

This review by Mohammad and Helin focuses on oncohistones in pediatric cancers. They provide an overview of histone mutations that have been identified and discuss the possible mechanisms by which they contribute to tumor development as well as proposed targeted therapies.
Keywords: cancer, chromatin, oncohistone
Abstract
One of the most striking results in the area of chromatin and cancer in recent years has been the identification of recurrent mutations in histone genes in pediatric cancers. These mutations occur at high frequency and lead to the expression of mutant histones that exhibit oncogenic features. Thus, they are termed oncohistones. Thus far, mutations have been found in the genes encoding histone H3 and its variants. The expression of the oncohistones affects the global chromatin landscape through mechanisms that have just begun to be unraveled. In this review, we provide an overview of histone mutations that have been identified and discuss the possible mechanisms by which they contribute to tumor development. We further discuss the targeted therapies that have been proposed to treat cancers expressing oncohistones.
Pertinent questions
Why are oncohistones restricted to certain types of tumors and, in particular, pediatric tumors?
Why do histone mutations occur in specific genes?
What are the cells of origin for tumors harboring histone mutations?
Are there additional mutations giving rise to oncohistones other than those identified so far?
How do oncohistones affect global chromatin landscape and contribute to tumor development?
Gene expression in eukaryotic cells is a highly regulated process in which the binding of transcription factors and the activity of the basic transcriptional machinery are constrained and facilitated at the level of chromatin (for recent reviews, see Voss and Hager 2013; Venkatesh and Workman 2015; Buschbeck and Hake 2017). Chromatin, which consists of DNA and associated proteins, is organized by nucleosomes. The nucleosome contains DNA wrapped around an octamer containing four heterodimers of the core histone proteins H2A, H2B, H3, and H4 (Fig. 1A). Higher-order chromatin also contains the linker histone H1, which binds to linker DNA between two nucleosomes and stabilizes the more compact chromatin. In addition to “canonical” histones, eukaryotic cells also express histone variants that are incorporated at specific genomic loci and provide another layer of gene regulation.
Figure 1.

Nucleosomes, expression of H3 proteins, and their incorporation into nucleosomes. (A) Schematic diagram showing a nucleosome, which consists of a histone octamer (containing two copies each of H2A, H2B, H3, and H4) and 146 base pairs of DNA wrapped around the octamer. (B) Expression of canonical (H3.1 and H3.2) and variant (H3.3) histone H3 during the cell cycle. H3.1 and H3.2 are expressed only during the S phase of the cell cycle, and their incorporation into the chromatin is coupled with DNA replication, whereas H3.3 is expressed and incorporated throughout the cell cycle independently of DNA replication. (C) Sequence alignment of H3.1, H3.2, and H3.3 proteins. Residues that are not conserved are shown in red, and the residues that are found to be mutated in tumors are highlighted. (D) Differential incorporation of canonical and variant H3 into chromatin. During S phase, H3.1 and H3.2 are deposited into chromatin throughout the chromosome by CAF1 (chromatin-associated factor 1), while H3.3 is incorporated at distinct genomic loci by two dedicated histone chaperons. The ATRX/DAXX complex mediates the assembly of H3.3 at telomeres and pericentric heterochromatin, while HIRA mediates the incorporation of H3.3 at euchromatic regions, including promoters, gene bodies, and regulatory elements.
Canonical histones comprise the majority of histones in any given cell; are synthesized only during S phase, where DNA replication takes place; and are therefore termed “replication-coupled histones.” In contrast, histone variants are expressed throughout the cell cycle and are termed “replication-independent histones” (Fig. 1B). Canonical histones are the most abundant proteins in all nucleated cells and are expressed from multiple genes that are organized in gene clusters mostly located on human chromosomes 1 and 6 (Buschbeck and Hake 2017). Expression of canonical histones from gene clusters ensures the simultaneous expression of all the four core histones to similar levels and the efficient organization of newly replicated DNA into chromatin. Canonical histone genes are intronless and contain a conserved palindromic termination element instead of code for a polyA tail, whereas histone variants often contain introns and have polyA tails.
Histones carry a large number of post-translational modifications, which are deposited and removed by specific enzymes. In addition, histones and nucleosomes are subjected to remodeling by ATP-dependent chromatin remodeling complexes. The combination of these events impacts the chromatin state, making it permissible or restrictive to gene expression, and thus in turn plays an essential part in determining cellular phenotypes. Given the fundamental role of chromatin in regulating gene expression, many histone-modifying enzymes and chromatin remodeling complexes have been found to be essential for normal development, and their function is affected in many diseases, including cancer (Albert and Helin 2010; Kooistra and Helin 2012; Narlikar et al. 2013; Feinberg et al. 2016).
The role of chromatin in cancer has been further strengthened by the recent discovery of mutations in histone H3 genes in specific adolescent and pediatric tumors. Mutations have been found in both the canonical and variant forms of H3, involving amino acid residues that are known substrates or are adjacent to substrates of histone-modifying enzymes. The mutations in histone-coding genes occur at very high frequencies in these types of tumors, and, in some cases, the histone mutation is found to be the only recurrent mutation identified in the tumor, suggesting that the histone mutations act as driver mutations. Furthermore, missense, rather than nonsense, mutations occur at specific residues, indicating that they are oncogenic mutations, and thus mutant histones are termed as “oncohistones” (Schwartzentruber et al. 2012; Behjati et al. 2013). So far, the mutations have been found to occur in a single allele of one of the multiple histone H3 genes in the tumor cell. Since there are 15 different H3 genes, this indicates a highly dominant nature of oncohistones. Interestingly, the amino acid changes found in the histone H3 genes are highly specific to the cancer type as well as to the anatomical location of the tumor, suggesting that the transforming ability of the oncohistones is dependent on the cell of origin.
Histone H3 and its variants
H3.1 and H3.2 are the two canonical forms of H3 that are expressed from 10 and three genes clustered on human chromosomes 6 and 1, respectively. There are six replication-independent variants of H3: H3.3, centromere protein A (CENP-A), H3.X, H3.Y, and two testis-specific H3 variants (H3.1T and H3.5). Thus far, all of the histone mutations found in cancer have been identified in either the canonical forms of H3 or in H3.3. The relative abundance of H3.3 varies according to cell type and the proliferation status of the cell. While H3.3 in actively dividing cells constitutes up to 25% of total H3, it makes up close to 90% of total H3 in terminally differentiated neurons (Piña and Suau 1987). This is consistent with the fact that H3.1 and H3.2 can only incorporate into the chromatin during DNA replication, whereas, in nondividing cells, H3.3 continues to be incorporated and replaces H3.1 and H3.2 (Maze et al. 2015; Tvardovskiy et al. 2017).
