

Abstract
Movement is one of the central attributes of life, and a key feature in many technological processes. While artificial motion is typically provided by macroscopic engines powered by internal combustion or electrical energy, movement in living organisms is produced by machines and motors of molecular size that typically exploit the energy of chemical fuels at ambient temperature to generate forces and ultimately execute functions. The progress in several areas of chemistry, together with an improved understanding of biomolecular machines, has led to the development of a large variety of wholly synthetic molecular machines. These systems have the potential to bring about radical innovations in several areas of technology and medicine. In this Minireview, we discuss, with the help of a few examples, the multidisciplinary aspects of research on artificial molecular machines and highlight its translational character.
Keywords: molecular devices, molecular motors, nanoscience, rotaxanes, supramolecular chemistry
1. Introduction
In an analogy with translational medical research,1 in chemistry one can define as “translational” any activity in which concepts, results and techniques from any area of chemical research are combined with those from other disciplines to deal with problems of scientific or applicative importance.2
The translational potential of research on molecular machines emerges from its own origins. In the last decades of the past century, it was commonly accepted that the most creative art in chemistry is the creation of new molecules.3 In the same period, however, supramolecular chemistry4—particularly in its broader meaning of the chemistry of multicomponent species5, 6—started to trigger the curiosity and the creativity of the chemical community. In this context, the idea that the concepts of macroscopic device and machine, typical of engineering, could be transferred to the molecular level began to arise.7
During the past thirty years chemists have designed, constructed and investigated a large variety of molecular devices and machines by exploiting photonic, electronic and chemical inputs to stimulate appropriately designed supramolecular systems.8, 9, 10, 11, 12, 13, 14, 15 This flourishing research has been enabled by the marriage of synthetic, physical and analytical chemistry with an engineering mentality. Already from early studies it became clear that scientists engaged in the development of artificial molecular machines would have needed to cross the traditional boundaries of chemistry with physics and biology, as foreseen by Richard Feynman in his famous 1959 talk.16
The field has now reached maturity17 and its scientific value has been recognized by the award of the Nobel Prize in Chemistry 2016 to Jean‐Pierre Sauvage, Fraser Stoddart and Ben Feringa.18 Synthetic molecular machines hold great promise for major progress in several areas of technology and medicine, providing the bases for ground‐breaking applications limited only by imagination. Recent research shows that impact is expected in materials science, catalysis, energy conversion, information technology, drug delivery and medical therapies.19 As noted earlier, the old flasks of chemistry are being filled with the new scent of nanoscience and nanotechnology.20
2. Molecular versus Macroscopic Machines
A molecular machine can be defined as an assembly of a discrete number of molecular components that exhibit mechanical movements in response to an external stimulus. Molecular switches and motors are classes of molecular machines, which in turn are a type of molecular devices.10, 15
Molecular machines are present in all living organisms, where they perform essential tasks encompassing movement and locomotion, energy conversion, transport and regulating functions.21 It is estimated that around 10000 different types of nanomachines are at work in the human body.22 In fact, the existence of biomolecular machines and their importance in Nature provide a compelling demonstration of the utility and feasibility of nanotechnology, and strongly motivate the research on synthetic nanomachines.
The knowledge of the biophysical principles at the basis of the operation of natural molecular machines is of essential importance for the design and construction of artificial versions.23 In fact, the observation of biological systems has shown that molecular machines are not “shrunken” versions of the macroscopic counterparts, and that the direct one‐on‐one mapping of the properties of a macroscopic object when its size is reduced down to the nanoscale can lead to completely wrong conclusions.24
While macroscopic machines are made with hard materials and their operation can rely on temperature differences between different parts, molecular machines are made of floppy components and must operate at a constant temperature (dictated by their environment), because heat flows very rapidly over nanometer distances. Because of the tiny mass of molecules, gravity and inertial effects are negligible at the molecular scale, where viscous forces resulting from intermolecular interactions—including those with solvent molecules—dominate. This is one of the reasons why obtaining directed motion at the molecular scale is very challenging.
The most significant feature of the nanoworld as far as movement is concerned is the fact that molecular‐sized objects are subjected to the ceaseless random motion caused by thermal energy (Brownian motion). The second law of thermodynamics states that such a motion cannot be harnessed to produce useful work, and it cannot be eliminated, unless at 0 K. At ambient temperature Brownian motion has a disruptive effect on small objects: it can be estimated that a molecule experiences a thermal noise power of 10−8 W, that is, at least 8 orders of magnitude larger than the power provided by the fueling chemical reaction in a typical biomolecular motor.25 This means that obtaining controlled and directed molecular movement is like walking in a hurricane, or riding a bike during an earthquake. If the latter cannot be stopped, the only way to move forward is to exploit its random shakes. This is exactly what biomolecular machines do: they use an external chemical energy source to bias thermal agitation, making the movement in a given direction more likely than that in other directions.
This principle is schematically illustrated in (Figure 1) for a one dimensional system. When the initial, more stable state is destabilized by an energy input, the system is brought in a non‐equilibrium condition, and thermal motion will push the system over the (decreased) barrier to the lower energy state. Hence, thermal agitation has actually caused the motion, which has been rendered directional by the energy input. Reset occurs when the energy input ends, or an opposite input is activated.
Figure 1.
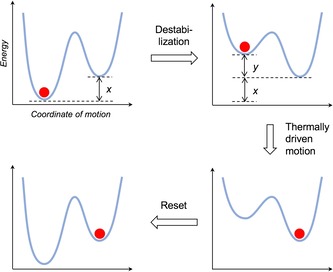
Schematic representation of the thermally driven motion of a chemical system biased by an energy input (y). The state of the system with respect to the motion coordinate is identified by the position of the red circle on the potential energy curve.
3. The Energy Issue
As discussed above, Brownian motion at thermal equilibrium cannot be exploited to achieve directed and controlled movement of a molecular machine. Thus, molecular machines need an external input of energy to operate.
3.1. Chemical Energy
The most obvious way to supply energy to a chemical system is through a reactant (“fuel”) that undergoes an exoergonic reaction. This is what happens in biomolecular machines, which are typically powered by ATP hydrolysis.21 In the case of ATP synthase, chemical energy is supplied in the form of a transmembrane proton gradient.
Chemically powered molecular machines require that the fuel be delivered where and when necessary. The reaction of the fuel will generate products (“waste”) which, even if they do not interfere with the operation of the machine, will accumulate in the reaction medium, unless they are appropriately removed. Remarkably, the waste of biomolecular machines (ADP and inorganic phosphate) is not only removed but also recycled into new fuel by ATP synthase, providing a beautiful example of circular economy at the nanoscale.
Another important quality of chemically powered molecular machines is the way in which they exploit the fuel. If the reaction of the fuel brings the system into a new equilibrium state, as it happens in thermodynamic switches, another reactant (“anti‐fuel”) must be added in sequence to reset the switching process and close the operation cycle (Figure 2 a). Such a behavior requires the alternate additions of fuel and anti‐fuel, resulting in an operator‐dependent working cycle. In other words, the machine is not capable of using the chemical energy input in an autonomous fashion. This is by far the most common operation mode of artificial molecular machines developed until now which are, in fact, mechanical switches.
Figure 2.
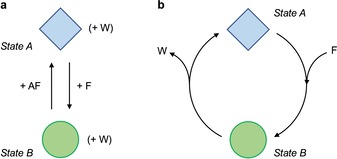
a) A mechanical molecular switch that uses a fuel (F) and an anti‐fuel (AF) to convert between two structurally different equilibrium states. Repeated switching (cycling) between the states requires the sequential addition of reactants. b) An autonomous chemically driven molecular machine that exploits the catalytic decomposition of a fuel to perform the transformation between two structurally different states. The process can continue indefinitely without the intervention of an operator as long as the fuel is available. Waste products (W) are formed in both cases.
An example of a system of this kind is the “molecular elevator” 1 H3 9+ shown in Figure 3 a.26 This compound consists of two mechanically interlocked components: 1) a framework‐like species with three legs, each endowed with two different sites—a dialkylammonium and a 4,4′‐bipyridinium—at different levels; 2) a platform‐like species in which three crown ether rings are connected trigonally to a central aromatic moiety. The platform, whose dissociation from the framework is prevented by the bulky stoppers placed at the end of each leg, can be made to stop at the two different levels. At the beginning, in acetonitrile solution, the platform is positioned at the “upper” level, that is, with its three macrocycles surrounding the ammonium sites (Figure 3 b, state A), as a result of strong N+‐H⋅⋅⋅O hydrogen bonds.
Figure 3.
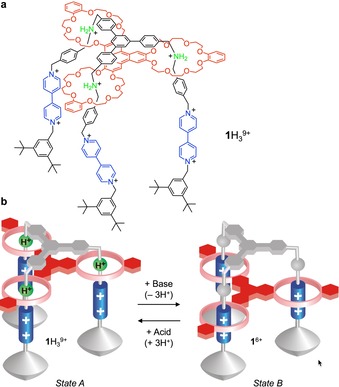
Structure formula (a) and base/acid‐triggered operation scheme (b) of molecular elevator 1 H3 9+.26
When an appropriate base is added, the ammonium sites of 1 H3 9+ are deprotonated and, as a consequence, the platform moves to the “lower” level, with the three rings surrounding the bipyridinium sites (Figure 3 b, state B). Such a structure of 1 6+ is stabilized by charge‐transfer interactions between the π‐electron‐rich aromatic units of the platform and the π‐electron‐poor‐ bipyridinium units of the framework. Upon successive addition of acid, the ammonium sites are restored and the platform returns to the “upper” level. The elevator motion, which can be followed by NMR spectroscopy, electrochemistry, and absorption and fluorescence spectroscopy, is quantitative and can be repeated by the sequential addition of a basic fuel and an acid anti‐fuel.26 Obviously, the one‐shot simultaneous addition of these reactants to a solution containing 1 H3 9+ would lead to their annihilation, thus frustrating the operation of the machine.
In stark contrast, biomolecular machines can autonomously repeat their operation cycle in a constant environment (apart from the energy input) as long as the fuel is available (Figure 2 b). This is because such systems exploit the fuel in a catalytic manner; in other words, they act as catalysts for the decomposition of the fuel. While performing the catalytic cycle during which the fuel is transformed, the molecular machine travels along different mechanical states, ultimately exhibiting controlled motion. In fact, most biomolecular machines catalyze the hydrolysis of ATP and use the resulting energy to bias Brownian motion.21, 22, 23, 24, 25
A step forward towards the development of fully artificial molecular machines that can exploit autonomously a chemical fuel is the system shown in Figure 4.27 In solution, [2]catenane 2 assumes a co‐conformation in which the two bulky and rigid phenanthroline moieties are located away from each other (Figure 4, state A).28 The addition of 2‐cyano‐2‐phenylpropanoic acid (3) causes the protonation of the catenane (2 H+), which is accompanied by a rearrangement of the molecular rings (Figure 4, state B). The resulting phenylpropanoate anion undergoes quantitative decarboxylation and is converted into a carbanion intermediate which eventually takes a proton back from 2 H+, forming the waste product 4 and regenerating the starting compound 2. The catenane thus acts as a catalyst for the base‐promoted decarboxylation of 3, and completes a mechanical switching cycle between states A and B for each processed fuel molecule.27
Figure 4.
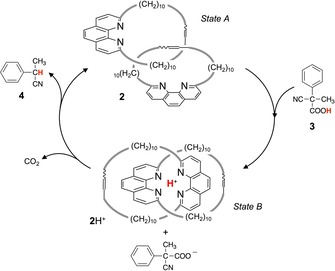
Schematic representation of the switching cycle of catenane 2, activated by the decomposition of the fuel 3 which is converted into waste products carbon dioxide and 4.27.
Unfortunately, the decomposition of the fuel is profitably coupled with the catenane switching only if the fuel is present in a (sub)stoichiometric amount. In fact, when acid 3 is in excess, an alternative decomposition mechanism not involving deprotonation of 2 H+ is activated. The Scheme shown in Figure 4 is restored only when the acid in excess has been decomposed.27a Hence, this molecular machine cannot repeat its operation cycle autonomously in the presence of an excess of fuel. Very recently, the same principle has been used to drive directional rotary and linear motions in catenanes and rotaxanes, respectively, by using pulses of an acid chemical fuel.29
The chemically powered autonomous and repetitive unidirectional rotation of a molecular ring in a [2]catenane was reported by the Leigh group.30 The system relies on an information ratchet mechanism15b based on the fact that the reaction of the fuel with two chemically equivalent sites along the larger “track” ring is affected by the position of the smaller “moving” ring.
A key feature of the catalytic operation mode is that the different mechanical states of the machine are not equilibrium states for the whole system (machine + fuel). The machine operates away from thermodynamic equilibrium, which is eventually reached when the fuel is over. It is worthwhile to recall, however, that out‐of‐equilibrium conditions can also be obtained with carefully designed molecular machines that consume a fuel and an anti‐fuel in a non‐autonomous fashion. This is the case for the chemically driven supramolecular pump illustrated in Figure 5,31 whose components are the tetracationic macrocycle 5 4+ and the tricationic axle 6 3+. In water, the chemical reduction of the bipyridinium units of the ring and the axle promotes their threading by passage of the ring over the positively charged 3,5‐dimethylpyridinium end group (Figure 5 b). Successive oxidation causes the buildup of an electrostatic repulsion between the 5 4+ ring and the bipyridinium site on 6 3+. The ring, however, cannot dethread because of the now increased electrostatic barrier represented by the 3,5‐dimethylpyridinium group; it can only move towards the ring collecting alkyl chain. In the successive reduction step, the presence of the isopropylphenylene group makes the return of the threaded macrocycle slower than the entrance of a second ring which, upon oxidation, is pushed on the ring collecting chain. Such a reduction‐oxidation cycle can be repeated indefinitely, although only two rings have been pumped so far experimentally.31
Figure 5.
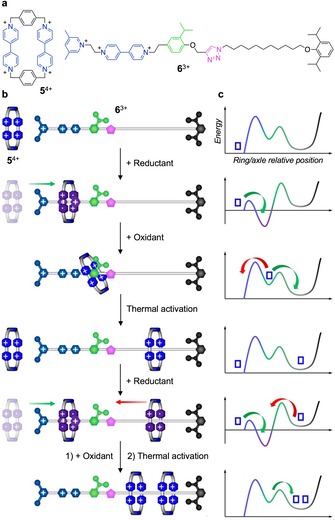
a) Structure formula of the components of a chemically driven artificial molecular pump.31 b) Schematic operation mechanism. c) Idealized potential energy profiles for each structure in (b) as a function of the ring‐axle relative position. Adapted with permission from Ref. 32.
Overall, as shown in Figure 5 c, the potential energy landscape for the translation of the ring along the axle is modulated by the alternated effects of the fuel (reductant) and the anti‐fuel (oxidant) in solution. Such a ratcheting effect prevents the system from reaching equilibrium at the end of each switching cycle, thus enabling the accumulation of molecular rings on a portion of the axle devoid of recognition sites.32
3.2. Light Energy
Although solar radiation is the only energy source for our planet, in Nature sunlight—or, in general, light energy—is not directly converted into mechanical movements. Rather, it is used to produce chemical fuels (e.g., ATP) suitable for powering biomolecular machines.33 Light energy, however, can cause reactions that involve large structural changes in molecular systems. A simple example of this kind of photochemical reactions is the light‐induced transformation between the E and Z isomers of molecules that contain a ‐N=N‐ or ‐C=C‐ double bond.9, 11 In multicomponent species, photoinduced electron‐ or proton‐transfer reactions can also cause a major displacement of the molecular parts.34
The use of light to power artificial molecular machines has several advantages in comparison with chemical fuels.35 First of all, by relying on reversible and clean photochemical reactions, one can design machines powered solely by photons that do not generate waste products during operation. Moreover, the amount of energy transferred to chemical systems through light can be carefully controlled by its wavelength and intensity, in relation to the absorption spectrum of the targeted species. Such an energy can be transferred to the molecules without any physical connection to the source, the only requirement being the transparency of the matrix at the excitation wavelength. Fiber optics, however, can provide solutions in cases light has to be delivered in particular places (e.g. inside an instrument or in the human body). With modern light sources one can investigate very small spaces and extremely short time domains, and near‐field techniques allow excitation with nanometer resolution. Conversely, the parallel addressing of a very large number of individual molecular machines can be afforded upon irradiation of large areas and volumes. Another significant difference between chemically and light driven molecular machines36 is that photoinduced processes can directly result into motion and generation of force, thus deterministically relating the energy input with the “power stroke” of the machine.
By taking advantage of reversible photochemical processes, molecular machines capable of operating autonomously by consuming only photons of light can be designed. For instance, the operation of the machine could be based on a photoinduced sequence of processes that lead the system through transient electronic and (co‐)conformational (mechanical) states; the final deactivation of the system to the ground state provides an automatic reset and closes the cycle of operation.37 An example of this kind is [2]rotaxane 7 6+ shown in Figure 6.38
Figure 6.
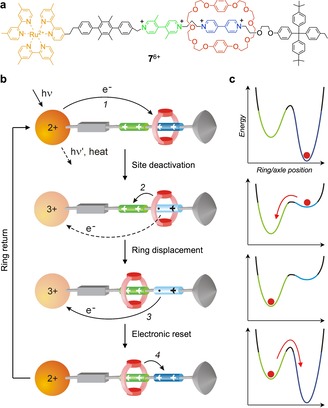
a) Structure formula of rotaxane 7 6+.38 b) Operation mechanism of 7 6+ as a light‐driven linear molecular machine; processes 1 and 2 compete with undesirable processes (dashed lines). c) Idealized potential energy profiles for each structure in (b) as a function of the ring‐axle relative position.
In the stable co‐conformation of the rotaxane, the ring component—an electron donor crown ether—encircles the primary electron acceptor site—a 4,4′‐bipyridinium‐type unit—because this site is a better electron acceptor than the other one—a 3,3′‐dimethyl‐4,4′‐bipyridinium‐type unit (Figure 6 a). The mechanism for photoinduced ring displacement consists of four steps (Figure 6 b). In solution at room temperature, excitation of the photoactive RuII‐based moiety with visible light causes the transfer of an electron from this unit to the primary electron acceptor site (step 1), which becomes deactivated. As a consequence, the ring moves to encircle the secondary site (step 2). A back electron transfer from the free reduced site to the oxidized Ru‐based unit (step 3) restores the electron acceptor ability of the former site. As a consequence of such an electronic reset, the ring returns to the original primary acceptor site (step 4). The switching cycle is thus completed and the rotaxane is ready to process another photon. The automatic thermally driven reset of the molecular machine enables its autonomous operation under the supply of light energy.
As it happens for macroscopic motors, the successful operation of this nanomachine relies on a correct synchronization of the processes at the basis of the mechanism (Figure 6 b). Such a result implies a fine control on kinetics and can be achieved by careful structural design. Because of competition with undesirable energy‐wasting processes (Figure 6 b), the overall quantum yield for ring shuttling is only 2 %. Despite this disappointing figure, 7 6+ is a remarkable example of an artificial nanomachine that operates autonomously by consuming only photons of visible light (i.e., sunlight); no waste products are formed.
Alternatively, the ability of a molecular machine to process light energy in autonomous fashion could arise from the photochemically triggered switching between two states, as in photochromic systems. This is the case for the well‐known family of molecular rotary motors developed by Ben Feringa and co‐workers,18c which exploit the photoisomerization of sterically hindered alkenes.
The photoisomerization of azobenzene between E and Z form, combined with the self‐assembly of a pseudorotaxane species, was recently exploited to obtain an autonomous supramolecular pump powered by light energy.39 The components (Figure 7 a) are the non‐symmetric molecular axle 8H+, comprising an azobenzene photoswitchable extremity (P), an ammonium recognition site (A) and a photoinactive cyclopentyl end (S), and the crown ether ring 9. In a dichloromethane solution at room temperature, there is a driving force for the assembly of a pseudorotaxane in which 9 encircles the ammonium site of E‐8H+ on account of hydrogen bonding interactions. As the P‐end in its E configuration exhibits a much smaller hindrance than the S‐end for the transit of the ring, there is a strong kinetic preference for threading through the P‐end, thus dictating the direction of the first step (Figure 7 b).
Figure 7.
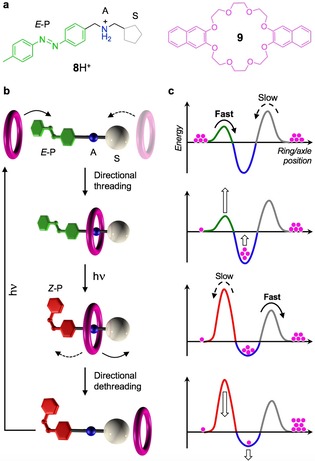
a) Structure formula and cartoon representation of the components of a photochemical autonomous supramolecular pump.39 b) Schematic operation mechanism of the light‐driven relative unidirectional transit of the ring along the axle. c) Idealized potential energy profiles for each structure in (b) as a function of the ring‐axle relative position. The processes represented by dashed lines are too slow to occur.
Absorption of a photon in the UV or blue region causes the isomerization of the P‐end to the Z form. Such a transformation has two key consequences: 1) the threading barrier at the P‐end is dramatically increased, making the S‐end the kinetically preferred extremity for the transit of the ring, and 2) the stability of the complex is decreased. As a result, the pseudorotaxane species are partially dethreaded, and the rings must escape from the S‐end (Figure 7 b). Because both isomeric forms of azobenzene are photoreactive and absorb in the same spectral region, another photon, identical to the first one, can trigger the transformation of Z‐8H+ back to E‐8H+, thereby closing the switching cycle. Hence, what is usually a drawback for azobenzene as a photoswitch, because it prevents complete conversion between the isomers, becomes an advantage, because it enables autonomous cycling.
As shown in Figure 7 c, the system uses photons to rectify Brownian fluctuations, and can repeat its working cycle under photostationary conditions, that is, under the action of a single optical stimulus with constant wavelength and intensity. The device takes advantage of light energy to operate repetitively away from equilibrium, thus providing a significant case of a dissipative self‐assembling system, and the first and sole example of an autonomous artificial molecular pump reported to date.40 Further elements of interest of the system are its minimalist design, structural simplicity, stability and reversibility.
3.3. Electrical Energy
It has long been known that redox reactions, triggered by the application of electrical potentials, can cause substantial structural changes in supramolecular systems.41 If a reversible redox couple is utilized, one can promote the forward reaction, and later return to the reactants by inverting the potential; in other words, a switching process can be performed without forming waste products. Electrochemical inputs, unlike chemical redox inputs, can be readily turned on and off, and electrodes are a most convenient way for interfacing molecular‐scale systems with the macroscopic world. Furthermore, electrochemical methods offer useful tools for observing the operation of the machine.
Electrical energy can also be conferred to chemical systems without causing redox processes. Indeed, individual molecules lying on a surface can be electronically and/or vibrationally excited by the application of an appropriate voltage with a local probe, such as the tip of a scanning tunneling microscope (STM). In recent years, this approach was followed to accomplish directed motion of single molecules on surfaces.42
An impressive example of this kind is the “nanocar” molecule 10 (Figure 8 a), that consists of four overcrowded alkene rotary motor units as the wheels, mounted on a rigid bis(phenylene ethynylene) chassis.43 In each wheel, electronic (e.g., light) and vibronic (e.g., thermal) excitation can induce, respectively, configurational and conformational changes that ultimately cause the unidirectional rotation of the fluorene moiety around the C=C bond.18c It was thus hypothesized that the STM tip, by inducing electronic and vibronic excitations, could trigger the isomerization processes necessary for the rotary motion of the wheels of 10, and concurrently provide single molecule imaging. To achieve directed motion on the surface, the four wheels need to move in a conrotatory manner; as the direction of rotation of each motor unit is dictated by its chirality, the meso‐(R,S‐R,S) isomer of 10 is the appropriate nanocar candidate (Figure 8 b). Another necessary condition for translation is that the energy dissipated in the isomerization steps be greater than that associated with the adsorption of the molecule on the surface. In fact, the adsorption energy of 10 was expected to be modest because the strained helical structure of the wheels prevents the aromatic moieties from aligning parallel to the surface.
Figure 8.
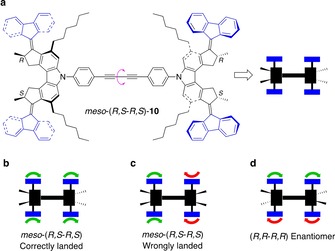
a) Structure formula and cartoon representation of the meso‐(R,S‐R,S) isomer of nanocar 10.43 b–d) Schematics of the directionality of the motion induced by the concerted rotation of the four motor units. The green and red arrows indicate the direction in which the rotary action of the individual motor units propels the molecule on the surface. Two distinct “landing geometries” of the meso‐isomer lead to either directional movement (b) or to no movement at all (c), whereas (R,R‐R,R) (d) or (S,S‐S,S) enantiomers move randomly.
The species were deposited on Cu{111} by sublimation, and investigated by STM at 7 K. Under mild conditions (scanning potential <60 mV), single 10 molecules could be imaged several times without affecting their position. Conversely, the application of a voltage pulse of 600 mV by means of the STM tip caused the excitation of the target molecule; a subsequent scan performed under mild conditions revealed its position after the energy dissipation.43
Upon ten excitation steps, a translation of 6 nm across the surface was observed for some of the imaged molecules. Not all nanocars, however, were found to move directionally. Because of the rotational freedom around the bis‐alkyne C−C bond of the chassis (pink arrow in Figure 8 a), which is locked upon deposition, some molecules became adsorbed in a geometry such that the rotations of the wheels cancel out, thereby preventing translation (Figure 8 c).
The electrically driven movement of 10 was further supported by the investigation of the individual (R,R‐R,R) or (S,S‐S,S) enantiomers, deposited on the surface from the racemic mixture,43 which were found to spin and wander on the surface. Such a behavior can be explained considering that the disrotatory motion of the motor units on opposite sides of the nanocar ideally causes the molecule to spin (Figure 8 d); in a non‐ideal case, the spinning motion occurs along with random translational motion.
The successful construction and operation of a motorized single‐molecule vehicle not only had a strong symbolic impact on the nanoscience community44 but it also demonstrated that an external electrical energy input can be converted into directed nanoscale motion across a surface. The value for chemistry is significant, because 1) the motion of the nanocar arises from its intrinsic molecular motor functions, and not from tip‐related manipulation, and 2) the type of movement is a direct consequence of the molecular design.
4. Towards Advanced Applications
Although market applications of artificial molecular machines are not around the corner, recent outstanding experiments have highlighted their potential to revolutionize current approaches to technological problems. For space reasons we will briefly discuss only a few prominent examples, and omit artificial molecular devices based on biomolecules (e.g., DNA). Interested readers can consult references 10–15.
4.1. Information Technology: Molecular Machine‐Based Ultradense Electronic Memories
Current computer technology is based on George Boole's binary algebraic formulation. Boolean variables can assume only two values—for example, 0 or 1. Bistable mechanical molecular switches, such as the one shown in Figure 3, can be associated with binary logic.8, 9, 10, 11, 12 As the application of an external stimulus (input) can change their state (output), molecular machines can be viewed as logic devices. Rotaxanes and catenanes that, depending on the applied electrical potential, can exist in two structurally different states for the relative position of their molecular components, have been used to realize electrically addressable solid‐state memory devices.45
The geometry of the device is shown schematically in Figure 9. A monolayer of bistable rotaxanes (or catenanes), such as 11 4+, was deposited on a photolithographically patterned poly‐Si electrode consisting of parallel lines. Orthogonally oriented metal wires were then constructed on top of the first set of lines, thus creating a “crossbar” architecture in which each individual junction is electrically addressable. The conductance of the junction is determined by electron tunneling through the molecular monolayer trapped between the electrodes. Thus, any change in the electronic properties of the sandwiched molecules is expected to affect the tunneling efficiency and change the resistance of the junction.
Figure 9.
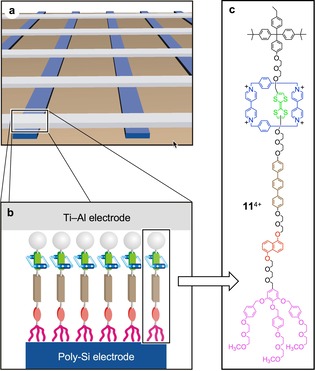
Schematic representation of solid‐state crossbar electrical junctions (a) containing a monolayer (b) of the electroactive bistable rotaxane 11 4+ (c), which were used to construct high‐density electronic memory devices.45 The different parts of the device are not to scale.
It was found experimentally that the junctions can exist in either low‐ or high‐conductance states, depending on the applied voltage; moreover, the current–voltage curves exhibit a marked hysteresis, making the device potentially interesting for random access memory (RAM) data storage applications. With the aid of electrochemical experiments performed in solution or on films,46 these properties were correlated with the electrically induced relative motion of the molecular components of the embedded rotaxanes (or catenanes).45
In a first setup the wire electrodes were a few μm across; the scalability of the method, however, allowed the fabrication of wires less than 100 nm wide, providing junctions with areas of 0.005 to 0.01 μm2 that contained about 5000 rotaxane molecules.45 Later, a molecular electronic memory with an amazingly high density −1011 bits cm−2—was realized with the same strategy using nanowire electrodes in a crossbar arrangement.47 Each memory element—i.e., a junction—contained about only 100 rotaxane molecules.
A circuit with 160000 memory cells (400×400 nanowires) was made, but only 128 of them (16×8 contacts) were investigated for practical reasons. Although only 25 % of the tested cells displayed good and reproducible switching, this work is a compelling demonstration that the combination of engineering top‐down and chemical bottom‐up nanofabrication methods can lead to outstanding technological achievements. Real industrial applications, however, require the optimization of several aspects, such as stability, reliability and ease of fabrication.
4.2. Catalysis: Molecular Machines for Making Molecules
As magnificently shown by DNA polymerases and ribosomes, one of the main tasks of biomolecular machines is the making of other molecules.22, 24 Recent experiments have demonstrated the potential utility of synthetic molecular machines for controlling catalytic effects.48
An artificial molecular machine that can assemble a tripeptide, whose sequence is pre‐programmed in the machine itself, has been described.49 The system (Figure 10) consists of the [2]rotaxane 12, comprising a macrocycle endowed with a catalytic arm and a peptide elongation site, and a stoppered axle that contains a sequence of three amino acid building blocks separated by stiff spacers. As the rotaxane is prepared by an active template catalytic methodology, there are no ring‐axle interactions. Under appropriate conditions, the thiol group present in the catalytic arm first reacts with the ester connecting the amino acid to the axle, then transfers the activated amino acid to the peptide elongation site. Before the reaction the motion of the ring along the axle is limited by the steric hindrance of the amino acid; when the latter has been removed, the ring is free to reach the second amino acid and the transfer sequence can be repeated. After the third and final amino acid has been cleaved, the ring dethreads and the synthesized oligopeptide can be isolated.
Figure 10.
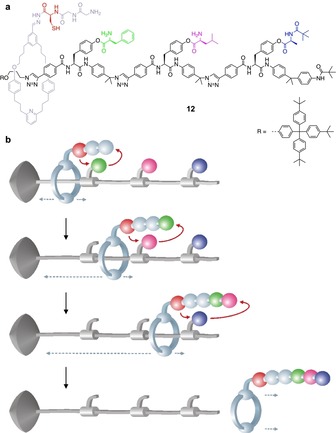
a) Structure formula of the rotaxane 12, and b) cartoon representation of its operation as an artificial molecular peptide synthesizer.49
Experiments showed that this molecular machine proceeds autonomously and processively with a high fidelity. As the rigidity of the axle prevents folding, the reactive arm can only encounter the amino acids because of the translational motion of the ring, thus ensuring the accurate transfer of the sequence from the axle to the ring. Besides the relatively slow reaction rate, a drawback of this design is that the chemical information present on the axle (i.e., the sequence of amino acids) is destroyed upon reading.
Very recently, by relying on a different molecular design, the same group described an artificial molecular machine that can be programmed to afford different product outcomes by moving a substrate between different activating sites.50
4.3. Materials Science and Engineering: Towards Artificial Muscles Capable of Macroscopic Actuation
Skeletal muscles show that motion can be amplified from the molecular to the macroscopic scale if molecular motors are appropriately organized and integrated within polymeric structures.21, 22, 24 Along the same line, concepts and systems pertaining to artificial molecular machines can be applied in polymer science to develop mechanically active materials. Research efforts in this direction51, 52 have shown that the action of large numbers of artificial molecular machines incorporated in polymers can be collected, leading to macroscopic actuation. Bistable [c2]daisy‐chain rotaxanes that can reversibly change their length in response to external stimuli—the so‐called “molecular muscles”14, 53—are particularly amenable for the construction of polymeric actuators.
A representative example of a daisy chain molecular muscle, derived from the principles of the molecular machine described in Figure 3, is compound 13 H2 4+ shown in Figure 11 a.54 In acidic media, the macrocycles surround the ammonium sites and the muscle assumes an extended structure. Upon increasing the pH, the ammonium sites are deprotonated and the rings move to interact with the secondary recognition site (a triazolium unit), thus causing the contraction of the system.
Figure 11.
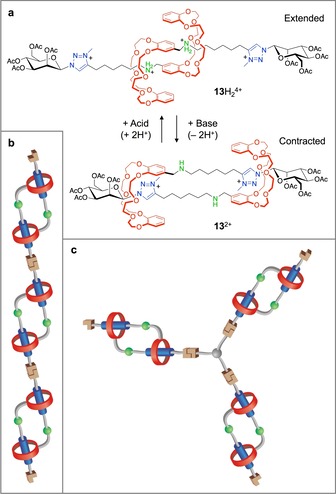
a) The chemically driven contraction/extension of the rotaxane dimer 13H2 4+/13 2+, characterized by a [c2]daisy‐chain topology.54 b, c) Cartoon representation of a main chain (b) and a branched (c) polymer incorporating daisy chain molecular muscle elements.51.
In principle, a main chain polymer could be obtained by joining together 13 H2 4+ (or its deprotonated form 13 2+) monomers (Figure 11 b). Initial attempts involving the covalent end‐to‐end connection of daisy chains led to relatively short oligomers.55 Later, high degrees of polymerization were obtained by coordination51a or supramolecular51b approaches. In these systems, chemically driven microscopic extension/contraction of single chain polymers was observed. Three‐dimensional covalent polymer networks incorporating photoswitchable daisy chains were also obtained, and the corresponding xerogel was shown to undergo photoinduced deformation.56
Very recently, daisy chains of the type shown in Figure 11 a have been incorporated in branched covalent polymers, leading to gel materials consisting of cross‐linked networks with extensible/contractible edges (Figure 11 c).51c Interestingly, upon pH‐triggered operation of the molecular machines the gel undergoes a large (ca. 50 %) and reversible volume change. In principle higher order daisy chains could also be utilized.57 These results are significant because they show that materials capable of macroscopic actuation can be constructed by relying on molecular machines. Such soft materials are of high interest for technological applications, e.g., in robotics and prosthetics.
4.4. Medical Therapy: Mechanized Nanovalves for Targeted Drug Delivery
Ideally, drug delivery systems should carry pharmaceutical compounds through the body and administer them to a target tissue in a specific and controlled manner. These systems are a formidable tool for medical therapy because they can increase the efficacy of drugs by protecting the latter from degradation during transport, and focusing their release to the diseased tissue, thereby limiting adverse effects.
Mesoporous silica nanoparticles have large surface exteriors and porous interiors that can be harnessed as reservoirs for small molecules. These features, together with other properties such as ease of synthesis and functionalization, chemical stability, resistance to enzymatic degradation, efficient cellular uptake and low inherent toxicity, make them attractive containers for drug delivery applications.58 To afford targeted delivery, however, the release of the drug molecules stored in the nanoparticles should occur controllably in response to either external or cellular stimuli. This goal has been achieved by using the movement of molecular machines to close and open the pore entrances, thus regulating the access to the internal voids.
As shown schematically in Figure 12, rotaxane‐based molecular shuttles were covalently attached to the nanoparticles.59 The pores could be closed and opened by moving the mechanically interlocked ring component of the rotaxane closer to and away from the pores’ orifices, respectively. The properties of these rotaxane‐based nanovalves can be fine‐tuned by changing the length of the linkers between the surface and the rotaxane molecules, and the location of the movable components on the nanoparticles.60 Mechanized nanoparticles operated by light, redox stimuli, pH, and enzymes, capable of delivering payloads including metal complexes, fluorescent dyes, and antitumor drugs, have been constructed. The stimulated release of cargos of different size in succession and dual‐input operation have also been demonstrated.14
Figure 12.
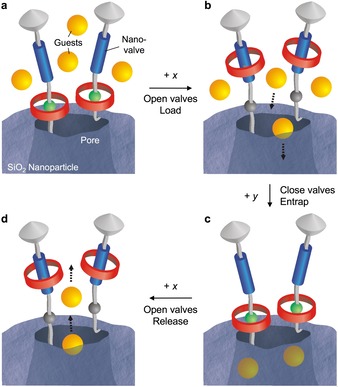
Schematic operation of a drug‐delivery system consisting of a mesoporous silica nanoparticle decorated with rotaxane‐based molecular machines acting as nanovalves.59 a) With the nanovalves closed, the access of external drug molecules to the empty pore is prevented. b) Upon opening of the nanovalves induced by a stimulus (x), guests can enter the pore. c) Switching of the nanovalves with another stimulus (y) and dialysis afford a drug‐loaded nanoparticle. d) The successive stimuli‐triggered opening causes the release of the guest. The different parts of the device are not to scale.
5. Conclusions
In the past three decades, research on molecular machines has stimulated the ingenuity and creativity of scientists with various backgrounds. It has also placed them in front of fundamental physical problems, hard synthetic routes, sophisticated modelling and mechanistic issues, (bio)chemical enigmas, conceptually and technically challenging characterization experiments, and unexplored issues related to the interface of molecular devices with their environment in order to perform useful tasks. Although the artificial molecular machines reported so far—some of which have been described in this article—are incredibly rudimentary in comparison with biological counterparts, their investigation has brought about a tremendous scientific progress, as recognized last year by the Nobel committee for chemistry.
The growth of the field of molecular machines has greatly benefited from the permeation of the concepts of chemistry towards other disciplines (mainly, physics and biology) and vice versa. The next step towards a real exploitation of such nanoscale devices demands a truly multidisciplinary effort for using the concepts and techniques of molecular machines to solve problems in materials science and engineering, information technology, environmental science and medicine. Until now, the design and investigation of synthetic molecular machines have been essentially pursued by following a basic science approach, in which operating conditions can be stretched at will to reach the final goal—the proof of principle. The time is ripe for researchers to face application‐dependent technological constraints, so that the next generation of molecular machines will be designed to operate in practical settings under real world conditions. In this regard, parameters such as long‐term stability, efficiency and cost, as well as toxicity and environmental effects, will need to be considered.
Aside from practical applications, translational efforts in chemistry—nicely represented by the field of molecular machines—implies that scientists belonging to different disciplines, ranging from mathematics to robotics, cooperate and learn a common language. The resulting cultural leaps forward will surely be beneficial for the progress of our society.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Alberto Credi is Professor of Chemistry at the University of Bologna (Italy) and director of the Center for Light Activated Nanostructures (CLAN), a University‐National Research Council joint laboratory for research in photochemistry, supramolecular chemistry, materials science, and nanoscience. He received his “Laurea” (1994) from the University of Bologna, where, after a research period in the USA, he later earned his PhD (1999). He has co‐authored 3 books and over 260 scientific publications, and he is the PI of an ERC Advanced Grant for the development of light driven molecular motors.

Acknowledgements
Financial support from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 692981) is gratefully acknowledged.
M. Baroncini, L. Casimiro, C. de Vet, J. Groppi, S. Silvi, A. Credi, ChemistryOpen 2018, 7, 169.
Dedicated to Margherita Venturi on her retirement
References
- 1. Butler D., Nature 2008, 453, 840–842. [DOI] [PubMed] [Google Scholar]
- 2. Lodeiro C., Capelo J. L., ChemistryOpen 2017, 7, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Breslow R., Chemistry Today and Tomorrow—The Central, Useful, and Creative Science American Chemical Society, Washington DC, 1997. [Google Scholar]
- 4. Lehn J.-M., Supramolecular Chemistry: Concepts and Perpectives, Wiley-VCH, Weinheim, 1995. [Google Scholar]
- 5.
- 5a. Balzani V., Credi A., Venturi M., Proc. Natl. Acad. Sci. USA 2002, 99, 4814–4817; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Balzani V., Credi A., Venturi M., Chem. Eur. J. 2002, 8, 5524–5532. [DOI] [PubMed] [Google Scholar]
- 6. From Non-Covalent Assemblies to Molecular Machines (Eds. J.-P. Sauvage, P. Gaspard), Wiley-VCH, Weinheim, 2011. [Google Scholar]
- 7.
- 7a. Balzani V., Moggi L., Scandola F., in Supramolecular Photochemistry (Ed.: V. Balzani), Reidel, Dordrecht, 1987, pp. 1–28; [Google Scholar]
- 7b. Lehn J.-M., Angew. Chem. Int. Ed. Engl. 1988, 27, 89–112; [Google Scholar]; Angew. Chem. 1988, 100, 91–116. [Google Scholar]
- 8. Balzani V., Credi A., Venturi M., Molecular Devices and Machines—A Journey into the Nanoworld, Wiley-VCH, Weinheim, 2003. [Google Scholar]
- 9. Molecular Switches (Eds.: B. L. Feringa, W. R. Browne), Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 10. Balzani V., Credi A., Venturi M., Molecular Devices and Machines—Concepts and Perspectives for the Nanoworld, Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 11. Molecular Switches, Second Edition (Eds.: B. L. Feringa, W. R. Browne), Wiley-VCH, Weinheim, 2011. [Google Scholar]
- 12. de Silva A. P., Molecular Logic-based Computation, RSC Publishing, Canbridge, 2012. [Google Scholar]
- 13. Wang J., Nanomachines—Fundamentals and Applications, RSC Publishing, Cambridge, 2012. [Google Scholar]
- 14. Bruns C., Stoddart J. F., The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, Hoboken, 2016. [Google Scholar]
- 15.
- 15a. Balzani V., Credi A., Raymo F. M., Stoddart J. F., Angew. Chem. Int. Ed. 2000, 39, 3348–3391; [DOI] [PubMed] [Google Scholar]
- 15b. Erbas-Cakmak S., Leigh D. A., McTernan C. T., Nussbaumer A. L., Chem. Rev. 2015. , 115, 10081–10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feynman R. P., Eng. Sci. 1960, 23, 22–36. [Google Scholar]
- 17.
- 17a. Kassem S., van Leeuwen T., Lubbe A. S., Wilson M. R., Feringa B. L., Leigh D. A., Chem. Soc. Rev. 2017, 46, 2592–2621; [DOI] [PubMed] [Google Scholar]
- 17b. Pezzato C., Cheng C., Stoddart J. F., Astumian R. D., Chem. Soc. Rev. 2017, 46, 5491–5507; [DOI] [PubMed] [Google Scholar]
- 17c. Baroncini M., Credi A., Science 2017, 356, 906–907. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Sauvage J.-P., Angew. Chem. Int. Ed. 2017, 56, 11080–11093; [DOI] [PubMed] [Google Scholar]
- 18b. Stoddart J. F., Angew. Chem. Int. Ed. 2017, 56, 11094–11125; [DOI] [PubMed] [Google Scholar]
- 18c. Feringa B. L., Angew. Chem. Int. Ed. 2017, 56, 11060–11078. [DOI] [PubMed] [Google Scholar]
- 19. Kay E. R., Leigh D. A., Angew. Chem. Int. Ed. 2015, 54, 10080–10088; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10218–10226. [Google Scholar]
- 20.
- 20a. Stimulating Concepts in Chemistry (Eds. F. Vögtle, J. F. Stoddart, M. Shibasaki), Wiley-VCH, Weinheim, 2000; [Google Scholar]
- 20b. Balzani V., Credi A., Venturi M., Chem. Eur. J. 2008, 14, 26–39. [DOI] [PubMed] [Google Scholar]
- 21. Schliwa M., Molecular Motors, Wiley-VCH, Weinheim, 2003. [Google Scholar]
- 22. Goodsell D. S., Bionanotechnology: Lessons from Nature Wiley-Liss, Hoboken, NJ, 2004. [Google Scholar]
- 23. Astumian R. D., Chem. Sci. 2017, 8, 840–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones R. A. L., Soft Machines—Nanotechnology and Life, Oxford University Press, Oxford, 2004. [Google Scholar]
- 25.
- 25a. Astumian R. D., Hänggi P., Phys. Today 2002, 55, 33–39; [Google Scholar]
- 25b. Astumian R. D., Phys. Chem. Chem. Phys. 2007, 9, 5067–5083. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Badjic J. D., Balzani V., Credi A., Silvi S., Stoddart J. F., Science 2004, 303, 1845–1849; [DOI] [PubMed] [Google Scholar]
- 26b. Badjic J. D., Ronconi C. M., Stoddart J. F., Balzani V., Silvi S., Credi A., J. Am. Chem. Soc. 2006, 128, 1489–1499. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Berrocal J. A., Biagini C., Mandolini L., Di Stefano S., Angew. Chem. Int. Ed. 2016, 55, 6997–7001; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7111–7115; [Google Scholar]
- 27b. Biagini C., Albano S., Caruso R., Mandolini L., Berrocal J. A., Di Stefano S., Chem. Sci. 2018, 9, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cesario M., Dietrich-Buchecker C. O., Edel A., Guilhem J., Kintzinger J.-P., Pascard C., Sauvage J.-P., J. Am. Chem. Soc. 1986, 108, 6250–6254. [Google Scholar]
- 29. Erbas-Cakmak S., Fielden S. D. P., Karaca U., Leigh D. A., McTernan C. T., Tetlow D. J., Wilson M. R., Science 2017, 358, 340–343. [DOI] [PubMed] [Google Scholar]
- 30. Wilson M. R., Solà J., Carlone A., Goldup S. M., Lebrasseur N., Leigh D. A., Nature 2016, 534, 235–240. [DOI] [PubMed] [Google Scholar]
- 31. Cheng C., McGonigal P. R., Schneebeli S. T., Li H., Vermeulen N. A., Ke C., Stoddart J. F., Nat. Nanotechnol. 2015, 10, 547–553. [DOI] [PubMed] [Google Scholar]
- 32. Cheng C., Stoddart J. F., ChemPhysChem 2016, 17, 1780–1793. [DOI] [PubMed] [Google Scholar]
- 33. Balzani V., Credi A., Venturi M., ChemSusChem 2008, 1, 26–58. [DOI] [PubMed] [Google Scholar]
- 34. Credi A., Silvi S., Venturi M., in Supramolecular Chemistry: From Molecules to Nanomaterials (Eds.: P. A. Gale, J. W. Steed), Wiley, Chichester, 2012, pp. 3719–3750. [Google Scholar]
- 35. Silvi S., Venturi M., Credi A., Chem. Commun. 2011, 47, 2483–2489. [DOI] [PubMed] [Google Scholar]
- 36. Astumian R. D., Faraday Discuss. 2016, 195, 583–579. [DOI] [PubMed] [Google Scholar]
- 37. Credi A., Venturi M., Balzani V., ChemPhysChem 2010, 11, 3398–3403. [DOI] [PubMed] [Google Scholar]
- 38. Balzani V., Clemente-Leon M., Credi A., Ferrer B., Venturi M., Flood A. H., Stoddart J. F., Proc. Natl. Acad. Sci. USA 2006, 103, 1178–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ragazzon G., Baroncini M., Silvi S., Venturi M., Credi A., Nat. Nanotechnol. 2015, 10, 70–75. [DOI] [PubMed] [Google Scholar]
- 40. Sevick E., Nat. Nanotechnol. 2015, 10, 18–19. [DOI] [PubMed] [Google Scholar]
- 41.For a recent review, see: Ragazzon G., Baroncini M., Ceroni P., Credi A., Venturi M., in Comprehensive Supramolecular Chemistry II, (Ed.: J. L. Atwood), Vol. 2, Elsevier, Oxford, 2017, pp. 343–368. [Google Scholar]
- 42.
- 42a. Vives G., Tour J. M., Acc. Chem. Res. 2009, 42, 473–487; [DOI] [PubMed] [Google Scholar]
- 42b. Hess H., Annu. Rev. Biomed. Eng. 2011, 13, 429–450; [DOI] [PubMed] [Google Scholar]
- 42c. Joachim C., Rapenne G., ACS Nano 2013, 7, 11–14; [DOI] [PubMed] [Google Scholar]
- 42d. García-López V., Chiang P.-T., Chen F., Ruan G., Martí A. A., Kolomeisky A. B., Wang G., Tour J. M., Nano Lett. 2015, 15, 8229–8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kudernac T., Ruangsupapichat N., Parschau M., Maciá B., Katsonis N., Harutyunyan S. R., Ernst K.-H., Feringa B. L., Nature 2011, 479, 208–211. [DOI] [PubMed] [Google Scholar]
- 44. Peplow M., Nature 2015, 525, 18–21. [DOI] [PubMed] [Google Scholar]
- 45.
- 45a. Collier C. P., Mattersteig G., Wong E. W., Luo Y., Beverly K., Sampaio J., Raymo F. M., Stoddart J. F., Heath J. R., Science 2000, 289, 1172–1175; [DOI] [PubMed] [Google Scholar]
- 45b. Luo Y., Collier C. P., Jeppesen J. O., Nielsen K. A., Delonno E., Ho G., Perkins J., Tseng H.-R., Yamamoto T., Stoddart J. F., Heath J. R., ChemPhysChem 2002, 3, 519–525. [DOI] [PubMed] [Google Scholar]
- 46. Choi J. W., Flood A. H., Steuerman D. W., Nygaard S., Braunschweig A. B., Moonen N. N. P., Laursen B. W., Luo Y., DeIonno E., Peters A. J., Jeppesen J. O., Xu K., Stoddart J. F., Heath J. R., Chem. Eur. J. 2006, 12, 261–279. [DOI] [PubMed] [Google Scholar]
- 47. Green J. E., Choi J. W., Boukai A., Bunimovich Y., Johnston-Halperin E., DeIonno E., Luo Y., Sheriff B. A., Xu K., Shin Y. S., Tseng H.-R., Stoddart J. F., Heath J. R., Nature 2007, 445, 414–417. [DOI] [PubMed] [Google Scholar]
- 48. Blanco V., Leigh D. A., Marcos V., Chem. Soc. Rev. 2015, 44, 5341–5370. [DOI] [PubMed] [Google Scholar]
- 49. Lewandowski B., De Bo G., Ward J. W., Papmeyer M., Kuschel S., Aldegunde M. J., Gramlich P. M. E., Heckmann D., Goldup S. M., D′Souza D. M., Fernandes A. E., Leigh D. A., Science 2013, 339, 189–193. [DOI] [PubMed] [Google Scholar]
- 50. Kassem S., Lee A. T. L., Leigh D. A., Marcos V., Palmer L. I., Pisano S., Nature 2017, 549, 374–378. [DOI] [PubMed] [Google Scholar]
- 51.
- 51a. Du G., Moulin E., Jouault N., Buhler E., Giuseppone N., Angew. Chem. Int. Ed. 2012, 51, 12504–12508; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12672–12676; [Google Scholar]
- 51b. Goujon A., Du G., Moulin E., Fuks G., Maaloum M., Buhler E., Giuseppone N., Angew. Chem. Int. Ed. 2016, 55, 703–707; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 713–717; [Google Scholar]
- 51c. Goujon A., Lang T., Mariani G., Moulin E., Fuks G., Raya J., Buhler E., Giuseppone N., J. Am. Chem. Soc. 2017, 139, 14825–14828. [DOI] [PubMed] [Google Scholar]
- 52.
- 52a. Li Q., Fuks G., Moulin E., Maaloum M., Rawiso M., Kulic I., Foy J. T., Giuseppone N., Nat. Nanotechnol. 2015, 10, 161–165; [DOI] [PubMed] [Google Scholar]
- 52b. Foy J. T., Li Q., Goujon A., Colard-Itté J.-R., Fuks G., Moulin E., Schiffmann O., Dattler D., Funeriu D. P., Giuseppone N., Nat. Nanotechnol. 2017, 12, 540–545. [DOI] [PubMed] [Google Scholar]
- 53.
- 53a. Jiménez M. C., Dietrich-Buchecker C., Sauvage J.-P., Angew. Chem. Int. Ed. 2000, 39, 3284–3287; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 3422–3425; [Google Scholar]
- 53b. Dawson R. E., Lincoln S. F., Easton C. J., Chem. Commun. 2008, 3980–3982; [DOI] [PubMed] [Google Scholar]
- 53c. Bruns C. J., Stoddart J. F., Acc. Chem. Res. 2014, 47, 2186–2199. [DOI] [PubMed] [Google Scholar]
- 54. Coutrot F., Romuald C., Busseron E., Org. Lett. 2008, 10, 3741–3744. [DOI] [PubMed] [Google Scholar]
- 55.
- 55a. Fang L., Hmadeh M., Wu J., Olson M. A., Spruell J. M., Trabolsi A., Yang Y. W., Elhabiri M., Albrecht-Gary A. M., Stoddart J. F., J. Am. Chem. Soc. 2009, 131, 7126–7134; [DOI] [PubMed] [Google Scholar]
- 55b. Clark P. G., Day M. W., Grubbs R. H., J. Am. Chem. Soc. 2009, 131, 13631–13633. [DOI] [PubMed] [Google Scholar]
- 56. Iwaso K., Takashima Y., Harada A., Nat. Chem. 2016, 8, 625–632. [DOI] [PubMed] [Google Scholar]
- 57. Chang J.-C., Tseng S.-H., Lai C.-C., Liu Y.-H., Peng S.-M., Chiu S.-H., Nat. Chem. 2017, 9, 128–134. [Google Scholar]
- 58. Li Z., Barnes J. C., Bosoy A., Stoddart J. F., Zink J. I., Chem. Soc. Rev. 2012, 41, 2590–2605. [DOI] [PubMed] [Google Scholar]
- 59. Nguyen T. D., Tseng H.-R., Celestre P. C., Flood A. H., Liu Y., Stoddart J. F., Zink J. I., Proc. Natl. Acad. Sci. USA 2005, 102, 10029–10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ambrogio M. W., Thomas C. R., Zhao Y.-L., Zink J. I., Stoddart J. F., Acc. Chem. Res. 2011, 44, 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]