios are calculated from mean total AUC tissue versus AUC plasma for each route. Comparison of Cmax from the IN route for each tissue (n = 3) was performed by t test,
p < 0.05,
p < 0.01, versus the IV route for corresponding tissue. Comparison of AUC from IN for each tissue was conducted using Bailer’s SEM method versus AUC from the IV route each corresponding tissue,
p < 0.05.
Intact prodrug 1 was quantifiable in plasma with a Cmax of 22 nmol/mL (Figure 2B) and AUC of 8.04 h·nmol/mL. The exposures from the intact prodrug in plasma were almost 17-fold lower than 2-PMPA. No intact prodrug was quantifiable in the brain or sciatic nerve at any time point.
Following IV administration, compound 1 delivered 2-PMPA in plasma with a Cmax of 87 nmol/mL (Figure 2C). In selected brain regions, compound 1 delivered peak 2-PMPA concentrations (Cmax) of 1.2 ± 0.0 nmol/g, 2.8 ± 0.9 nmol/g, and 2.0 ± 0.3 nmol/g to the OB, cortex, and cerebellum, respectively (Figure 2C) at 15 min post IV administration (Tmax). Overall exposures of 2-PMPA from compound 1 in plasma were high (76 ± 9 h·nmol/mL) but substantially lower in OB, cortex, and cerebellum (2.7 ± 0.3 h·nmol/g, 3.3 ± 0.5 h·nmol/g, and 3.2 ± 0.3 h·nmol/g, respectively), resulting in low brain-to-plasma ratios (AUC0−t,brain/AUC0−t,plasma = 0.03–0.04; Table 2). In the sciatic nerve, compound 1 delivered 2-PMPA at a Cmax of 12 ± 4 nmol/g and AUC0−t of 15.8 ± 16 h·nmol/g (Figure 2C, Table 2) resulting in a peripheral nerve to plasma ratio of 0.21. Intact prodrug 1 was quantifiable at low levels in plasma with a Cmax of 0.2 nmol/mL (Figure 2D). No intact prodrug was quantifiable in the brain or sciatic nerve at any time point, confirming rapid biotransformation.
Statistical analysis showed no significant difference in the plasma exposure (based on AUC values) of 2-PMPA following either IN or IV administration of compound 1.
When compared to IV delivery, IN administration of 1 improved Cmax delivery of 2-PMPA to nervous tissues including OB (t4 = 4.42, p = 0.0115), cerebellum (t4 = 5.35, p = 0.0059), and sciatic nerve (t4 = 3.02, p = 0.0393). Statistical analysis based on AUC comparison for each corresponding tissue between the IV and IN routes showed no significant difference in the plasma exposure (p > 0.5), but significant differences (p < 0.05) in brain and sciatic AUC of 2-PMPA following IN administration of compound 1 versus the IV route.
IN Delivery of Compound 1 to Nonhuman Primates Resulted in 1:1 CSF/Plasma Ratio of 2-PMPA
IN delivery of compound 1 (2.5 mg/kg 2-PMPA equivalent dose) in rhesus macaques (Macaca mulatta) resulted in significant 2-PMPA exposure in the plasma and CSF (Figure 3A,B). Specifically, at 30 min post dose compound 1 delivered 0.2 ± 0.06 nmol/mL and 0.3 nmol/mL 2-PMPA in CSF and plasma, respectively, resulting in a CSF/plasma ratio of greater than unity. Concentrations of the intact prodrug 1 in CSF were 0.2 nmol/mL, but were undetectable in plasma.
Figure 3.
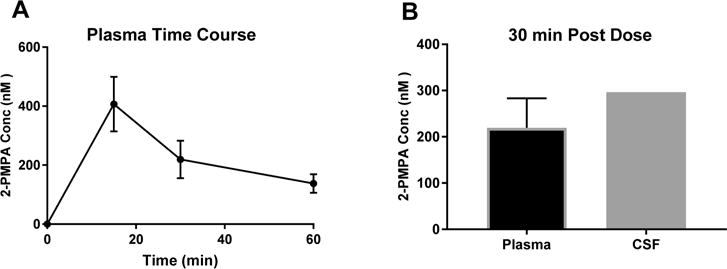
IN delivery of compound 1 to nonhuman primates resulted in 1:1 CSF/plasma ratio of 2-PMPA. (A) Time course of 2-PMPA delivery to plasma after IN administration of compound 1 to nonhuman primate shows that the prodrug resulted in delivery of high nM concentrations of 2-PMPA and significant systemic exposure. (B) At 30 min post dose, compound 1 achieved CSF delivery of high nM 2-PMPA, resulting in a CSF/plasma ratio of near unity, indicative of favorable distribution into the CNS. Data are presented as mean ± SEM (n = 3) for plasma and as mean for CSF (n = 2).