The amino acid sequences of H3.1, H3.2, and H3.3 are highly similar, with H3.1 and H3.2 differing in only a single amino acid, whereas H3.3 differs from H3.1 and H3.2 in five and four amino acids, respectively (Fig. 1C). All except one of these differences are found in the core histone fold domain and provide specificity for different histone chaperones: While H3.1 and H3.2 are incorporated all over the genome during DNA replication by CAF1 (chromatin-associated factor 1), the incorporation of H3.3 is mediated by two separate chaperones (HIRA and DAXX–ATRX) and is enriched at distinct genomic regions (Fig. 1D; Tagami et al. 2004; Drané et al. 2010; Goldberg et al. 2010). Remarkably, a single divergent residue, Gly90, in H3.3 is required for the specific H3.3–DAXX interaction (Elsässer et al. 2012). H3.3 is specifically enriched over transcription start sites (TSSs) of both active and repressed genes as well as over the body of active genes (Goldberg et al. 2010). Additionally, H3.3 is enriched over regulatory elements, including pericentric heterochromatin and telomeres (Drané et al. 2010; Goldberg et al. 2010). While enrichment of H3.3 over genes is mediated by HIRA, the ATRX–DAXX complex incorporates H3.3 at pericentric heterochromatin and telomeres (Fig. 1D; Drané et al. 2010; Goldberg et al. 2010; Lewis et al. 2010; Wong et al. 2010).
H3.3 is expressed by two genes, H3F3A and H3F3B, and, in contrast to canonical histones that are present in clusters, H3F3A and H3F3B are located separately on human chromosomes 1 and 17, respectively. Both H3F3A and H3F3B are expressed ubiquitously but at variable levels in different tissues (Jang et al. 2015). Genetic studies in mice have shown that deletion of both H3F3A and H3F3B leads to early embryonic lethality (Jang et al. 2015). Mice lacking either H3F3A or H3F3B have distinct phenotypes, and the severity of the phenotypes varies between different studies (Couldrey et al. 1999; Tang et al. 2013; Yuen et al. 2014; Jang et al. 2015). Since H3F3A and H3F3B have identical amino acid sequences, the different phenotypes might reflect the difference in expression of the two genes during development (Jang et al. 2015).
Oncohistones
Histone mutations in cancer were first reported in histone H3 genes encoding p.Lys27Met (K27M) or p.Gly34Arg/Val (G34R/V) in pediatric high-grade gliomas (pHGGs) (Khuong-Quang et al. 2012; Schwartzentruber et al. 2012; Wu et al. 2012; Fontebasso et al. 2014; Taylor et al. 2014). Later, mutations in histone H3 genes encoding p.Lys36Met (K36M) and p.Gly34Trp/Leu (G34W/L) were found in chondroblastomas and giant cell tumors of bone (GCTBs), respectively (Behjati et al. 2013). Chondroblastomas are rare bone tumors affecting mostly children and young adults of <20 yr of age (De Mattos et al. 2013), whereas GCTBs are relatively common bone tumors that are typically seen in early adulthood. The histone mutations occur at very high frequency, and, in the case of diffuse intrinsic pontine gliomas (DIPGs), H3K27M mutation is observed across all of the anatomical regions in the primary and metastatic tumor sites analyzed, including radiologically and histologically normal areas, suggesting that H3K27M mutation is the initial oncogenic event (Hoffman et al. 2016; Nikbakht et al. 2016). Moreover, H3K27M mutation is maintained throughout the course of the disease, from diagnosis to the end stage, suggesting that H3K27M is required for tumor maintenaince (Nikbakht et al. 2016).
Strikingly, all of the driver histone mutations are found in the tumors of children or young adults. Thus, even though adult HGGs are histologically very similar to pHGGs, K27M and G34R/V mutations are rarely found in adult HGGs, suggesting that the impact of histone mutations in cancer initiation and progression is limited to specific developmental settings.
One of the remarkable features of histone mutations is that these mutations are highly tumor type-specific and restricted to defined anatomical locations. For example, K27M and G34R/V mutations are mutually exclusive and restricted to specific locations in the central nervous system (CNS). K27M mutations are restricted to midline (thalamus, cerebellum, spine, and pons) tumors with the highest frequency (up to 93%) (Fontebasso et al. 2014) found in DIPGs, an aggressive and diffusely infiltrating tumor located in the pons (Mackay et al. 2017). In contrast, G34R/V mutations are found exclusively in pHGGs of cerebral cortex with the frequency of 14%–15% (Fig. 2; Table 1). The restricted occurrence of K27M and G34R/V mutations to specific anatomical sites suggests a distinct cell of origin for midline and cortical pediatric tumors as compared with adult brain tumors.
Figure 2.

Different mutations in histone H3 are found in tumors located in distinct anatomical locations. Depicted here is the occurrence of G34R/V and K27M mutations in different anatomical regions in the brain. G34R/V mutants are restricted to the tumors located in cerebral cortex, while the K27M mutant is found only in tumors of the midline structure (including the thalamus, cerebellum, brain stem, spine, and fourth ventricle).
Table 1.
Histone mutations identified in pediatric tumors
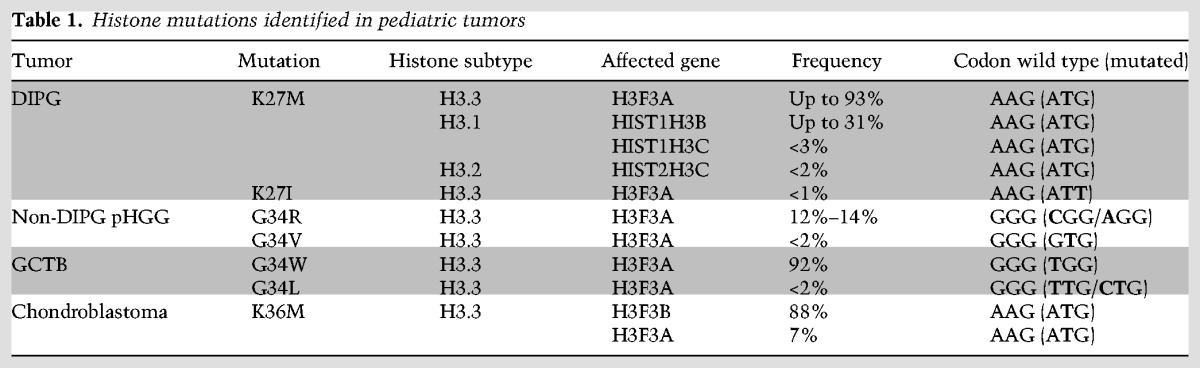
Another feature of histone mutations is that they preferentially affect a specific histone H3 variant. The majority of histone mutations found in cancer occurs in the H3.3 variant, except for the K27M mutation, which has also been found in canonical H3.1 and H3.2, albeit with lower frequency. We still do not understand the significance of the preferential prevalence of histone mutations in the H3.3 variant. While H3.1 and H3.2 are incorporated into nucleosomes throughout the genome with no specificity, H3.3 is enriched at discrete genomic regions, including active genes, regulatory elements, pericentric heterochromatin, and telomeres (Goldberg et al. 2010). Thus, mutation in canonical and variant histones may well have distinct effects on the cellular phenotype. Consistent with this, tumors harboring K27M mutations in H3.1 and H3.3 are clinically and molecularly distinct. H3.1K27M tumors occur in younger patients and are restricted to the pons, and these patients show better prognosis than patients with H3.3K27M-positive tumors (Fontebasso et al. 2014; Taylor et al. 2014; Wu et al. 2014; Castel et al. 2015). Moreover, H3.1K27M and H3.3K27M tumors show distinct gene expression profiles. Gene expression in H3.1K27M tumors is similar to the expression profiles in adult tumors with a mesenchymal/astrocytic phenotype, whereas H3.3K27M tumors are more similar to tumors with a proneural/oligodendroglial phenotype (Castel et al. 2015). Last, H3.1K27M and H3.3K27M mutant tumors have distinct cosegregating mutations. For example, mutations in Activin A receptor type 1 (ACVR1) and BCL6 corepressor (BCOR) genes cosegregate with H3.1K27M mutation, while platelet-derived growth factor receptor α (PDGFRA) and DNA topoisomerase III α (TOP3A) amplification and TP53 mutation cosegregate with H3.3K27M mutation (Taylor et al. 2014; Wu et al. 2014; Mackay et al. 2017).
