

Abstract
Arrhythmogenic cardiomyopathy (AC) is an inherited disorder that is predominantly present in the right ventricular myocardium. Mutations in the genes encoding the desmosomal protein are thought to underlie the pathogenesis of AC. Since AC is genetically heterogeneous and phenotypically diverse, modifier genes and environmental factors have an important role in disease expression. The aim of the present study was to identify AC-associated desmosomal gene variations, and examine the expression levels of intercalated disc proteins in AC patients who carry the variations (DSG2 p.Leu797Gln, PKP2 p.Ser249Thr and p.E808fsX30). The results of the present investigation provided information on the search for modifier genes and desmosomal gene mutations, and improved our understanding of the mechanism underlying these AC mutations. Genetic screening of five desmosomal genes (DSG2, DSC2, JUP, PKP2, and DSP) in 23 patients with AC who underwent heart transplantation was performed and the expression levels and localizations of intercalated disc proteins were assessed using western blotting and immunohistochemistry, respectively. The results enabled the identification of three desmosomal gene variations (DSG2 L797Q, PKP2 S249T, and E808fsX30), two of which are reported for the first time. DSG2 L797Q was identified in one patient. The protein expression levels of DSG2 in the L797Q carrier were unchanged compared with the healthy controls, and the expression levels of the other proteins (JUP and Cx43) in the intercalated disc were also similar between the healthy controls, the variation carrier and the case controls. Two variations (S249T and E808fsX30) in PKP2 were identified in one patient, the protein expression levels of PKP2 in this patient were significantly decreased, and the expression levels of the other proteins in the intercalated disc was also decreased. The data suggest that there may be modifier genes and other AC-associated mutations requiring identification, in order to further our understanding of the disease mechanism induced by these mutations.
Keywords: arrhythmogenic cardiomyopathy, gene variation, desmosomal protein, intercalated disc, protein expression
Introduction
Arrhythmogenic cardiomyopathy (AC) is a clinically and genetically heterogeneous heart muscle disease estimated to affect ~1/5,000 individuals (1). The pathological hallmark of AC consists of progressive myocardial atrophy followed by fibro-fatty replacement (2). Palpitations, syncope, ventricular tachycardia, right heart failure and even sudden cardiac death (SCD) are typical clinical manifestations of AC (2).
In recent years, great advances have been made in the understanding of the pathogenesis of AC (3). Mutations in genes that encode desmosomal proteins (desmoglein-2, desmocollin-2, plakoglobin, plakophilin and desmoplakin) are thought to cause AC. Approximately 50% of patients with AC have one or more mutations in their desmosomal genes; thus, AC is referred to as ‘a disease of the desmosome’ (4). In addition to desmosomal genes, mutations in non-desmosomal genes such as TGFβ3, TMEM43, RYR2, DES, and TTN are also involved in AC (5–9).
Mutations in disease-causing genes have been identified in AC patients with a variable frequency (10,11), and there is limited knowledge regarding the molecular disease mechanisms leading to the condition in individuals with pathogenic gene mutations. Since it is difficult to obtain myocardial tissue for protein studies, the majority of previous studies investigated the molecular disease mechanisms by culturing the keratinocytes of patients with AC (12,13). These studies revealed two disease mechanisms induced by the pathogenic mutations: Haploinsufficiency and dominant-negative effects.
In the present study, a systematic sequence analysis was performed for five selected desmosomal genes obtained from patients with AC who underwent heart transplantation. The aim of the study was to identify novel AC-associated variations or known mutations of the five desmosomal genes and to observe their effects on desmosomal protein expression, in order to further our understanding of the mechanism underlying the effects of desmosomal gene mutations on AC pathogenesis.
Materials and methods
Study patients
Patients enrolled in the present study included 23 individuals, 70% of whom were male, aged 41.4±13.8 years (range, 18–68 years) with AC who underwent heart transplantation at Fuwai Hospital (Beijing, China) between 2005 and 2012. Subjects were diagnosed from the pathological results of a biopsy. All patients gave written informed consent for this investigation, which was approved by the Ethical Committee of Fuwai Hospital. Routine medical history, physical examinations and laboratory tests were collected.
Tissue samples
Cardiac tissue samples were collected from the patients with AC who underwent heart transplantation. Healthy control hearts were obtained from autopsies or deceased donors with no history of heart disease.
Electrocardiography
Twelve-lead electrocardiogram was carried out during echocardiographic examination. Data analyses (Epsilon wave, T wave inversion and ventricular tachycardia with left bundle branch block) were performed.
Echocardiographic studies
Two-dimensional echocardiographic studies were performed and data were analyzed with EchoPAC version 112 (GE Healthcare, Chicago, IL, USA). From 2-dimensional echocardiography, we assessed proximal right ventricular outflow tract (RVOT) diameter in the parasternal short-axis view and right ventricular basal diameter (RV diameter). Ventricular wall structure was assessed through RVOT, RV diameter, left ventricular (LV) end-diastolic volume, LV end-systolic volume, and left ventricular ejection fraction.
Genetic analysis
Genomic DNA from the cohort of 23 patients with AC was extracted from the myocardial tissue samples using a QIAamp DNA Mini kit (Qiagen China Co., Ltd., Shanghai, China). The DNA sequence of these patients was screened by target capture sequencing, then verified by Sanger sequencing (14,15). All coding exons of the desmosomal genes (DSG2, NM_001943.3, DSC2, NM_024422.3, JUP, NM_021991.2, PKP2, NM_004572.3 and DSP, NM_004415.2) were enriched using custom-made SureSelectXT Target Enrichment arrays (Agilent Technologies, Inc., Santa Clara, CA, USA). HiSeq 2000 was used to perform next-generation sequencing as previously described (16).
Common polymorphisms were excluded based on their presence in the 1,000 genome databases (ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/), yhSNP (http://yh.genomics.org) and Hapmap (http://www.ncbi.nlm.nih.gov/snp/). Variants were considered to be a possible AC-associated variation if they exhibited the following (3): i) Absence from the 1,000 genome database; ii) the variation was expected to have a drastic effect on the protein (nonsense mutation, frame shift mutation or the mutated residue was highly conserved among species); iii) the variation was predicted to be damaging by polymorphism phenotyping and sorting intolerant from tolerant.
Sanger sequencing
Sanger sequencing were performed according to method described previously (17). A 600 base pair fragment containing the candidate variant was polymerase chain reaction-amplified from genomic DNA of the index case. Base sequences of the primer pairs and annealing temperature are listed in Table I. Each amplicon was bi-directionally sequenced, followed by electrophoresis on an ABI 3730XL Genetic Analyzer (Perkin-Elmer; Applied Biosystems Inc., Foster City, CA, USA).
Table I.
Base sequence of the primer pairs.
| Location | Forward primer (5′-3′) | Reverse primer (5′-3′) | TM (°C) |
|---|---|---|---|
| DSG2 | |||
| c.2390T>A | CAGTTGTGTCGTAGGATCTC | ACTGCCAGAGGAGTAGGTAT | 55.4 |
| c.593A>G | TCTAATGCCAGATACTCTTGTG | GAGAGTTTAAAGATGGGCAAC | 57.0 |
| PKP2 | |||
| c.746G>C | AAGTGCCAGCTCATGCTGTC | CTACACGCACAGCGATTACC | 57.4 |
| c.2422delC | TAGCGATTTCTTCCCAGGGTC | AGCAGTTGAGGAGCGAAGAG | 57.5 |
DSG2, desmoglein 2; PKP2, plakophilin 2; TM, temperature.
Immunohistochemistry
When available, dependent on the patients willingness to donate cardiac tissue, the formalin-fixed cardiac tissue samples obtained from the patients with AC were examined for the distribution of intercellular junction proteins. Antigens were exposed in the paraffin-embedded tissue sections using high pressure antigen-recovery techniques. Following washing three times with PBS and blocking with goat serum at room temperature for 20 min, the tissue sections were subsequently treated with commercially-available rabbit antibodies targeting connexin-43 (Cx43; 3512; 1:100; Cell Signaling Technology, Inc., Danvers, MA, USA), N-cadherins (ab18203; 1:1,000; Abcam, Cambridge, UK), junction plakoglobin (JUP; 2309; 1:50; Cell Signaling Technology, Inc.), plakophilin 2 (PKP2; ab151402; 1:200; Abcam) and desmoglein 2 (DSG2; ab85632; 1:200; Abcam) overnight at 4°C. Following washing with PBS three times, tissue sections were treated with anti-rabbit (pv6001) and anti-mouse (pv6002; both Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China) secondary antibodies and visualized with 3,3′-diaminobenzidine (ZLI-9017; Zhongshan Golden Bridge Biotechnology Co., Ltd.) via light microscopy (DMI-600B; Leica Microsystems, Inc., Buffalo Grove, IL, USA). In addition, myocardial tissue sections obtained from two age-matched controls with no clinical or autopsy evidence of heart disease were also examined by immunohistochemistry. Only two age-matched controls were used to the valuable nature of the tissues. Although there were only two controls, repeated independent experiments were performed to ensure the reliability of the data.
Western blotting
Snap frozen tissue samples (80 µg) were homogenized with a buffer (8 M Urea, 50 mM Tris-HCL, 100 mM NaCl, 1 mM DTT, 1% Triton-X-100, pH 7.5) supplemented with protease inhibitors (1:100) as previously described (12). Following centrifugation for 15 min at 4°C (3,000 × g), the supernatant was extracted and the protein concentration was determined via bicinchoninic acid assay. Protein (30 µg) was loaded onto 4–12% NuPAGE Bis-Tris Protein Gels (NP0321BOX; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and electrotransferred to a polyvinylidene difluoride membrane. The membrane was subsequently blocked by 5% bovine serum albumin for 1 h at room temperature. Assessment of the cytoskeletal proteins was performed using the following primary antibodies: Rabbit anti-DSG2 (ab85632; 1:1,000), anti-JUP (2309; 1:50), anti-Cx43 (3512; 1:100) and mouse anti-PKP2 (ab151402; 1:200), and the loading control was β-tubulin (2146; 1:1,000; Cell Signaling Technology, Inc.) was used as the internal reference. The membrane was incubated for 2 h at room temperature and was subsequently washed three times (5 min each) with 10 ml TBST. Following this, the membrane was incubated with horseradish peroxidase-conjugated anti-rabbit (A0208; 1:1,000) and anti-mouse secondary antibodies (A0216; 1:1,000; both Beyotime Institute of Biotechnology, Beijing, China). Following washing three times with TBST, the blots were visualized with a chemiluminescence imaging instrument (ProteinSimple, Santa, Clara, CA, USA).
Statistical analyses
Each independent experiment was repeated at least three times, and at least three independent experiments were performed. Expression levels were quantified using ImageJ software (V2.1.4.7; National Institutes of Health, Bethesda, MA, USA). Data were expressed as the mean ± standard deviation. GraphPad Prism software (version 5; GraphPad Software, Inc., La Jolla, CA, USA) was used for the statistical analyses. P<0.05 was considered to indicate a statistically significant difference.
Results
Genetic analysis
Six variations (four in DSG2, two in PKP2) were identified in the desmosome genes (Table II). Two of the variations were single nucleotide polymorphisms, and one of the variations was not verified by Sanger sequencing; thus, it was a false positive from next generation sequencing. The three variations, DSG2 L797Q, PKP2 S249T and E808fsX30 with possible functional alteration were absent in the 1,000 genome database. Their position was highly conserved and function prediction indicated that the variations were damaging. Therefore, the three variants were considered to be unique to the patients with AC and determined to be AC-associated variants (Table III). The DNA sequencing results of the three variations are shown in Fig. 1.
Table II.
Variations in the desmosome genes of the 23 patients with AC.
| Gene | Variation type | Nucleotide change | Amino acid change | PolyPhena | SIFTb |
|---|---|---|---|---|---|
| DSG2 | SNP | c.618T>A | p.Ala206Ala | − | 0.60 |
| DSG2 | Missense | c.2390T>A | p.Leu797Gln | 0.998 (++) | 0.95 |
| DSG2 | Missense | c.593A>G | p.Tyr198Cys | 0.995 | 0.95 |
| DSG2 | SNP | c.161C>T | p.Ala54Val | Benign | 0.55 |
| PKP2 | Missense | c.746G>C | p.Ser249Thr | 0.97 (++) | 1 |
| PKP2 | Frameshift | c.2422delC | p.Glu808fsX30 |
PolyPhen prediction: ++, likely damaging; +, possibly damaging; -, benign
SIFT prediction: Amino acids with scores >0.95 are predicted to be deleterious. PolyPhen, polymorphism phenotyping; SIFT, sorting intolerant from tolerant; SNP, single nucleotide polymorphism; DSG2, desmoglein 2; PKP2, plakophilin 2.
Table III.
Potential AC-associated variations identified in the 23 patients.
| Gene | Type of variation | Nucleotide change | Amino acid change | Patient index no. | PolyPhena | SIFTb |
|---|---|---|---|---|---|---|
| DSG2 | Missense | c.2390T>A | p.Leu797Gln | 1 | 0.998 (++) | 0.95 |
| PKP2 | Missense | c.746G>C | p.Ser249Thr | 1 | 0.97 (++) | 1 |
| PKP2 | Frameshift | c.2422delC | p.Glu808fsX30 | 1 | N/Ac | N/Ac |
PolyPhen prediction: ++, likely damaging; +, possibly damaging
SIFT prediction: Amino acids with scores >0.95 are predicted to be deleterious.
PolyPhen and SIFT predict SNP and point mutations, not nonsense mutations and frame shift mutations. PolyPhen, polymorphism phenotyping; SIFT, sorting intolerant from tolerant; DSG2, desmoglein 2; PKP2, plakophilin 2; N/A, not applicable.
Figure 1.
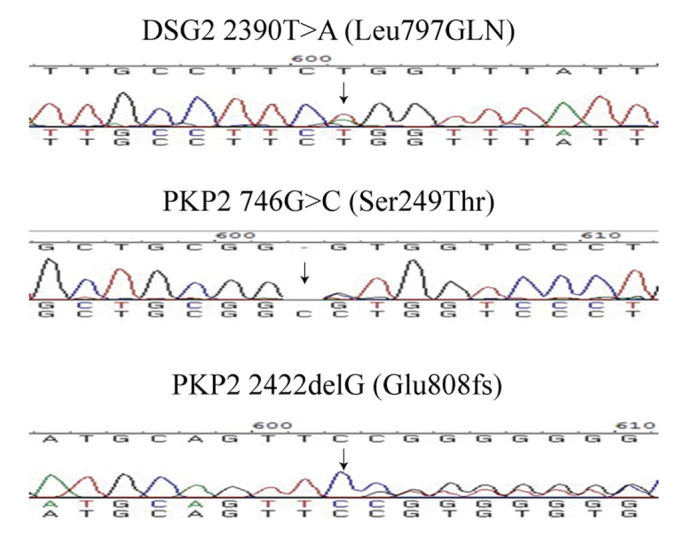
Sequencing results of the three desmosomal gene mutations identified. PKP2, plakophilin 2.
Pathological and clinical characteristics
Among the 23 patients diagnosed with AC who received heart transplantation, two patients carried the three desmosomal gene variations. The clinical characteristics of the patients are summarized in Table IV. The first symptoms of the two patients were dyspnea and arrhythmia, respectively. Patient 1 had a family history of SCD, with the patient's mother having suffered from SCD at 52 years of age, and also had atrial fibrillation and regional RV dyskinesia. Patient 2 also had a family history of SCD (the patient's brother suffered SCD when 14 years old), along with ventricular tachycardia. The two patients were both in end-stage heart failure (NYHA III–IV). The pathological characteristic of the right ventricular myocardium in the two patients is shown in Fig. 2. Compared with the control, the number of myocardiocytes in the myocardium of the patients with AC had markedly decreased, and numerous fibro-fatty had infiltrated into the myocardium.
Table IV.
Clinical characteristics of the two patients with arrhythmogenic cardiomyopathy-associated variations.
| No. | Age (yr)/sex | Presentation | FH | Epsilon wave | T wave inversion | VT with LBBB | IMG |
|---|---|---|---|---|---|---|---|
| 1 | 38/M | Dyspnea | + | − | − | − | + |
| 2 | 30/M | Arrhythmia | + | − | − | + | + |
−, benign; ++, likely damaging; +, possibly damaging; FH, family history; VT, ventricular tachycardia; LBBB, left bundle branch block; IMG, imaging by echocardiography.
Figure 2.
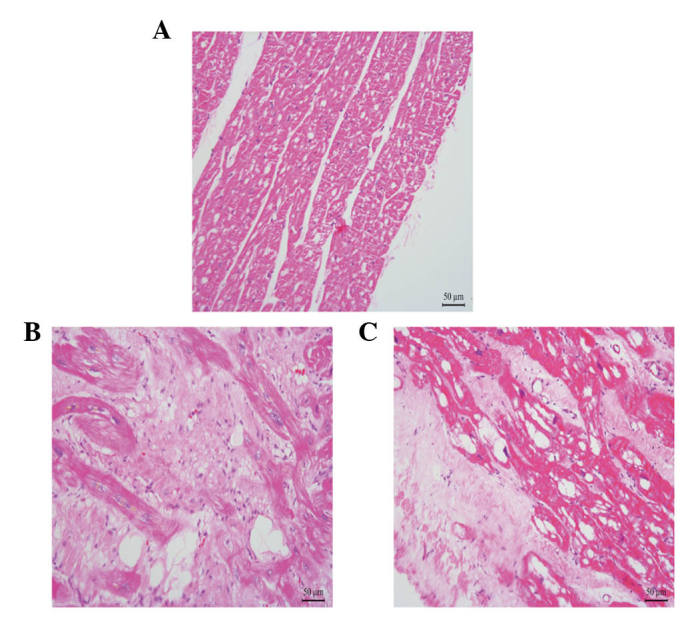
Representative pathological features of the right ventricular myocardium from patients with arrhythmogenic cardiomyopathy. (A) Right ventricular myocardium of the healthy control; (B) right ventricular myocardium of patient 1; (C) right ventricular myocardium of patient 2. Magnification, ×400. Scale bar, 50 µm.
Change in location of the intercalated disc proteins in the two patients
Myocardial samples were obtained from the two individuals. These myocardial tissue samples were sectioned and stained for various junctional proteins, including N-cadherin, DSG2, PKP2, and JUP, as well as Cx43.
The primary component of the adherens junction within the intercalated disc, which was N-cadherin, was present and comparable with the controls in all samples (Fig. 3). In the myocardial tissue of patient 1 (Fig. 3A), who carried DSG2 L797Q, the staining of DSG2 produced no change compared with the control. Staining of samples from this patient was comparable to the controls for other antibodies, including Cx43 and JUP.
Figure 3.

Immunohistochemistry of the right ventricular myocardium from desmosomal gene variation carriers. (A) Immunostaining of intercalated disc protein in patients with DSG2 L797Q (magnification, ×400); (B) immunostaining of intercalated disc protein in patients with PKP2 S249T and G808fsX30 (magnification, ×400). Scale bar, 50 µm. DSG2, desmoglein 2; N-Cad, N-cadherin; JUP, junction plakoglobin; Cx43, connexin 43; PKP2, plakophilin 2.
Immunohistochemical staining performed in the myocardial tissue of patient 2 (Fig. 3B), who carried the PKP2 point mutation and deletion in exon 12 demonstrated that the positive expression of PKP2 is reduced compared with the control. The other intercalated disc proteins, JUP and Cx43, also showed a marked reduction in positive expression at the intercalated disc.
Change in expression levels of intercalated disc protein in the two patients
Immunoblotting was performed on the right ventricle to identify potential AC-specific cardiac protein expression patterns.
As shown in Fig. 4A, the total expression levels of DSG2 in the right ventricle remained unchanged in the heterozygous patient, compared with the healthy controls and case controls without this variation. The expression levels of the other proteins in the intercalated disc (JUP, Cx43) were similar in the myocardium of the three groups. In the carrier with the PKP2 variation, the expression levels of PKP2 were markedly decreased compared with the healthy controls and non-carriers. Notably, the expression levels of JUP and Cx43 were also decreased in patient 2 compared with the healthy controls and case controls.
Figure 4.

Representative western blotting of intercalated disc proteins and β-tubulin in the right ventricular myocardium from patients with AC and healthy controls. (A) Western blotting of myocardial intercalated disc proteins in the controls, and AC patients with and without DSG2 variation, along with the quantification of the protein expression; (B) western blotting of myocardial intercalated disc proteins in the controls, and AC patients with and without PKP2 variation, along with the quantification of the protein expression. DSG2, desmoglein 2; JUP, junction plakoglobin; Cx43, connexin 43; PKP2, plakophilin 2.
Discussion
AC is an inheritable structural heart disease first described by Frank et al (18) in 1978. Since 50% of the diagnosed patients carried the desmosome gene mutation (19), AC became known as a ‘desmosome disease’. To date, 12 genes have been identified as AC-causing genes (20), and ~300 genetic variants of these genes have been classified as damaging (21). The majority of pathogenic mutations have been identified in genes encoding desmosomal proteins, with the PKP2 gene being responsible for ~35–40% of cases (3,22). Mutations in the genes DSP, DSG2, and DSC2 are responsible for ~15–20% of AC cases (20). Thus, genetic counseling is important to identify patients who are at high risk of the disease.
The discovery of novel genes has been limited by the reduced penetrance and variable expressivity associated with AC (23). A previous study reported that in individuals with a desmosomal mutation, only ~30–50% will fulfill the diagnostic criteria for AC (24). Modifier genes are hypothesized to account for much of the variation between individuals, even within the same family. In addition, numerous environmental factors have a role in disease expression, including age, gender, strenuous exercise, drugs, hormones, infection/inflammation and emotional stress (25).
In the present study, we identified three variations and amongst these variations, a heterozygous variant of DSG2, L797Q, was newly found. In the myocardium of patients with the L797Q variant, the protein expression levels of DSG2 are similar in both the variation carrier and the controls. The expression levels of other proteins in intercalated discs remained unchanged. These results, combined with the results of the location of DSG2 observed in the myocardial tissue, indicated that the intracellular localization and expression level of proteins in intercalated disc were normal. Therefore, we speculated that this variant may be non-pathogenic, fir example it may be a disease modifier gene, and other pathogenic genes remain unrecognized. Furthermore, these findings indicated that even this position is a highly conserved intracellular anchor domain, and the predicted functions indicated that the variation is damaging. It is unreasonable to consider it as a disease-causing mutation.
Since family members with identical genotypes and even monozygotic twins show significant differences in symptoms, presence and distribution of structural changes, and rate of disease progression, modifier genes are likely responsible for phenotypic heterogeneity (26). Therefore, detection of modifier genes is helpful to further understand the disease mechanism underlying AC.
Two variants pertaining to PKP2, S249T and E808fsX30 were also observed, which were all carried by one patient.
PKP2 is an important desmosomal protein. Dominant mutations in PKP2 were reported in ~25–50% of patients with AC, suggesting that PKP2 is the major disease gene (27). The mutation rates of PKP2 in Chinese subjects were reported to be 18–39% (28,29) and 58% (30). Unexpectedly, in the present study, the mutation rate of PKP2 was observed to be 4%, which may be due to the small sample size.
The PKP2 S249T variant has been previously identified (17,30). Variants were considered to be a possible AC-related variation if they were: i) Absent in 1000 genome databases; ii) a drastic effect was expected in the protein (nonsense mutation, frame shift mutation, or the mutated residue was highly conserved among species); or iii) predicted to be damaging by PolyPhen. As SIFT.PKP2 S249T fuifilled the above-mentioned conditions via a change in amino acid sequence, its location in a highly conserved domain and the prediction of its mutational function, this variant is considered to be an AC-associated mutation.
There is a 10% chance of missense mutation of PKP2, resulting in truncated proteins (20,27). In the present study, the heterozygous point mutation S249T and frameshift mutation E808fsX30 were demonstrated to be associated with decreased PKP2 protein expression. Although E808fsX30 resulted in a truncated PKP2 protein, the protein expression levels decreased >50%. Therefore, the two mutations may result in protein instability and degradation, similarly to other PKP2 mutations (Q59L, Q62K, R79X, C796R), which have exhibit a similar mode of action (31–33). In addition to the decreased expression levels of PKP2, the expression levels of JUP and Cx43 also decreased in the mutation carrier. This suggests that since PKP2, JUP, and Cx43 are structural proteins in the intercalated disc, the degradation of PKP2 could influence the expression of JUP and Cx43. The possible mechanism underlying this process requires further investigation.
A previous study reported that loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes (34); thus, decreased sodium current due to the loss of PKP2 expression may result in arrhythmia. Future experiments will be necessary to address this possibility.
Further research in to the disease-associated mutations and protein expression pattern associated with desmosomal gene mutations will provide a better understanding of the molecular mechanisms underlying AC. To date, the effects of desmosomal gene mutations have most commonly been studied in artificial expression systems using transfected cell cultures and transgenic mice that overexpress the mutant proteins (12). The present study examined both the location and expression levels of these proteins in the myocardial tissue of patients with mutations, and revealed the potential molecular mechanism underlying these mutations in AC.
As the most common variants, desmosomal gene mutations are closely related to several clinical characteristics, including T wave inversion, epsilon wave, age at AC onset and ventricular arrhythmias. A definite genotype-phenotype relationship in patients with AC caused by desmosomal gene mutations is helpful in the diagnosis of AC. Although considerable progress has been made in the research of genotype-phenotype relationship in patients with AC, numerous challenges remain due to the genetic and clinical heterogeneity in these patients. The current study did not include genotype-phenotype correlation analysis due to the small sample size; as a result, it could not describe the genetic background of the AC index of patients who received heart transplantation comprehensively. In addition, the use of in vitro functional tests to reveal the pathogenic mechanism underlying these mutations is required.
This report described variations of five desmosomal genes in the AC index of patients who underwent heart transplantation. The results enabled the identification of three potential AC-associated gene variations, including DSG2 L797Q, PKP2 S249T, and E808fsX30; two of them [PKP2 S249T (Ser249Thr) and E808fsX30 (Glu808fsX30)] may reduce protein stability and promote degradation. Conversely, DSG2 L797Q exhibited modifying effects in AC. The data uncovered one desmosomal gene variation in AC which has modifying effect and two potential AC-related mutations, this provides clues for early diagnosis of AC patients in the future. Furthermore, we detected the expression pattern of related protein in intercalated disc and provided potential disease mechanism of the mutations.
Acknowledgements
The authors of the present study are grateful to the patients and their families for their support and collaboration, to Dr Zhigang Wang for her advice on data analyses, and to Dr Bi Huang for his valuable suggestions in the preparation of this manuscript. The present study was supported by research grants from the CAMS Innovation Fund for the Medical Sciences (grant no. 2016-I2M-1-015) and the National Natural Science Foundation of China (grant no. 81470424).
Glossary
Abbreviations
- AC
arrhythmogenic cardiomyopathy
- SCD
sudden cardiac death
- DSG2
desmoglein2
- DSC2
desmocollin2
- PKP2
plakophilin2
- DSP
desmoplakin
References
- 1.Awad MM, Calkins H, Judge DP. Mechanisms of disease: Molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5:258–267. doi: 10.1038/ncpcardio1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, et al. Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation. 2005;112:3823–3832. doi: 10.1161/CIRCULATIONAHA.105.542266. [DOI] [PubMed] [Google Scholar]
- 3.Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, Klug D, Dubourg O, Delacretaz E, Cosnay P, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Spectrum of mutations and clinical impact in practice. Europace. 2010;12:861–868. doi: 10.1093/europace/euq104. [DOI] [PubMed] [Google Scholar]
- 4.Saffitz JE. Arrhythmogenic cardiomyopathy and abnormalities of cell-to-cell coupling. Heart Rhythm. 2009;6(8 Suppl):S62–S65. doi: 10.1016/j.hrthm.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 5.Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–373. doi: 10.1016/j.cardiores.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–821. doi: 10.1016/j.ajhg.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (arvd2) Hum Mol Genet. 2001;10:189–194. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 8.van Tintelen JP, Van Gelder IC, Asimaki A, Suurmeijer AJ, Wiesfeld AC, Jongbloed JD, van den Wijngaard A, Kuks JB, van Spaendonck-Zwarts KY, Notermans N, et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the des gene. Heart Rhythm. 2009;6:1574–1583. doi: 10.1016/j.hrthm.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 9.Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 2011;124:876–885. doi: 10.1161/CIRCULATIONAHA.110.005405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauce B, Nava A, Beffagna G, Basso C, Lorenzon A, Smaniotto G, De Bortoli M, Rigato I, Mazzotti E, Steriotis A, et al. Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm. 2010;7:22–29. doi: 10.1016/j.hrthm.2009.09.070. [DOI] [PubMed] [Google Scholar]
- 11.Lorenzon A, Beffagna G, Bauce B, De Bortoli M, Li Mura IE, Calore M, Dazzo E, Basso C, Nava A, Thiene G, Rampazzo A. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;111:400–405. doi: 10.1016/j.amjcard.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasmussen TB, Hansen J, Nissen PH, Palmfeldt J, Dalager S, Jensen UB, Kim WY, Heickendorff L, Mølgaard H, Jensen HK, et al. Protein expression studies of desmoplakin mutations in cardiomyopathy patients reveal different molecular disease mechanisms. Clin Genet. 2013;84:20–30. doi: 10.1111/cge.12056. [DOI] [PubMed] [Google Scholar]
- 13.Rasmussen TB, Palmfeldt J, Nissen PH, Magnoni R, Dalager S, Jensen UB, Kim WY, Heickendorff L, Mølgaard H, Jensen HK, et al. Mutated desmoglein-2 proteins are incorporated into desmosomes and exhibit dominant-negative effects in arrhythmogenic right ventricular cardiomyopathy. Hum Mutat. 2013;34:697–705. doi: 10.1002/humu.22289. [DOI] [PubMed] [Google Scholar]
- 14.Liang WC, Tian X, Yuo CY, Chen WZ, Kan TM, Su YN, Nishino I, Wong LC, Jong YJ. Comprehensive target capture/next-generation sequencing as a second-tier diagnostic approach for congenital muscular dystrophy in Taiwan. PLoS One. 2017;12:e0170517. doi: 10.1371/journal.pone.0170517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang B, Chen Y, Xu B, Hong N, Liu R, Qi M, Shen L. Identification of a novel missense mutation of MIP in a Chinese family with congenital cataracts by target region capture sequencing. Sci Rep. 2017;7:40129. doi: 10.1038/srep40129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyanaga A, Masuda M, Tsuta K, Kawasaki K, Nakamura Y, Sakuma T, Asamura H, Gemma A, Yamada T. Hippo pathway gene mutations in malignant mesothelioma: Revealed by RNA and targeted exon sequencing. J Thorac Oncol. 2015;10:844–851. doi: 10.1097/JTO.0000000000000493. [DOI] [PubMed] [Google Scholar]
- 17.Kinnear C, Glanzmann B, Banda E, Schlechter N, Durrheim G, Neethling A, Nel E, Schoeman M, Johnson G, van Helden PD. Exome sequencing identifies a novel TTC37 mutation in the first reported case of Trichohepatoenteric syndrome (THE-S) in South Africa. BMC Med Genet. 2017;18:26. doi: 10.1186/s12881-017-0388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank R, Fontaine G, Vedel J, Mialet G, Sol C, Guiraudon G, Grosgogeat Y. Electrocardiology of 4 cases of right ventricular dysplasia inducing arrhythmia. Arch Mal Coeur Vaiss. 1978;71:963–972. (In French) [PubMed] [Google Scholar]
- 19.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 20.Campuzano O, Alcalde M, Allegue C, Iglesias A, García-Pavía P, Partemi S, Oliva A, Pascali VL, Berne P, Sarquella-Brugada G, et al. Genetics of arrhythmogenic right ventricular cardiomyopathy. J Med Genet. 2013;50:280–289. doi: 10.1136/jmedgenet-2013-101523. [DOI] [PubMed] [Google Scholar]
- 21.van der Zwaag PA, Jongbloed JD, van den Berg MP, van der Smagt JJ, Jongbloed R, Bikker H, Hofstra RM, van Tintelen JP. A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum Mutat. 2009;30:1278–1283. doi: 10.1002/humu.21064. [DOI] [PubMed] [Google Scholar]
- 22.Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ, Cox MG, Bhuiyan Z, Bikker H, Wiesfeld AC, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol. 2011;57:2317–2327. doi: 10.1016/j.jacc.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murray B. Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C): A review of molecular and clinical literature. J Genet Couns. 2012;21:494–504. doi: 10.1007/s10897-012-9497-7. [DOI] [PubMed] [Google Scholar]
- 24.Towbin JA. Arrhythmogenic right ventricular cardiomyopathy: A paradigm of overlapping disorders. Ann Noninvasive Electrocardiol. 2008;13:325–326. doi: 10.1111/j.1542-474X.2008.00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sen-Chowdhry S, Syrris P, Pantazis A, Quarta G, McKenna WJ, Chambers JC. Mutational heterogeneity, modifier genes and environmental influences contribute to phenotypic diversity of arrhythmogenic cardiomyopathy. Circ Cardiovasc Genet. 2010;3:323–330. doi: 10.1161/CIRCGENETICS.109.935262. [DOI] [PubMed] [Google Scholar]
- 26.Calkins H. Arrhythmogenic right ventricular dysplasia. Curr Probl Cardiol. 2013;38:103–123. doi: 10.1016/j.cpcardiol.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. doi: 10.1038/ng1461. [DOI] [PubMed] [Google Scholar]
- 28.Qiu X, Liu W, Hu D, Zhu T, Li C, Li L, Guo C, Liu X, Wang L, Zheng H, et al. Mutations of plakophilin-2 in Chinese with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Cardiol. 2009;103:1439–1444. doi: 10.1016/j.amjcard.2009.01.356. [DOI] [PubMed] [Google Scholar]
- 29.Wu SL, Wang PN, Hou YS, Zhang XC, Shan ZX, Yu XY, Deng M. Mutation of plakophilin-2 gene in arrhythmogenic right ventricular cardiomyopathy. Chin Med J (Engl) 2009;122:403–407. [PubMed] [Google Scholar]
- 30.Bao J, Wang J, Yao Y, Wang Y, Fan X, Sun K, He DS, Marcus FI, Zhang S, Hui R, et al. Correlation of ventricular arrhythmias with genotype in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013;6:552–556. doi: 10.1161/CIRCGENETICS.113.000122. [DOI] [PubMed] [Google Scholar]
- 31.Joshi-Mukherjee R, Coombs W, Musa H, Oxford E, Taffet S, Delmar M. Characterization of the molecular phenotype of two arrhythmogenic right ventricular cardiomyopathy (arvc)-related plakophilin-2 (pkp2) mutations. Heart Rhythm. 2008;5:1715–1723. doi: 10.1016/j.hrthm.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall C, Li S, Li H, Creason V, Wahl JK., III Arrhythmogenic right ventricular cardiomyopathy plakophilin-2 mutations disrupt desmosome assembly and stability. Cell Commun Adhes. 2009;16:15–27. doi: 10.1080/15419060903009329. [DOI] [PubMed] [Google Scholar]
- 33.Kirchner F, Schuetz A, Boldt LH, Martens K, Dittmar G, Haverkamp W, Thierfelder L, Heinemann U, Gerull B. Molecular insights into arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 missense mutations. Circ Cardiovasc Genet. 2012;5:400–411. doi: 10.1161/CIRCGENETICS.111.961854. [DOI] [PubMed] [Google Scholar]
- 34.Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patino GA, Taffet SM, Isom LL, Delmar M. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105:523–526. doi: 10.1161/CIRCRESAHA.109.201418. [DOI] [PMC free article] [PubMed] [Google Scholar]