DISCUSSION
In the present study, we demonstrate that brain, peripheral nerve, and systemic exposure of the GCPII inhibitor 2-PMPA can be improved using a prodrug strategy combined with the IN route of administration. These findings have important implications for the future treatment of neurological and psychiatric disorders. 2-PMPA has demonstrated efficacy in many animal models of neurological disorders wherein abnormal glutamatergic transmission has been implicated,33 but its clinical translation has been hindered by poor pharmacokinetic properties including low brain and nerve exposures. Recently, we reported that 2-PMPA brain penetration can be improved by using an IN route of administration.24 In another report, we showed that prodrugs of 2-PMPA could improve oral bioavailability.23 The current results expand on these findings, showing that additive improvement of 2-PMPA brain and nerve delivery can be achieved by pairing these two approaches.
The prodrug strategy has been successfully employed for other drugs with poor brain penetration and low bioavailability.34–36 For example, esterification of the carboxylic acid on L-DOPA has produced prodrugs with greater levels of CNS exposure following IN delivery.34 Using a similar approach, with the intent of enhancing permeability to biological membranes and further improving brain penetration after IN dosing, we strategically masked the glutarate γ-carboxylic acid functionality on 2-PMPA with PAB, POC, n-butyl, benzyl, and n-octyl promoieties to obtain ester prodrugs.
The resulting compounds 1–5 were then tested for GCPII inhibitory activity. While not as potent as 2-PMPA (IC50 = 300 pM),33 all five prodrugs exhibited low nanomolar IC50 values (IC50 = 1–20 nM). This finding was not unexpected as previous SAR studies with other 2-PMPA analogues showed that the γ-carboxylate position could be modified with moderate loss of activity.23 All five prodrugs also exhibited increased cLogP values versus 2-PMPA, suggesting enhanced lipophilicity that could improve brain penetration after IN administration. We next tested the in vivo stability of compounds 1–5 after IN administration in rats. The benzyl and alkyl chain ester promoieties present in compounds 4 and 5, respectively, conferred the highest cLogP values and promoted increased distribution of the intact prodrugs into the OB and plasma relative to equimolar doses of 2-PMPA. However, these prodrugs were too stable and failed to liberate 2-PMPA, resulting in low 2-PMPA plasma and brain concentrations. Shortening of the alkyl chain promoiety in compound 3 resulted in lower cLogP and reduced OB penetration, although distribution into the plasma remained high. Like compounds 4 and 5, compound 3 was also found to be too stable in vivo, failing to liberate significant concentrations of 2-PMPA. By contrast, IN administration of compound 1, γ-PAB-2-PMPA, readily released high concentrations of 2-PMPA in the OB and plasma concomitant with a complete disappearance of the parent prodrug.
