

Abstract
Next-generation sequencing (NGS) has enabled whole-exome and whole-genome sequencing of tumors for causative mutations, allowing for more accurate targeting of therapies. In the process of sequencing the tumor, comparisons to the germline genome may identify variants associated with susceptibility to cancer as well as other hereditary diseases. Already, the combination of massively parallel sequencing and selective capture approaches has facilitated efficient simultaneous genetic analysis (multiplex testing) of large numbers of candidate genes. As the field of oncology incorporates NGS approaches into tumor and germline analyses, it has become clear that the ability to achieve high-throughput genotyping surpasses our current ability to interpret and appropriately apply the vast amounts of data generated from such technologies. A review of the current state of knowledge of rare and common genetic variants associated with cancer risk or treatment outcome reveals significant progress, as well as a number of challenges associated with the clinical translation of these discoveries. The combined efforts of oncologists, genetic counselors, and cancer geneticists will be required to drive the paradigm shift toward personalized or precision medicine and to ensure the incorporation of NGS technologies into the practice of preventive oncology.
INTRODUCTION
The last three decades have witnessed significant strides in our understanding of the genetic basis of cancer susceptibility. In the 1980s and 1990s, rare but highly penetrant cancer predisposition genes were identified by studying cancer-prone families showing Mendelian modes of inheritance. These investigations successfully implicated genes such as BRCA1 and BRCA2 in hereditary breast-ovarian cancer syndrome, DNA mismatch repair genes in Lynch syndrome, p53 in Li-Fraumeni syndrome, and APC in familial adenomatous polyposis.1 Identification of the genetic basis of such syndromes has had a powerful impact on the practice of preventive oncology. The incorporation of cancer genetic testing into oncology marked one of the first applications of personalized genomics in medicine, because it allowed tailored cancer screening, prevention, and, in some cases, therapeutic measures.2–5
Recently, the applications of next-generation sequencing (NGS) technology have led to multiplex gene-panel testing and genome-wide sequencing, posing broad new challenges to clinical oncologists (definitions of important terms involved in NGS are listed in Table 1). As genotyping costs continue to decrease, and computational abilities improve, there will be increasing demand for all patients with cancer to undergo tumor genome sequencing to guide targeted therapies.6,7 In the process, patients' normal or germline DNA may also be scanned, thrusting oncologists into the position of providing genome-based risk assessment to patients and their families. This genomic information will include not only cancer-associated risk but also pharmacogenomic markers to guide treatment choices as well as noncancer disease risks. The task of integrating and translating this genomic information for patients with cancer may require consultation with genetic counselors and clinical cancer geneticists. As in the early days of BRCA testing, oncologists will again be called on to take on new challenges in the emerging field of genomic prevention and personalized medicine.
Table 1.
Definition of Terms
| Term | Definition |
|---|---|
| NGS or MPS | High-throughput DNA sequencing technique that allows for parallel sequencing of thousands to millions of simultaneous sequences, producing vast amounts of data at a fraction of the cost of traditional Sanger sequencing |
| WGS | Sequencing of the complete DNA sequence of an individual |
| WES | Sequencing limited to protein-coding regions of the genome, constituting approximately 1% of the total human genome (approximately 30 Mb) |
| Multiplex gene-panel testing | Targeted analysis of multiple genes of interest simultaneously using NGS/MPS technologies |
| GWAS | Systematic hypothesis-free search for genetic variations, usually in the form of SNPs, across the genome to identify genetic association with disease or trait |
| VUS | Genetic sequence change whose association with disease risk is currently unknown |
| Clinical utility | Degree to which use of test informs clinical decision making and leads to improved health outcomes |
| Clinical validity | Accuracy with which genetic test can identify or predict presence or absence of a particular clinical condition, taking into account specificity, sensitivity, and penetrance of genetic variation |
| Incidentalome | Incidental and/or unexpected genomic findings that may result from genomic evaluation of an individual's DNA sequence |
| Pharmacogenomics | Identification of genetic factors associated with a specific response or side effect for a particular drug/treatment |
Abbreviations: GWAS, genome-wide association study; MPS, massively parallel sequencing; NGS, next-generation sequencing; SNP, single-nucleotide polymorphism; VUS, variant of unknown significance; WES, whole-exome sequencing; WGS, whole-genome sequencing.
From Rare to Common Variants Associated With Risk for Cancer
Although known cancer susceptibility syndromes now number > 100, mutations in high-penetrance genes explain only a fraction of the heritability of human cancers.8 Largely on the basis of knowledge of tumor genomes and pathways, a candidate gene approach has also been applied to the study of cancer susceptibility. As an example, mutations in candidate genes in DNA damage response pathways (ATM, CHEK2, BRIP1, PALB2) were found to be associated with a modest increase in breast cancer risk and are now considered cancer susceptibility genes.9–13
Coinciding with the introduction of high-resolution genotyping arrays in approximately 2005, under the common disease–common variant hypothesis, wherein heritability is presumed to be determined by the joint action of multiple common genes, the genetic architecture of complex diseases, such as cancer, began to be dissected. Using these high-resolution genotyping platforms, genome-wide association studies (GWASs) were rapidly completed for nearly all common cancers.14,15 Although hundreds of statistically robust risk variants, largely in the form of single-nucleotide polymorphisms (SNPs), were identified, each genetic variant was associated with only a modest increase in disease risk (relative risk, approximately 1.1 to 1.5). With > 90% of risk variants residing in noncoding introns, the causal variants in most implicated risk loci have remained elusive, and the biologic basis of most associations remains unclear, although recent mapping of genetic switches to noncoding regions promises greater insight into some variants.16 Given the modest effect size for most risk variants identified, the clinical utility of genomic profiling for risk stratification based on GWAS data has been limited for most common cancers.17–25 However, the clinical utility of common genetic variants in risk assessment continues to evolve. For example, as a result of large international consortia studies, 49 new loci were recently identified for breast cancer, 26 for prostate cancer, and eight for ovarian cancer.26–33 With such additional discoveries, the incorporation of genetic susceptibility into models of risk stratification for public health programs and cancer screening may eventually be feasible.34
NGS Technology
Recently, a shift toward identifying rare genomic variants was made possible by the emergence of NGS using massively parallel sequencing (MPS) platforms that enable whole-exome sequencing (WES) and whole-genome sequencing (WGS) of tumors as well as normal tissue (Table 2; Fig 1 and Appendix Fig A1, online only). NGS technology may directly identify causative mutations, which can then be studied at a functional level. Although promising, NGS gives rise to significant computational and analytic hurdles.
Table 2.
Commercially Available MPS Machines Compared With Traditional Sanger Sequencing
| Platform | Amplification Step | Sequencing Chemistry | Average Read Length (bases) | Run Time | Reads per Run | Bases per Run |
|---|---|---|---|---|---|---|
| Sanger 330XL* | Yes (PCR) | Chain termination by dideoxynucleotides | 400 to 900 | 20 minutes to 3 hours | 96 | 1,900 to 84,000 |
| Pacific Biosciences/PacBio RS† | No (single molecule) | Real-time single-molecule sequencing reactions | 1,500 (C1 chemistry) | 2 hours | 45,000 | 100,000,000 |
| 454 GS FLX* | Yes (emulsion PCR) | Pyrosequencing detection of pyrophosphate release with incorporation of correct complementary base | 700 | 24 hours | 1,000,000 | 700,000,000 |
| Ion Torrent (Personal Genome Machine, 318 Chip)† | Yes (emulsion PCR) | Detection of pH change by semiconductor technology with incorporation of correct complementary base | 200 | 2 hours | 5,000,000 | 1,000,000,000 |
| SOLiD 4* | Yes (emulsion PCR) | Sequencing by ligation using four fluorescently labeled di-base probes | 50 paired end | 14 days | 1,400,000,000 | 120,000,000,000 |
| Illumina/HiSeq 2000* | Yes (bridge amplification PCR) | Sequencing by synthesis using base-specific fluorophores and cyclic reversible-chain termination | 100 paired end | 3 to approximately 10 days | 3,000,000,000 | 600,000,000,000 |
Fig 1.