Although canonical and variant forms of H3 are expressed by multiple genes, mutations in histones occur only in specific genes (Table 2). For example, among the two genes encoding H3.3, K27M and G34R/V mutations occur exclusively in H3F3A, whereas the majority of K27M mutation in H3.1 occurs in HIST1H3B (one of the 10 H3.1 genes). However, rare mutations in HIST1H3C (H3.1) and HIST2H3C (H3.2) have also been found in midline tumors (Fontebasso et al. 2014; Castel et al. 2015). This specificity can be explained partially by the fact that Lys27 in H3F3A and H3F3B is encoded by “AAG” and “AAA,” respectively. Thus, mutation of Lys27 to methionine (coded by “ATG”) in H3F3A would require substitution of only one residue, while two residues need to be mutated in H3F3B. However, this does not explain why the majority of H3.1K27M mutations occurs in the HIST1H3B gene, since Lys27 is encoded by “AAG” in six out of 10 genes expressing H3.1, including HIST1H3B. Similarly, the preferential occurrence of G34R/V or G34W/L mutation in H3F3A or K36M mutation in H3F3B cannot be explained by the codon sequence because Gly34 and Lys36 are encoded by identical codons in H3F3A and H3F3B. The specificity of histone mutations to different genes might reflect the differential expression of histone genes in different tissues (Krimer et al. 1993; Bramlage et al. 1997; Jang et al. 2015). H3F3A has more uniform expression across different tissues, whereas H3F3B shows more restricted expression (Krimer et al. 1993; Bramlage et al. 1997; Jang et al. 2015). Although H3F3A and H3F3B mRNAs code for identical proteins, they have very different 5′ and 3′ UTRs (untranslated regions). Thus, H3F3A and H3F3B mRNAs could be subject to different post-transcriptional regulation such as by microRNA, which would further lead to differential contributions of the H3F3A and H3F3B genes to total H3.3 in different tissues. Estimating the contribution of H3F3A and H3F3B to total H3.3 protein at different developmental stages and in different tissues might help in understanding the biased occurrence of histone mutations in different H3.3 genes.
Table 2.
Codon sequences of amino acid residues of human histone H3 and its variants that are found to be mutated in tumors
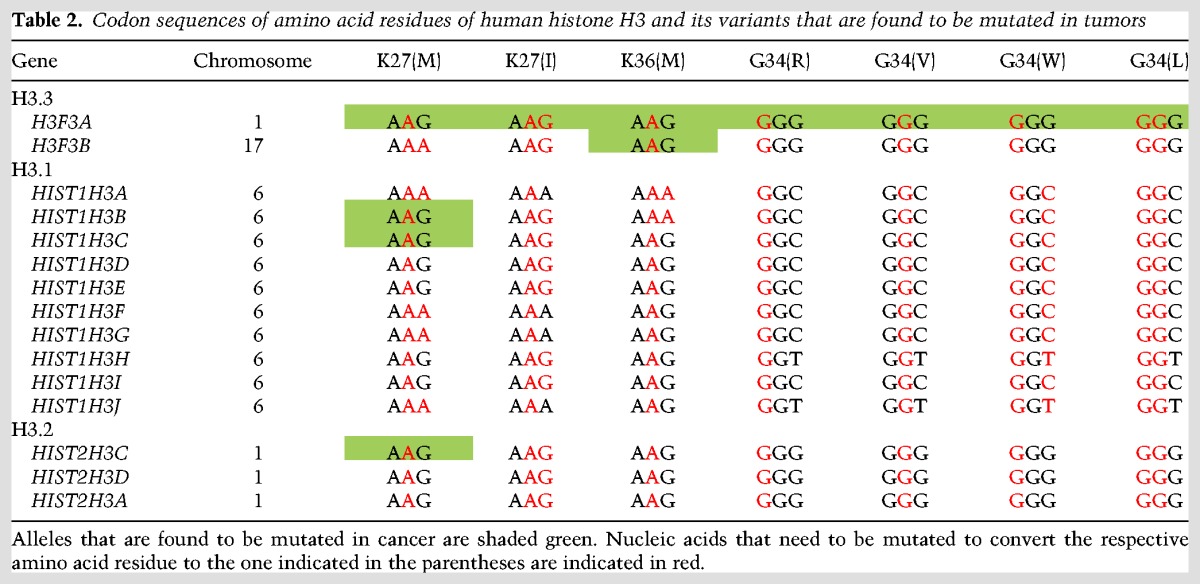
The three amino acids (K27, K36, and G34) that are found as mutants in tumors lie within the unstructured N-terminal tail of histone H3. This region contains many different types of post-translational modifications, which are involved in regulating different biological processes, including gene expression, nuclear organization, DNA repair, and DNA replication (Kouzarides 2007). In the following sections, we discuss how the known histone mutants may contribute to tumorigenesis through disruption of normal H3 modification patterns and cellular functions.
H3K27M mutation in pHGGs
H3K27 can be monomethylated (H3K27me1), dimethylated (H3K27me2), and trimethylated (H3K27me3) as well as acetylated (H3K27ac). Methylation of K27 is catalyzed by Polycomb-repressive complex 2 (PRC2), a multiprotein complex with three core components: enhancer of zeste homolog 1 (EZH1) or EZH2 (catalytic subunit), suppressor of zeste 12 (SUZ12), and embryonic ectoderm development (EED) (Di Croce and Helin 2013). They are members of the Polycomb group (PcG) protein family and work together with the functionally related PRC1 complex to maintain gene repression. PRC2 can have both oncogenic and tumor-suppressing functions, and both activating and loss-of-function mutations in PRC2 genes are frequently found in various cancers (Comet et al. 2016).
pHGGs, including DIPGs, expressing mutant H3K27M display globally reduced H3K27me3 and increased H3K27ac levels (Bender et al. 2013; Lewis et al. 2013; Venneti et al. 2013). The global effect of H3K27M on chromatin is surprising given the fact that the histone H3.1/3 K27M is expressed from a single allele, and the mutant protein therefore constitutes only a small fraction (3.63%–17.61%) of total H3 in the tumor cells (Lewis et al. 2013). This suggests a strong dominant effect of H3K27M over wild-type H3. The dominant effect of H3.1K27M is even more remarkable considering that H3.1 is incorporated into chromatin only during DNA replication and that H3.1K27M is expressed from one single allele of a total of 20 different H3.1 alleles.
