

Azizoglu et al. identified mechanisms underlying pancreatic lumen formation and remodeling and show that central lumen network morphogenesis impacts pancreatic endocrine mass. Codepletion of the actomyosin regulator RhoA and Afadin results in defects in the central lumen and arrests lumen remodeling.
Keywords: apical polarity, Rab GTPases, actomyosin, vesicular trafficking, islet, progenitor niche
Abstract
Proper lumen morphogenesis during pancreas development is critical to endocrine and exocrine cell fate. Recent studies showed that a central network of lumens (termed core), but not the surrounding terminal branches (termed periphery), produces most islet endocrine cells. To date, it remains unclear how pancreatic lumens form and remodel and which aspects of lumen morphogenesis influence cell fate. Importantly, models testing the function of the central lumen network as an endocrine niche are lacking. Here, we identify mechanisms underlying lumen formation and remodeling and show that central lumen network morphogenesis impacts pancreatic endocrine mass. We show that loss of the scaffolding protein Afadin disrupts de novo lumenogenesis and lumen continuity in the tip epithelium. Codepletion of the actomyosin regulator RhoA and Afadin results in defects in the central lumens and arrests lumen remodeling. This arrest leads to prolonged perdurance of the central lumen network over developmental time and expansion of the endocrine progenitor population and, eventually, endocrine mass. Our study uncovers essential roles of Afadin and RhoA in pancreatic central lumen morphogenesis, which subsequently determines endocrine cell mass.
Extensive efforts have been directed toward developing β-cell replacement therapies for diabetic patients. Although β-cell (or endocrine cell) generation has been accomplished by recapitulating in vivo developmental steps, these protocols have yet to be optimized and become therapeutically viable (Pagliuca et al. 2014; Massumi et al. 2016). Several studies suggest that a three-dimensional microenvironment enhances endocrine differentiation (Jiang et al. 2007; Kesavan et al. 2009; Kelly et al. 2011; Greggio et al. 2013; Bankaitis et al. 2015). To date, what this environment entails and how it forms remain unknown. Therefore, a deeper understanding of the endocrine differentiation “niche” will likely propel development of novel treatments for diabetes.
Differentiation of pancreatic progenitors into the endocrine lineage has been thoroughly characterized (Pan and Wright 2011). Endodermal progenitors first transition through a ducto–endocrine bipotential state, identified by expression of the transcription factor Sox9. A fraction of Sox9+ progenitors transiently expresses Neurogenin3 (Neurog3), and a subset of these becomes endocrine progenitors (Pan and Wright 2011). These progenitors then delaminate from the epithelium and differentiate into endocrine cells, including glucagon-producing α and insulin-producing β cells (Pan and Wright 2011). These differentiation events coincide with major morphological changes of the developing pancreatic progenitor epithelium, including lumen morphogenesis.
Close spatiotemporal association between pancreatic lumen formation and cell fate determination brings up the question of whether lumen morphogenesis impacts pancreatic fate. Indeed, loss of Cdc42 in the murine pancreas leads to failure to initiate pancreatic lumens and a concomitant increase in acinar cells at the expense of endocrine differentiation (Kesavan et al. 2009). The investigators noted that the resulting abnormal epithelial morphology increased exposure to laminin, which in turn promoted acinar fate. This work suggests that epithelial morphology and lumen formation profoundly influence pancreatic fate determination.
We and others previously characterized pancreatic lumen morphogenesis in the mouse embryo (Hick et al. 2009; Kesavan et al. 2009; Villasenor et al. 2010). This process can be categorized into four steps. First, microlumens initiate between epithelial cells as isolated foci in the embryonic day 10.5 (E10.5) stratified bud. Next, lumens progressively fuse into longer channels throughout the epithelium by E12.5. These channels take on a three-dimensional (3D) net-like morphology, called a plexus, that undergoes remodeling and extension between E13 and E18.5. By birth, the lumenal plexus resolves into a hierarchical tree. Recent work by Bankaitis et al. (2015) characterized two distinct compartments in the pancreas at E13.5–E18.5 based on their lumenal/tubular morphology. Regions at the center of the pancreas, which contain a lumenal plexus in the process of remodeling, were termed the core, while regions surrounding the core, which display ramifying branches, were termed the periphery. The core plexus progressively resolves into a ramifying tree at perinatal stages (Bankaitis et al. 2015). Furthermore, the investigators identified the core region as the site of endocrine differentiation. Hence, the core is a transiently formed region distinguished by lumen morphology and differs from the periphery in its cellular composition.
Although the core and periphery are morphologically distinct, it is unknown how these two regions form during pancreas development or how their morphogenesis is influenced by initial lumen formation or subsequent remodeling. The functional relevance of morphogenesis of either region in endocrine differentiation also remains to be determined, as a model that alters either the core or the periphery has not yet been reported. Elucidation of these questions requires a better understanding of pancreatic lumen development.
We took a genetic approach to study pancreatic lumen development and its influence on endocrine cell fate. Given the function of the scaffolding molecule Afadin in kidney tubulogenesis, we hypothesized that Afadin may play a similar role during pancreas development (Takai et al. 2008; Yang et al. 2013; Marciano 2017). We further hypothesized that the Afadin mutant model could be used as a tool to manipulate pancreatic lumens to assess impact on cell fate, including endocrine cells. In this study, we found that pancreas-specific ablation of Afadin results in delayed and discontinuous lumen formation, most prominently in the tip epithelium. We show that Afadin regulates de novo lumen formation through Rab-dependent vesicular trafficking, which can be observed in the tip epithelium during midgestation. Interestingly, Afadin mutants form and resolve the central plexus relatively normally. Through time-lapse imaging and ex vivo studies, we show that, following initial lumen formation, lumen morphogenesis differs in the center versus the tips. In contrast to continuing tip de novo lumenogenesis, the central plexus takes shape by lumen extension and anastomosis, processes regulated by actomyosin. Codeletion of Afadin and the master actomyosin regulator RhoA leads to an arrest of central lumenogenesis, resulting in a discontinuous plexus that fails to resolve. This resolution failure causes perdurance of the pancreatic core plexus and an increase in endocrine differentiation of bipotential progenitors, leading to a dramatic increase in endocrine mass by birth. With these findings, we demonstrate that Afadin and RhoA functionally interact to direct morphogenesis of the pancreatic core, which determines endocrine mass.
Results
Afadin is required to form a continuous lumenal network in the developing pancreas
Given the requirement of Afadin in renal vesicle lumen formation, we asked whether it plays a similar role in pancreatic lumenogenesis. Afadin is expressed in many mouse tissues and localizes to the apical membrane in epithelial cells of the renal vesicle (Yang et al. 2013). However, its expression has not been reported in the pancreas. We examined Afadin in the embryonic pancreatic bud and found that it localized to cell–cell boundaries, marked by the adherens junction protein E-cadherin, throughout development (Fig. 1A–D′). At E10.5, Afadin localized along the primary central lumen (data not shown) and showed enrichment at scattered membrane domains within the stratified epithelium prior to formation of lumens (Fig. 1A,A′). Afadin was also observed adjacent to foci expressing the lumenal glycoprotein Mucin1 (Muc1), suggesting that it marked nascent lumens (Fig. 1B,B′).
Figure 1.
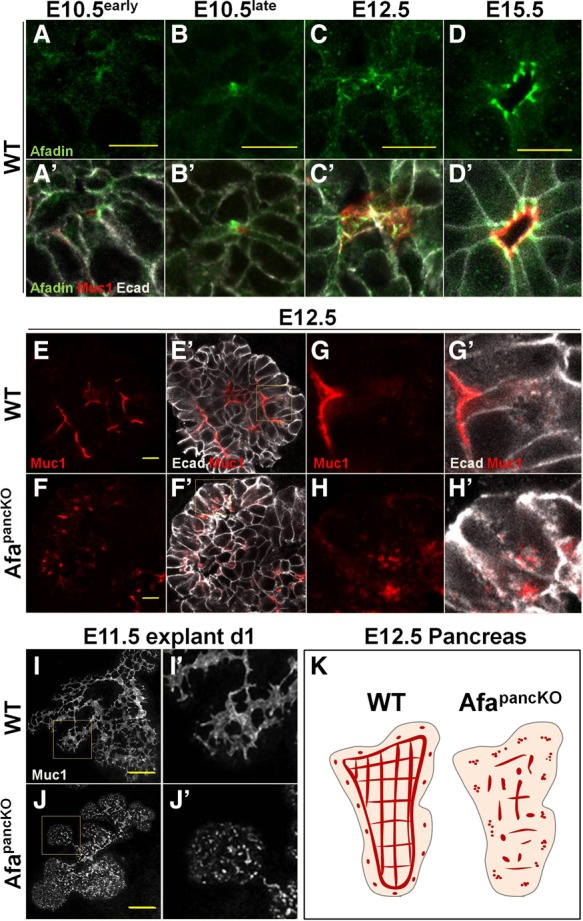
The early Afadin mutant pancreas fails to form a continuous lumenal network. (A–D′) Paraffin sections from wild-type embryonic pancreata at the indicated stages were immunostained for Afadin, Muc1 (lumens), and E-cadherin (Ecad; epithelial membranes). “Early” and “late” refer to the timing with respect to microlumen formation based on Muc1 enrichment. Bars, 10 µm. (E–H′) Paraffin sections from E12.5 wild-type and Afadinf/fPdx1Cre (AfapancKO) embryonic pancreata were immunostained for Muc1 and E-cadherin. Representative images from five independent experiments are shown. n = 5 embryos per genotype. The insets in E′ and F′ are shown at higher magnification in G–H′. Bars, 10 µm. (I–J′) E11.5 wild-type and AfapancKO pancreatic explants were cultured for 1 d (d1) and whole-mount-immunostained for Muc1. Representative images from six independent experiments are shown. n = 7 embryos per genotype. The insets in I and J are shown at higher magnification in I′ and J′, respectively. Bars, 100 µm. (K) Schematic illustrating lumen discontinuity in the AfapancKO pancreas compared with the continuous lumen network (or plexus) in wild type at E12.5. Red lines represent continuous or semicontinuous lumens, while red dots indicate discontinuous lumen foci. The gray-outlined pink region represents the pancreatic epithelium. See also Supplemental Figure S1.
At E12.5, Afadin became enriched at junctions flanking the apical membrane (Fig. 1C,C′) and restricted to subapical junctions at lumenal boundaries at later stages (Fig. 1D,D′). Superresolution microscopy revealed localization of Afadin at sites with progressive enrichment of E-cadherin or the tight junction (TJ) protein ZO-1, pointing to a function in junction formation in the pancreas, as described previously in other systems (Supplemental Fig. S1A–B′; Takahashi et al. 1998; Ikeda et al. 1999; Zhadanov et al. 1999). Interestingly, a subset of Afadin foci did not colocalize with either junctional protein, suggesting potential junction-independent functions prior to lumen formation (Supplemental Fig. S1A–B′, arrows). Thus, Afadin localizes to scattered membrane domains and nonjunctional foci prior to lumen formation and becomes restricted to apical junctions after lumen formation in the developing pancreas. Such dynamics of subcellular localization suggested a potential function of Afadin in pancreatic lumen formation.