Given the favorable profile of compound 1, a full time-dependent pharmacokinetic analysis after IN and IV administration in rats was performed. Following IN administration, compound 1 resulted in high 2-PMPA exposures in multiple brain regions and sciatic nerve as well as plasma. Encouragingly, IN administration of compound 1 resulted in significant 2-PMPA exposures in the cortex, a site of action with potential therapeutic importance for several neurological and psychiatric disorders in which GCPII is implicated. Despite being more distal to the OB dosing site, compound 1 also surprisingly delivered high concentrations of 2-PMPA to the cerebellum. This parity may be due to the ability of compound 1 to permeate the blood–brain barrier from systemic circulation and distribute evenly into the CNS both directly from the olfactory bulb and from the periphery. Indeed, compound 1 exhibits enhanced distribution into the plasma via the respiratory epithelium, which may be similarly mediated by improved lipophilicity and cellular permeability. In fact, plasma exposure was higher (99 h·nmol/mL) for 1 relative to equivalent 2-PMPA administration both by IN and intraperitoneal (IP) routes (79.1 and 77.1 h·nmol/mL, respectively) as previously shown.24 In addition to improving brain exposure, compound 1 also increased peripheral nerve delivery of 2-PMPA, resulting in a sciatic nerve to plasma ratio of 0.84, the therapeutic target for utility of GCPII inhibition in neuropathic and inflammatory pain. Notably, this represents a significant improvement (7-fold) over free 2-PMPA, which achieved a sciatic nerve to plasma ratio of 0.12 following an equivalent dose.37 When compared head to head with the IV route, IN administration of compound 1 in rats showed similar systemic exposure of 2-PMPA, but provided enhanced nervous tissue exposure. Specifically, the levels of 2-PMPA in the OB, cortex, cerebellum, and sciatic nerve following IV dosing was 40-, 4.5-, 4-, and 5-fold lower as compared to IN administration. The brain/plasma ratio following IV administration ranged between 0.03 and 0.04, similar to what we have previously reported for the systemic IP route.37
Although the study results are promising, rodent and human olfactory anatomy differs in key ways that complicate direct translation of the rodent findings. For example, rodent olfactory epithelium covers a broader region of the nasal cavity than that of humans.27 We therefore conducted a pilot study using a device specifically designed for targeting the olfactory epithelium in nonhuman primates which possess nasal anatomy very similar to that of humans.38 When administered IN to primates, compound 1 (2.5 mg/kg) delivered 2-PMPA concentrations of 0.296 ± 0.002 μM to the CSF at 30 min post dose. We had shown previously that IN administration of 2-PMPA (30 mg/kg) to primates led to 1.5 μM in the CSF.24 Although not performed as a head-to-head comparison, the increased CSF exposure of 2-PMPA delivery by IN compound 1 (>2-fold) relative to equimolar IN 2-PMPA supports the rodent findings. Although CSF exposure is not equivalent to brain exposure, it is often used as a surrogate compartment for CNS penetration in nonhuman primates, a practice supported by recent preclinical findings of comparable drug concentration measurements when sampled from either CSF or brain interstitial fluid in a within-subjects study design.39 In addition, CSF is a clinically relevant tissue compartment, lending higher translational value to the present results.
CONCLUSIONS
These findings support the hypothesis that the CNS can be preferentially targeted by combining prodrug delivery with IN administration. By pairing these two approaches we have demonstrated additive improvement in brain delivery of a highly active GCPII inhibitor. Collectively, our findings support the potential clinical utility of administering 2-PMPA prodrugs IN for the treatment of nervous system disorders. More broadly, the results of this study indicate that prodrugs with enhanced lipophilicity can be delivered IN to improve CNS exposures of highly hydrophilic compounds.
Supplementary Material
Acknowledgments
This research was supported by an NIH R01CA161056 and P30MH075673-S1 and National Multiple Sclerosis Society grant (to B.S.S.) and the Institute of Organic Chemistry and Biochemistry of the Academy of Sciences of the Czech Republic v.v.i. (RVO 61388963).
ABBREVIATIONS
- 2-PMPA
2-(phosphonomethyl)pentanedioic acid
- AUC
area under the curve
- Cmax
maximum plasma concentration
- CNS
central nervous system
- CSF
cerebrospinal fluid
- GCPII
glutamate carboxypeptidase-II
- IN
intranasal
- IP
intraperitoneal
- IV
intravenous
- NAAG
N-acetyl-aspartyl-glutamate
- SAR
structure–activity relationship
- PAB
p-acetoxybenzyl
- TFA
trifluoroacetic acid
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.7b00231.
Detailed synthesis and characterization for each compound and mass transitions used for LC–MS/MS analysis of 1–5 (PDF)
ORCID
Barbara S. Slusher: 0000-0001-9814-4157
Rana Rais: 0000-0003-4059-2453
Notes
The authors declare no competing financial interest.