Principles of next-generation sequencing (NGS) technology. For NGS library preparation, DNA is randomly fragmented into desired size ranges. Adaptors containing the universal priming sites are ligated to the target ends of the fragments. After ligation, the template is immobilized to a solid support. Immobilization strategies for clonally amplified templates include either using emulsion polymerase chain reaction (emPCR)37 or a solid-phase amplification.38 In emPCR, an oil-aqueous emulsion reaction mixture is created to encapsulate bead-DNA complexes into single aqueous droplets. PCR is then performed within the droplets to create beads that contain several thousand copies of the same template sequence. The emPCR beads can then be attached to a glass slide or loaded into PicoTiterPlate (Roche Applied Science, Indianapolis, IN) wells. Solid-phase amplification relies on bridge PCR, where both forward and reverse PCR primers are tethered to a solid substrate by a flexible linker such that the clonally amplified clusters remain immobilized, thereby localizing to a single physical location on an array. At the conclusion of bridge PCR, each clonal cluster contains approximately 1,000 copies of a single member of the template library. Regardless of platform used, amplification is a necessary step because it allows the sequencing reactions to produce sufficient signal for detection by the imaging system of the instrument. Single-molecule as opposed to clonally amplified templates can also be accomplished using a number of possible approaches for immobilization. On the basis of whether clonally amplified or single-molecule templates are used, different sequencing and imaging strategies need to be applied. Although the DNA sequencing reactions vary among platforms, the most popular technologies, such as cyclic reversible termination, pyrosequencing, and the pH-based/semiconductor sequencing, perform sequencing by synthesis to sequence the template. The ability to move away from optically based detection systems to more scalable semiconductor technology has drastically reduced the costs associated with sequencing. Other sequencing technologies exist, such as the sequencing by ligation method, referred to as the SOLiD (support oligonucleotide) platform (Table 2).39
As opposed to conventional Sanger-based capillary sequencing methods,40 NGS allows for MPS through a series of repeating sequencing reactions, performed and detected automatically, with the production of thousands to millions of simultaneous sequences. MPS, thereby, generates drastically more sequence reads per instrument run, at a significantly lower expense. Applications of NGS include WGS of tumor and germline DNA, as well as targeted sequencing of specific regions of interest, including WES or multigene (multiplex) gene-panel testing. In multiplex testing, the simultaneous interrogation of target genes of interest allows for an efficient and cost-effective method of screening panels of cancer genes concurrently, as opposed to screening on a gene-by-gene basis as occurs in Sanger sequencing.
The basic principles of NGS technology are shown in Figure 1; an in-depth review of NGS technologies can be found elsewhere.41–44 Although each NGS technology is distinct (Table 2), all manufacturers aim to increase the amount of sequence output per run, increase the number of nucleotides per sequence read (or read lengths), lower cost, and improve base-calling accuracy. Although cost effective and highly efficient, disadvantages of NGS include higher error rates and shorter read lengths, enrichment of rare variants, and a large proportion of missing values. A comparison of the accuracy and completeness of variant calling for two commonly used sequencing platforms found that although both technologies achieved a relatively high concordance for unique single-nucleotide variants of 88%, for indels, concordance was only 27%.45 Technologic advancements may improve base-calling accuracy; however, for now, all NGS-acquired data require analytic validation using alternate technologies. These technical limitations also constitute important caveats to regulators and clinicians seeking to certify or use NGS-based diagnostic panels for cancer predisposition or prognostic evaluation.
Cancer Genetics Through NGS Technologies
In the last 4 years, WES of unrelated individuals or families with multiple affected members with the same rare disorder has identified the genetic basis of diseases such as Freeman-Sheldon syndrome, Kabuki syndrome, Miller syndrome, and autosomal dominant spinocerebellar ataxias.46–51 In 2010, the first report of WGS of a patient with Charcot-Marie-Tooth disease was published and was followed by a number of WGS studies of rare phenotypes.52–54 The rapidly growing catalogue of NGS studies can be found in the NGS catalogue maintained by Vanderbilt University.55,56 In cancer, the use of WES and WGS has enabled a dramatic improvement on the classical methods for gene discovery, such as linkage analysis.
Evaluation of Cancer Susceptibility Using WES and WGS Technologies
One of the first applications of NGS for cancer susceptibility was use of WES to identify a germline mutation in PALB2, a gene previously implicated in breast cancer risk, in an individual with familial pancreatic cancer.57,58 WGS approaches also identified heterozygous variants in ATM in families with rare pancreatic cancer.59 Homozygous mutations in ATM are known to cause ataxia telangiectasia,60,61 whereas heterozygous mutations have previously been linked to breast cancer susceptibility.62 However, these PALB2- and ATM-linked pancreatic cancer clusters have proven to be exceedingly rare.
NGS was also used to identify a novel gene involved in familial pheochromocytoma (PCC).63 Although the RET, VHL, SDHA, SDHB, SDHC, SDHD, SDHAF2, NF1, and TMEM127 genes have already been linked to familial PCC,64–67 approximately 10% of hereditary PCCs with autosomal-dominant pattern of inheritance are not explained by mutations within these genes.68,69 WES in individuals with familial PCC identified mutations of MAX.63 MAX, the MYC-associated factor X gene, is a transcription factor that regulates cell proliferation, differentiation, and apoptosis.70 Similar to the sex-linked transmission of familial PCC cases associated with SDHD and SHAF2 mutations, MAX mutations associated with familial PCC seem to have a preferential paternal transmission pattern. Taken in aggregate, these NGS discoveries set the stage for a panel of nearly a dozen genes that could be simultaneously tested in familial PCC.
The theme of transcription factor–associated cancer predisposition as revealed by WES also applies to acute myelogenous leukemia (AML). Inherited predisposition to myelodysplastic syndrome/AML had previously been linked to mutations within hematopoietic transcription factors RUNX1 and CEBPA.71,72 NGS studies linked mutations of the transcription factor GATA2 to familial AML and, occasionally, Emberger syndrome, an autosomal-dominant primary lymphedema associated with widespread cutaneous warts, deafness, and predisposition to myelodysplastic syndrome.73–75 Recently, our group demonstrated that inherited mutations of another transcription factor, PAX5, lead to childhood acute lymphoblastic leukemia.76 The pedigrees in both GATA2- and PAX5-mutant kindreds demonstrate multiple mutation carriers who remain unaffected, illustrating the incomplete penetrance and variable expressivity associated with these NGS discoveries.
Genetic alterations in KLHDC8B have been associated with Hodgkin lymphoma risk,77,78 and combining WES from one family member with genome-wide linkage data, a truncating germline mutation in nuclear protein ataxia telangiectasia locus (NPAT) was identified in familial nodular lymphocyte–predominant Hodgkin lymphoma.79 With an important role in cell-cycle regulation and promotion of ATM activation, NPAT is a promising cancer susceptibility gene.80
WGS in an individual with familial melanoma (without identified CDKN2A or CDK4 mutations) showed a single-nucleotide variant in the melanoma lineage–specific oncogene microphthalmia-associated transcription factor (MITF) to be segregating with melanoma in the family.81 The variant had an odds ratio of 2.3 (95% CI, 1.2 to 4.7) in epidemiologic studies, and functional analysis demonstrated that MITF encoded by the E318K variant allele had impaired sumoylation and differentially regulated MITF targets. The E318K variant has since been implicated in susceptibility to both melanoma and renal cancers.82 As in the case of the leukemia-associated transcription factors we have mentioned, this study illustrates a clinical conundrum common to many NGS-based gene discovery studies: The MITF risk variant did not segregate fully with disease status. The reduced penetrance and expressivity are more marked than those observed, for example, in BRCA mutation carriers, thereby complicating the genetic counseling process. Presumably, in these patients, cancer risk is affected by other genetic or environmental risk factors. Targeted resequencing of other melanoma families identified mutations in the promoter of telomerase reverse transcriptase (TERT), which create novel binding sites for Ets/TCF transcription factors.83
Despite extensive research, only approximately 30% of familial breast cancer risk is explained by known genetic factors,14 and it was expected that NGS of unexplained kindreds would rapidly reveal the so-called low-hanging fruit of novel candidate genes. Such fruit, however, have not always ripened. WES in selected affected family members from 13 breast cancer families identified two families with mutations in x-ray repair cross-complementing 2 (XRCC2), including a protein-truncating change and a probable deleterious missense mutation.84 WES in families with early-onset breast cancer also revealed variants in the Fanconi pathway gene FAN1.85 Unfortunately, subsequent large-scale studies did not confirm the FAN1 or XRCC2 variants as associated with breast cancer risk.85,86 We implicated another Fanconi pathway gene, SLX4, in only one of > 700 BRCA-negative breast cancer kindreds, suggesting that additional such mutations may in rare cases contribute to breast cancer risk.87 Most recently, a large pooled NGS study focusing on DNA repair pathways identified mutations in the p53-inducible protein phosphatase PPM1D as occurring mosaically in individuals with predisposition to breast and ovarian cancers.88 Using imputed-risk SNPs identified via WGS in an Icelandic population, a novel frameshift mutation was discovered in the BRIP1 Fanconi pathway gene, with an odds ratio of 8.1 for ovarian cancer.89 Such testing for genes associated with the homologous recombination DNA-repair pathways, in addition to preventive applications, has an emerging role in screening for targeted therapies, such as poly (ADP-ribose) polymerase inhibitors.