To test whether Afadin is required for formation of pancreatic lumens, we deleted Afadin using Pdx1Cre (AfapancKO) in the early embryonic pancreas (Gu et al. 2002). Deletion efficiency was assessed by Afadin immunostaining and was mostly complete by E11.5 (Supplemental Fig. S1C–D″). In the absence of Afadin, the developing pancreas failed to form continuous lumens, as shown by Muc1 immunostaining, while control tissue exhibited fine continuous lumens (Fig. 1E–F′). Furthermore, Muc1 was frequently observed in intracellular punctae within mutant epithelial cells but not in controls (Fig. 1G–H′). Whole-mount immunostaining revealed discontinuous Muc1 foci in the E12.5 AfapancKO pancreas, in contrast to a mostly continuous network or a plexus in controls (Supplemental Fig. S1E,F, arrows). Ex vivo, lumen defects of the mutants were evident in E11.5 pancreatic explants following 1 d in culture (Fig. 1I–J′; Supplemental Fig. S1E,F). Unlike lumen formation failure, epithelial branching occurred normally in the AfapancKO pancreas (Supplemental Fig. S1G,H). The schematic in Figure 1K illustrates lumen discontinuity in the AfapancKO pancreas at E12.5 as opposed to a continuous plexus in wild type. Thus, Afadin is required for lumen continuity in the developing pancreas.
Tip, but not central, lumens develop via Afadin-dependent mechanisms
To determine whether lumen defects in AfapancKO persisted, we analyzed later stages of pancreatic development. We examined epithelial branch tips as well as the center of the epithelium. AfapancKO pancreas failed to form normal lumens by E15.5 in the tip epithelium (Fig. 2A,B). Most cells exhibited extensive intracellular Muc1 and discontinuous Muc1 foci between cells, whereas control tissue almost always displayed a single continuous lumen per tip (Fig. 2A,B). Quantification of lumen continuity using E14.5 thick sections revealed that ∼80% of control branch tips had a single lumen per tip, while only ∼20% of the AfapancKO tips had one lumen, and the rest had at least two discontinuous Muc1 foci per tip (Supplemental Fig. S2A). Consistent with the E15.5 results, the mutant pancreas failed to form normal lumens at epithelial tips and developed abnormally enlarged lumens after birth (Fig. 2C,D; Supplemental Fig. S2B,C).
Figure 2.
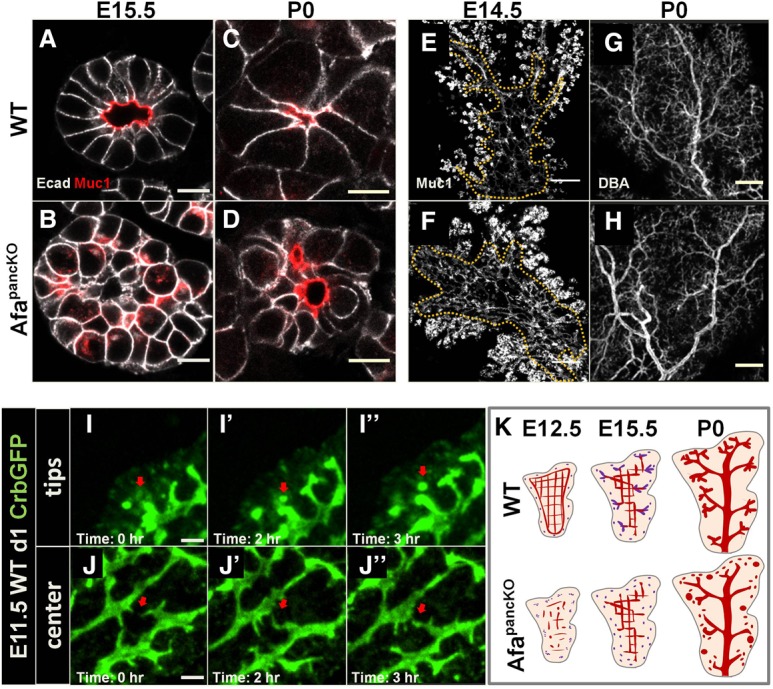
Afadin regulates de novo lumenogenesis in the tips but is dispensable for remodeling in the center. (A–D) Paraffin sections of wild-type and AfapancKO pancreata at the indicated stages were immunostained for Muc1 (lumens) and E-cadherin (Ecad; epithelial membranes). Bars, 10 μm. (A,B) Representative images from five independent experiments are shown. n = 5 embryos per genotype. (C,D) Representative images from three independent experiments are shown. n = 3 embryos per genotype. (E–H) E14.5 and postnatal day 0 (P0) wild-type and AfapancKO pancreata were whole-mount-immunostained for Muc1 to mark lumens and for Dolichos biflorus agglutinin (DBA) to mark large and mid-sized branching ducts. Summed slices of the same thickness are shown for each whole mount. A dotted line shows the central region in E and F. Head regions are shown in G and H. Representative images from three independent experiments are shown. n = 3 embryos per genotype. Bars: E,F, 100 µm; G,H, 200 µm. (I–J″) Snapshots from time-lapse imaging of E11.5 wild-type pancreatic explants on day 1 (d1) in culture. The pancreata express Crumbs3GFP (Crb3-GFP) that localizes to lumens and apical vesicles. A red arrow indicates a de novo lumen formation event in I–I″ and a lumen fusion event in J–J″. Representative images from eight independent experiments are shown. n = 8 embryos. Bars, 10 µm. (K) Schematic illustrating lumen morphogenesis events throughout development in wild type and defects in AfapancKO. At E12.5, central lumens (red) in the wild-type pancreas are in plexus conformation, while de novo lumens (purple) are forming in the tip epithelium. At midgestation, central lumens remodel into branches, while tip lumens fuse with these branches (purple branches). De novo lumens continue to form in the tip epithelium (purple dots). By birth, a hierarchical tree is formed. The AfapancKO pancreas fails to form continuous lumens at E12.5 in both central and tip epithelia. By midgestation, the mutant central lumens remodel, while de novo lumens continue to form but remain discontinuous. This gives rise to an AfapancKO prenatal pancreas with a relatively normal central lumenal tree but discontinuous and enlarged tip lumens. See also Supplemental Figure S2 and Supplemental Movies S2 and S3.
Defects in the center of the AfapancKO epithelium evolved differently than in the tips despite Afadin being expressed equally in both regions (Supplemental Fig. S1C). Whole-mount immunostaining revealed a continuous but denser plexus in the bud center at E14.5 relative to controls, suggesting remodeling defects (Fig. 2E,F). By birth, the pancreatic center recovered from these defects in most cases (Fig. 2G,H). Furthermore, intracellular Muc1 accumulation was never observed in the AfapancKO epithelial center, unlike in the epithelial tips (Supplemental Fig. S2D–E′). Therefore, the AfapancKO pancreas fails to establish continuous lumens in the tip epithelium by perinatal stages, while the central lumens largely recover from discontinuities and remodeling defects by late gestation.
The striking difference between the tip and central lumen phenotypes in AfapancKO pancreata suggested that center and tip epithelia might undergo different lumen morphogenesis events at a given time. Previous studies have shown that lumen formation in other systems can occur de novo through biogenesis of a new apical membrane and/or via extension of existing lumens (Sigurbjornsdottir et al. 2014). The presence of intracellular Muc1 accumulation in the tip, but not the central, epithelial cells in the AfapancKO pancreas suggested de novo lumen formation in epithelial tips at the time of deletion (E11.5 onward) (Fig. 2A–D; Supplemental Fig. S2D–E′). We further hypothesized that central lumen morphogenesis might instead rely on lumen extension following initial lumen formation. To test this idea, we performed time-lapse imaging on pancreatic explants using the Crumbs3-GFP (Crb3-GFP) reporter line that marks the apical membrane and apically targeted vesicles (Pan et al. 2015).
Time-lapse imaging of E11.5 day 1 Crb3-GFP pancreata revealed striking differences between the central and tip regions. Extensive vesicular trafficking was directed toward the apical membrane in cap cells of the pancreatic tips and indicated biogenesis of a new apical membrane (Supplemental Movie S1). Furthermore, we observed gradual accumulation of Crb3-GFP at discrete foci in the pancreatic tips that eventually became isolated lumens, suggestive of de novo lumenogenesis events (Fig. 2I–I″, arrows; Supplemental Movie S2). In contrast, the central epithelium exhibited little noticeable vesicular trafficking at the stages examined (Supplemental Movie S1). Additionally, existing central lumens underwent dynamic changes, where they extended and made connections with each other (Fig. 2J–J″, arrows; Supplemental Movie S3). These observations suggest that once lumens have appeared and coalesced into a plexus, central lumen morphogenesis occurs primarily through lumen extension as opposed to continuing de novo lumenogenesis.
These findings are in line with a model in which the developing pancreas undergoes a centripetal wave of lumen formation and remodeling. Following initial lumen formation in the early E10.5 bud, the central epithelium forms new lumens by extending existing lumens, while the tip epithelium continues to form lumens de novo as it grows outward. This gives rise to a central plexus and transiently disconnected tip lumens (E12.5). The central plexus then remodels and resolves into branching lumens as tip lumens fuse with extending branches from the center (E13.5–18.5). This way, the central plexus and tip lumens remodel into a ramifying tree (postnatal day 0 [P0]). In the absence of Afadin, tip lumens fail to form in a timely manner and remain discontinuous. However, the central lumens can remodel and resolve relatively normally (Fig. 2K; Supplemental Fig. S2F).
Afadin is required for coordinated apical membrane formation in epithelial tips
Lumen formation requires apico–basal polarization, and integrity of the apical membrane relies on correct positioning of TJs (Sigurbjornsdottir et al. 2014). In the pancreas, as cells undergo apico–basal polarization, they adopt a bottle shape via apical constriction and organize into rosette structures prior to lumen initiation (Villasenor et al. 2010). Given persistent lumen discontinuity in the AfapancKO tip epithelium, we asked whether apico–basal polarization or apical membrane formation was affected in the absence of Afadin.
To examine apical polarity, we assessed the Par complex proteins Par3 and aPKC in the E12.5 pancreas and found that they localized to the apical membrane at the center of rosettes (Fig. 3A,C). In contrast, at the onset of the Afadin mutant phenotype (E12.5), the AfapancKO epithelium exhibited enrichment of these molecules at scattered membrane domains and in cytoplasmic punctae (Fig. 3B,D). We also evaluated TJ localization in AfapancKO and observed ZO1 mislocalized at lateral membranes, in contrast to its normal apical enrichment (Fig. 3E–F′). Furthermore, we observed large vesicles carrying the apical glycoprotein Podocalyxin (Podxl) localized in close proximity to ZO1 enrichment sites in AfapancKO as opposed to its normal lumenal localization (Fig. 3F, arrow). Interestingly, E-cadherin was cleared from the membrane between ectopic ZO1 enrichment sites and apposed vesicles, suggesting that this apico–lateral domain is aberrantly determined as the apical membrane in the absence of Afadin (Fig. 3F′, arrow).
Figure 3.