References
- 1.Mesters JR, Barinka C, Li W, Tsukamoto T, Majer P, Slusher BS, Konvalinka J, Hilgenfeld R. Structure of glutamate carboxypeptidase II, a drug target in neuronal damage and prostate cancer. EMBO J. 2006;25(6):1375–84. doi: 10.1038/sj.emboj.7600969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Slusher BS, Vornov JJ, Thomas AG, Hurn PD, Harukuni I, Bhardwaj A, Traystman RJ, Robinson MB, Britton P, Lu XC, Tortella FC, Wozniak KM, Yudkoff M, Potter BM, Jackson PF. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat Med. 1999;5(12):1396–402. doi: 10.1038/70971. [DOI] [PubMed] [Google Scholar]
- 3.Adedoyin MO, Vicini S, Neale JH. Endogenous N-acetylaspartylglutamate (NAAG) inhibits synaptic plasticity/transmission in the amygdala in a mouse inflammatory pain model. Mol Pain. 2010;6:60. doi: 10.1186/1744-8069-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kozikowski AP, Zhang J, Nan F, Petukhov PA, Grajkowska E, Wroblewski JT, Yamamoto T, Bzdega T, Wroblewska B, Neale JH. Synthesis of urea-based inhibitors as active site probes of glutamate carboxypeptidase II: efficacy as analgesic agents. J Med Chem. 2004;47(7):1729–38. doi: 10.1021/jm0306226. [DOI] [PubMed] [Google Scholar]
- 5.Nagel J, Belozertseva I, Greco S, Kashkin V, Malyshkin A, Jirgensons A, Shekunova E, Eilbacher B, Bespalov A, Danysz W. Effects of NAAG peptidase inhibitor 2-PMPA in model chronic pain -relation to brain concentration. Neuropharmacology. 2006;51(7–8):1163–71. doi: 10.1016/j.neuropharm.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto T, Hirasawa S, Wroblewska B, Grajkowska E, Zhou J, Kozikowski A, Wroblewski J, Neale JH. Antinociceptive effects of N-acetylaspartylglutamate (NAAG) peptidase inhibitors ZJ-11, ZJ-17 and ZJ-43 in the rat formalin test and in the rat neuropathic pain model. Eur J Neurosci. 2004;20(2):483–94. doi: 10.1111/j.1460-9568.2004.03504.x. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto T, Nozaki-Taguchi N, Sakashita Y, Inagaki T. Inhibition of spinal N-acetylated-alpha-linked acidic dipeptidase produces an antinociceptive effect in the rat formalin test. Neuroscience. 2001;102(2):473–9. doi: 10.1016/s0306-4522(00)00502-9. [DOI] [PubMed] [Google Scholar]
- 8.Carpenter KJ, Sen S, Matthews EA, Flatters SL, Wozniak KM, Slusher BS, Dickenson AH. Effects of GCP-II inhibition on responses of dorsal horn neurones after inflammation and neuropathy: an electrophysiological study in the rat. Neuropeptides. 2003;37(5):298–306. doi: 10.1016/j.npep.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W, Murakawa Y, Wozniak KM, Slusher B, Sima AA. The preventive and therapeutic effects of GCPII (NAALADase) inhibition on painful and sensory diabetic neuropathy. J Neurol Sci. 2006;247(2):217–23. doi: 10.1016/j.jns.2006.05.052. [DOI] [PubMed] [Google Scholar]
- 10.Zhang W, Slusher B, Murakawa Y, Wozniak KM, Tsukamoto T, Jackson PF, Sima AA. GCPII (NAALADase) inhibition prevents long-term diabetic neuropathy in type 1 diabetic BB/Wor rats. J Neurol Sci. 2002;194(1):21–8. doi: 10.1016/s0022-510x(01)00670-0. [DOI] [PubMed] [Google Scholar]
- 11.Olszewski RT, Bzdega T, Neale JH. mGluR3 and not mGluR2 receptors mediate the efficacy of NAAG peptidase inhibitor in validated model of schizophrenia. Schizophrenia research. 2012;136(1–3):160–1. doi: 10.1016/j.schres.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 12.McKinzie DL, Li TK, McBride WJ, Slusher BS. NAALADase inhibition reduces alcohol consumption in the alcohol-preferring (P) line of rats. Addict Biol. 2000;5(4):411–6. doi: 10.1111/j.1369-1600.2000.tb00209.x. [DOI] [PubMed] [Google Scholar]
- 13.Shippenberg TS, Rea W, Slusher BS. Modulation of behavioral sensitization to cocaine by NAALADase inhibition. Synapse (Hoboken, NJ, U S) 2000;38(2):161–6. doi: 10.1002/1098-2396(200011)38:2<161::AID-SYN7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 14.Xi ZX, Kiyatkin M, Li X, Peng XQ, Wiggins A, Spiller K, Li J, Gardner EL. N-acetylaspartylglutamate (NAAG) inhibits intravenous cocaine self-administration and cocaine-enhanced brain-stimulation reward in rats. Neuropharmacology. 2010;58(1):304–13. doi: 10.1016/j.neuropharm.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ha D, Bing SJ, Ahn G, Kim J, Cho J, Kim A, Herath KH, Yu HS, Jo SA, Cho IH, Jee Y. Blocking glutamate carboxypeptidase II inhibits glutamate excitotoxicity and regulates immune responses in experimental autoimmune encephalomyelitis. FEBS J. 2016;283(18):3438–56. doi: 10.1111/febs.13816. [DOI] [PubMed] [Google Scholar]
- 16.Hollinger KR, Alt J, Riehm AM, Slusher BS, Kaplin AI. Dose-dependent inhibition of GCPII to prevent and treat cognitive impairment in the EAE model of multiple sclerosis. Brain Res. 2016;1635:105–12. doi: 10.1016/j.brainres.2016.01.035. [DOI] [PubMed] [Google Scholar]
- 17.Rahn KA, Watkins CC, Alt J, Rais R, Stathis M, Grishkan I, Crainiceau CM, Pomper MG, Rojas C, Pletnikov MV, Calabresi PA, Brandt J, Barker PB, Slusher BS, Kaplin AI. Inhibition of glutamate carboxypeptidase II (GCPII) activity as a treatment for cognitive impairment in multiple sclerosis. Proc Natl Acad Sci U S A. 2012;109(49):20101–6. doi: 10.1073/pnas.1209934109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng JF, Gurkoff GG, Van KC, Song M, Lowe DA, Zhou J, Lyeth BG. NAAG peptidase inhibitor reduces cellular damage in a model of TBI with secondary hypoxia. Brain Res. 2012;1469:144–52. doi: 10.1016/j.brainres.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao Y, Xu S, Cui Z, Zhang M, Lin Y, Cai L, Wang Z, Luo X, Zheng Y, Wang Y, Luo Q, Jiang J, Neale JH, Zhong C. Mice lacking glutamate carboxypeptidase II develop normally, but are less susceptible to traumatic brain injury. J Neurochem. 2015;134(2):340–53. doi: 10.1111/jnc.13123. [DOI] [PubMed] [Google Scholar]
- 20.Neale JH. N-acetylaspartylglutamate is an agonist at mGluR(3) in vivo and in vitro. J Neurochem. 2011;119(5):891–5. doi: 10.1111/j.1471-4159.2011.07380.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao Y, Gao Y, Xu S, Bao J, Lin Y, Luo X, Wang Y, Luo Q, Jiang J, Neale JH, Zhong C. Glutamate carboxypeptidase II gene knockout attenuates oxidative stress and cortical apoptosis after traumatic brain injury. BMC Neurosci. 2016;17:15. doi: 10.1186/s12868-016-0251-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barinka C, Rojas C, Slusher B, Pomper M. Glutamate carboxypeptidase II in diagnosis and treatment of neurologic disorders and prostate cancer. Curr Med Chem. 2012;19(6):856–70. doi: 10.2174/092986712799034888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majer P, Jancarik A, Krecmerova M, Tichy T, Tenora L, Wozniak K, Wu Y, Pommier E, Ferraris D, Rais R, Slusher BS. Discovery of Orally Available Prodrugs of the Glutamate Carboxypeptidase II (GCPII) Inhibitor 2-Phosphonomethylpentanedioic Acid (2-PMPA) J Med Chem. 2016;59:2810–9. doi: 10.1021/acs.jmedchem.6b00062. [DOI] [PubMed] [Google Scholar]
- 24.Rais R, Wozniak K, Wu Y, Niwa M, Stathis M, Alt J, Giroux M, Sawa A, Rojas C, Slusher BS. Selective CNS Uptake of the GCP-II Inhibitor 2-PMPA following Intranasal Administration. PLoS One. 2015;10(7):e0131861. doi: 10.1371/journal.pone.0131861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson PF, Tays KL, Maclin KM, Ko YS, Li W, Vitharana D, Tsukamoto T, Stoermer D, Lu XC, Wozniak K, Slusher BS. Design and pharmacological activity of phosphinic acid based NAALADase inhibitors. J Med Chem. 2001;44(24):4170–5. doi: 10.1021/jm0001774. [DOI] [PubMed] [Google Scholar]
- 26.Stoermer D, Liu Q, Hall MR, Flanary JM, Thomas AG, Rojas C, Slusher BS, Tsukamoto T. Synthesis and biological evaluation of hydroxamate-Based inhibitors of glutamate carboxypeptidase II. Bioorg Med Chem Lett. 2003;13(13):2097–100. doi: 10.1016/s0960-894x(03)00407-4. [DOI] [PubMed] [Google Scholar]
- 27.Dhuria SV, Hanson LR, Frey WH. 2nd Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–73. doi: 10.1002/jps.21924. [DOI] [PubMed] [Google Scholar]
- 28.Hin B, Majer P, Tsukamoto T. Facile synthesis of alpha-substituted acrylate esters. J Org Chem. 2002;67(21):7365–8. doi: 10.1021/jo026101s. [DOI] [PubMed] [Google Scholar]
- 29.Barinka C, Rinnova M, Sacha P, Rojas C, Majer P, Slusher BS, Konvalinka J. Substrate specificity, inhibition and enzymological analysis of recombinant human glutamate carboxypeptidase II. J Neurochem. 2002;80(3):477–87. doi: 10.1046/j.0022-3042.2001.00715.x. [DOI] [PubMed] [Google Scholar]
- 30.Bailer AJ. Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biopharm. 1988;16(3):303–9. doi: 10.1007/BF01062139. [DOI] [PubMed] [Google Scholar]
- 31.Nedelman JR, Gibiansky E, Lau DTW. Applying Bailer’s Method for AUC Confidence Intervals to Sparse Sampling. Pharm Res. 1995;12(1):124–128. doi: 10.1023/a:1016255124336. [DOI] [PubMed] [Google Scholar]
- 32.Yuan J. Estimation of variance for AUC in animal studies. J Pharm Sci. 1993;82(7):761–3. doi: 10.1002/jps.2600820718. [DOI] [PubMed] [Google Scholar]
- 33.Vornov JJ, Hollinger KR, Jackson PF, Wozniak KM, Farah MH, Majer P, Rais R, Slusher BS. Still NAAG’ing After All These Years: The Continuing Pursuit of GCPII Inhibitors. Adv Pharmacol. 2016;76:215–55. doi: 10.1016/bs.apha.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Kao HD, Traboulsi A, Itoh S, Dittert L, Hussain A. Enhancement of the systemic and CNS specific delivery of L-dopa by the nasal administration of its water soluble prodrugs. Pharm Res. 2000;17(8):978–84. doi: 10.1023/a:1007583422634. [DOI] [PubMed] [Google Scholar]
- 35.Aungst BJ, Hussain MA. Use of prodrugs of 3-hydroxymorphinans to prevent bitter taste upon buccal, nasal or sublingual administration. Google Patents. 1987 [Google Scholar]
- 36.Hussain AA, AL-Bayatti AA, Dakkuri A, Okochi K, Hussain MA. Testosterone 17β-N, N-dimethylglycinate hydrochloride: A prodrug with a potential for nasal delivery of testosterone. J Pharm Sci. 2002;91(3):785–789. doi: 10.1002/jps.10083. [DOI] [PubMed] [Google Scholar]
- 37.Rais R, Rojas C, Wozniak K, Wu Y, Zhao M, Tsukamoto T, Rudek MA, Slusher BS. Bioanalytical method for evaluating the pharmacokinetics of the GCP-II inhibitor 2-phosphonomethyl pentanedioic acid (2-PMPA) J Pharm Biomed Anal. 2014;88:162–9. doi: 10.1016/j.jpba.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lochhead JJ, Thorne RG. Intranasal delivery of biologics to the central nervous system. Adv Drug Delivery Rev. 2012;64(7):614–628. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Nagaya Y, Nozaki Y, Takenaka O, Watari R, Kusano K, Yoshimura T, Kusuhara H. Investigation of utility of cerebrospinal fluid drug concentration as a surrogate for interstitial fluid concentration using microdialysis coupled with cisternal cerebrospinal fluid sampling in wild-type and Mdr1a(−/−) rats. Drug Metab Pharmacokinet. 2016;31(1):57–66. doi: 10.1016/j.dmpk.2015.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.