Exogenous expression of H3.3K27M or H3.1K27M in mouse and human cells results in global reduction in H3K27me2/3 levels (Bender et al. 2013; Chan et al. 2013; Lewis et al. 2013; Funato et al. 2014; Mohammad et al. 2017). Interestingly, comprehensive testing of various H3K27 mutant proteins has shown that the global decrease in H3K27me2/3 levels is specific to K27M and, to a lesser extent, K27I (Lewis et al. 2013). Consistent with this, a rare K27I mutation, which is also associated with reduced H3K27me3 levels, has been identified in DIPGs (Castel et al. 2015). Substitution of K27 with any other amino acid residue does not lead to a global reduction in H3K27me3 levels (Lewis et al. 2013), indicating the remarkable specificity of K27M/I mutations. The exclusive finding of K27M/K27I mutants indicates a causal role of the global effect on H3K27 methylation.
Although a reduction in H3K27me3 levels is a hallmark of pHGGs expressing H3K27M, genome-wide analyses of H3K27me3 show significant locus-specific enrichment of H3K27me3 in H3K27M tumor cells (Bender et al. 2013; Chan et al. 2013). Interestingly, the regions that remain enriched for H3K27me3 in H3K27M cells span larger chromatin domains and have higher H3K27me3 density when compared with wild-type cells, indicating significant PRC2 activity in H3K27M cells (Chan et al. 2013).
Locus-specific changes in H3K27me3 caused by H3K27M have also been analyzed in DIPG models, where wild-type or K27M mutant H3.3 was exogenously expressed in isogenic cells. Funato et al. (2014) described a DIPG model in which exogenous expression of H3.3K27M along with overexpression of PDGFRA and p53 knockdown transformed human embryonic stem cell (ESC)-derived neural stem cells (NSCs) and led to the resetting of NSCs to a more primitive stem cell state. Interestingly, the transforming ability of H3.3K27M was highly cell type-specific, as expression of H3.3K27M in differentiated somatic cells such as astrocytes or fibroblasts led to senescence (Funato et al. 2014). Compared with NSCs expressing wild-type H3.3, NSCs expressing H3.3K27M showed genomic redistribution of H3K27me3 with both loss and gain of gene-specific H3K27me3 (Funato et al. 2014).
Recently, we described a DIPG model in which coexpression of H3.3K27M and PDGFB in mouse NSCs lead to tumor development in immunodefficient mice (Mohammad et al. 2017). Analysis of the genomic distribution of H3K27me3 showed that it was lost or reduced at the majority of genomic loci and unaffected at a significant number of loci. Interestingly, a few loci showed increased H3K27me3 levels in H3.3K27M-expressing cells when compared with cells expressing wild-type H3.3. The distribution of H3K27me3 in mouse NSCs expressing H3.3K27M was significantly overlapping with the H3K27me3 profile in human H3K27M mutant DIPG cell lines. Interestingly, there was a very strong correlation between the initial H3K27me3 enrichment in wild-type NSCs and the effect of H3K27M expression. Loci with low initial H3K27me3/PRC2 enrichment showed a complete loss of or reduced H3K27me3, while loci with high initial H3K27me3/PRC2 enrichment showed no change in H3K27me3 levels. However, loci with increased H3K27me3 in H3K27M cells showed no apparent correlation with the initial H3K27me3. Thus, the majority of differential effects of H3K27M on different genomic loci can be explained by the level of PRC2 activity (Mohammad et al. 2017).
The cyclin-dependent kinase inhibitor 2a (Cdkn2a) locus, coding for the p19Arf and p16Ink4a (p16) tumor suppressor proteins, was one of the loci with increased H3K27me3 in H3K27M cells. The increased H3K27me3 levels at the Cdkn2a locus was associated with reduced expression of p16 in H3K27M cells. Consistent with this, DIPG cell lines harboring the H3K27M mutant also showed high H3K27me3 levels at the CDKN2A locus and, correspondingly, very low p16 expression (Mohammad et al. 2017). Homozygous deletion of the CDKN2A locus is very frequent (>55%) in adult HGGs (Brennan et al. 2013) but rarely found in pHGGs (Mackay et al. 2017). Thus, H3K27M-mediated repression of the CDKN2A locus likely reflects an alternative approach to silence the CDKN2A locus in pHGGs. In addition to the effect on the CDKN2A locus, cells expressing H3K27M show expression changes in several other genes, including potential oncogenes and tumor suppressor genes when compared with cells expressing wild-type H3. The contribution of these genes to the development of H3K27M tumors is currently not known.
How the expression of H3K27M can lead to increased H3K27me3 at specific loci is not yet completely understood. Since the association of H3K27M with a locus should lead to a reduction and not an increase of H3K27me3 at the locus, the increased levels of H3K27me3 at specific loci are most likely occurring through a selection process in which the cell of origin for DIPGs already has high levels of H3K27me3 associated with these loci, which are maintained and/or selected for during the development of the tumor. Importantly, however, it is also not yet clear whether the global changes in H3K27me3 enrichment observed in H3K27M tumors are the cause or the consequence of cellular transformation.
One of the caveats of DIPG models described so far is that they are made without prior knowledge regarding the cell of origin for DIPGs, which is yet unknown. Candidates for the cell of origin for DIPGs include precursor cells in the ventral pons (Nestin+/Vimentin+/Olig2+) (Monje et al. 2011) or the fourth ventricle floor (Nestin+/Pax3+) (Misuraca et al. 2016). Despite this, the NSC-based DIPG models show features similar to patient tumors and have provided useful mechanistic insights. H3K27M-driven tumors in these models show gene expression profiles overlapping with H3K27M patient tumors (Funato et al. 2014; Mohammad et al. 2017). Moreover, NSCs expressing H3K27M show H3K27me3 enrichment profiles similar to patient-derived DIPG cell lines (Mohammad et al. 2017).
Recently, an in utero DIPG model has been described in which expression of H3.3K27M along with the loss of p53 in the hindbrain and the cortex of mouse embryos lead to the development of serially transplantable diffuse tumors (Pathania et al. 2017). H3.3K27M can induce tumorigenesis only during a specific stage of development, as expression of H3.3K27M was lethal during post-zygotic embryo development, whereas its expression in the postnatal mouse brain failed to induce tumors. Interestingly, expression of H3.3K27M from Nestin (Nes) or glial fibrillary acidic protein (Gfap) promoters that are active in embryonic and postnatal NSCs also did not lead to tumor development, and thus the cell of origin for H3K27M tumors remains elusive.
The mechanistic basis for the global loss of H3K27me3 upon expression of H3K27M is incompletely understood. Several results have suggested that the H3K27M histone tail directly interferes with PRC2 activity. For instance, in vitro studies show that an H3K27M mutant peptide inhibits PRC2 activity through interaction with its catalytic subunit, EZH2, by competing with the substrate (Lewis et al. 2013; Brown et al. 2014). However, it is interesting to note that the modes of inhibition of PRC2 by H3K27M and currently available small molecule PRC2 inhibitors, which act by competing with the SAM cofactor for binding to EZH2, are very different, and small molecule PRC2 inhibitors do not induce molecular and phenotypic effects similar to H3K27M (Mohammad et al. 2017).
The mode of interaction of H3K27M with PRC2 was resolved by crystal structures of human and fungal PRC2 in a complex with the H3K27M peptide, showing tight binding of H3K27M to the catalytic site of EZH2 (Jiao and Liu 2015; Justin et al. 2016). Compared with the wild-type H3 peptide, the H3K27M peptide binds PRC2 with 16-fold higher affinity (Justin et al. 2016). Based on these observations, it has been proposed that strong binding of PRC2 to H3K27M mutant histones at the chromatin leads to sequestration and inactivation of PRC2 at H3K27M-containing nucleosomes, thereby leading to global reduction of H3K27me3 levels (Fig. 3A). However, several laboratories have analyzed the interaction between H3K27M and PRC2 using various assays, including immunoprecipitation and electrophoretic mobility shift assay (EMSA), and have reached different conclusions. Some studies (Bender et al. 2013; Chan et al. 2013; Justin et al. 2016) have shown a tighter interaction between PRC2 and H3K27M as compared with wild-type H3, whereas other studies (Herz et al. 2014; Wang et al. 2017) have not observed the same. The reason for these different results is not clear.
Figure 3.

Proposed mechanisms by which oncohistones affect the activity of histone methyltransferases (HMTs). (A) K-to-M mutant histones bind more tightly to the catalytic site within the SET domain of HMTs than wild-type histones, leading to sequestration of the HMT complex to mutant nucleosomes and rendering the HMT nonproductive due to the absence of the substrate. (B) The G34 residue of histone H3 is buried in a narrow tunnel within the active site of SETD2. Mutation of G34 to residues with a larger side chain (R/V/W/L) might result in distortion of the catalytic site that would render SETD2 inactive.
In potential contrast to the sequestration model, recent results suggest that H3.3K27M is enriched specifically in actively transcribed regions of the genome and that PRC2 is precluded from these regions (Piunti et al. 2017). These results would be in line with the fact that H3.3 shows significantly higher enrichment over the bodies of active genes (Goldberg et al. 2010). Thus, reduced enrichment of H3.3K27M over the repressed genes that are strong PRC2 targets might explain the retained H3K27me3 levels over these genes in H3.3K27M tumors. However, this reasoning does not explain the retained gene-specific H3K27me3 in cells expressing H3.1K27M, considering that H3.1 and H3.2 are uniformly distributed over the genome (Goldberg et al. 2010). Taken together, the available results show that H3K27M inhibits PRC2 in vitro; however, H3K27M expression does not lead to complete loss of PRC2 activity in vivo, and further studies are required to elucidate whether differential enrichment of H3K27M is responsible for residual PRC2 activity in the tumor cells.
The H3K36M mutant in chondroblastomas
Mutations in H3 genes leading to the expression of H3K36M have been identified with high frequency (∼95%) in chondroblastomas (Behjati et al. 2013). The majority of K36M mutations occurs in the H3F3B gene encoding H3.3 (Behjati et al. 2013). The K36 residue on histone H3 can be monomethylated (H3K36me1), dimethylated (H3K36me2), or trimethylated (H3K36me3). In mammals, several histone methyltransferases (HMTs) catalyze H3K36 methylation. Nuclear receptor SET domain-containing 1 (NSD1), NSD2, NSD3, and ASH1-like (ASH1L) are the main enzymes that catalyze monomethylation and dimethylation of H3K36, whereas SETD2 is the only known enzyme that catalyzes the conversion of H3K36me2 to H3K36me3 (Wagner and Carpenter 2012). H3K36me2 and H3K36me3 are enriched over the gene bodies of transcriptionally active genes, where they are required for the repression of aberrant transcriptional initiation within coding sequences (Carrozza et al. 2005). In addition, H3K36me2 and H3K36me3 have been shown to play important roles in DNA repair, dosage compensation, and splicing (Wagner and Carpenter 2012). Similar to H3K27M mutation in pHGGs, H3K36M mutant chondroblastomas show global loss of H3K36me2/3 (Fang et al. 2016; Lu et al. 2016). The K36M peptide or K36M mononucleosomes inhibit NSD2 and SETD2 with no effect on NSD1 and ASH1L (Fang et al. 2016; Lu et al. 2016). Ectopic expression of H3.3K36M in mesenchymal progenitor cells (MPCs) blocks the differentiation of MPCs into different lineages, including chondrocytes, adipocytes, and osteocytes (Lu et al. 2016), and MPCs expressing H3.3K36M generate sarcomas when transplanted into immunodefficient mice (Lu et al. 2016). These results demonstrate that H3K36M can be a driving oncoprotein in MPCs.
Genome-wide enrichment analysis has shown that ectopic expression of H3K36M leads to a global reduction of H3K36me2/3 in MPCs, with the strongest effect observed on H3K36me2 (Lu et al. 2016). H3K36me2 was completely lost at the intergenic loci, while it was significantly reduced at the genic regions in H3K36M cells. Moreover, H3K36M-expressing cells showed significantly reduced H3K36me3 enrichment over gene bodies, which was associated with gene expression changes (Lu et al. 2016). Similar results were obtained in human immortalized chondrocytes harboring a H3K36M mutation in the endogenous H3F3B gene (Fang et al. 2016). The genome-wide distribution of H3K36M histones has been analyzed in primary cells expressing H3K36M by immunoprecipitating tagged histones or using an H3K36M-specific antibody (Fang et al. 2016; Lu et al. 2016). The distribution of histone H3 was found to be unaffected by the H3K36M mutation, suggesting that the mutation does not affect how nucleosomes are positioned.
Interestingly, Lu et al. (2016) observed a global increase in H3K27me3 in primary cells expressing H3K36M as well as in H3K36M mutant chondroblastomas, suggesting that the loss of H3K36 methylation provides a new nucleosomal substrate for PRC2. This is consistent with previous observations showing that PRC2 activity is inhibited by H3K36 methylation (Schmitges et al. 2011; Yuan et al. 2011). The increased H3K27me3 in H3K36M cells was observed primarily in the intergenic regions and was associated with a decrease in PRC1 enrichment in the genic regions. Based on these observations, Lu et al. (2016) proposed that an increase in H3K27me3 at the intergenic regions in H3K36M cells leads to redistribution and dilution of PRC1 in genic regions. A similar model has been proposed to explain chromatin changes in multiple myeloma, where increased activity of NSD2 results in a global increase in H3K36me2 levels and a concurrent decrease in H3K27me3 levels (Popovic et al. 2014). In contrast to the report by Lu et al. (2016), another study did not observe increased H3K27me3 in human chondrocytes expressing H3K36M (Fang et al. 2016). At present, the reason for these contrasting observations is not clear.