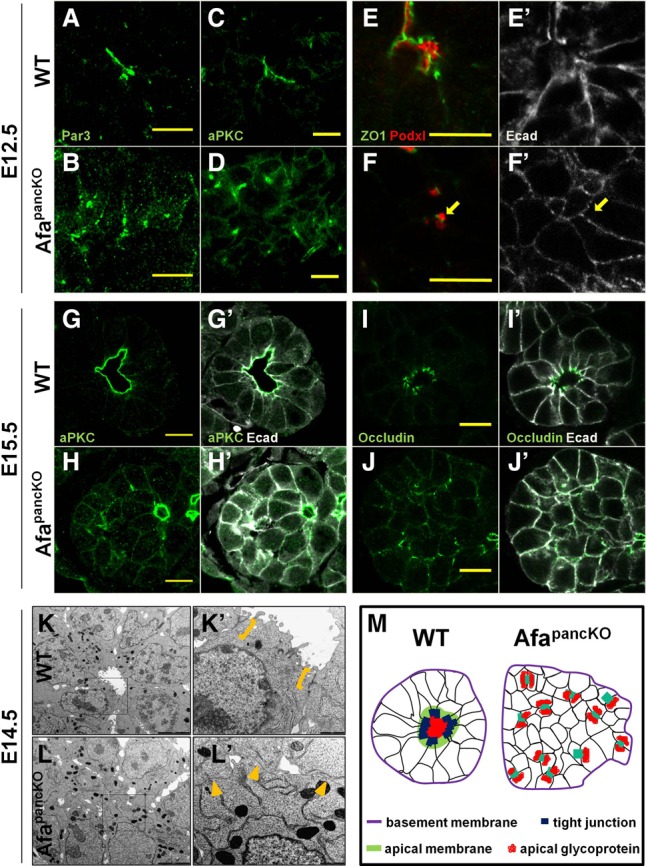
Apical membrane formation is not coordinated in the Afadin mutant tip epithelium. (A–J′) Paraffin sections of wild-type and AfapancKO embryonic pancreata at the indicated stages immunostained for apical determinants Par3 and aPKC, TJ proteins ZO1 and occludin, and apical glycoprotein Podxl. The arrow in F′ indicates the membrane region cleared from E-cadherin. Bars, 10 μm. (A–H′) Representative images from three independent experiments are shown. n = 3 embryos per genotype. (I–J′) Representative images from six independent experiments are shown. n = 6 embryos per genotype. (K–L′) Transmission electron microscopy images of sections of E14.5 wild-type and AfapancKO pancreata. The insets in K and L are shown at high magnification in K′ and L′. Brackets indicate electron-dense TJ structures at the subapical membrane. Arrowheads designate regions of cell–cell contact in the rosette center lacking TJ structures. Representative images from two independent experiments are shown. n = 2 embryos per genotype. Bars: K,L, 5 µm; K′,L′, 1 µm. (M) Schematic of wild-type and AfapancKO tip epithelia. (Purple) Basement membrane; (black) epithelial membranes; (red) lumenal glycoproteins; (green) apical determinant enrichment (wild type only); (dark blue) TJ enrichment (wild type only); (light blue) colocalization of TJ proteins and apical determinants (AfapancKO only). See also Supplemental Figure S3.
At later stages, both apical determinants Par3 and aPKC retained their discontinuous membrane/cytoplasmic localization in AfapancKO tips (Fig. 3G–H′; Supplemental Fig. S3A–B′). Likewise, the transmembrane TJ molecule occludin exhibited mislocalization to lateral membranes, similar to the early ZO1 pattern (Fig. 3I–J′). Apical constriction and rosette structure were also largely absent in AfapancKO tip cells (Fig. 3G–J′). Importantly, the basal lamina appeared grossly normal in mutants, as depicted by laminin staining (Supplemental Fig. S3C,D). These findings suggest that AfapancKO cells are able to determine basal polarity; however, they fail to properly segregate apical from lateral/junctional membranes.
Transmission electron microscopy (TEM) analysis at E14.5 confirmed that AfapancKO tip epithelial cells fail to form a single lumen per tip and revealed an absence of electron-dense TJ structures at cell–cell boundaries facing the rosette centers (Fig. 3K–L′, arrowheads). Moreover, similar to immunostainings, abnormal TJ localization could be observed at scattered membrane domains in AfapancKO, which occasionally flanked small lumen-like slits between cells (Supplemental Fig. S3E, arrowheads). The schematic in Figure 3M illustrates mislocalized apical membranes, loss of rosette structure, and lumen discontinuity in AfapancKO pancreatic tips compared with controls. Thus, AfapancKO pancreata fail to align central TJs and coordinate apical membrane formation across cells of the tip epithelium.
Afadin regulates lumen formation through Rab8-mediated apical membrane docking
To investigate how coordination of apical membrane formation fails in the absence of Afadin, we examined apical membrane biogenesis in the AfapancKO tips. Intracellular glycoprotein accumulation in close proximity to the membrane (Figs. 1H,H′, 3F,F′) suggested that AfapancKO tip cells may incur failure of apical membrane biogenesis. In fact, we frequently observed abnormally large vesicles upon loss of Afadin, which were positive for Muc1 (Fig. 4A–D′) and ZO1 (Fig. 4E–H′). Notably, these vesicles localized to the membrane domain near the putative center of rosettes, where apical membranes normally form. These observations indicated that apical vesicle docking/tethering at the plasma membrane is likely to be defective in the absence of Afadin.
Figure 4.
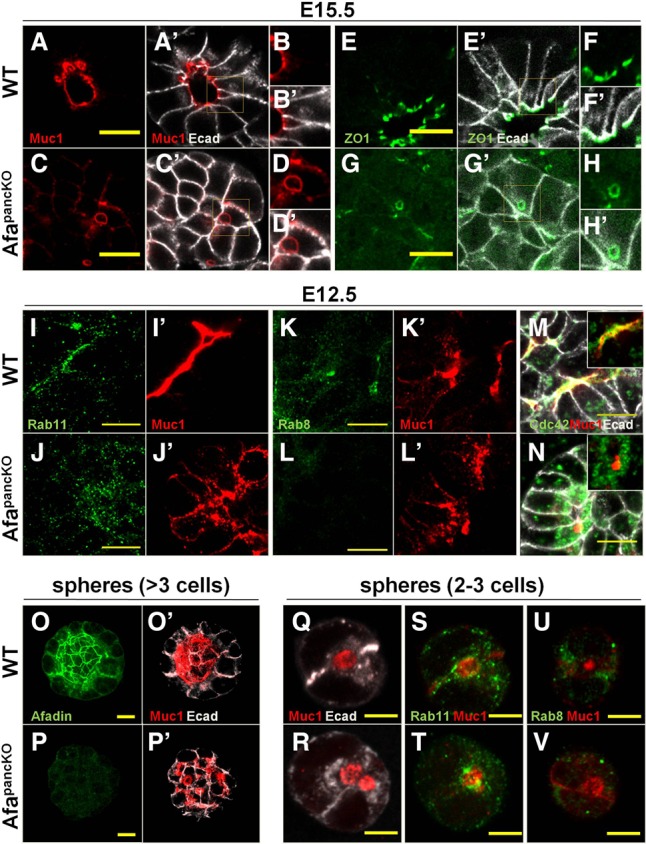
Afadin regulates Rab8-mediated apical docking during de novo lumen formation. (A–N) Paraffin sections of E15.5 and E12.5 wild-type and AfapancKO pancreata immunostained for lumenal Muc1; TJ protein ZO1; vesicular GTPases Rab11, Rab8, and Cdc42; and E-cadherin (Ecad; epithelial membranes). Insets in A, A′, C, C′, E, E′, G, and G′ are shown at higher magnification in B, B′, D, D′, F, F′, H, and H′, respectively. Insets in M and N show Cdc42 and Muc1 staining, excluding E-cadherin. Bars, 10 μm. (A–D′) Representative images from five independent experiments are shown. n = 5 embryos per genotype. (E–H′) Representative images from four independent experiments are shown. n = 4 embryos per genotype. (I–N) Representative images from three independent experiments are shown. n = 3 embryos per genotype. (O–V) Pancreatic sphere assays seeded with cells from E10.5–E11.5 wild type and AfapancKO. After 5 d (O–P′) or 1 d (Q–V) in culture, spheres were immunostained. Representative images from four independent experiments are shown. n = 4 embryos per genotype. Bars: O–P′, 10 µm; Q–V, 5 µm. See also Supplemental Figure S4 and Supplemental Movies S4 and S5.
Apical membrane docking relies on the small GTPases Rab8, Rab11, and Cdc42 in various systems, including in the developing intestine and pancreas (Sato et al. 2007; Kesavan et al. 2009; Bryant et al. 2010; Khandelwal et al. 2013). In vitro studies indicate that Rab11 activates Rab8, which in turn induces Cdc42 activity required for apical vesicle trafficking (Bryant et al. 2010). To determine whether Afadin regulates apical docking machinery, we tested subcellular localization of these factors in the control and AfapancKO developing pancreas. In line with their roles, Rab8, Rab11, and Cdc42 localized to the apical membrane in control tip epithelia at E14.5–E15.5 (Supplemental Fig. S4A–A′,C–C′,E). In contrast, apical membranes in AfapancKO displayed no enrichment of Rab11 or Cdc42 (Supplemental Fig. S4B–B′,F). Although Rab8 did mark the plasma membrane in the putative rosette center, it was not present on Muc1-positive vesicles in AfapancKO, indicating its loss from apically targeted vesicles (Supplemental Fig. S4D,D′).
As later defects may result secondarily from failure in lumen formation as opposed to directly from Afadin loss, we analyzed localization of the docking machinery at the onset of the AfapancKO phenotype. Similar to later stages, early lumens were enriched for Rab11, Rab8, and Cdc42 in controls (Fig. 4I,I′,K,K′,M). In AfapancKO, Rab11 grossly localized to the same cellular domain as Muc1 vesicles (Fig. 4J,J′), in contrast to its absence at later stages. This suggested that the later apical absence of Rab11 is likely to be a result of apical membrane defects and not directly caused by Afadin loss. In contrast, Rab8 failed to apically enrich and Cdc42 did not colocalize with Muc1-positive vesicles in early mutant cells (Fig. 4L,L′,N). Analysis of immunofluorescence intensity revealed no overall decrease in levels of these proteins (data not shown). These findings suggest that Afadin is needed for proper localization of Rab8 and Cdc42 but not their upstream regulator, Rab11.
To dissect the role of Afadin in localizing the apical docking machinery with high spatial detail and temporal control, we made use of an ex vivo cell-based system. Pancreatic sphere assays developed by Greggio et al. (2013) provide a simplified 3D progenitor expansion system that allows the study of lumen formation. We validated the suitability of this system by analyzing Afadin expression and lumen formation in wild-type and AfapancKO pancreas-derived spheres. AfapancKO spheres that lacked Afadin expression failed to form proper continuous lumens, recapitulating the in vivo phenotype (Fig. 4O–P′). At the two-cell sphere stage, a single Muc1-positive lumen formed between cells in wild-type spheres (Fig. 4Q). We termed this the nascent lumen. AfapancKO spheres failed to form a proper nascent lumen and localized Muc1-positive vesicles to the cell–cell boundaries, similar to the in vivo phenotype (Fig. 4R). Rab11 showed enrichment at the nascent lumen and Muc1-positive vesicles in wild-type and AfapancKO spheres, respectively (Fig. 4S,T). Rab8 localized to cell–cell boundaries flanking the nascent lumen in wild-type spheres; however, it failed to enrich in AfapancKO spheres (Fig. 4U,V). Together, these findings suggest that Afadin is necessary for Rab8-mediated vesicular docking during lumen initiation.
To gain insight into vesicle accumulation in Afadin mutants, we performed time-lapse imaging of control and mutant pancreatic explants. We induced deletion of Afadin in the early pancreatic epithelium using Sox9CreERT2 at E8.5 (AfapancindKO) and analyzed Crb3-GFP-positive E11.5 pancreata following 1 d in culture (Kopp et al. 2011). The control epithelium maintained lumens >3 h (Supplemental Fig. S4G–G″; Supplemental Movie S4). In contrast, AfapancindKO pancreata showed intracellular apical cargo accumulation in the form of large vesicles, which often dissociated into smaller vesicles over time (Supplemental Fig. S4H–H″; Supplemental Movie S5). Taken together, our data suggest that the Afadin mutant tip epithelium fails to carry out exocytosis due to missing components of the vesicle docking machinery during de novo lumen formation (Supplemental Fig. S4I).
Central lumen morphogenesis depends on cooperative activity of Afadin and RhoA
Although we show that Afadin directs de novo lumen formation in the pancreas tips, its loss within central lumens resulted in only mild morphogenesis defects. Furthermore, AfapancKO central lumens did not exhibit apical vesicle accumulation. Therefore, our findings indicated that once lumens have formed in the central pancreas, they do not rely on further de novo lumenogenesis. These lumens continue to develop via extension and interconnection of existing lumens. RhoA-directed actomyosin machinery is critical to the lumen extension process, and we showed recently that endothelial lumen formation relies on actomyosin activity (Lee and Kolodziej 2002; Denker et al. 2015; Barry et al. 2016). We thus hypothesized that lumen morphogenesis within the central epithelium would also require actomyosin contractility.
To test the function of actomyosin in pancreatic lumen formation, we inhibited components of this machinery (actin and myosin II) in pancreatic explant cultures and assessed lumens. Indeed, lumen connections were not maintained when myosin II activity was inhibited using blebbistatin (Fig. 5A–B′, arrowsheads; Supplemental Movies S6, S7). Longer exposure to blebbistatin caused lumens to become discontinuous and enlarged (Fig. 5C,D). Inhibiting actin polymerization resulted in similar but more robust defects, likely due to actomyosin-independent functions of actin (Supplemental Fig. S5A,A′).
Figure 5.
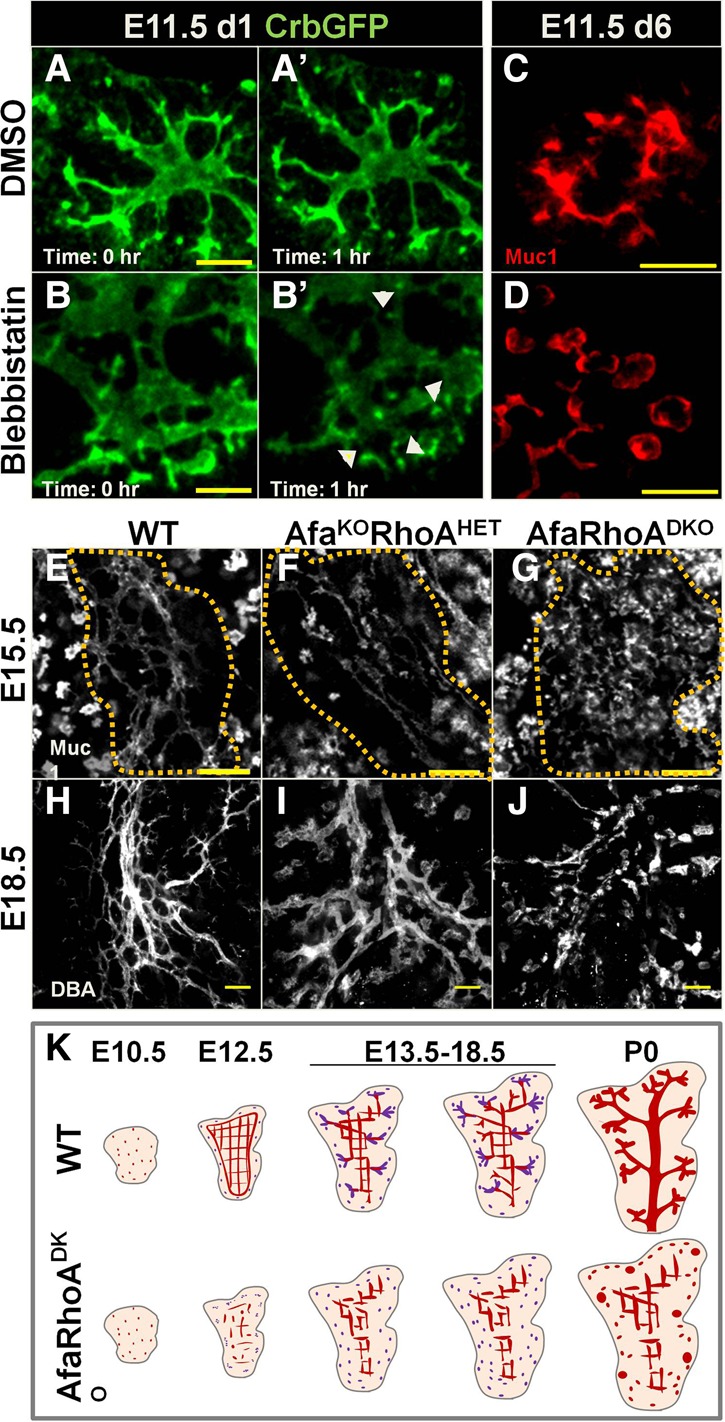
Lumen morphogenesis in the pancreas center depends on the cooperative activity of Afadin and RhoA. (A–B′) Snapshots from time-lapse imaging of DMSO- or blebbistatin-treated Crb3-GFP E11.5 pancreatic explants on day 1 (d1) in culture. Arrowheads in B′ indicate points of disconnection in lumens. Representative images from three independent experiments are shown. n = 3 embryos. Bars, 20 µm. (C,D) E11.5 wild-type pancreatic explants treated with DMSO or blebbistatin for 5 d, fixed, and immunostained for Muc1. Representative images from two independent experiments are shown. n = 8 embryos. Bars, 20 µm. (E–J) E15.5 wild-type, Afadinf/fRhoAf/+Pdx1Cre (AfaKORhoHET), and Afadinf/fRhoAf/fPdx1Cre (AfaRhoDKO [DKO]) pancreata whole-mount-stained for Muc1 (all lumens) or DBA (large and mid-sized branching ducts, excluding tips). Dotted lines in E–G delineate the central region. (H–J) Head regions at E18.5. Summed slices of the same thickness were used for each whole mount. (E–G) Representative images from three independent experiments are shown. n = 5 embryos per genotype. (H–J) Representative images from three independent experiments are shown. n = 4 embryos per genotype. Bars: E–G, 50 µm; H–J, 100 µm. (K) The schematic illustrates lumen morphogenesis defects in DKO. At E10.5, lumens are discontinuous in both wild-type and DKO pancreata. Later (E12.5), the DKO pancreas fails to form a continuous plexus. While wild-type central lumens remodel at midgestation, DKO central lumens remain discontinuous and fail to undergo remodeling into branches. As a result, central lumens of the prenatal DKO pancreas remain in plexus conformation. In the DKO tip epithelium, de novo lumens (purple) remain discontinuous and become enlarged perinatally. See also Supplemental Figure S5 and Supplemental Movies S6 and S7.
Since the AfapancKO pancreas largely recovered from central lumen defects by birth and since lumen continuity depended on actomyosin, we asked whether actomyosin-dependent mechanisms could compensate for Afadin loss in central lumen remodeling. To test this, we ablated the actomyosin regulator RhoA simultaneously with Afadin in a pancreas-specific manner (Afadinf/f;RhoAf/f;Pdx1Cre [DKO]). This double depletion led to death by P9, in contrast to normal survival of Afadin or RhoA single mutants. At birth, DKO pancreata were smaller compared with controls (Supplemental Fig. S5B,C′). Glucose measurements in newborns revealed that blood glucose levels of DKOs were significantly lower than their control littermates (Supplemental Fig. S5D).
We next asked whether DKO mice suffered from worsened pancreatic lumen discontinuity. Indeed, DKO pancreata displayed exacerbated lumen defects compared with AfapancKO. Starting as early as E12.5, DKOs exhibited severe lumen discontinuity, incomplete epithelial destratification, and defective apical membrane formation (Supplemental Fig. S5E–M′). Similar to AfapancKO, DKOs exhibited no initial branching defects (data not shown). Unlike AfapancKO, DKO pancreata showed persistent central lumen morphogenesis failure. Central lumens of E15.5 DKO pancreata were discontinuous and enlarged, in contrast to the continuous plexus in controls or AfapancKO (Fig. 5E–G).
AfaKORhoAHET pancreata also exhibited a worsened central lumen phenotype compared with AfapancKO, albeit considerably milder than DKO (Fig. 5F). Notably, AfaHETRhoAKO pancreata displayed no abnormalities (Supplemental Fig. S5N,O). In line with this, no effect on lumen formation or continuity was observed upon depletion of either RhoA (data not shown) or the Rho kinase ROCK (Supplemental Fig. S5P,Q), both of which are activators of myosin II. Thus, the actomyosin machinery is required for pancreatic lumen continuity but is likely regulated by additional upstream factors independent of Rho signaling.
At E18.5, a hierarchical ductal tree could be distinguished in controls where Dolichos biflorus agglutinin (DBA) staining labeled large and mid-sized branching ducts (Fig. 5H). AfaKORhoAHET pancreata displayed slight discontinuity as well as nonhierarchical arrangement, with all lumens increased in diameter compared with controls (Fig. 5H,I). E18.5 DKO pancreata had severe discontinuity and failed to form proper ducts (Fig. 5J). Therefore, Afadin and RhoA act together to ensure central lumen morphogenesis and remodeling in the developing pancreas (Fig. 5K).
Arrest of lumen morphogenesis results in perdurance of the pancreatic core
Our analysis of lumen development in DKOs revealed a failure of continuous lumen formation throughout the pancreas and, as a result, widespread arrest in central lumen morphogenesis and remodeling. Importantly, recent work by Bankaitis et al. (2015) described two distinct and transiently formed regions in the developing pancreas: the core and the periphery. The periphery consists of terminal branch tips, where lumens have completed remodeling, while the core consists of lumens interconnected as a plexus with ongoing lumen remodeling. As the plexus remodels into hierarchical ducts and branches (plexus-to-duct remodeling), the core region disappears (Bankaitis et al. 2015). The schematic in Figure 6A illustrates this transient core–periphery distinction. An example of a core region is shown by DBA staining on the E18.5 wild-type pancreas, where the core can be architecturally distinguished by looping of nonhierarchical lumens (of similar or irregular thickness) (Fig. 6B). Looping can be identified by tracing lumens: Whenever a lumen being traced connects with an already traced lumen, this architecture is defined as looping. To understand how lumen morphogenesis arrest affected core–periphery regionalization, we examined DKO pancreata stained for Muc1 and DBA. At E18.5, the wild-type pancreatic trunk consisted of one main duct and smaller hierarchically organized branches; however, a narrow region of core lumens around the main duct could be distinguished (Fig. 6C). In contrast, the DKO pancreatic trunk consisted of nonhierarchical longitudinal tubes connected by horizontal lumens with a few discontinuities (Fig. 6D). The DKO core region was therefore expanded compared with the wild type (Fig. 6C,D). Likewise, the control pancreatic head consisted of a main duct, hierarchically organized branching lumens, and only a few distributed core regions (Fig. 6E). In contrast, DKO lumens appeared nonhierarchical and mostly discontinuous (Fig. 6F). Analysis with Filament Tracer (Imaris) enabled tracking and generation of a continuous counterpart of the lumens in the DKO pancreatic head (Fig. 6E–F″). This analysis showed that most DKO lumens were organized into loops, although they remained discontinuous (Fig. 6F′,F″). Based on the lack of hierarchical/ramifying organization and the presence of loop conformation, we quantified the area of the core region and found that it is dramatically expanded in the DKO (Fig. 6G). Ex vivo pancreatic cultures showed a similar expansion of the core region (Fig. 6H–I′). Thus, DKO lumens fail to undergo plexus remodeling and remain as nonhierarchical lumens, leading to prolonged maintenance of the core region.
Figure 6.
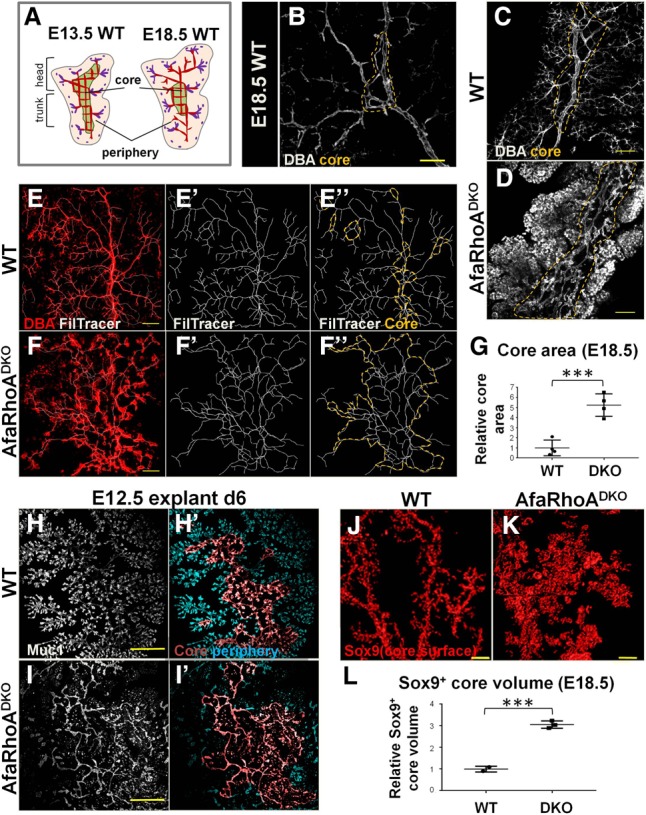
Arrest of lumen morphogenesis leads to ectopic perdurance of the core. (A) The schematic illustrates the distinction between the core and the periphery in the murine embryonic pancreas and the transient nature of the core–periphery regionalization. The pink region corresponds to the periphery with lumen branches, while the green region represents the core with an interconnected plexus (red) mid-remodeling. The central plexus undergoes remodeling to form branches as the tips continue to form de novo lumens and fuse with the lumen branches arising from the center. Red lumens in the periphery represent lumens arising from the remodeling of central lumens, and purple lumens represent lumens arising de novo. Purple dots indicate the most recent transiently discontinuous de novo lumens. Plexus remodeling takes place gradually, encompassing inward to the center of the tissue as more central plexus lumens become hierarchical branches. Thus, the core region becomes gradually smaller relative to the periphery. (B) E18.5 wild-type pancreata whole-mount-immunostained for DBA to mark large and mid-sized ducts. A maximum projection of the z-stack is shown. The dotted line outlines core lumens. Bar, 40 µm. (C,D) E18.5 wild-type and DKO pancreata whole-mount-immunostained for DBA. The pancreatic trunk is shown. Summed slices (50 µm) through z-stack are shown. The dotted line outlines core lumens. Representative images from four independent experiments are shown. n = 4 embryos per genotype. Bars, 100 µm. (E–F″) E18.5 wild-type and DKO pancreata whole-mount-immunostained for DBA. A pancreatic head is shown. (E,F) The maximum projection of z-stacks is shown; the Filament Tracer function in Imaris tracks lumens and creates a continuous counterpart of lumens. (E″,F″) The orange dotted line indicates the core region. Representative images from four independent experiments are shown. n = 4 embryos per genotype. Bars, 150 µm. (G) Quantification of the core area using Filament Tracer analysis on pancreata from wild-type (n = 3 embryos) and DKO (n = 3 embryos). Representative images are shown in E–F″. (H–I′) E12.5 wild-type and DKO pancreatic explants cultured for 6 d (d6), fixed, and immunostained for Muc1. Lumens were traced using Filament Tracer. Core and peripheral lumens are highlighted in different colors by using the Surface function in Imaris. Representative images from three independent experiments are shown. n = 4 embryos per genotype. Bars, 300 µm. (J,K) E18.5 wild-type and DKO pancreata whole-mount-immunostained for Sox9. The Surface function in Imaris was used to visualize the total Sox9+ surface area in the core region. Head region of the pancreas is shown. Representative images from three independent experiments are shown. n = 4 embryos per genotype. Bars, 150 µm. (L) E18.5 core volume calculated on Sox9 whole-mount-stained wild-type (n = 2) and DKO (n = 3) pancreas head regions using surface function in Imaris. Representative images are shown in J and K. See also Supplemental Figure S6.
Developing pancreatic lumens are lined by a Sox9+ bipotential population that gives rise to endocrine and mature ductal cells (Pan and Wright 2011). Given the aberrant maintenance of the core plexus in DKOs due to blocked lumen resolution, we predicted an expansion in the Sox9+ population associated with core lumens. Indeed, using whole-mount Sox9/DBA costaining, we found a dramatic expansion of the Sox9+ population associated with the core plexus (Fig. 6J–L). Therefore, arrest in lumen morphogenesis leads to ectopic maintenance of the core and expansion of the core-resident bipotential progenitor population.
Core perdurance promotes endocrine commitment and increases endocrine mass
Next, we asked how defects in the DKO pancreas affected differentiation of pancreatic lineages. Strikingly, DKOs displayed a significant increase in endocrine mass by birth. Both whole-mount and section insulin/glucagon stainings revealed dramatic expansion of endocrine cells in the DKO pancreas at P0 (Fig. 7A–D). Both β and α cells were increased in number, and islet morphology was severely disrupted (Fig. 7B,D). DKO explants similarly exhibited increased endocrine mass (Fig. 7E,F). Quantification of endocrine volume in vivo and ex vivo revealed a significant increase in DKOs (Fig. 7G,H). Notably, the relative contribution of each endocrine lineage to the islet mass was not altered compared with controls (Supplemental Fig. S6A–D). Unlike endocrine mass, acinar and ductal differentiation remained unaffected in DKOs (Supplemental Fig. S6E,F).
Figure 7.
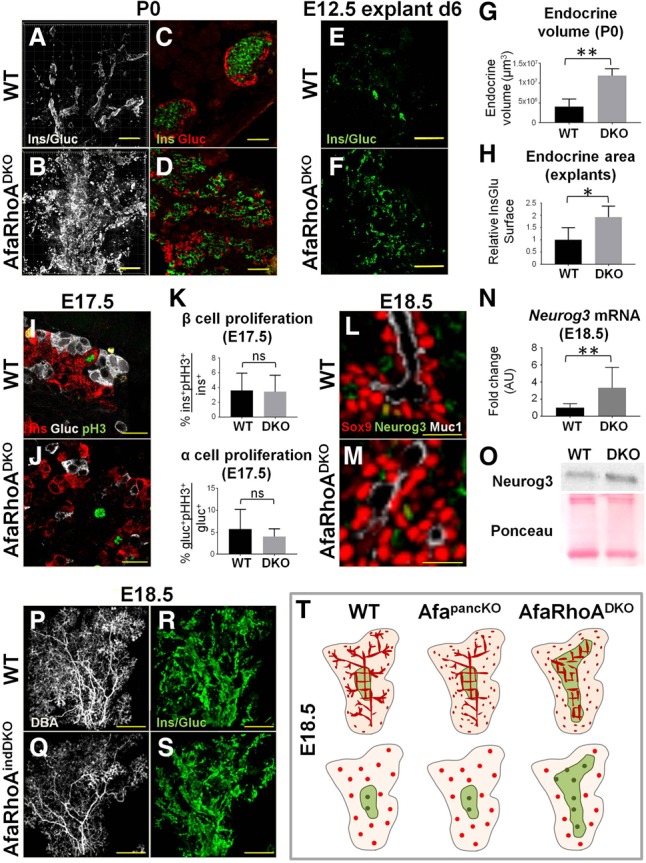
Core perdurance promotes endocrine commitment in bipotential progenitors. (A,B) P0 wild-type and DKO pancreata whole-mount-immunostained for insulin and glucagon in the same channel as a proxy for endocrine content. Maximum projections of the head regions are shown. Representative images from four independent experiments are shown. n = 6 embryos per genotype. Bars, 200 µm. (C,D) P0 wild-type and DKO pancreas paraffin sections immunostained for insulin and glucagon to label β and α cells, respectively. Representative images from three independent experiments are shown. n = 3 embryos per genotype. Bars, 50 µm. (E,F) E12.5 wild-type and DKO pancreatic explants cultured for 6 d (d6), fixed, and immunostained for insulin/glucagon in the same channel. Representative images from three independent experiments on pancreata from wild-type (n = 11) and DKO (n = 3) embryos are shown. Bars, 300 µm. (G) Quantification of endocrine volume at P0 using the Surface function in Imaris. n = 4 embryos per genotype. Representative images are shown in A and B. (H) The Surface function in Imaris was used to quantify endocrine area on E12.5 wild-type and DKO pancreatic explants cultured for 6 d from wild-type (n = 3) and DKO (n = 3) embryos. Representative images are shown in E and F. (I,J) Paraffin sections of E17.5 wild-type and DKO pancreata immunostained for insulin (Ins), glucagon (Gluc), and phosphorylated histone H3 (pH3) to assess endocrine cell proliferation. Representative images from three independent experiments are shown. n = 3 embryos per genotype. Bars, 20 µm. (K) α-Cell and β-cell proliferation assessed on E17.5 wild-type and DKO pancreas sections stained for insulin, glucagon, and pH3. Data are represented as mean (SD). (ns) Nonsignificant. (L,M) E18.5 wild-type and DKO pancreata whole-mount-immunostained for Sox9 (bipotential progenitors), Neurog3 (endocrine progenitors), and Muc1 (lumens). A visual section through the whole mount is shown. Representative images from three independent experiments are shown. n = 3 embryos per genotype. Bars, 20 µm. (N) Quantitative PCR (qPCR) data from E18.5 wild-type and DKO pancreata show fold change in mRNA levels for Neurog3 wild-type (n = 10) and DKO (n = 4) embryos. Data are shown as mean (SD). (O) Western blotting from E18.5 wild-type and DKO pancreata using a Neurog3 antibody. Ponceau staining is shown as a loading control. Representative images of membranes from two independent experiments are shown. n = 3 embryos per genotype. (P–S) E18.5 wild-type and tamoxifen-induced Afadinf/fRhoAf/fPdx1CreERT2 (AfaRhoindDKO) pancreata whole-mount-stained for DBA (large and mid-sized branching ducts) and insulin/glucagon (in the same channel). Maximum projections of the head regions are shown. Representative images from three independent experiments are shown. n = 3 embryos per genotype. Bars, 300 µm. (T) The schematic illustrates core–periphery regionalization (top row) and the resulting cellular differentiation phenotype at E18.5 (bottom row) in AfapancKO and DKO pancreata compared with the wild-type. (Top row) The core region (green) and the periphery (pink). Peripheral lumen discontinuity is designated by isolated apical/lumenal foci (red). The lumen plexus in the core is normal in AfapancKO but remains discontinuous and fails to resolve in DKO. As a result, the core region perdures in the DKO pancreas. (Bottom row) Bipotential (red) and endocrine (green) progenitors. Progenitor distribution is similar in wild-type and AfapancKO pancreata. In contrast, most bipotential progenitors reside in the core in DKOs due to perdurance of this region. As a result, DKOs generate more endocrine progenitors. See also Supplemental Figures S6 and S7.
To understand how the DKO pancreas promotes endocrine differentiation, we asked what causes the increase in endocrine cell numbers. We tested whether this increase derives from an increase in the number of Sox9+ bipotential progenitors or Neurog3+ committed endocrine progenitors or the proliferation of differentiated endocrine cells. DKO pancreata showed no significant increase in endocrine cell proliferation as assessed by insulin/glucagon/phosphorylated histone H3 (pH3) immunostaining on sections at E16.5 and E17.5 (Fig. 7I–K; data not shown). Quantification of whole-mount stains and sections at E16.5, E17.5, and E18.5 also showed no difference in the total (core- and periphery-resident) number of Sox9+ cells in DKO pancreata compared with controls (data not shown). Neurog3 immunostaining showed grossly normal numbers of endocrine progenitors in any given field of view within the core area in DKOs (Fig. 7L,M). However, the overall core area was expanded in DKOs.
Given that the core has been identified as the birthplace of endocrine cells, we asked whether core perdurance in DKOs can promote endocrine fate. We reasoned that if the increase in endocrine mass is due to the core acting as a supportive microenvironment, we might expect no difference in Neurog3+ cell numbers in a given area but an increase in overall Neurog3+ cell number in DKOs. To quantify gross Neurog3 expression in the pancreas, we assessed mRNA levels by quantititaive PCR (qPCR). Indeed, qPCR analysis revealed significant elevation in Neurog3, and not Sox9, expression in DKO pancreata (Fig. 7N; Supplemental Fig. S6G). Neurog3 protein levels were also elevated in the E18.5 DKO pancreas (Fig. 7O), in line with increased overall endocrine progenitor numbers.
In order to assess the commitment capacity of Sox9+ bipotential progenitors toward endocrine lineage in DKOs, we calculated the percentage of Neurog3+ cells out of the total Sox9+ population (termed endocrine yield). When analyzed separately for the core and periphery, the endocrine yield was not altered in DKOs relative to controls at E18.5 (representative images in Fig. 7L,M; Supplemental Fig. S6H). As reported previously (Bankaitis et al. 2015), core bipotential progenitors did show a higher endocrine yield than the periphery-resident population in both controls and DKOs (Supplemental Fig. S6H). The endocrine yield (or volume) was not altered before E18.5 in the DKOs (Supplemental Fig. S6I–L; data not shown). Together, these data suggest that the observed endocrine increase (Fig. 7A–H) is not due to the altered differentiation capacity of either the core-resident or the periphery-resident bipotential population but the perdurance of the high-endocrine-yielding core-resident bipotential cells.
To confirm that core perdurance results in an endocrine mass increase, we examined the small percentage of AfapancKO pancreata with core perdurance (AfapancKO severe) resembling the DKO phenotype (Supplemental Fig. S6M–O). Supporting our hypothesis, only those AfapancKO with core perdurance, but not those without, displayed an increase in endocrine mass (Supplemental Fig. S6P–R). This correlation strongly suggested that core perdurance leads to increased endocrine differentiation. Altogether, these findings indicate that progenitors have a higher potential to give rise to endocrine cells when exposed to the core environment rather than the periphery, thereby attributing niche properties to the core.
To rule out the possibility that Afadin and/or RhoA act cell-intrinsically to direct endocrine differentiation, and independent of core morphogenesis, we carried out inducible deletion of Afadin and RhoA at the onset of endocrine differentiation (E14.5) using Pdx1CreErt2 (AfaRhoAindDKO). Deletion of Afadin in epithelial cells was evident by the loss of Afadin from Muc1-positive lumens (Supplemental Fig. S6S,T). AfaRhoAindDKO pancreata exhibited lumen defects only in the outermost tip epithelium (i.e., the growing tips) (Supplemental Fig. S6U–V′) and displayed normal core lumen morphology (Fig. 7P,Q). AfaRhoAindDKO pancreata had normal endocrine mass, suggesting that Afadin and RhoA do not function cell-autonomously to regulate endocrine mass (Fig. 7R,S; Supplemental Fig. S6W–X′). Therefore, Afadin or RhoA do not regulate endocrine mass through an endocrine progenitor cell-intrinsic mechanism but through core morphogenesis.
Together, these findings uncover the reliance of endocrine cell differentiation on its cellular context. We propose a model for lumen morphogenesis defects caused by the absence of Afadin and RhoA and the resulting phenotypes in cellular differentiation (Fig. 7T). Only the DKO model exhibits a persistent central lumen morphogenesis failure. The resulting distribution of bipotential progenitors (Fig. 7T, red) and the endocrine progenitor population therein (Fig 7T, green) is critical to ultimate endocrine mass. Notably, the total number of bipotential progenitors remains the same in all models, as depicted by the equal summed number of Sox9+ and Neurog3+ cells. However, failure of central lumen morphogenesis in DKO leads to remodeling arrest and prolonged progenitor exposure to the core region relative to controls. This in turn leads to increased endocrine mass, as the core provides a niche for endocrine differentiation. Altogether, Afadin and RhoA control endocrine mass through remodeling of the core niche.
Discussion
Recent work by Bankaitis et al. (2015) distinguished core and periphery regions in the embryonic pancreas and characterized the core as the birthplace for endocrine progenitors. Here, we identify Afadin and RhoA as key regulators of pancreatic core and peripheral lumen formation and remodeling (Supplemental Fig. S7A). By generating novel mouse models of pancreatic lumen remodeling failure, we show that perdurance of the pancreatic core can drive increased endocrine differentiation. Overall, we show that Afadin and RhoA are required for normal remodeling dynamics of the pancreas core, which in turn functions as a niche for endocrine differentiation.
Our studies reveal that Afadin is a critical regulator of lumen morphogenesis during pancreas development. Previously, we showed that Afadin is required for kidney tubulogenesis (Yang et al. 2013). Together, these findings suggest that the role of Afadin in lumen formation is conserved across tissues. Furthermore, the delay in continuous lumen formation that we identified in the center of Afadin mutants closely resembles the lumen formation/fusion delay seen in the renal vesicle. These data indicate that Afadin is required for coordination of lumen initiation and fusion in both tissues. However, most cells can compensate for the loss of Afadin and eventually proceed through developmental events. Our double deletion of Afadin and RhoA shows that this compensation occurs through a RhoA-dependent cellular mechanism.
We also show that early branch formation is not affected by depletion of Afadin or of Afadin and RhoA combined. Our previous work showed that branch formation results from the splitting of branch tips and remodeling of the underlying epithelium (Villasenor et al. 2010). We found that, following the emergence of lumen defects in both AfapancKO and DKO pancreata, mutant branches progressively and abnormally enlarge. These findings suggest that underlying lumen continuity is required for proper branch formation.
In this study, we also show that formation of continuous pancreatic lumens occurs via apical membrane biogenesis in a manner that is exquisitely coordinated between epithelial cells. Afadin loss causes mislocalized apical domains relative to epithelial rosette structures. Interestingly, single cells are able to generate distinct apical and basal membrane regions but fail to synchronize and align with neighboring cells. Afadin thus promotes coordinated apical membrane formation across multicellular tubules. Such coordination may depend in part on timely lumen initiation. Indeed, Afadin loss leads to a delay in lumen initiation, which may result from subapical vesicle accumulation due to loss of Rab8 and Cdc42 recruitment to the preapical domain (Supplemental Fig. S7B). Thus, our data suggest a novel role for Afadin in Rab8-dependent apical vesicular trafficking.
In line with these findings, Rab8 mutant epithelia display intracellular accumulation of apical components, very similar to AfapancKO (Sato et al. 2007). Further studies are needed to determine how Afadin regulates apical docking. Given that Afadin localizes subapically, binds F-actin, and carries a myosin-like Dilute domain, it may act as an anchor for apically directed vesicles at the subapical actin network (Mandai et al. 2013). Alternatively, it might help regulate exocytic Rab activation or expression levels. Interestingly, ADIP (Afadin DIL domain-interacting protein), which binds Afadin, has been shown to be essential for Rab8 enrichment during cilia formation and therefore might provide a link between Afadin and Rab8 recruitment (Klinger et al. 2014). We note that Afadin is the epithelial counterpart of Rasip1, which we showed to be an endothelial-specific molecule required for continuous vascular lumen formation (Xu et al. 2011). Our previous findings also suggest that Rasip1 is involved in vesicular trafficking (Barry et al. 2016). Therefore, Afadin and Rasip1 may represent analogous molecules that carry out similar cellular functions to direct coordinated lumenogenesis in the epithelium and endothelium, respectively.
Recent work by Bankaitis et al. (2015) characterized the core and the periphery as distinct regions in the developing pancreas based on lumen architecture. Here, we further demonstrate that prior to core–periphery regionalization, early central and tip lumens start using distinct cellular and molecular mechanisms to develop once initial lumens are formed. We note that de novo lumens can also be observed at earlier stages (E10.0–E11.5) in the bud center (data not shown), suggesting a temporal centripetal wave of de novo lumen formation as the bud grows. However, at the stages that we focused on (E11.5–E18.5), during which endocrine cell differentiation accelerates, lumens in the central region form mainly through extension and fusion of existing lumens.
The spatiotemporal distinction of de novo lumen formation versus extension of lumens throughout pancreas development is also made evident by subapical vesicle trapping exclusively in epithelial tips of the E18.5 Afadin-RhoA-ablated pancreas (AfaRhoAindDKO). This suggests that timely and proper de novo lumen formation clearly requires Afadin-mediated vesicular trafficking. On the other hand, central lumen extension and remodeling depend on RhoA-mediated actomyosin contractility. Afadin activity can compensate for this function in the RhoA mutant pancreas. The molecular role of Afadin in this compensation remains to be determined. Afadin has been implicated in the regulation of actomyosin in other systems, indicating that it may regulate central lumen morphogenesis through actomyosin contractility, similar to its ortholog, Rasip1 (Sawyer et al. 2009, 2011). In line with this idea, blebbistatin treatment of pancreatic explants disrupts lumen continuity, while depletion of RhoA or ROCK does not, indicating likely contribution of other actomyosin regulators.
Proper lumen morphogenesis has been linked previously to pancreatic differentiation. (Kesavan et al. 2009; Bankaitis et al. 2015). However, which aspect of lumenogenesis regulates endocrine differentiation has remained elusive. Importantly, the study of the pancreas-specific Cdc42 mutant model revealed that loss of this molecule leads to a near-complete loss of lumens in the pancreas epithelium, disintegration of the epithelium, and suppression of endocrine fate. This morphogenetic phenotype appears severe enough to abolish core region formation in the Cdc42 mutant altogether. In contrast, the DKO model is able to initiate lumenogenesis with a delay but fails to undergo lumen extension, fusion, and, later, remodeling. Additionally, the core region displays distinct phenotypes in our two models (mild AfapancKO and DKO), enabling us to ask how core–periphery regionalization impacts cell fate. Our results suggest that peripheral lumen morphogenesis is dispensable for proper cellular differentiation, including acinar or ductal lineage. Given that endocrine cells emerge within the core, it is not unexpected that peripheral morphogenesis has little impact on this lineage. In contrast, arrest in lumen remodeling leads to perdurance of the core region in severe AfapancKO and DKO and results in increased endocrine differentiation. Other cells types, such as exocrine acinar and ductal cells, display no gross abnormalities in DKOs. Furthermore, lumen continuity per se is not required for differentiation of pancreatic lineages, as lumens display discontinuity in AfapancKO and DKO, but cell differentiation is not hampered in these models.
It remains to be determined how exactly the core supports endocrine differentiation: What cellular neighbors are required within the niche? What signals do they transmit? Importantly, continuous plexus conformation is not a required property of the core to act as a niche, suggesting that the core is unlikely to act through a lumen-derived signal. Our data indicate that total bipotential progenitor number or endocrine cell proliferation was not altered upon core perdurance in DKOs, while Neurog3 levels were elevated. We propose that this is due to prolonged exposure of progenitors to the core niche, likely inducing/stabilizing Neurog3 expression in bipotential progenitors. Supplemental Figure S7C illustrates the distinction between cellular organizations in a core versus a periphery epithelial unit. Closed conformation of lumens in the core may provide concentrated signaling that promotes endocrine fate. In DKOs, lumens are discontinuous, leading to a block in core plexus remodeling; however, overall cellular organization in the core unit is maintained.
Altogether, our study uncovers the pancreatic core as a functional niche for endocrine progenitors and advances our understanding of endocrine differentiation during pancreas development. Importantly, we uncouple lumen formation from lumen extension and plexus remodeling and show that endocrine progenitors are influenced by cellular context created by the latter processes. Elucidating properties of the pancreatic core will provide a better understanding of in vivo β-cell differentiation, which will improve in vitro β-cell differentiation attempts to treat diabetes patients.
Materials and methods
Mice and embryo handling
All animal husbandry was performed in accordance with protocols approved by the Univerity of Texas Southwestern Medical Center Institutional Animal Care and Use Committee. E10.5–E18.5 embryos were collected and dissected in PBS buffer. Pancreata were fixed in 4% paraformaldehyde (PFA) in PBS for 3 h at 4°C for section or whole-mount immunostaining. Tissues were washed in PBS, dehydrated, and stored in 70% ethanol at −20°C. Postnatal tissues were collected and fixed in a similar manner.
CD1 mice were used as wild types. Afadinf/f (Tanaka-Okamoto et al. 2011), Crb3-GFP (Pan et al. 2015), RhoAf/f (Barry et al. 2016), Pdx1Cre-early, Pdx1CreERT2, Sox9CreERT2, and R26-tdTomato+/+ were used for experiments in this study. Inducible simultaneous deletion of Afadin and RhoA was achieved through mating Afadinf/fRhoAf/f females with Afadinf/+RhoAf/+Pdx1CreERT2 males and gavaging of tamoxifen at a dose of 3 mg/40 kg to pregnant mothers at E14.5.
Sectioning
For paraffin sectioning, tissues were rinsed twice in 100% ethanol, twice in xylene for 30 min at room temperature, a mixture of 1:1 paraplast:xylene for 10 min at 60°C, and then a series of 100% paraplast at 60°C (McCormick Scientific). The tissues were then embedded in paraplast and sectioned at 10 µm with a Biocut 2030 microtome. SuperfrostPlus glass slides (Fisher) were used. For cryosectioning, tissues were rinsed in PBS and incubated in 30 % sucrose overnight at 4°C for cryoprotection. The next day, the tissues were rinsed in OCT twice for 30 min each at room temperature. The tissues were embedded in OCT, snap-frozen on dry ice, and sectioned at 30 µm using a Leica CM-3050S cryostat. SuperfrostPlus glass slides (Fisher) were used.
Immunostaining on sections
Paraffin sections were deparaffinized with xylene, rehydrated through ethanol wash series into PBS, permeabilized in PBS + 0.3%Triton X-100 for 10 min, and then treated with R buffer A (nuclear antigens) or R buffer B (cytoplasmic antigens) in a 2100 Retriever (Electron Microscopy Sciences). Slides were incubated in 5% normal donkey serum in PBS for at least 2 h and then in primary antibody overnight at 4°C, washed in PBS, and then incubated in secondary antibody for 2 h at room temperature. Slides were then washed again in PBS and mounted using Prolong Gold (with or without DAPI). For nuclear staining, slides were also incubated in DAPI in PBS for 10 min at room temperature prior to mounting. For cryosections, slides were baked for 10 min at 55°C, rinsed in PBS, and treated for antigen retrieval as above. The following steps were identical to paraffin section staining except that CAS Block (Invitrogen) was used for blocking and antibody steps. For thick cryosections (30 µm), all washes were performed in 0.1% TritonX in PBS. Slides were permeabilized in 0.3% TritonX in PBS for 30 min, blocked in CAS-Block, and incubated in primary antibody overnight at 4°C. The next day, slides were washed, incubated in secondary antibody for 2 h at room temperature, and washed again. Mounting and nuclear staining were performed as above.
To recognize Rab8, Rab8A and Rab8B antibodies were used in combination. Immunostaining for Cdc42 was performed using Tyramide Signal Amplification, where TSA kit 12 (Invitrogen) was used according to the manufacturer's directions.
The antibodies used were as follows: Afadin (1:100), E-cadherin (1:100), Muc1 (1:200), ZO1 (both antibodies at 1:100), DBA (1:300), Par3 (1:100), aPKC (1:100), Podxl (1:100), occludin (1:100), laminin (1:100), Rab11 (1:50), Rab8A (1:50), Rab8B (1:50), Cdc42 (1:50), Sox9 (1:300), insulin (sections: 1:200 [DAKO]; whole-mount: 1:200 [Cell Signaling]), glucagon (1:200), somatostatin (1:200), ghrelin (1:50), pH3 (1:100), Neurog3 (1:100), and amylase (1:300).
Ex vivo pancreas cultures and inhibitors
E11.5–E12.5 embryonic pancreata were dissected and explanted as described before (Petzold and Spagnoli 2012). Fibronectin-coated dishes were used. Culture medium consisted of 5% FBS, 1% penicillin/streptomycin, and gentamicin at 5 µg/mL. Blebbistatin, cytochalasin D, and Y-27632 were used at 10 µM, and 0.1% DMSO (carrier for blebbistatin and cytochalasin D) or dH20 (carrier for Y-27632) in culture medium was used as control. The day of dissection was taken as day 0. Drug treatments were initiated at day 1 and carried out for 3 h for explants fixed and stained at day 1 or for 5 d by supplying fresh medium every other day. For staining, explants were fixed in 4% PFA in PBS for 15 min at room temperature, washed in PBS, permeabilized in 0.3% Triton X-100 in PBS for 1 h, washed in PBS, and blocked using CAS-Block. Next, samples were incubated in primary antibody overnight at 4°C, washed in PBS the next day, incubated in secondary antibody overnight at 4°C, washed in PBS the next day, and mounted with Prolong Gold with DAPI on regular slides.
Superresolution microscopy
Paraffin sections immunostained as usual were imaged using a Nikon N-SIM superresolution system.
Whole-mount immunostaining
For embryonic tissues, immunostaining was carried out as described before (Marty-Santos and Cleaver 2016). For postnatal pancreata, tissues were washed in PBS following fixation and stored in PBS overnight at 4°C. Next, tissues were dehydrated into 50% methanol, incubated in 50% methanol for 1 h at room temperature, rehydrated back into PBS, and permeabilized in PBS + 1% Triton X-100 for 2 h at room temperature. Tissues were blocked in CAS-Block for at least 2 h and incubated in primary antibody overnight at 4°C. Tissues were then washed in PBS six times for 1 h each, incubated in secondary antibody overnight at 4°C, and washed in PBS five times for 30 min each. Next, tissues were dehydrated to 100% methanol, washed three times, and mounted in BABB (one-third benzyl alcohol and two-thirds benzyl benzoate) on slides using coverslip spacers.
Live imaging of ex vivo cultures
Afadinf/f females were mated with Afadinf/+Crumbs3GFP/+ Pdx1Cre-early males. For the indicated experiments, Afadinf/f females were mated with Afadinf/+Crumbs3GFP/+Sox9CreERT2 males, and pregnant mothers were gavaged with tamoxifen at a dose of 3 mg/40 kg at E8.5. E11.5 pancreatic explants positive for the reporter transgene Crumbs3GFP were used for visualization of apically directed vesicles, the apical membrane, and lumens (Pan et al. 2015). The explants were cultured on fibronectin-coated 35-mm glass-bottomed dishes (Greiner Bio-One, 627860). After 16–24 h in culture (indicated as day 1 [d1]), explants were time-lapse-imaged (with the z-stack covering the entire tissue) for 3 h every 3 min using an Andor spinning disk confocal microscopy. Data are shown as summed projections of the z stacks obtained using Imaris.
Hematoxylin and eosin (H&E) staining
Paraffin sections were incubated twice in xylene for 6 min, twice in 100 % ethanol for 2 min, and twice in 95% ethanol for 2 min and washed under running distilled water for 2 min and then with hematoxylin for 7 min. Next, slides were washed under running distilled water for 2 min, submerged in acid alcohol for 5 sec, washed under running distilled water for 5 min, and then stained with eosin for 3 min. The slides were then incubated twice in 95% ethanol for 2 min, twice in 100% ethanol for 2 min, and twice in xylene for 6 min and mounted using Permount.
TEM
TEM was carried out by the University of Texas Southwestern Electron Microscopy Core Facility per their standard protocols.
Pancreatic sphere assay and immunostaining
Sphere assays were carried out by mating Afadinf/fR26-tdTomato+/+ females with Afadinf/fPdx1Cre males, where all and only mutants express tdTomato, eliminating the genotyping step. E10.5–E11.5 pancreata were dissociated into single cells and cultured to form spheres in Matrigel as described by Greggio et al. (2013) with the following exceptions: Pancreata of the same genotypes were pooled together in each dissection. Dispase treatment and subsequent mesenchyme removal in PBS were each performed in a small drop of liquid in a regular dissection dish. The pancreata were transferred into 100 µL of DMEM + trypsin + EDTA using forceps, and 500 µL of DMEM + FBS was used to stop trypsinization. Cell clumps were broken by pipetting up and down, and cells were centrifuged at 300g for 5 min. Cells were then resuspended in an appropriate volume of culture medium described in the original protocol. The Matrigel–cell mix was seeded on coverslips in 96-well plates. Spheres were fixed after 5.5 d in culture. Immunostaining was performed as follows: Spheres were rinsed with PBS on ice and fixed in 4% PFA in PBS for 10 min at room temperature. Next, spheres were washed in PBS + 0.1% NP40 (PBSN) and permeabilized in PBSN for 15 min at room temperature. Spheres were blocked in CAS-Block for 30 min, incubated in primary antibody for 1 h, washed in PBSN, incubated in secondary antibody for 1 h, washed in PBSN again, and mounted upside down onto slides using Prolong Gold with DAPI.
Blood glucose measurement in newborns
P0 DKO litters were decapitated to obtain blood. A TRUEbalance glucose meter and test strips were used for plasma glucose level measurements (n = 30 wild type; n = 6 DKO; n = 4 AfaKORhoAHET; three litters). Statistical analysis was performed using nonparametric one-way ANOVA in GraphPad Prism software. P < 0.05 was considered statistically significant.
qPCR on E18.5 pancreata
Real-time qPCR was performed as described previously (Marty-Santos and Cleaver 2016). Briefly, 2 µg of total RNA was isolated from individual E18.5 mouse pancreata (dissected on ice) using RNeasy microkit (Qiagen). cDNA was synthesized using SuperScript III (Invitrogen). One microliter of cDNA in Power SYBR Green Master mix (Applied Biosystems) was used for real-time qPCR analysis (CFX96, Bio-Rad) of gene expression. Primers for Cyclophilin, GAPDH, Sox9, and Neurog3 have been described previously (Das et al. 2013; Caprioli et al. 2015; Marty-Santos and Cleaver 2016). Gene expression levels were determined by PCR reactions (30 sec at 95°C, 30 sec at 62°C, and 30 sec at 72°C for 35 cycles), and fluorescence was measured at 72°C. Gene expression levels were normalized to Cyclophilin or GAPDH, and the ΔΔCt method was used to calculate fold change. Data were collected from individual embryos (n = 3–10 per genotype), and samples were analyzed in triplicate. Data are presented as mean (SD).
Western blot on E18.5 pancreata
Individual E18.5 pancreata were dissected on ice under a dissection scope and homogenized in PBS with 10 µg/mL aprotinin, 10 µg/mL leupeptin, and 10 µg/mL pepstatin. Triton X-100 was added to each tube to a final concentration of 1%. Samples were frozen to –80°C, thawed, and centrifuged at 10,000g for 5 min. Supernatants containing protein were analyzed for protein quantification using Pierce BCA protein assay (ThermoScientific) on a 96-well plate. Thirty micrograms of total protein from individual pancreatic lysates was run on a Western blot. Neurog3 primary antibody (F25A1B3-c from Developmental Studies Hybridoma Bank) was used at 1:500.
Quantification and statistical analysis
Data are presented as mean (SD). E12.5 lumen discontinuity quantifications were performed on whole-mount-immunostained tissues, where pancreata were analyzed for isolated foci of Muc1 covering ≈80% of the tissue (n = 2 per genotype). Statistical analysis for lumen discontinuity assessment was performed using one-way ANOVA in GraphPad Prism software. E14.5 lumen discontinuity quantifications were performed on 30-µm cryosections, where at least 25 tips per pancreas were analyzed for isolated foci of Muc1 (n = 3 per genotype). The E18.5 core area was measured on head regions of pancreata whole-mount-stained for DBA. The core region was identified by using the Filament Tracer function in Imaris, and the area of this region was calculated using ImageJ measurements (n = 4 per genotype). The core area on E12.5 explants cultured for 6 d was identified by using Filament Tracer. The Surface function was used to create a separate channel for the core region with different color assignment, and this channel was overlaid with the nonmasked surface to visualize the core and the periphery in different colors. The E18.5 Sox9+ core volume was calculated on head regions of pancreata whole-mount-stained for DBA and Sox9. The core region was identified by using the Filament Tracer function in Imaris, and the Surface function was used to create a separate channel for the core region within the Sox9 channel. The core Sox9+ volume was calculated using the Surface function within this channel created for the core (n = 2 wild-type; n = 3 DKO). Quantification of endocrine volume at P0 was performed on whole-mount insulin/glucagon-immunostained tissues. Head regions of pancreata were analyzed. To account for the size difference between wild-type and DKO pancreata, z-stack images of whole head regions were taken regardless of the area that they occupied (two z-stacks at 10× magnification and 0.5× digital zoom for wild type and one z-stack at 10× magnification and 0.5× digital zoom for DKO). The Surface function of Imaris was used to calculate the overall volume of insulin/glucagon-positive cells. The summed volume of two z-stacks for wild type was compared with one z-stack for DKO. The endocrine area in explants was analyzed in whole-mount-immunostained tissue using the Surface function in Imaris (n = 11 wild type; n = 3 DKO). E17.5 endocrine proliferation was assessed on pH3/endocrine marker-costained sections, where at least five sections per pancreas were analyzed (n = 3 per genotype). The E18.5 endocrine yield in the core versus the periphery was analyzed on pancreata whole-mount-immunostained for Neurog3 and Sox9, where at least three different areas for each region (the core or periphery) were analyzed per tissue (n = 2 per genotype). Sox9+ cells were counted using the Spots function in Imaris. Neurog3+ cells were counted manually. The endocrine yield was calculated as Neurog3+ cell number divided by Sox9+ cell number multiplied by 100 to represent percentage. The overall endocrine yield at E15.5 or E17.5 was assessed on sections where Neurog3+ and Sox9+ were counted manually. Neurog3+ cell numbers were analyzed by counting Neurog3+ cells on sections and normalizing these numbers to the DAPI+ pancreatic surface area (the obtained numbers were multiplied by 106 for convenience). The endocrine volume at E15.5 was assessed using the same method as at E18.5 except the entire pancreas was analyzed. Unless multiple comparisons were made, all statistical analyses were performed using two-tailed unpaired Student's t-test in GraphPad Prism software. Multiple comparison analyses are described in the relevant Materials and Methods sections. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Thomas Carroll for Crb3GFP, Sox9CreERT2, and R26-tdTomato+/+ mice, and Doug Melton for Pdx1Cre-early and Pdx1CreERT2 mice. We are grateful to the MacDonald, Carroll, Olson, and Cleaver laboratories for invaluable discussions and assistance, and Michael Dellinger, Michael Buszczak, Arnaldo Carreira Rosario, and George Davis for critical reading of the manuscript. This work was supported by a Center for Regenerative Science and Medicine predoctoral fellowship and National Institutes of Health Institutional National Research Service Award (T32) 2T32GM008203-26A1 to D.B.A., R01DK099478 to D.K.M., and Cancer Prevention Research Institute of Texas RP110405, R01HL113498, and R01DK079862 to O.C.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.307637.117.
References
- Bankaitis ED, Bechard ME, Wright CVE. 2015. Feedback control of growth, differentiation, and morphogenesis of pancreatic endocrine progenitors in an epithelial plexus niche. Genes Dev 29: 2203–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry DM, Koo Y, Norden PR, Wylie LA, Xu K, Wichaidit C, Azizoglu DB, Zheng Y, Cobb MH, Davis GE, et al. 2016. Rasip1-mediated Rho GTPase signaling regulates blood vessel tubulogenesis via nonmuscle myosin II. Circ Res 119: 810–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, Datta A, Rodriguez-Fraticelli AE, Peranen J, Martin-Belmonte F, Mostov KE. 2010. A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol 12: 1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caprioli A, Villasenor A, Wylie LA, Braitsch C, Marty-Santos L, Barry D, Karner CM, Fu S, Meadows SM, Carroll TJ, et al. 2015. Wnt4 is essential to normal mammalian lung development. Dev Biol 406: 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Tanigawa S, Karner CM, Xin M, Lum L, Chen C, Olson EN, Perantoni AO, Carroll TJ. 2013. Stromal–epithelial crosstalk regulates kidney progenitor cell differentiation. Nat Cell Biol 15: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker E, Sehring IM, Dong B, Audisso J, Mathiesen B, Jiang D. 2015. Regulation by a TGFβ–ROCK–actomyosin axis secures a non-linear lumen expansion that is essential for tubulogenesis. Development 142: 1639–1650. [DOI] [PubMed] [Google Scholar]
- Greggio C, De Franceschi F, Figueiredo-Larsen M, Gobaa S, Ranga A, Semb H, Lutolf M, Grapin-Botton A. 2013. Artificial three-dimensional niches deconstruct pancreas development in vitro. Development 140: 4452–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. 2002. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 129: 2447–2457. [DOI] [PubMed] [Google Scholar]
- Hick AC, van Eyll JM, Cordi S, Forez C, Passante L, Kohara H, Nagasawa T, Vanderhaeghen P, Courtoy PJ, Rousseau GG, et al. 2009. Mechanism of primitive duct formation in the pancreas and submandibular glands: a role for SDF-1. BMC Dev Biol 9: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda W, Nakanishi H, Miyoshi J, Mandai K, Ishizaki H, Tanaka M, Togawa A, Takahashi K, Nishioka H, Yoshida H, et al. 1999. Afadin: a key molecule essential for structural organization of cell–cell junctions of polarized epithelia during embryogenesis. J Cell Biol 146: 1117–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JJ, Au M, Lu KH, Eshpeter A, Korbutt G, Fisk G, Majumdar AS. 2007. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells 25: 1940–1953. [DOI] [PubMed] [Google Scholar]
- Kelly OG, Chan MY, Martinson LA, Kadoya K, Ostertag TM, Ross KG, Richardson M, Carpenter MK, D'Amour KA, Kroon E, et al. 2011. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat Biotechnol 29: 750–756. [DOI] [PubMed] [Google Scholar]
- Kesavan G, Sand FW, Greiner TU, Johansson JK, Kobberup S, Wu X, Brakebusch C, Semb H. 2009. Cdc42-mediated tubulogenesis controls cell specification. Cell 139: 791–801. [DOI] [PubMed] [Google Scholar]
- Khandelwal P, Prakasam HS, Clayton DR, Ruiz WG, Gallo LI, van Roekel D, Lukianov S, Peranen J, Goldenring JR, Apodaca G. 2013. A Rab11a–Rab8a–Myo5B network promotes stretch-regulated exocytosis in bladder umbrella cells. Mol Biol Cell 24: 1007–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger M, Wang W, Kuhns S, Bärenz F, Dräger-Meurer S, Pereira G, Gruss OJ. 2014. The novel centriolar satellite protein SSX2IP targets Cep290 to the ciliary transition zone. Mol Biol Cell 25: 495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, Ma J, Sander M. 2011. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 138: 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kolodziej PA. 2002. The plakin Short Stop and the RhoA GTPase are required for E-cadherin-dependent apical surface remodeling during tracheal tube fusion. Development 129: 1509–1520. [DOI] [PubMed] [Google Scholar]
- Mandai K, Rikitake Y, Shimono Y, Takai Y. 2013. Afadin/AF-6 and canoe: roles in cell adhesion and beyond. Prog Mol Biol Transl 116: 433–454. [DOI] [PubMed] [Google Scholar]
- Marciano DK. 2017. A holey pursuit: lumen formation in the developing kidney. Pediatr Nephrol 32: 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty-Santos L, Cleaver O. 2016. Pdx1 regulates pancreas tubulogenesis and E-cadherin expression. Development 143: 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massumi M, Pourasgari F, Nalla A, Batchuluun B, Nagy K, Neely E, Gull R, Nagy A, Wheeler MB. 2016. An abbreviated protocol for in vitro generation of functional human embryonic stem cell-derived β-like cells. PLoS One 11: e0164457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. 2014. Generation of functional human pancreatic β cells in vitro. Cell 159: 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan FC, Wright C. 2011. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn 240: 530–565. [DOI] [PubMed] [Google Scholar]
- Pan XC, Schnell U, Karner CM, Small EV, Carroll TJ. 2015. A cre-inducible fluorescent reporter for observing apical membrane dynamics. Genesis 53: 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzold KM, Spagnoli FM. 2012. A system for ex vivo culturing of embryonic pancreas. J Vis Exp 10.3791/3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Mushiake S, Kato Y, Sato K, Sato M, Takeda N, Ozono K, Miki K, Kubo Y, Tsuji A, et al. 2007. The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature 448: 366–369. [DOI] [PubMed] [Google Scholar]
- Sawyer JK, Harris NJ, Slep KC, Gaul U, Peifer M. 2009. The Drosophila afadin homologue Canoe regulates linkage of the actin cytoskeleton to adherens junctions during apical constriction. J Cell Biol 186: 57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer JK, Choi W, Jung KC, He L, Harris NJ, Peifer M. 2011. A contractile actomyosin network linked to adherens junctions by Canoe/afadin helps drive convergent extension. Mol Biol Cell 22: 2491–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurbjornsdottir S, Mathew R, Leptin M. 2014. Molecular mechanisms of de novo lumen formation. Nat Rev Mol Cell Biol 15: 665–676. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Matsuo T, Katsube T, Ueda R, Yamamoto D. 1998. Direct binding between two PDZ domain proteins Canoe and ZO-1 and their roles in regulation of the Jun N-terminal kinase pathway in Drosophila morphogenesis. Mech Dev 78: 97–111. [DOI] [PubMed] [Google Scholar]
- Takai Y, Ikeda XA, Ogita H, Rikitake Y. 2008. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu Rev Cell Dev Biol 24: 309–342. [DOI] [PubMed] [Google Scholar]
- Tanaka-Okamoto M, Hori K, Ishizaki H, Itoh Y, Onishi S, Yonemura S, Takai Y, Miyoshi J. 2011. Involvement of afadin in barrier function and homeostasis of mouse intestinal epithelia. J Cell Sci 124: 2231–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villasenor A, Chong DC, Henkemeyer M, Cleaver O. 2010. Epithelial dynamics of pancreatic branching morphogenesis. Development 137: 4295–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Sacharidou A, Fu S, Chong DC, Skaug B, Chen ZJ, Davis GE, Cleaver O. 2011. Blood vessel tubulogenesis requires Rasip1 regulation of GTPase signaling. Dev Cell 20: 526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZF, Zimmerman S, Brakeman PR, Beaudoin GM, Reichardt LF, Marciano DK. 2013. De novo lumen formation and elongation in the developing nephron: a central role for afadin in apical polarity. Development 140: 1774–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhadanov AB, Provance DW, Speer CA, Coffin JD, Goss D, Blixt JA, