

Abstract
A number of culture-dependent and -independent studies have reported that the number and significance of bacterial species in semen may have been underestimated. The aim of the present study was to profile the seminal microbiome in patients with obstructive or non-obstructive azoospermia. A high-throughput sequencing method was adopted to sequence genomic DNA extracted from the semen of healthy people (C group), patients with obstructive azoospermia (OA group) and patients with non-obstructive azoospermia (NOA group). The results revealed that Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria species comprised the majority of bacteria in the C (98.14%), OA (98.26%) and NOA (90.96%) groups. Patients in the OA and NOA groups exhibited an increase in Bacteroidetes and Firmicutes, whereas the number of Proteobacteria and Actinobacteria were decreased compared with the C group. A total of 398 common operational taxonomic units were identified, of which 27 belonged to the genus Lactobacillus. Furthermore, Phylogenetic Investigation of Communities by Reconstruction of Unobserved States analysis indicated that the pathogenic species and reduced biodiversity in the semen of patients with azoospermia may result in an increased risk of metabolic, infectious and immune diseases. In the present study, the seminal microbiome of patients with obstructive or non-obstructive azoospermia was explored, which may be useful for developing novel treatments against azoospermia as well as for its diagnosis.
Keywords: high-throughput sequencing, obstructive azoospermia, non-obstructive azoospermia, microbial diversity, Lactobacillus sp.
Introduction
Azoospermia, which affects ~1% of the male population and may be responsible for up to 20% of male infertility situations, is a medical condition in which the semen contains no sperm (1–3). Azoospermia may be caused by a lack of spermatozoa production in the testes (secretory or non-obstructive azoospermia; NOA) or due to spermatozoa being unable to reach the ejaculate (excretory or obstructive azoospermia; OA) (1). Previous studies have indicated that genetic factors, including chromosomal abnormalities, are responsible for azoospermia; however, few studies have explored the association between microorganisms and azoospermia (1–3).
Microbiomes in the human skin, intestines, oral cavity, vagina and urethra serve a key role in human health, disease and other etiologies (4–6). It has been reported that several bacteria, including Lactobacillus iners, Gardnerella vaginalis, Escherichia faecalis, E. coli and Staphylococcus aureus, are associated with male infertility as demonstrated using polymerase chain reaction (PCR) or culture-based methods (7–12). However, the majority of previous studies focused on a few types of bacteria and relied on qualitative analysis to discover associations between semen microbiomes and semen quality; as such, at present there is no comprehensive understanding of bacterial communities in semen (13).
Recent technological advances in next generation sequencing technology have enabled the pleiotropic effects of microorganisms in the human host to be elucidated, providing an excellent platform to explore the relationship between human and semen microbiota (14–20). In the present study, the high-throughput sequencing method was used to compare seminal bacterial composition in patients with obstructive and non-obstructive azoospermia. The results may be useful for developing clinical therapies for the prevention and treatment of azoospermia.
Materials and methods
Ethical statement
The present study was approved by the Ethical Committee of the Jiangxi Maternal and Child Health Hospital (Nanchang, China) and all participants provided written informed consent. All experiments were carried out in accordance with the guidelines provided by the Jiangxi Maternal and Child Health Hospital.
Patients and sampling
Samples of seminal plasma were collected at the Jiangxi Maternal and Child Health Hospital between October and December 2016 from three groups of men without leukocytospermia as follows: The control (C) group (22–36 years old), the obstructive azoospermia (OA) group (24–38 years old) and the non-obstructive azoospermia (NOA) group (25–37 years old). A total of 5 fertile donors with normal sperm parameters (seminal volume, 2–6 ml; sperm count, 0.6–1.5×1012/l; progressive motile spermatozoa ≥30% or progressive motile spermatozoa + motile spermatozoa ≥50%) (4) were recruited as the C group. A total of 12 patients with azoospermia who underwent surgical sperm retrieval via percutaneous epididymal sperm aspiration or testicular sperm extraction were recruited and were divided into the OA group (n=6) and the NOA group (n=6). In OA, sperm are produced normally inside the testicle, but are prevented from reaching the ejaculate due to a blockage or obstruction in the reproductive tubing. In non-obstructive azoospermia group, the tubes are not blocked; however, there is a spermatogenesis problem whereby there is a very low level of sperm production or a total lack of production (8). Patients with abnormal karyotypes and those who had previously suffered from an injury to the genitals were excluded.
Prior to sampling, the glans penis of each patient was washed using soap and water. Semen was obtained by masturbation, ejaculated into a sterile collection tube and incubated at 37°C for 30 min for liquefaction. Basic semen parameters and leukocytospermia were detected as previously described and the samples were stored at −80°C prior to DNA extraction (20).
Extraction of genomic DNA and high-throughput sequencing
Genomic DNA was extracted from each semen sample using a TIANamp Genomic DNA kit (Tiangen Biotech Co., Ltd., Beijing, China) and combined with bead beating as previously described (14). The concentration and quality of extracted genomic DNA were tested prior to sequencing using a spectrophotometer at 230 nm (A 260) and 260 nm (A 260) (NanoDrop; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The extracted genomic DNA was used as a template to amplify the V4 region of the 16S rRNA genes of all samples using 515F/806R primers (515F, 5′-GTGCCAGCMGCCGCGGTAA-3′; 806R, 5′-GGACTACVSGGGTATCTAAT-3′). The PCR reaction conditions were as follows: 2 min initial denaturation at 94°C and 30 cycles of 30 sec denaturation at 94°C, 30 sec annealing at 52°C, 30 sec extension at 72°C, followed by a 5 min final extension at 72°C. The samples were maintained at 4°C when the PCR reaction finished. Each 25 µl reaction made up of 2.5 µl 10X Ex Taq buffer (Mg2+ free; Takara Biotechnology Co., Ltd., Dalian, China), 2 µl (2.5 mM) dNTP mixture, 1.5 µl Mg2+ (2.5 mM), 0.25 µl Ex-Taq DNA polymerase (Takara Biotechnology Co., Ltd.), 0.5 µl barcode primer V4F (10 µM), 0.5 µl barcode primer V4R252 (10 µM), 2 µl of fecal DNA and 15.75 µl milli-Q water. These PCR products were sequenced using an Illumina HiSeq 2000 platform (21).
Bioinformatics and multivariate statistics
Paired-end reads from the original DNA fragments were merged using FLASH (version 1.2.7; ccb.jhu.edu/software/FLASH/) when any of the reads overlapped that generated from the opposite end of the same DNA fragment. Paired-end reads were assigned to each sample according to the unique barcodes using Trimmomatic (version 0.33) (22). Sequence analysis was subsequently performed using the UPARSE software package (version 7.0.100) (23) with the UPARSE-OTU and Chao1, the Chao1 estimator and the Shannon index (24) with UPARSE-OTUref algorithms. In-house Perl scripts were used to analyze α (within samples) and β (among samples) diversity. Sequences with ≥97% similarity were assigned to the same OTUs. A representative sequence was selected for each OTU and the RDP classifier (version 2.2) (25); was used to annotate taxonomic information for each representative sequence. Cluster analysis was preceded by weighted UniFrac distance using QIIME software package (version 1.8.0) (26), partial least squares discriminate analysis (PLS-DA) was preceded using SIMCA-P software version 11.5 (Umetrics; Sartorius Stedim Biotech, Malmö, Sweden) and the species that were differentially abundant were characterized for their metabolic capacity using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt), Version 1.0.0 (27,28). The statistical significance was set at P<0.05 for correction of multiple comparisons.
Results
Seminal bacterial composition at phylum level
To compare microbes in the OA and NOA groups, the V4 hypervariable region of bacterial genomic DNA was sequenced using high-throughput sequencing and the effective tags with ≥97% similarity were considered as one OTU. In total, 675,539 usable sequences and 5,867 OTUs were obtained from the samples, with a mean of 345 OTUs per group (data not shown). Furthermore, the Chao1 and Shannon indices were nearly saturated and the rarefaction curve of every sample entered the plateau phase, which indicated that the sequencing data was reasonable (OTU number was sufficient for further analysis) and species composition was highly uniform in each group (data not shown).
At the phylum level, data from the 20 microorganisms that were most abundant in each group was analyzed. Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria were the predominant phyla, accounting for 98.14% in the C group, 98.26% in the OA group and 90.96% in the NOA group (Fig. 1A).
Figure 1.

(A) Bacterial composition (percentage) and (B) relative abundance of phyla (number) in semen from the C, OA and NOA groups. *P<0.05 vs. the C group. C, control; OA, obstructive azoospermia; NOA, non-obstructive azoospermia.
Compared with the C group, azoospermia reduced the OTU number of Cyanobacteria, Acidobacteria, Gemmatimonadetes, Planctomycetes, Chloroflexi, Crenarchaeota, Armatimonadetes, Elusimicrobia, Nitrospirae, Euryarchaeota, Spirochaetes, Chlorobi, Synergistetes, Chlamydiae and Verrucomicrobia, whereas the number of Bacteroidetes was increased (P<0.05; Fig. 1B).
Seminal bacterial composition at the genus level
Lactobacillus, Prevotella, Proteus, Pseudomonas, Veillonella, Corynebacterium, Rhodococcus, Staphylococcus and Bacillus were the dominant genera detected in the C (55.02%), OA (62.54%) and NOA (59.74%) groups (Fig. 2A). The distribution map indicated that Alicyclobacillus, Amaricoccus, Anaeromyxobacter, Aquicella, Arsenicicoccus, Azospirillum, Chitinimonas, Chlorobaculum, Coprococcus, Desulfovibrio, Dokdonella, Gallionella, Geobacter, Helicobacter, Idiomarina, Kaistia and Kribbella were significantly reduced in the C group compared with the OA and NOA groups (P<0.05; Fig. 2B).
Figure 2.

(A) Bacterial composition (percentage) and (B) relative abundance of genera (number) in semen from the C, OA and NOA groups. *P<0.05 vs. the C group. C, control; OA, obstructive azoospermia; NOA, non-obstructive azoospermia.
The diversity of the microbial community in the C, OA and NOA groups
There were 1,093, 925 and 840 OTUs detected in the C, OA and NOA groups, respectively (Fig. 3A). A total of 398 common OTUs were identified in the C, OA and NOA groups, of which 27 belonged to the Lactobacillus phylum (Fig. 3A). In addition, the partial least squares discriminant analysis (PLS-DA) indicated that patients in the OA and NOA groups scattered on the left, whereas the C group gathered together on the right of the coordinate axis (Fig. 3B). No overlapping of the C group with the OA or NOA groups was observed; however, some samples in OA and NOA groups overlapped (Fig. 3B), indicting the microbial diversity between the OA or NOA groups had a higher degree of similarity than the C group.
Figure 3.
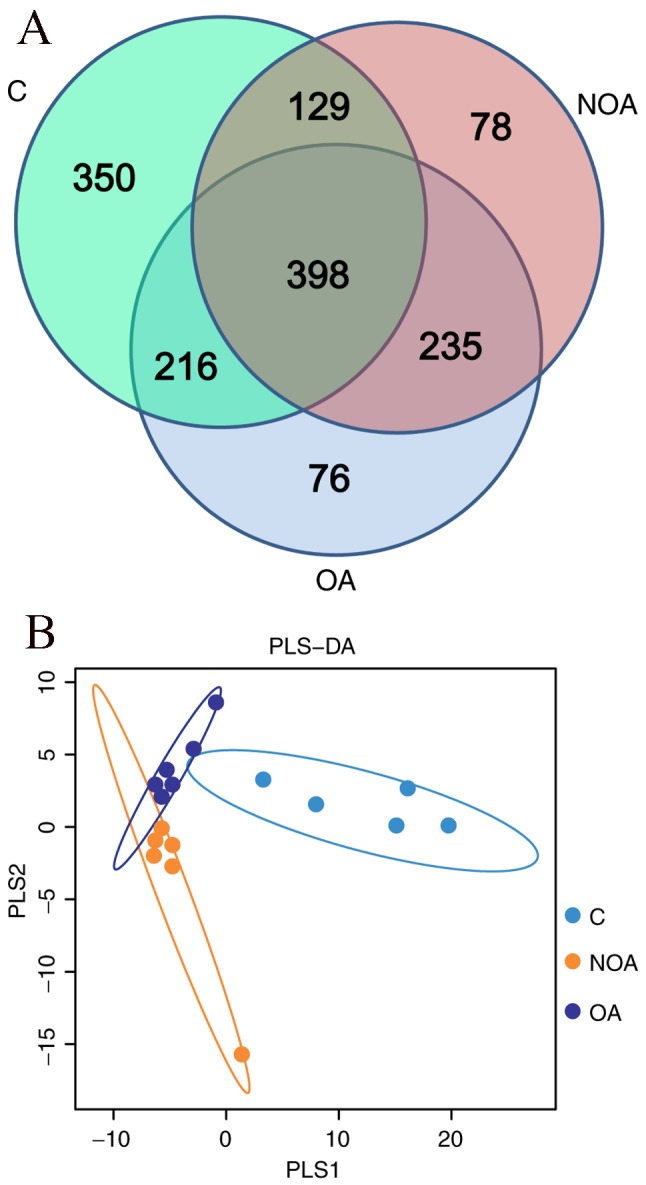
(A) Scalar-Venn representation and (B) PLS-DA of shared genera among microbiomes in the C, OA and NOA groups. PLS-DA, partial least squares discriminant analysis; C, control; OA, obstructive azoospermia; NOA, non-obstructive azoospermia.
Composition of the bacterial communities in C, OA and NOA groups
Significantly more Sneathia and Lysobacter sp. were present in the NOA group compared with the C and OA groups (P<0.05; Fig. 4). The number of Solibacillus, Campyiobacter, Campyiobacteraceae and Plesiomonas OTUs in the OA group was significantly higher compared with the NOA and C groups (P<0.05; Fig. 4). The number of bacteria in red-labeled genera was significantly higher in the C group compared with the OA and NOA groups (P<0.05; Fig. 4).
Figure 4.

Taxonomic representation of statistically significant differences between semen microbiomes of the C, OA and NOA groups. Differences are represented by color of the most abundant classes. Red indicates the C group, blue indicates the OA group and green indicates the NOA group. Yellow indicates non-significant data. C, control; OA, obstructive azoospermia; NOA, non-obstructive azoospermia.
Prediction of bacterial functions in C, OA and NOA groups
Correlation analyses were performed for various Kyoto Encyclopedia of Genes and Genomes pathways using the PICRUSt software. The association between seminal bacteria and health was investigated (Fig. 5A). The results indicated that patients in the OA and NOA groups were at an increased risk of having metabolic, infectious and immune diseases (Fig. 5A). Furthermore, the results indicate that azoospermia enhances the protein expression of genes associated with nucleotide metabolism, metabolism of cofactors and vitamins, glycan biosynthesis and metabolism, enzyme families and energy metabolism; however, the expression of genes associated with xenobiotic biodegradation and metabolism, metabolism of terpenoids and polyketides, lipid metabolism and amino acid metabolism was downregulated (Fig. 5B).
Figure 5.

The potential effects of bacteria in the seminal fluid samples of the C, OA and NOA groups on (A) diseases and (B) metabolism. C, control; OA, obstructive azoospermia; NOA, non-obstructive azoospermia; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Discussion
In the present study, semen samples were obtained from patients with azoospermia and healthy control subjects, following which the 16S rRNA gene fragments were sequenced using high-throughput sequencing method. The results revealed the composition of polymicrobial communities in semen samples and the most prevalent phyla were Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria, comprising 98.14, 98.26 and 90.96% of total phyla in the C, OA and NOA groups, respectively. Patients in the OA and NOA groups had a greater number of Bacteroidetes and Firmicutes phyla, whereas the number of Proteobacteria and Actinobacteria was decreased compared with the C group. Firmicutes is a phylum of bacteria, the majority of which produce endospores, are resistant to desiccation, are able to survive extreme conditions and are typically found in various environments; this group includes some notable pathogens (29). In addition, the phylum Bacteroidetes comprises three large classes of Gram-negative, non-spore-forming, anaerobic or aerobic and rod-shaped bacteria; furthermore, some members of the genus Bacteroides are opportunistic pathogens (29). The increased number of Bacteroidetes and Firmicutes species in patients with azoospermia may therefore increase the risk of gynecological inflammation in their sexual partners.
At the genus level, nine dominant bacteria of the Lactobacillus, Prevotella, Proteus, Pseudomonas, Veillonella, Corynebacterium, Rhodococcus, Staphylococcus and Bacillus genera accounted for 55.02, 62.54 and 59.74% of total OTUs in the C, OA and NOA groups, respectively. The OTU number of Lactobacillus was 6.79% in the C group, whereas it accounted for 17.98 and 17.24% in the OA and NOA groups, respectively. The vagina and its microflora form a balanced ecosystem; vaginal dysbacteriosis therefore results in obstetric and gynecological diseases, as well as complications characterized by a reduced number or total absence of Lactobacilli (30,31). It may therefore be of use to further study the role of Lactobacillus in the semen of patients with azoospermia.
To further compare the microbial diversity across groups, PLS-DA and Venn methods were used. The Venn results indicated that there were 398 OTUs common to the C, OA and NOA groups, of which 27 were Lactobacillus. Furthermore, PLS-DA indicated that the bacterial community was more similar when comparing the OA and NOA groups than when comparing either azoospermia group with control subjects. This suggests that sperm may be an important factor in determining microbial composition.
Patients with azoospermia exhibited a reduction in many seminal bacteria compared with healthy controls; however, the OTU number of Solibacillus, Campylobacter, Campyiobacteraceae and Plesiomonas in the OA group, as well as Sneathia and Lysobacter in the NOA group, were increased. The majority of Campylobacter species cause diseases that affect humans and other animals, so this increase poses a potential health risk to patients with azoospermia (32).
PICRUSt software as used to assess bacterial functions and the results indicated that patients with azoospermia had an increased expression of genes associated with nucleotide metabolism, metabolism of cofactors and vitamins, glycan biosynthesis and metabolism, enzyme families and energy metabolism, as well as a reduced expression of genes associated with xenobiotic biodegradation and metabolism, metabolism of terpenoids and polyketides, lipid metabolism and amino acid metabolism. These changes in expression increase the risk of metabolic, infectious and immune diseases in patients with azoospermia compared with the C group.
In the present study, the seminal bacterial composition in patients with OA and NOA was explored using high-throughput sequencing and the results indicate that the semen of patients with azoospermia contains less beneficial microbes compared with healthy controls. The increased number of Bacteroidetes and Firmicutes species in patients with azoospermia puts them at a greater risk of metabolic, infectious and immune diseases. To the best of our knowledge, this is the first study comparing seminal microbiomes in patients with OA or NOA and healthy men using Illumina sequencing. The small sample size used in the present study may be insufficient to fully reflect the actual microbiota composition of semen from patients with OA or NOA and, as such, future studies are required.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant nos. 81501317, 81503364, 31560264), Excellent Youth Foundation of Jiang Xi Scientific Committee (grant nos. 20171BCB23028).
References
- 1.Luo T, Zou QX, He YQ, Wang HF, Wang T, Liu M, Chen Y, Wang B. Matrine compromises mouse sperm functions by a [Ca(2+)]i-related mechanism. Reprod Toxicol. 2016;60:69–75. doi: 10.1016/j.reprotox.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 2.He Y, Zou Q, Li B, Chen H, Du X, Weng S, Luo T, Zeng X. Ketamine inhibits human sperm function by Ca(2+)-related mechanism. Biochem Biophys Res Commun. 2016;478:501–506. doi: 10.1016/j.bbrc.2016.04.144. [DOI] [PubMed] [Google Scholar]
- 3.He Y, Zou Q, Chen H, Weng S, Luo T, Zeng X. Lead inhibits human sperm functions by reducing the levels of intracellular calcium, cAMP and tyrosine phosphorylation. Tohoku J Exp Med. 2016;238:295–303. doi: 10.1620/tjem.238.295. [DOI] [PubMed] [Google Scholar]
- 4.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson MH, Diven MA, Huff LW, Paulos CM. Harnessing the microbiome to enhance cancer immunotherapy. J Immunol Res. 2015;2015:368736. doi: 10.1155/2015/368736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, Shafiq F, Kotol PF, Bouslimani A, Melnik AV, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med. 2017;9:pii: eaah4680. doi: 10.1126/scitranslmed.aah4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Croo I, Van der Elst J, Everaert K, De Sutter P, Dhont M. Fertilization, pregnancy and embryo implantation rates after ICSI in cases of obstructive and non-obstructive azoospermia. Hum Reprod. 2000;15:1383–1388. doi: 10.1093/humrep/15.6.1383. [DOI] [PubMed] [Google Scholar]
- 8.Franasiak JM, Scott RT., Jr Reproductive tract microbiome in assisted reproductive technologies. Fertil Steril. 2015;104:1364–1371. doi: 10.1016/j.fertnstert.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 9.Javurek AB, Spollen WG, Ali AM, Johnson SA, Lubahn DB, Bivens NJ, Bromert KH, Ellersieck MR, Givan SA, Rosenfeld CS. Discovery of a novel seminal fluid microbiome and influence of estrogen receptor alpha genetic status. Sci Rep. 2016;6:23027. doi: 10.1038/srep25216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tuddenham S, Ghanem KG. A microbiome variable in the HIV-prevention equation. Science. 2017;356:907–908. doi: 10.1126/science.aan6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Francesco MA, Negrini R, Ravizzola G, Galli P, Manca N. Bacterial species present in the lower male genital tract: A five-year retrospective study. Eur J Contracept Reprod Health Care. 2011;16:47–53. doi: 10.3109/13625187.2010.533219. [DOI] [PubMed] [Google Scholar]
- 12.Younes JA, Lievens E, Hummelen R, van der Westen R, Reid G, Petrova MI. Women and their microbes: The unexpected friendship. Trends Microbiol. 2018;26:16–32. doi: 10.1016/j.tim.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Weng SL, Chiu CM, Lin FM, Huang WC, Liang C, Yang T, Yang TL, Liu CY, Wu WY, Chang YA, et al. Bacterial communities in semen from men of infertile couples: Metagenomic sequencing reveals relationships of seminal microbiota to semen quality. PLoS One. 2014;9:e110152. doi: 10.1371/journal.pone.0110152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu X, Wu X, Qiu L, Wang D, Gan M, Chen X, Wei H, Xu F. Analysis of the intestinal microbial community structure of healthy and long-living elderly residents in Gaotian village of liuyang city. Appl Microbiol Biot. 2015;99:9085–9095. doi: 10.1007/s00253-015-6888-3. [DOI] [PubMed] [Google Scholar]
- 15.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, Mujagic Z, Vila AV, Falony G, Vieira-Silva S, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 2016;352:565–569. doi: 10.1126/science.aad3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Derrien M, van Hylckama Vlieg JE. Fate, activity and impact of ingested bacteria within the human gut microbiota. Trends Microbiol. 2015:1–366. doi: 10.1016/j.tim.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Xin F, Xin W, Yang S, Meng F, Wang X, Wei H, Chen T. Evaluation of the microbial diversity in amyotrophic lateral sclerosis using high-throughput sequencing. Front Microbiol. 2016;7:1479. doi: 10.3389/fmicb.2016.01479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui M, Xiao H, Li Y, Zhou L, Zhao S, Luo D, Zheng Q, Dong J, Zhao Y, Zhang X, et al. Faecal microbiota transplantation protects against radiation-induced toxicity. EMBO Mol Med. 2017;4:448–461. doi: 10.15252/emmm.201606932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen T, Wu Q, Zhou H, Deng K, Wang X, Meng F, Yang S, Wang X, Shah NP, Wei H. Assessment of commercial probiotic products in China for labelling accuracy and probiotic characterisation of selected isolates. Int J Dairy Technol. 2017;70:119–126. doi: 10.1111/1471-0307.12331. [DOI] [Google Scholar]
- 21.Xu J, Lian F, Zhao L, Zhao Y, Chen X, Zhang X, Guo Y, Zhang C, Zhou Q, Xue Z, et al. Structural modulation of gut microbiota during alleviation of type 2 diabetes with a Chinese herbal formula. Isme J. 2015;9:552–562. doi: 10.1038/ismej.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolger A, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edgar R. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 24.Steege HT. Measuring biological diversity. Environ Ecol Stat. 2004;1:95–103. [Google Scholar]
- 25.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. Ribosomal database project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42(Database Issue):D633–D642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Javurek AB, Spollen WG, Johnson SA, Bivens NJ, Bromert KH, Givan SA, Rosenfeld CS. Consumption of a high-fat diet alters the seminal fluid and gut microbiomes in male mice. Reprod Fert Devel. 2017;29:1602–1612. doi: 10.1071/RD16119. [DOI] [PubMed] [Google Scholar]
- 28.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolf M, Müller T, Dandekar T, Pollack JD. Phylogeny of Firmicutes with special reference to Mycoplasma (Mollicutes) as inferred from phosphoglycerate kinase amino acid sequence data. Int J Syst Evol Microbiol. 2004;54:871–875. doi: 10.1099/ijs.0.02868-0. [DOI] [PubMed] [Google Scholar]
- 30.Donders GG, Bosmans E, Dekeersmaeckerb A, Vereecken A, Van Bulck B, Spitz B. Pathogenesis of abnormal vaginal bacterial flora. Am J Obstet Gyneco. 2000;182:872–878. doi: 10.1016/S0002-9378(00)70338-3. [DOI] [PubMed] [Google Scholar]
- 31.Kovachev SM. Obstetric and gynecological diseases and complications resulting from vaginal. dysbacteriosis. 2014;68:1–184. doi: 10.1007/s00248-014-0414-5. [DOI] [PubMed] [Google Scholar]
- 32.Bae HS, Im WT, Lee ST. Lysobacter concretionis sp. nov., isolated from anaerobic granules in an upflow anaerobic sludge blanket reactor. Int J Syst Evol Microbiol. 2005;55:1155–1161. doi: 10.1099/ijs.0.63399-0. [DOI] [PubMed] [Google Scholar]