A recent crystal structure shows that H3K36M binds strongly to the catalytic site of SETD2 (Yang et al. 2016; Zhang et al. 2017). Based on this, a model similar to the one discussed for H3K27M-mediated inhibition of PRC2 has been proposed, in which H3K36M mutant nucleosomes strongly bind and sequester H3K36 methyltransferases (Fig. 3A). However, it is not clear why H3K36M inhibits only NSD2 and SETD2 but not NSD1 even though residues interacting with H3K36M are highly conserved in these H3K36 methyltransferases. Moreover, similar to NSD2 and SETD2, NSD1 also preferentially associates with H3K36M nucleosomes (Lu et al. 2016), indicating that mechanisms other than sequestering of methyltransferases might be involved. Structural comparison of NSD1 and NSD2 bound to K36M could provide more insight into the differences.
H3G34 mutants in pHGGs and GCTB
One of the most interesting observations regarding the H3G34 oncohistone mutants is the fact that the G34R/V mutant is specifically found in pHGGs located in the cerebral cortex, while the G34W/L mutant is specifically found in GCTB. pHGGs harboring the G34R/V mutant are clinically and biologically distinct from H3K27M mutant DIPGs. H3G34 mutant pHGGs show better prognosis and occur in patients with a higher median age than H3K27M tumors (Schwartzentruber et al. 2012; Sturm et al. 2012; Wu et al. 2012). H3G34 mutant pHGGs show reduced levels of DNA methylation, which is more prominent at telomeric regions (Sturm et al. 2012). This observation is consistent with the frequently observed alternative lengthening of telomeres (ALT) in H3G34 mutant pHGGs (Schwartzentruber et al. 2012). The Gly34 residue of H3 has not yet been shown to be associated with any modification, and, unlike the H3K27M mutant, the H3G34R/V mutant has not been shown to change the global post-translational modification level of any amino acid on H3. However, G34 is located close to K36 on the H3 tail, and ectopic expression of H3.3G34R/V leads to reduced H3K36me2 and H3K36me3 levels on mutant nucleosomes, with no effect on nucleosomes containing wild-type histones, suggesting that, unlike H3K27M, H3G34R/V does not act as a dominant-negative mutant (Lewis et al. 2013).
Crystallographic studies of the SET domain of SETD2 bound to an H3K36M peptide have provided insights into the molecular mechanisms by which H3G34 mutants can affect H3K36 methylation. The H3G34 residue is buried deep in the SET domain in a very narrow tunnel with no space to accommodate residues with large side chains (Zhang et al. 2017). In agreement with this, the introduction of mutations in G34 in one of the endogenous H3 genes in fission yeast—leading to the expression of oncogenic R, V, L, or W residues or other residues with longer side chains (A, C, S, D, N, and T)—has been shown to affect H3K36me3 on the mutant histone H3, most likely by affecting the conformation of the SET domain of SETD2 (Fig. 3B; Zhang et al. 2017). These results suggest that the effect of H3G34R/V or H3G34W/L mutants on H3K36me3 is not specific and can be mimicked by several other H3G34 mutants. The reason why H3G34 mutants identified in tumors are limited to specific residues may therefore be explained by the codon sequence: In the H3F3A gene, H3G34 is coded by “GGG,” and mutation of single nucleic acids can convert it to Arg/R (“AGG” and “CGG”), Trp/W (“TGG”), Val/V (“GTG”), Glu/E (“GAG”), and Ala/A (“GCG”), while two or more nucleic acid residues need to be mutated to convert H3G34 to any other amino acid residue. This also explains why the H3G34R mutant is more frequent than the H3G34V mutant in pHGGs and why the H3G34L mutant, identified in only one sample of GCTB, is so rare. However, this does not explain why the H3G34R/V and H3G34W/L mutants are present only in pHGGs and GCTB, respectively, or why the H3G34E and H3G34A mutants have not yet been identified in pHGGs or GCTB. These observations suggest that the H3G34 mutants do more than just affect H3K36me2/3. Indeed, the H3G34R/V mutant has also been shown to impair specific binding of ZMYND11, a tumor suppressor, to H3.3K36me3 (Wen et al. 2014). Whether H3G34W/L similarly affects ZMYND11 binding and how interference in chromatin binding of factors such as ZMYND11 by H3G34 mutations would contribute to tumorigenesis are some of the open questions.
Thus far, only a few reports have been published on the biological effects of H3G34 mutant expression. Ectopic expression of H3.3G34V in normal human astrocytes or transformed human fetal glial cells has been shown to result in increased MYCN expression, which was corroborated by expression analysis of H3G34 mutant pHGGs (Bjerke et al. 2013). The effects of H3G34R expression have been studied in fission yeast, where H3G34R expression from the only H3 gene led to a global decrease in H3K36me3 and H3K36ac without detectable effects on H3K36me2 (Yadav et al. 2017). Consistent with the role of H3K36me3 in DNA repair, H3G34R mutant fission yeast showed genomic instability and defects in DNA repair (Yadav et al. 2017). However, these results were obtained in an organism engineered to express only mutant H3, which is in contrast to tumors where just one out of four H3.3 alleles is mutated. Thus, these mechanisms will need to be investigated in a more relevant system, where the correlation between the biological effects of H3G34 mutants with locus-specific changes in H3K36me2/3 enrichment can also be analyzed.
Targeted epigenetic therapies for oncohistone-expressing tumors
The fact that the majority of pHGGs harbors histone mutations that lead to alterations in the chromatin landscape of tumor cells has prompted several investigators to test the efficacy of different epigenetic therapies. In an attempt to reverse the reduced H3K27me3 levels observed in H3K27M tumors, Hashizume et al. (2014) analyzed the effect of inhibiting the H3K27me3 demethylases in H3K27M mutant pHGGs. Inhibition of H3K27me3 histone demethylases JMJD3 (KDM6B) and UTX (KDM6A) by GSK-J4 resulted in increased H3K27me3 in H3K27M mutant cell lines and showed in vitro and in vivo anti-tumor activity (Hashizume et al. 2014). Interestingly, GSK-J4 showed anti-tumor activity specifically against pHGGs expressing H3K27M, with no significant effect on cells expressing wild-type or G34V mutant H3. The mechanism by which GSK-J4 would be able to rescue the reduced H3K27me3 in H3K27M cells is not clear, considering that the tumor cells continue to express H3K27M. It is also not clear whether the effect of GSK-J4 on tumor cells is mediated by changes in H3K27me3 level particularly, since GSK-J4 can also inhibit the activity of other histone demethylases (Heinemann et al. 2014).
In another study, Grasso et al. (2015) performed a chemical screen on DIPG cell lines and identified panobinostat as a drug that strongly affects in vitro and in vivo tumor cell growth. Panobinostat is a nonselective histone deacetylase (HDAC) inhibitor that has been approved for the treatment of multiple myeloma. Interestingly, along with the increase in the level of acetylated histone H3, panobinostat treatment of H3K27M DIPG cells also leads to an increase in H3K27me3 levels and thus the partial rescue of H3K27M-induced decrease in H3K27me3. This is consistent with the previous observation that acetylation of lysine residues in the N-terminal tail of H3 counteracts the PRC2 inhibitory effect of H3K27M (Brown et al. 2014). Mechanistically, the use of panobinostat in H3K27M mutant pHGGs appears counterintuitive, considering that H3K27M mutant pHGGs show increased acetylation of H3K27, a histone mark associated with active chromatin (Lewis et al. 2013; Piunti et al. 2017). It is not clear how panobinostat treatment affects the H3K27ac levels in H3K27M mutant tumors, and thus the mechanism by which panobinostat affects tumor growth remains unknown. Considering that panobinostat inhibits multiple HDACs and that each HDAC can have multiple histone and nonhistone targets (Yang and Seto 2008), identification of targets that mediate the effects of panobinostat on tumor cells will be challenging.
The increased H3K27ac levels in H3K27M mutant pHGGs lead to increased association of bromodomain-containing proteins (BRD2 and BRD4), which bind to acetylated histones (Herz et al. 2014; Piunti et al. 2017). This observation led Piunti et al. (2017) to show that an inhibitor of bromodomain and extraterminal domain (BET) family members, including BRD2 and BRD4, affects the growth of H3K27M mutant cells.
Despite the global loss of H3K27me3, H3K27M mutant cells show significant residual PRC2 activity. The genes that remain associated with H3K27me3 in H3K27M cells are classical Polycomb targets whose transcriptional regulation is crucial for cell fate decisions during development, suggesting that their repression is required to maintain cellular identity (Mohammad et al. 2017). Consistent with this, inhibiting PRC2 activity in H3K27M cells through EZH2 inhibition or genetic deletion of EZH2 strongly affects tumor cell growth (Mohammad et al. 2017; Piunti et al. 2017). Interestingly, reduced cell proliferation induced by EZH2 inhibition is mediated by p16, whose expression is significantly increased upon EZH2 inhibitor treatment. Moreover, deletion of p16 in a DIPG cell line renders tumor cells insensitive to EZH2 inhibition, suggesting that in DIPGs, p16 is the primary target of PRC2 inhibitors (Mohammad et al. 2017). p16 inhibits CDK4 and CDK6, which in turn results in activation of pRB and cellular growth arrest. Considering that DIPG cells are sensitive to high p16 levels, these cells might also respond to CDK4/6 inhibitors. Given that that several CDK4/6 inhibitors have already been approved for the treatment of advanced breast cancer, CDK4/6 inhibition could be a viable alternative for the treatment of DIPGs. Indeed, several clinical trials are under way to test the efficacy of CDK4/6 inhibitors in pediatric CNS tumors, including DIPGs.
Perspectives
The role of epigenetic regulators and chromatin-associated proteins in tumor etiology has been increasingly appreciated, especially in the last decade, with frequent mutations in the genes encoding epigenetic factors being discovered in almost every cancer type. The discovery of highly recurrent mutations in histone genes affecting the global chromatin landscape in pediatric tumors has further reinforced the role of epigenetic changes in tumor development. Mechanistic details of oncohistone-mediated molecular changes in tumor cells have just started to emerge, and these observations may lead to the development of therapeutic strategies, especially for H3K27M mutant pHGGs. K-to-M mutants of H3 act as dominant mutants, and their expression leads to global changes in the histone modification levels in the tumor cells, primarily through inhibition of specific HMTs. It is noteworthy that K-to-M mutant cells display significant residual activity of inhibited HMTs, indicating that the inhibition of HMTs by K-to-M mutant H3 is not absolute. In some cases, complete inhibition of the HMT is deleterious for the K-to-M mutant tumor cells (Mohammad et al. 2017). Moreover, no loss-of-function mutation in the inhibited HMTs has been identified in pediatric tumors, and pharmacological inhibition of HMT does not mimic the effects of K-to-M mutation (Mohammad et al. 2017). These observations indicate that the simple inhibition of HMTs cannot explain the molecular effects of K-to-M mutations in H3, and future studies will be required to determine the actual mode of inhibition of HMTs by K-to-M mutants. Additionally, an understanding of how mutation in canonical or variant histone H3 leads to distinct molecular profile in tumors will have potential implications in developing targeted therapies.
Mutations in histones are limited to specific genes even though histone H3 and its variants are expressed from multiple genes. This specificity of histone mutations might be due to differential expression of histone H3 from individual genes in the tumor cell of origin. Mutations leading to H3K27M expression occur in both canonical H3.1 and H3.2 as well as in histone variant H3.3, whereas mutations leading to expression of H3K36 and H3G34 occur only in H3.3 (Table 2). The functional importance of the preferential prevalence of mutations in histone variants is not clear, and it would be interesting to test whether mutations in H3.1 leading to the expression of K36 and G34 have effects similar to those of mutations in H3.3, which has been shown for H3K27M (Chan et al. 2013; Lewis et al. 2013; Mohammad et al. 2017).
In vitro studies have shown that, similar to K27M and K36M mutations, the methionine substitution at other lysine residues (e.g. K9 and K4) in the H3 tail affects the global methylation levels of the associated residues (Lewis et al. 2013). While this observation could suggest that mutants of these residues contribute to tumorigenesis, no such mutations have been identified in disease. They may be infrequent or exist only in rare tumors, and it will be exciting to see whether such mutations will be identified.
Acknowledgments
We thank members of the Helin laboratory for discussions, and Helene Damhofer and Anne Laugesen for comments and critical reading of the manuscript. F.M. was supported by a post-doctoral fellowship from EMBO (874-2011). The work in the Helin laboratory was supported by the European Research Council (294666_DNAMET), the Danish Medical Research Council (DFF-4004-00081), the Brain Tumour Charity (GN-000358), the Danish National Research Foundation (DNRF 82), and a center grant from the Novo Nordisk Foundation (NNF17CC0027852).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.309013.117.
References
- Albert M, Helin K. 2010. Histone methyltransferases in cancer. Semin Cell Dev Biol 21: 209–220. [DOI] [PubMed] [Google Scholar]
- Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et al. 2013. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 45: 1479–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DTW, Kool M, Zapatka M, Northcott PA, Sturm D, Wang W, et al. 2013. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24: 660–672. [DOI] [PubMed] [Google Scholar]
- Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, Bax DA, Carvalho D, Taylor KR, Vinci M, et al. 2013. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov 3: 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramlage B, Kosciessa U, Doenecke D. 1997. Differential expression of the murine histone genes H3.3A and H3.3B. Differentiation 62: 13–20. [DOI] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. 2013. The somatic genomic landscape of glioblastoma. Cell 155: 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown ZZ, Müller MM, Jain SU, Allis CD, Lewis PW, Muir TW. 2014. Strategy for ‘detoxification’ of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. J Am Chem Soc 136: 13498–13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschbeck M, Hake SB. 2017. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol 18: 299–314. [DOI] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia W-J, Anderson S, Yates J, Washburn MP, et al. 2005. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123: 581–592. [DOI] [PubMed] [Google Scholar]
- Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, Pagès M, Taylor KR, Saulnier P, Lacroix L, et al. 2015. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol 130: 815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K-MM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al. 2013. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 27: 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comet I, Riising EM, Leblanc B, Helin K. 2016. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat Rev Cancer 16: 803–810. [DOI] [PubMed] [Google Scholar]
- Couldrey C, Carlton MB, Nolan PM, Colledge WH, Evans MJ. 1999. A retroviral gene trap insertion into the histone 3.3A gene causes partial neonatal lethality, stunted growth, neuromuscular deficits and male sub-fertility in transgenic mice. Hum Mol Genet 8: 2489–2495. [DOI] [PubMed] [Google Scholar]
- De Mattos CBR, Angsanuntsukh C, Arkader A, Dormans JP. 2013. Chondroblastoma and chondromyxoid fibroma. J Am Acad Orthop Surg 21: 225–233. [DOI] [PubMed] [Google Scholar]
- Di Croce L, Helin K. 2013. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol 20: 1147–1155. [DOI] [PubMed] [Google Scholar]
- Drané P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. 2010. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 24: 1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsässer SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. 2012. DAXX envelops a histone H3.3–H4 dimer for H3.3-specific recognition. Nature 491: 560–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Gan H, Lee J-H, Han J, Wang Z, Riester SM, Jin L, Chen J, Zhou H, Wang J, et al. 2016. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 352: 1344–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, Koldobskiy MA, Göndör A. 2016. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 17: 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset P-O, Bechet D, Faury D, De Jay N, Ramkissoon LA, et al. 2014. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet 46: 462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V. 2014. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 346: 1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Banaszynski LA, Noh K-MM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al. 2010. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140: 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily M-A, Quist MJ, Davis LE, Huang EC, Woo PJ, et al. 2015. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 21: 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume R, Andor N, Ihara Y, Lerner R, Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, et al. 2014. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med 20: 1394–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann B, Nielsen JM, Hudlebusch HR, Lees MJ, Larsen DV, Boesen T, Labelle M, Gerlach L-O, Birk P, Helin K. 2014. Inhibition of demethylases by GSK-J1/J4. Nature 514: E1–E2. [DOI] [PubMed] [Google Scholar]
- Herz H-M, Morgan M, Gao X, Jackson J, Rickels R, Swanson SK, Florens L, Washburn MP, Eissenberg JC, Shilatifard A. 2014. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science 345: 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, DeWire M, Ryall S, Buczkowicz P, Leach J, Miles L, Ramani A, Brudno M, Kumar SS, Drissi R, et al. 2016. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun 4: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C-W, Shibata Y, Starmer J, Yee D, Magnuson T. 2015. Histone H3.3 maintains genome integrity during mammalian development. Genes Dev 29: 1377–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao L, Liu X. 2015. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 350: aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, et al. 2016. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 7: 11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuong-Quang D-AA, Buczkowicz P, Rakopoulos P, Liu X-YY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, et al. 2012. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124: 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra SM, Helin K. 2012. Molecular mechanisms and potential functions of histone demethylases. Nat Rev Mol Cell Biol 13: 297–311. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. 2007. Chromatin modifications and their function. Cell 128: 693–705. [DOI] [PubMed] [Google Scholar]
- Krimer DB, Cheng G, Skoultchi AI. 1993. Induction of H3.3 replacement histone mRNAs during the precommitment period of murine erythroleukemia cell differentiation. Nucleic Acids Res 21: 2873–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Elsaesser SJ, Noh K-M, Stadler SC, Allis CD. 2010. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci 107: 14075–14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. 2013. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340: 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, et al. 2016. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352: 844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, Bjerke L, Clarke M, Vinci M, Nandhabalan M, et al. 2017. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32: 520–537.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I, Wenderski W, Noh K-M, Bagot RC, Tzavaras N, Purushothaman I, Elsässer SJ, Guo Y, Ionete C, Hurd YL, et al. 2015. Critical role of histone turnover in neuronal transcription and plasticity. Neuron 87: 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misuraca KL, Hu G, Barton KL, Chung A, Becher OJ, Cells P, Misuraca KL, Hu G, Barton KL, Chung A, et al. 2016. A novel mouse model of diffuse intrinsic pontine glioma initiated in Pax3-expressing cells. Neoplasia 18: 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F, Weissmann S, Leblanc B, Pandey DP, Højfeldt JW, Comet I, Zheng C, Johansen JV, Rapin N, Porse BT, et al. 2017. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med 23: 483–492. [DOI] [PubMed] [Google Scholar]
- Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, Attema JL, Li G, Haddix T, Edwards MSB, et al. 2011. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci 108: 4453–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. 2013. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 154: 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M, Ho C-Y, Kambhampati M, Hwang EI, Faury D, et al. 2016. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun 7: 11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, Henderson S, Mikael LG, Richard-Londt A, Zhang Y, et al. 2017. H3.3(K27M) cooperates with Trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell 32: 684–700.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piña B, Suau P. 1987. Changes in histones H2A and H3 variant composition in differentiating and mature rat brain cortical neurons. Dev Biol 123: 51–58. [DOI] [PubMed] [Google Scholar]
- Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi Y-H, Woodfin AR, et al. 2017. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med 23: 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, Shah MY, Zheng Y, Will CM, Small EC, et al. 2014. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet 10: e1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitges FW, Prusty AB, Faty M, Stützer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, et al. 2011. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell 42: 330–341. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang D-AK, Tönjes M, et al. 2012. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482: 226–231. [DOI] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang D-AA, Jones DTW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, et al. 2012. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22: 425–437. [DOI] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. 2004. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116: 51–61. [DOI] [PubMed] [Google Scholar]
- Tang MCW, Jacobs SA, Wong LH, Mann JR. 2013. Conditional allelic replacement applied to genes encoding the histone variant H3.3 in the mouse. Genesis 51: 142–146. [DOI] [PubMed] [Google Scholar]
- Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, et al. 2014. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46: 457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tvardovskiy A, Schwämmle V, Kempf SJ, Rogowska-Wrzesinska A, Jensen ON. 2017. Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res 33: 5005–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Workman JL. 2015. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 16: 178–189. [DOI] [PubMed] [Google Scholar]
- Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A, Santi M, Thompson CB, Judkins AR. 2013. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol 23: 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Hager GL. 2013. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet 15: 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EJ, Carpenter PB. 2012. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 13: 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Paucek RD, Gooding AR, Brown ZZ, Ge EJ, Muir TW, Cech TR. 2017. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat Struct Mol Biol 24: 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, et al. 2014. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 508: 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, Hannan RD, George AJ, Morgan KA, Mann JR, Choo KHA. 2010. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res 20: 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. 2012. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44: 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, et al. 2014. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 46: 444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav RK, Jablonowski CM, Fernandez AG, Lowe BR, Henry RA, Finkelstein D, Barnum KJ, Pidoux AL, Kuo Y-M, Huang J, et al. 2017. Histone H3G34R mutation causes replication stress, homologous recombination defects and genomic instability in S. pombe. Elife 6: e27406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X-J, Seto E. 2008. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 31: 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Zheng X, Lu C, Li G-M, Allis CD, Li H. 2016. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev 30: 1611–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. 2011. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem 286: 7983–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen BTK, Bush KM, Barrilleaux BL, Cotterman R, Knoepfler PS. 2014. Histone H3.3 regulates dynamic chromatin states during spermatogenesis. Development 141: 3483–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shan C-M, Wang J, Bao K, Tong L, Jia S. 2017. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci Rep 7: 43906. [DOI] [PMC free article] [PubMed] [Google Scholar]