Predisposition to colon polyposis and colorectal cancer is only partially explained by known cancer susceptibility genes. WGS in families with multiple adenomas and/or colorectal cancer recently identified heterozygous POLE and POLD1 germline variations.90 These mutations, mapping to the exonuclease domains of DNA polymerases, are predicted to cause defective correction of mispaired bases and seem to be high-penetrance cancer susceptibility genes, with POLD1 mutations also increasing endometrial cancer risk. Using a combination of linkage analysis as well as high-throughput sequencing, GREM1 is the first gene to have been implicated in the genetic etiology of hereditary mixed polyposis syndrome.91 In prostate cancer, NGS of linkage regions on chromosome 17 helped to identify a particular mutation in the homeobox gene HOXB13, an important driver of prostate cancer risk.92
Somatic mutations in BAP1, a nuclear-localized, ubiquitin carboxy-terminal hydrolase that binds to the RING finger domain of BRCA1, were previously identified in mesothelioma and uveal and cutaneous melanomas.93,94 More recently, the identification of germline mutations in BAP1 in families with mesothelioma, melanoma, and renal cell cancer, in an autosomal-dominant manner, suggests a new BAP1-related cancer susceptibility syndrome.95–98 The frequency, penetrance, and spectrum of malignancies associated with BAP1-related cancer susceptibility remain to be elucidated.
The handful of studies published to date (Table 3), with many more in progress, highlights the power of NGS technologies in elucidating the genetic basis of hereditary cancers. In addition to providing important insights into the molecular mechanisms underlying carcinogenesis, even where there is no preventive strategy evident (eg, leukemia- or lymphoma-associated risks), this testing can be used in reproductive planning.100 In breast and colon cancers, these findings have already produced high-throughput screens for panels of genes.
Table 3.
Identification of Cancer Susceptibility Genes Using NGS
| Cancer/Cancer Syndrome | NGS | Gene Implicated | Cases Used for Identification of Cancer Susceptibility Gene |
|---|---|---|---|
| Familial pancreatic cancer | |||
| Jones et al57 | Exome | PALB2 | One affected with familial pancreatic cancer |
| Roberts et al59 | WGS and exome | ATM | WGS: 16 affecteds from six families; exome: 22 affecteds from 10 families |
| Familial pheochromocytoma | |||
| Comino-Méndez et al63 | Exome | MAX | Three affecteds from three families |
| Hematologic malignancies | |||
| AML with Emberger syndrome; Ostergaard et al73 | Exome | GATA2 | Three unrelated affecteds (two with familial, one with sporadic) |
| Familial HL; Saarinen et al79 | Exome | NPAT | One affected with familial nodular lymphocyte predominant HL, combined with genome-wide linkage data from family |
| Familial pre–B-cell ALL; Shah et al76 | Exome | PAX5 | Two families with exome sequencing of multiple affected and unaffected family members |
| Familial melanoma | |||
| Yokoyama et al81 | WGS | MITF | One affected with familial melanoma; also assessed in cases/controls |
| Horn et al83 | Targeted sequencing | TERT | Targeted sequencing of four affecteds and one unaffected in melanoma family |
| Familial mesothelioma, melanoma, and RCC | |||
| Testa et al95 | Exome | BAP1 | Two families with mesothelioma and uveal melanoma |
| Wiesner et al96 | Targeted sequencing | Two families with uveal and cutaneous melanocytic tumors | |
| Popova et al97 | Exome | Two affecteds from one RCC family | |
| Familial colorectal cancer and polyposis | |||
| HMPS; Jaeger et al91 | Targeted sequencing | GREM1 | Large Ashkenazi Jewish family with HMPS; additional HMPS families |
| Colorectal adenomas and colon cancer; Palles et al90 | WGS | POLE, POLD1 | 20 affecteds (colorectal adenomas ± colorectal cancer) from 15 families |
| Familial breast cancer | |||
| Park et al84 | Exome | XRCC2 | Five affecteds from two families; also assessed in case/controls |
| Park et al85 | Exome | FAN1* | Four early-onset multiple-case breast cancer families |
| Ruark et al88 | Targeted sequencing | PPM1D (mosaic) | 1,150 with breast cancer ± ovarian cancer; replication in large case/control |
| Ovarian cancer | |||
| Rafnar et al89 | WGS | BRIP1 | WGS of 457 Icelanders; case/control of imputed SNPs |
| Walsh et al99 | Targeted sequencing | Multiple genes† | 360 women with ovarian cancer |
Abbreviations: ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; HL, Hodgkin lymphoma; HMPS, hereditary mixed polyposis syndrome; NGS, next-generation sequencing; RCC, renal cell carcinoma; SNP, single-nucleotide polymorphism; WGS, whole-genome sequencing.
ldentified variants in FAN1 not found to independently influence breast cancer risk.85
Germline mutations found in BRCA1, BRCA2, BARD1, BRIP1, CHEK2, MRE11A, MSH6, NBN, PALB2, RAD50, RAD51C, and TP53.99
Multiplex Analysis for Cancer Syndromes Using NGS
In addition to gene discovery, another potential application of NGS technology is comprehensive and simultaneous (multiplex) mutational analysis of known cancer susceptibility genes and candidate genes. In a research context, this approach was demonstrated by screening 360 ovarian carcinomas for germline mutations in 21 tumor-suppressor genes.99 Of the 360 patient cases, 85 showed germline loss-of-function mutations in 12 genes: 40 (11.1%) in BRCA1, 23 (6.4%) in BRCA2, and 22 (6.1%) in 10 other genes, including BARD1, BRIP1, CHEK2, MRE11A, MSH6, NBN, PALB2, RAD50, RAD51C, and TP53. Multiplex testing using NGS also reidentified pathogenic mutations in 28 blinded specimens with known mutations in genes associated with Lynch and polyposis syndromes.101 Similarly, some of the newly implicated cancer susceptibility genes were identified partially via NGS, which provided an efficient method for screening large numbers of candidate genes (Table 3).83,88,91,95
As opposed to the traditional stepwise one-gene-at-a-time approach to genetic testing, the dramatic decreases in the cost of DNA sequencing make multiplex genetic testing an efficient and economically advantageous approach (Table 4). At the same time, recent court decisions have eroded intellectual property barriers to diagnostic genetic testing, further stimulating the proliferation of NGS-based multiplex gene-panel testing for genetic predispositions.103 However, multiplex testing will inevitably result in the identification of increased variants of uncertain significance. Although the technical ability for multiplex testing has arrived, mechanisms to provide meaningful counseling for multigene and possible variant results pose significant barriers to the responsible translation of these technologies.102
Table 4.
Commercially Available Multiplex Gene Panels
| Panel | Genes Included |
|---|---|
| Breast cancer | |
| BreastNext; Ambry Genetics, Aliso Viejo, CA | ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, MRE11A, MUTYH, NBN, PALB2, PTEN, RAD50, RAD51C, STK11, TP53 |
| Colorectal cancer | |
| ColoNext; Ambry Genetics | APC, BMPR1A, CDH1, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11, TP53 |
| ColoSeq; University of Washington Laboratory Medicine, Seattle, WA | APC, BMPR1A, CDH1, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11, TP53 |
| Mayo Medical Laboratories; Rochester, MN | APC, AXIN2, BMPR1A, CDH1, CHEK2, EPCAM, GREM1, MLH1, MLH3, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11, TP53 |
| Ovarian cancer | |
| OvaNext; Ambry Genetics | ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, EPCAM, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD50, RAD51C, STK11, TP53 |
| Multicancer panels | |
| CancerNext; Ambry Genetics | APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, EPCAM, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD50, RAD51C, SMAD4, STK11, TP53 |
| BROCA Cancer Risk Panel; University of Washington Laboratory Medicine | APC, ATM, ATR, BAP1, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A, CHEK1, CHEK2, FAM175A, GALNT12, GEN1, GREM1, HOXB13, MLH1, MRE11A, MSH2 (+EPCAM), MSH6, MUTYH, NBN, PALB2, PMS2, PRSS1, PTEN, RAD50, RAD51, RAD51C, RAD51D, RET, SMAD4, STK11, TP53, TP53BP1, VHL, XRCC2 |
| Myriad myRisk; Myriad Genetics, Salt Lake City, UT* | APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A (p16INKA and p14ARF), CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51C, RAD51D, SMAD4, STK11, TP53 |
NOTE. Listing of laboratory name here does not imply that testing meets regulatory criteria of relevant state health department. Data adapted.102
Expected to be commercially available later in 2013; published with permission from Myriad Genetics.
NGS of Cancer Genomes: Implications for Germline Cancer Risk Assessment
In cancer research, the most common application of NGS is in the identification of acquired (somatic) mutations in tumor genomes.6 Genetic changes specific to tumor cells may result in the discovery of novel genes and important pathways involved in carcinogenesis. Although initial approaches focused on assessment of an individual patient's tumor(s),104,105 rapid progress in deciphering the cancer genome is anticipated through ongoing international efforts, including the Cancer Genome Atlas and International Cancer Genome Consortium.106,107 These collaborative efforts aim to sequence up to 500 clinically well-annotated tumors for selected cancer types, with generation of an enormous amount of genomic data. To date, the Cancer Genome Atlas has already published initial analyses of glioblastomas and ovarian, colorectal, and breast cancers, among other tumor types, demonstrating the feasibility of this large-scale project and identifying a number of deregulated genes that may set the stage for the development of targeted therapies.108–111
Importantly, the study of somatic mutations in tumors using NGS may purposefully or inadvertently also shed light on the presence of corresponding germline mutations (Figs 2 and 3 summarize two clinical case scenarios).112
Fig 2.

Pedigree case 1.
Fig 3.

Pedigree case 2.
Case Scenarios
Patient Case 1. A 41-year-old female never-smoker with incidental bilateral ground glass opacities identified on computed tomography (CT) scan underwent thoracoscopic surgery, with removal of three distinct adenocarcinomas of the lung. Pathology demonstrated morphologically distinct, synchronous primary lung adenocarcinomas. For treatment purposes, somatic evaluation of the patient's tumors was undertaken. In two tumors, the presence of the L858R mutation in exon 21 of the epidermal growth factor receptor (EGFR) gene was noted, whereas the third tumor exhibited deletion of exon 19 of the EGFR gene. Although both of these activating mutations in EGFR have been associated with an enhanced response to EGFR tyrosine kinase inhibitors (TKIs),113–115 subsequent analysis demonstrated that all three tumors also exhibited the T790M EGFR mutation, associated with resistance to TKIs.116,117 Family history at the time of diagnosis was unrevealing (Fig 2; pedigree case 1; noncritical clinical features have been changed to preserve confidentiality). The patient was referred to clinical genetics for counseling and genetic testing, in light of the fact that the EGFR T790M germline mutation has been linked to lung cancer susceptibility, and the identification of this somatic mutation in all of her tumors was suspicious for a potential germline etiology.118 Germline analysis revealed the presence of the EGFR T790M mutation. Family testing revealed the EGFR T790M mutation in the patient's mother, in whom subsequent clinical workup confirmed a diagnosis of multifocal lung cancer. Currently, implications for unaffected germline EGFR mutation carriers remain unclear because of insufficient data regarding the risk of lung cancer (and/or other cancers) in such family members. Individualized counseling regarding the role of CT scan–based lung cancer screening and optional genetic testing for EGFR T790M were advised for relevant family members.
Patient Case 2. A 59-year-old man with a history of melanoma was being evaluated for oncologic treatment options for his newly diagnosed metastatic prostate cancer. In this setting, an outside laboratory performed targeted sequence analysis of > 200 cancer-related genes, using NGS technologies, on a sample of his prostate tumor. Germline DNA was not used for analysis. Results, after removal of known common germline genetic variants, revealed two genetic variations within the tumor, one in p53 and the other in BRCA1. The patient was subsequently referred to the clinical genetics service for further evaluation of the two presumed somatic genetic events. In addition to his cancer diagnosis, family history was significant for a strong family history of prostate cancer (Fig 3; pedigree case 2; noncritical clinical features have been changed to preserve confidentiality). Although the p53 nonsynonymous missense variation was noted to be a common somatic event in tumors, the identified, presumed somatic BRCA1 mutation was also known to be a deleterious (pathogenic) germline mutation, diagnostic of hereditary breast and ovarian cancer (HBOC) syndrome. After genetic counseling, germline genetic testing revealed the presence of the BRCA1 mutation within the germline, diagnostic of HBOC. Implications for family members, including the daughter, were explained. In this particular case, the presence of the BRCA1 mutation may also have had an impact on future treatment for the patient, with clinical trials suggesting increased efficacy of poly (ADP-ribose) polymerase inhibitors in the treatment of BRCA-associated malignancies.119,120
Case Scenarios: Points to Consider
Medical oncologists and other clinicians considering genomic analysis of tumors via NGS technologies (whole-exome and whole-genome sequencing, targeted cancer-panel testing) must understand that even without direct analysis of germline DNA, somatic (tumor) DNA analysis may reveal mutations in the constitutional (germline) genome that could have important implications for cancer (or other disease) risks for the affected individual as well as for at-risk family members.
For this reason, there is a strong rationale for pretesting informed consent to include discussion of potential tumor findings, incidental germline findings, and plans for disclosure of results.
Somatic genetic findings based on tumor analysis must be interpreted carefully within the context of the personal and family histories, preferably with the input of clinical cancer geneticists to identify situations in which interrogation of the germline is necessary.
Some commercial laboratories performing somatic (tumor) analyses for targeted cancer panels report the results of somatic analyses without indicating if such genetic events resulted from germline mutations. Some laboratories may filter the germline entirely. Although such approaches may not be appropriate for prospective tumor genomic analysis, they may be justified in a retrospective research setting where specific consent may not be obtainable. In such a setting, one alternative proposed by informaticians is simply to not subject the germline to variant calling algorithms.121
Maintaining Privacy in the Era of Genomics
Given the unique nature of an individual's DNA sequence, by definition, DNA data cannot truly be anonymized. A statistical approach was used to show that an individual's genomic data could be identified in large DNA data sets of aggregated SNP data.122 This finding resulted in the National Institutes of Health establishing barriers for review and approval of open-access databases.123 Recently, genomic and ancestry information was linked from online commercial databases to deduce a person's surname.124 A conclusion thus emerges that research participants must be informed that although all efforts can be made to de-identify DNA samples, there still exists potential for breach of privacy. At present, certain federal grants require primary genomic sequences to be available to the research community, requiring institutional and/or participant consent for such data release. A controversial potential revision to the Common Rule 45 CFR, which has governed such research, would no longer allow DNA research to be deemed eligible for waivers resulting from sample anonymization (ie, permanent removal of personal identifiers).125 Such a change would sharply limit the number of banked biospecimens eligible for federally funded research. Alternative solutions to this privacy concern would be regulatory deterrents to make it illegal to maliciously re-identify de-identified genomic data, as well as restrictions to data access ensuring that only credentialed researchers are able to use primary genomic data.
NGS Data Interpretation and Reporting of Results
With the profusion of WES and WGS data, a new challenge has been posed by the extent to which incidental or secondary genetic findings, termed incidentalomes,126 should be clinically transmitted to patients. Unexpected results may include identification of genetic predispositions to non-cancer-related diseases for which medical interventions may be available, such as cardiac or neurologic disease risks.127–129 Although there is emerging consensus that return of such actionable results should be offered to the patient,130,131 perhaps even more problematic is the challenge to inform patients of the possibility of detecting variants of uncertain significance. In an exploratory study evaluating the recommendations of specialists in clinical genetics and/or molecular medicine, 100% concordance was found in favor of disclosing incidental pathogenic mutations to adults in 21 conditions and/or genes; however, substantial discordance existed with respect to disclosure of mutations without proven pathogenicity.130
Recognizing the potential for rapid integration of WES and WGS into the clinical practice of medicine, in 2012, the American College of Medical Genetics and Genomics (ACMG) issued a policy statement on genomic sequencing that emphasized the importance of incidental results in pretest patient counseling, clinical testing, and results reporting.132 Subsequently, the ACMG specifically delineated a minimum list of genes in which germline mutations should be reported by the clinical laboratory, regardless of the indication for which the sequencing test was ordered.133 Although a majority of these must-be-reported genes were associated with cancer risk, there were no oncologists on the panel that selected them, and the report generated a firestorm of controversy within the medical genetics community. The controversy stemmed from the dichotomy between those arguing for autonomous right of choice to learn results of incidental findings and the ACMG requirement to disclose all results regardless of patient choice, which was perceived as paternalistic.134–136 However, this seems a false dichotomy, because the preferred outcome is for individuals to achieve a sufficient level of genomic knowledge to make an informed choice to specify which incidental findings they want to have transmitted. As evident to practitioners of cancer genetics, a substantial proportion of fully counseled patients do not wish, for example, to know inherited p53 mutation results,137 and it is possible to opt for effective cancer screening based on family history without knowing mutation results. As the earliest adopters of genomic scans in a clinical setting, oncologists will play an important role in mediating whether the personalized genome is perceived as a blanket liability to be imposed or an empowering choice for patients educated to understand precisely what health risks are at stake.121
Role of the Oncologist in Translation of NGS to Preventive Oncology
Oncologists as well as other specialized health care providers may rightly ask how they may be expected to approach the process of informed consent for testing entire genomes, both tumor and normal, when the meaning of the vast majority of variants uncovered is currently not defined. Because of these concerns, one incontrovertible conclusion of the ACMG and other guidelines is that oncologists should provide counseling before NGS testing of tumor samples, including a discussion of the possibility of findings of inherited genomic variants with as-yet-unknown significance (Fig 4).
Fig 4.
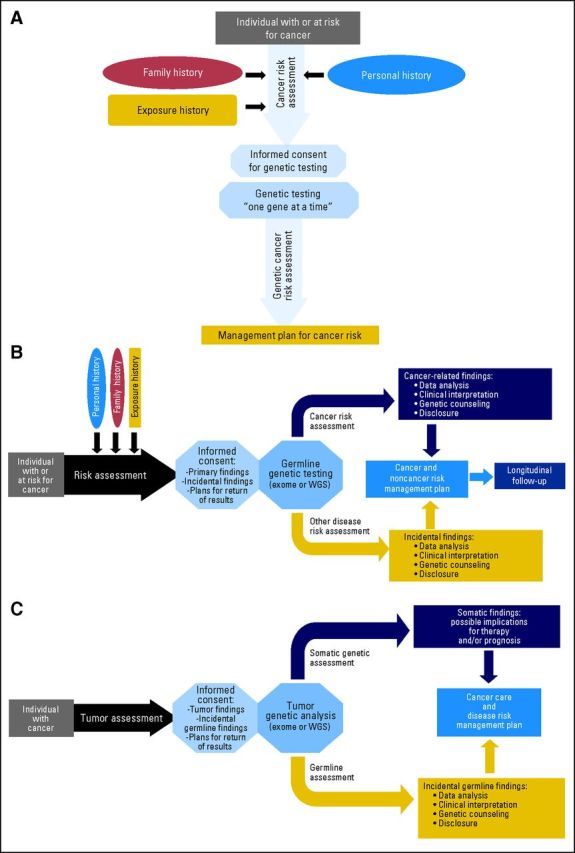
Challenging the traditional model of cancer genetic counseling. (A) Traditional model of clinical cancer genetics. (B) Incorporating next-generation sequencing (NGS) into genetic cancer risk assessment. (C) NGS of tumors with incorporation of incidental germline findings. WGS, whole-genome sequencing.
To address how oncologists can integrate the simultaneous findings of tumor and inherited genomes, in one model, pioneered at the University of Michigan, all patients undergoing NGS of their tumors had to meet with a genetic counselor before consenting to genomic analysis.138 At most major cancer centers, including our own, an advisory body, genomics review board, or other expert entity is constituted to advise on the need to return germline findings that emerge in the context of tumor genome analysis. This body may be constituted separately from or alongside the committee that advises on the suitability of particular tumor mutations as targets for therapy. Regardless of the approach used, findings to be reported need to be analytically validated and returned in compliance with state regulations.
Once a particular genomic variant uncovered by NGS is analytically validated, it may range in its clinical utility and suitability for communication to the patient as a primary or incidental finding. Various models have been proposed to guide the communication of incidental findings, ranging from tiered approaches to binning to full return. We favor, and are testing in a trial, the binning system, where genomic findings are categorized depending on clinical utility and clinical validity.139 One beneficial aspect of the binning process is that research participants would potentially have the ability to select upfront, at time of consent, which bin results they wish to receive at the results disclosure, allowing for a patient-directed approach. As the meaning of variants is defined over time, they can be deposited into bins for eventual transmission, depending on patient wishes. An important step forward in this regard is the creation by the National Human Genome Research Institute of the ClinVar database of genomic variation to provide a consensus of the biologic as well as clinical significance of genes and variants.140
Even if technically feasible, the premature translation of genomic information by oncologists also poses risks. These risks may be psychological, as a result of information overload. In addition, genetic information may create further health care disparities because of the high cost of these technologies or the medical interventions they motivate141,142 and even pose health care risks if results are prematurely translated by commercial marketing directly to consumers outside of regulatory protection.143
DISCUSSION
Although common genomic risk variants identified to date have had limited clinical utility, ongoing WES and WGS approaches have already provided striking insights for some cancer-prone individuals. However, it is clear that sequencing technologies have outpaced our ability to interpret and apply the vast amounts of data generated. Pending these advances, we suggest a model incorporating a tiered approach to NGS studies of tumor and normal tissues. In addition to germline cancer risk assessment, such models would include flexibility for the provision of analytically valid and clinically useful risk assessment for other incidental noncancer disease risk findings.
Oncology is now ground zero for a tectonic shift in paradigms regarding personalized medicine.144 Germline DNA profiles will be generated as references for DNA analysis of tumors to define therapeutic targets. As successfully accomplished during the prior era of cancer genetics, the practice of oncology will need to incorporate new concepts of genomic risk assessment. This will require new approaches to informed consent discussion with patients as well as decision algorithms to guide the communication of incidental germline findings that emerge from genome-wide tumor analyses.
Appendix
Fig A1.

Gene discovery: the potential of next-generation sequencing (NGS) technology. GWAS, genome-wide association study.
Footnotes
Supported by the Robert and Kate Niehaus Clinical Cancer Genetics Initiative at Memorial Sloan-Kettering Cancer Center, The Romeo Milio Lynch Syndrome Foundation, the Breast Cancer Research Foundation, and the Sharon Levine Corzine Fund for Cancer Research. Z.K.S. is a Damon Runyon Cancer Research Foundation Clinical Investigator Award recipient.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The author(s) indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: Zsofia K. Stadler, Kenneth Offit
Collection and assembly of data: Zsofia K. Stadler, Kasmintan A. Schrader, Kenneth Offit
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1. Garber JE, Offit K: Hereditary cancer predisposition syndromes J Clin Oncol 23: 276– 292,2005. [DOI] [PubMed] [Google Scholar]
- 2. Robson M, Offit K: Clinical practice: Management of an inherited predisposition to breast cancer N Engl J Med 357: 154– 162,2007. [DOI] [PubMed] [Google Scholar]
- 3. Lindor NM Petersen GM Hadley DW , etal: Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: A systematic review JAMA 296: 1507– 1517,2006. [DOI] [PubMed] [Google Scholar]
- 4. Galiatsatos P, Foulkes WD: Familial adenomatous polyposis Am J Gastroenterol 101: 385– 398,2006. [DOI] [PubMed] [Google Scholar]
- 5. Villani A Tabori U Schiffman J , etal: Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: A prospective observational study Lancet Oncol 12: 559– 567,2011. [DOI] [PubMed] [Google Scholar]
- 6. Macconaill LE, Garraway LA: Clinical implications of the cancer genome J Clin Oncol 28: 5219– 5228,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garraway LA: Genomics-driven oncology: Framework for an emerging paradigm J Clin Oncol 31: 1806– 1814,2013. [DOI] [PubMed] [Google Scholar]
- 8. Lichtenstein P Holm NV Verkasalo PK , etal: Environmental and heritable factors in the causation of cancer: Analyses of cohorts of twins from Sweden, Denmark, and Finland N Engl J Med 343: 78– 85,2000. [DOI] [PubMed] [Google Scholar]
- 9. Athma P, Rappaport R, Swift M: Molecular genotyping shows that ataxia-telangiectasia heterozygotes are predisposed to breast cancer Cancer Genet Cytogenet 92: 130– 134,1996. [DOI] [PubMed] [Google Scholar]
- 10. CHEK2*1100delC and susceptibility to breast cancer: A collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies Am J Hum Genet 74: 1175– 1182,2004. CHEK2 Breast Cancer Case-Control Consortium [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meijers-Heijboer H van den Ouweland A Klijn J , etal: Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations Nat Genet 31: 55– 59,2002. [DOI] [PubMed] [Google Scholar]
- 12. Seal S Thompson D Renwick A , etal: Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles Nat Genet 38: 1239– 1241,2006. [DOI] [PubMed] [Google Scholar]
- 13. Rahman N Seal S Thompson D , etal: PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene Nat Genet 39: 165– 167,2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stadler ZK Thom P Robson ME , etal: Genome-wide association studies of cancer J Clin Oncol 28: 4255– 4267,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stadler ZK Vijai J Thom P , etal: Genome-wide association studies of cancer predisposition Hematol Oncol Clin North Am 24: 973– 996,2010. [DOI] [PubMed] [Google Scholar]
- 16. Bernstein BE Birney E Dunham I , etal: An integrated encyclopedia of DNA elements in the human genome Nature 489: 57– 74,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wacholder S Hartge P Prentice R , etal: Performance of common genetic variants in breast-cancer risk models N Engl J Med 362: 986– 993,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gail MH: Value of adding single-nucleotide polymorphism genotypes to a breast cancer risk model J Natl Cancer Inst 101: 959– 963,2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Comen E Balistreri L Gönen M , etal: Discriminatory accuracy and potential clinical utility of genomic profiling for breast cancer risk in BRCA-negative women Breast Cancer Res Treat 127: 479– 487,2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Antoniou AC Beesley J McGuffog L , etal: Common breast cancer susceptibility alleles and the risk of breast cancer for BRCA1 and BRCA2 mutation carriers: Implications for risk prediction Cancer Res 70: 9742– 9754,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zheng SL Sun J Wiklund F , etal: Cumulative association of five genetic variants with prostate cancer N Engl J Med 358: 910– 919,2008. [DOI] [PubMed] [Google Scholar]
- 22. Salinas CA Koopmeiners JS Kwon EM , etal: Clinical utility of five genetic variants for predicting prostate cancer risk and mortality Prostate 69: 363– 372,2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Johansson M Holmström B Hinchliffe SR , etal: Combining 33 genetic variants with prostate-specific antigen for prediction of prostate cancer: Longitudinal study Int J Cancer 130: 129– 137,2012. [DOI] [PubMed] [Google Scholar]
- 24. Dunlop MG Tenesa A Farrington SM , etal: Cumulative impact of common genetic variants and other risk factors on colorectal cancer risk in 42 103 individuals Gut 62: 871– 881,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weitzel JN Blazer KR Macdonald DJ , etal: Genetics, genomics, and cancer risk assessment: State of the art and future directions in the era of personalized medicine CA Cancer J Clin [epub ahead of print on August 19, 2011] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michailidou K Hall P Gonzalez-Neira A , etal: Large-scale genotyping identifies 41 new loci associated with breast cancer risk Nat Genet 45: 353– 361,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garcia-Closas M Couch FJ Lindstrom S , etal: Genome-wide association studies identify four ER negative-specific breast cancer risk loci Nat Genet 45: 392– 398,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bojesen SE Pooley KA Johnatty SE , etal: Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer Nat Genet 45: 371– 384,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pharoah PD Tsai YY Ramus SJ , etal: GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer Nat Genet 45: 362– 370,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen H Fridley BL Song H , etal: Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer Nat Commun 4: 1628,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Permuth-Wey J Lawrenson K Shen HC , etal: Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21.31 Nat Commun 4: 1627,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eeles RA Olama AA Benlloch S , etal: Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array Nat Genet 45: 385– 391,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kote-Jarai Z Saunders EJ Leongamornlert DA , etal: Fine-mapping identifies multiple prostate cancer risk loci at 5p15, one of which associates with TERT expression Hum Mol Genet 22: 2520– 2528,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burton H Chowdhury S Dent T , etal: Public health implications from COGS and potential for risk stratification and screening Nat Genet 45: 349– 351,2013. [DOI] [PubMed] [Google Scholar]
- 35. Liu L Li Y Li S , etal: Comparison of next-generation sequencing systems J Biomed Biotechnol 2012: 251364,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Quail MA Smith M Coupland P , etal: A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers BMC Genomics 13: 341,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dressman D Yan H Traverso G , etal: Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations Proc Natl Acad Sci U S A 100: 8817– 8822,2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fedurco M Romieu A Williams S , etal: BTA, a novel reagent for DNA attachment on glass and efficient generation of solid-phase amplified DNA colonies Nucleic Acids Res 34: e22,2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Valouev A Ichikawa J Tonthat T , etal: A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning Genome Res 18: 1051– 1063,2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sanger F Air GM Barrell BG , etal: Nucleotide sequence of bacteriophage phi X174 DNA Nature 265: 687– 695,1977. [DOI] [PubMed] [Google Scholar]
- 41. Mardis ER: New strategies and emerging technologies for massively parallel sequencing: Applications in medical research Genome Med 1: 40,2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Metzker ML: Sequencing technologies: The next generation Nat Rev Genet 11: 31– 46,2010. [DOI] [PubMed] [Google Scholar]
- 43. Koboldt DC Ding L Mardis ER , etal: Challenges of sequencing human genomes Brief Bioinform 11: 484– 498,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shendure J, Ji H: Next-generation DNA sequencing Nat Biotechnol 26: 1135– 1145,2008. [DOI] [PubMed] [Google Scholar]
- 45. Lam HY Clark MJ Chen R , etal: Performance comparison of whole-genome sequencing platforms Nat Biotechnol 30: 78– 82,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ng SB Turner EH Robertson PD , etal: Targeted capture and massively parallel sequencing of 12 human exomes Nature 461: 272– 276,2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ng SB Bigham AW Buckingham KJ , etal: Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome Nat Genet 42: 790– 793,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hoischen A van Bon BW Gilissen C , etal: De novo mutations of SETBP1 cause Schinzel-Giedion syndrome Nat Genet 42: 483– 485,2010. [DOI] [PubMed] [Google Scholar]
- 49. Ng SB Buckingham KJ Lee C , etal: Exome sequencing identifies the cause of a Mendelian disorder Nat Genet 42: 30– 35,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang JL Yang X Xia K , etal: TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing Brain 133: 3510– 3518,2010. [DOI] [PubMed] [Google Scholar]
- 51. Ku CS, Naidoo N, Pawitan Y: Revisiting Mendelian disorders through exome sequencing Hum Genet 129: 351– 370,2011. [DOI] [PubMed] [Google Scholar]
- 52. Lupski JR Reid JG Gonzaga-Jauregui C , etal: Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy N Engl J Med 362: 1181– 1191,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sobreira NL Cirulli ET Avramopoulos D , etal: Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene PLoS Genet 6: e1000991,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rios J Stein E Shendure J , etal: Identification by whole-genome resequencing of gene defect responsible for severe hypercholesterolemia Hum Mol Genet 19: 4313– 4318,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vanderbilt University. Bioinformatics and Systems Medicine Laboratory: NGS catalog. http://bioinfo.mc.vanderbilt.edu/NGS/index.html.
- 56. Xia J Wang Q Jia P , etal: NGS catalog: A database of next generation sequencing studies in humans Hum Mutat 33: E2341– E2355,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jones S Hruban RH Kamiyama M , etal: Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene Science 324: 217,2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tischkowitz M, Xia B: PALB2/FANCN: Recombining cancer and Fanconi anemia Cancer Res 70: 7353– 7359,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roberts NJ Jiao Y Yu J , etal: ATM mutations in patients with hereditary pancreatic cancer Cancer Discov 2: 41– 46,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gatti RA Berkel I Boder E , etal: Localization of an ataxia-telangiectasia gene to chromosome 11q22-23 Nature 336: 577– 580,1988. [DOI] [PubMed] [Google Scholar]
- 61. Li A, Swift M: Mutations at the ataxia-telangiectasia locus and clinical phenotypes of A-T patients Am J Med Genet 92: 170– 177,2000. [DOI] [PubMed] [Google Scholar]
- 62. Ahmed M, Rahman N: ATM and breast cancer susceptibility Oncogene 25: 5906– 5911,2006. [DOI] [PubMed] [Google Scholar]
- 63. Comino-Méndez I Gracia-Aznárez FJ Schiavi F , etal: Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma Nat Genet 43: 663– 667,2011. [DOI] [PubMed] [Google Scholar]
- 64. Neumann HP Bausch B McWhinney SR , etal: Germ-line mutations in nonsyndromic pheochromocytoma N Engl J Med 346: 1459– 1466,2002. [DOI] [PubMed] [Google Scholar]
- 65. Hao HX Khalimonchuk O Schraders M , etal: SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma Science 325: 1139– 1142,2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yao L Schiavi F Cascon A , etal: Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas JAMA 304: 2611– 2619,2010. [DOI] [PubMed] [Google Scholar]
- 67. Burnichon N Brière JJ Libé R , etal: SDHA is a tumor suppressor gene causing paraganglioma Hum Mol Genet 19: 3011– 3020,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mannelli M Castellano M Schiavi F , etal: Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas J Clin Endocrinol Metab 94: 1541– 1547,2009. [DOI] [PubMed] [Google Scholar]
- 69. Cascón A Pita G Burnichon N , etal: Genetics of pheochromocytoma and paraganglioma in Spanish patients J Clin Endocrinol Metab 94: 1701– 1705,2009. [DOI] [PubMed] [Google Scholar]
- 70. Atchley WR, Fitch WM: Myc and Max: Molecular evolution of a family of proto-oncogene products and their dimerization partner Proc Natl Acad Sci U S A 92: 10217– 10221,1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Smith ML Cavenagh JD Lister TA , etal: Mutation of CEBPA in familial acute myeloid leukemia N Engl J Med 351: 2403– 2407,2004. [DOI] [PubMed] [Google Scholar]
- 72. Song WJ Sullivan MG Legare RD , etal: Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia Nat Genet 23: 166– 175,1999. [DOI] [PubMed] [Google Scholar]
- 73. Ostergaard P Simpson MA Connell FC , etal: Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome) Nat Genet 43: 929– 931,2011. [DOI] [PubMed] [Google Scholar]
- 74. Mansour S Connell F Steward C , etal: Emberger syndrome-primary lymphedema with myelodysplasia: Report of seven new cases Am J Med Genet A 152A: 2287– 2296,2010. [DOI] [PubMed] [Google Scholar]
- 75. Hahn CN Chong CE Carmichael CL , etal: Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia Nat Genet 43: 1012– 1017,2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shah S Schrader KA Waanders E , etal: A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia Nat Genet 45: 1226– 1231,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Enciso-Mora V Broderick P Ma Y , etal: A genome-wide association study of Hodgkin's lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3) Nat Genet 42: 1126– 1130,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Salipante SJ Mealiffe ME Wechsler J , etal: Mutations in a gene encoding a midbody kelch protein in familial and sporadic classical Hodgkin lymphoma lead to binucleated cells Proc Natl Acad Sci U S A 106: 14920– 14925,2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Saarinen S Aavikko M Aittomäki K , etal: Exome sequencing reveals germline NPAT mutation as a candidate risk factor for Hodgkin lymphoma Blood 118: 493– 498,2011. [DOI] [PubMed] [Google Scholar]
- 80. Medina R van der Deen M Miele-Chamberland A , etal: The HiNF-P/p220NPAT cell cycle signaling pathway controls nonhistone target genes Cancer Res 67: 10334– 10342,2007. [DOI] [PubMed] [Google Scholar]
- 81. Yokoyama S Woods SL Boyle GM , etal: A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma Nature 480: 99– 103,2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bertolotto C Lesueur F Giuliano S , etal: A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma Nature 480: 94– 98,2011. [DOI] [PubMed] [Google Scholar]
- 83. Horn S Figl A Rachakonda PS , etal: TERT promoter mutations in familial and sporadic melanoma Science 339: 959– 961,2013. [DOI] [PubMed] [Google Scholar]
- 84. Park DJ Lesueur F Nguyen-Dumont T , etal: Rare mutations in XRCC2 increase the risk of breast cancer Am J Hum Genet 90: 734– 739,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Park DJ Odefrey FA Hammet F , etal: FAN1 variants identified in multiple-case early-onset breast cancer families via exome sequencing: No evidence for association with risk for breast cancer Breast Cancer Res Treat 130: 1043– 1049,2011. [DOI] [PubMed] [Google Scholar]
- 86. Hilbers FS Wijnen JT Hoogerbrugge N , etal: Rare variants in XRCC2 as breast cancer susceptibility alleles J Med Genet 49: 618– 620,2012. [DOI] [PubMed] [Google Scholar]
- 87. Shah S Kim Y Ostrovnaya I , etal: Assessment of SLX4 mutations in hereditary breast cancers PLoS One 8: 366961,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ruark E Snape K Humburg P , etal: Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer Nature 493: 406– 410,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Rafnar T Gudbjartsson DF Sulem P , etal: Mutations in BRIP1 confer high risk of ovarian cancer Nat Genet 43: 1104– 1107,2011. [DOI] [PubMed] [Google Scholar]
- 90. Palles C Cazier JB Howarth KM , etal: Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas Nat Genet 45: 136– 144,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jaeger E Leedham S Lewis A , etal: Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1 Nat Genet 44: 699– 703,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ewing CM Ray AM Lange EM , etal: Germline mutations in HOXB13 and prostate-cancer risk N Engl J Med 366: 141– 149,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bott M Brevet M Taylor BS , etal: The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma Nat Genet 43: 668– 672,2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Harbour JW Onken MD Roberson ED , etal: Frequent mutation of BAP1 in metastasizing uveal melanomas Science 330: 1410– 1413,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Testa JR Cheung M Pei J , etal: Germline BAP1 mutations predispose to malignant mesothelioma Nat Genet 43: 1022– 1025,2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wiesner T Obenauf AC Murali R , etal: Germline mutations in BAP1 predispose to melanocytic tumors Nat Genet 43: 1018– 1021,2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Popova T Hebert L Jacquemin V , etal: Germline BAP1 mutations predispose to renal cell carcinomas Am J Hum Genet [epub ahead of print on May 14, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Farley MN Schmidt LS Mester JL , etal: Germline BAP1 mutation predisposes to familial clear-cell renal cell carcinoma Mol Cancer Res [epub ahead of print on May 24, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Walsh T Casadei S Lee MK , etal: Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing Proc Natl Acad Sci U S A 108: 18032– 18037,2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Offit K, Sagi M, Hurley K: Preimplantation genetic diagnosis for cancer syndromes: A new challenge for preventive medicine JAMA 296: 2727– 2730,2006. [DOI] [PubMed] [Google Scholar]
- 101. Pritchard CC Smith C Salipante SJ , etal: ColoSeq provides comprehensive Lynch and polyposis syndrome mutational analysis using massively parallel sequencing J Mol Diagn 14: 357– 366,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Domchek S Bradbury A Garber J , etal: Multiplex genetic testing for cancer susceptibility: Out on the high wire without a net? J Clin Oncol 31: 1267– 1270,2013. [DOI] [PubMed] [Google Scholar]
- 103. Offit K Bradbury A Storm C , etal: Gene patents and personalized cancer care: Impact of the myriad case on clinical oncology J Clin Oncol 31: 2743– 2748,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mardis ER Ding L Dooling DJ , etal: Recurring mutations found by sequencing an acute myeloid leukemia genome N Engl J Med 361: 1058– 1066,2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ding L Ellis MJ Li S , etal: Genome remodelling in a basal-like breast cancer metastasis and xenograft Nature 464: 999– 1005,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cancer Genome Atlas. http://cancergenome.nih.gov.
- 107.International Cancer Genome Consortium. https://www.icgc.org.
- 108. Comprehensive genomic characterization defines human glioblastoma genes and core pathways Nature 455: 1061– 1068,2008. Cancer Genome Atlas Research Network [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Integrated genomic analyses of ovarian carcinoma Nature 474: 609– 615,2011. Cancer Genome Atlas Research Network [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Comprehensive molecular characterization of human colon and rectal cancer Nature 487: 330– 337,2012. Cancer Genome Atlas Network [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Comprehensive molecular portraits of human breast tumours Nature 490: 61– 70,2012. Cancer Genome Atlas Network [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Oxnard GR Miller VA Robson ME , etal: Screening for germline EGFR T790M mutations through lung cancer genotyping J Thorac Oncol 7: 1049– 1052,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lynch TJ Bell DW Sordella R , etal: Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib N Engl J Med 350: 2129– 2139,2004. [DOI] [PubMed] [Google Scholar]
- 114. Paez JG Jänne PA Lee JC , etal: EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy Science 304: 1497– 1500,2004. [DOI] [PubMed] [Google Scholar]
- 115. Pao W Miller V Zakowski M , etal: EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib Proc Natl Acad Sci U S A 101: 13306– 13311,2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Kobayashi S Boggon TJ Dayaram T , etal: EGFR mutation and resistance of non-small-cell lung cancer to gefitinib N Engl J Med 352: 786– 792,2005. [DOI] [PubMed] [Google Scholar]
- 117. Pao W Miller VA Politi KA , etal: Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain PLoS Med 2: e73,2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Bell DW Gore I Okimoto RA , etal: Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR Nat Genet 37: 1315– 1316,2005. [DOI] [PubMed] [Google Scholar]
- 119. Sandhu SK Omlin A Hylands L , etal: Poly (ADP-ribose) polymerase (PARP) inhibitors for the treatment of advanced germline BRCA2 mutant prostate cancer Ann Oncol 24: 1416– 1418,2013. [DOI] [PubMed] [Google Scholar]
- 120. Balmāna J Domchek SM Tutt A , etal: Stumbling blocks on the path to personalized medicine in breast cancer: the case of PARP inhibitors for BRCA1/2-associated cancers Cancer Discov 1: 29– 34,2011. [DOI] [PubMed] [Google Scholar]
- 121. Bombard Y, Robson M, Offit K: Revealing the incidentalome when targeting the tumor genome JAMA 310: 795– 796,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Homer N Szelinger S Redman M , etal: Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays PLoS Genet 4: e1000167,2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zerhouni EA, Nabel EG: Protecting aggregate genomic data Science 322: 44,2008. [DOI] [PubMed] [Google Scholar]
- 124. Gymrek M McGuire AL Golan D , etal: Identifying personal genomes by surname inference Science 339: 321– 324,2013. [DOI] [PubMed] [Google Scholar]
- 125.US Department of Health and Human Services. ANPRM for revision to Common Rule. http://www.hhs.gov/ohrp/humansubjects/anprm2011page.html.
- 126. Kohane IS, Masys DR, Altman RB: The incidentalome: A threat to genomic medicine JAMA 296: 212– 215,2006. [DOI] [PubMed] [Google Scholar]
- 127. Ashley EA Butte AJ Wheeler MT , etal: Clinical assessment incorporating a personal genome Lancet 375: 1525– 1535,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Damani SB, Topol EJ: Emerging genomic applications in coronary artery disease JACC Cardiovasc Interv 4: 473– 482,2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Biskup S, Gasser T: Genetic testing in neurological diseases J Neurol 259: 1249– 1254,2012. [DOI] [PubMed] [Google Scholar]
- 130. Green RC Berg JS Berry GT , etal: Exploring concordance and discordance for return of incidental findings from clinical sequencing Genet Med 14: 405– 410,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Bombard Y, Offit K, Robson ME: Risks to relatives in genomic research: A duty to warn? Am J Bioeth 12: 12– 14,2012. [DOI] [PubMed] [Google Scholar]
- 132. Points to consider in the clinical application of genomic sequencing Genet Med 14: 759– 761,2012. ACMG Board of Directors [DOI] [PubMed] [Google Scholar]
- 133. Green RC Berg JS Grody WW , etal: ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing Genet Med 15: 565– 574,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Allyse M, Michie M: Not-so-incidental findings: The ACMG recommendations on the reporting of incidental findings in clinical whole genome and whole exome sequencing Trends Biotechnol 31: 439– 441,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Wolf SM, Annas GJ, Elias S: Point-counterpoint: Patient autonomy and incidental findings in clinical genomics Science 340: 1049– 1050,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. McGuire AL Joffe S Koenig BA , etal: Point-counterpoint: Ethics and genomic incidental findings Science 340: 1047– 1048,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lammens CR Aaronson NK Wagner A , etal: Genetic testing in Li-Fraumeni syndrome: Uptake and psychosocial consequences J Clin Oncol 28: 3008– 3014,2010. [DOI] [PubMed] [Google Scholar]
- 138. Roychowdhury S Iyer MK Robinson DR , etal: Personalized oncology through integrative high-throughput sequencing: A pilot study Sci Transl Med 3: 111ra121,2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Berg JS, Khoury MJ, Evans JP: Deploying whole genome sequencing in clinical practice and public health: Meeting the challenge one bin at a time Genet Med 13: 499– 504,2011. [DOI] [PubMed] [Google Scholar]
- 140.National Center for Biotechnology Information. ClinVar database. http://www.ncbi.nlm.nih.gov/clinvar/
- 141. McBride CM Bowen D Brody LC , etal: Future health applications of genomics: Priorities for communication, behavioral, and social sciences research Am J Prev Med 38: 556– 565,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Mardis ER: The $1,000 genome, the $100,000 analysis? Genome Med 2: 84,2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Offit K: Genomic profiles for disease risk: Predictive or premature? JAMA 299: 1353– 1355,2008. [DOI] [PubMed] [Google Scholar]
- 144. Offit K: Personalized medicine: New genomics, old lessons Hum Genet 130: 3– 14,2011. [DOI] [PMC free article] [PubMed] [Google Scholar]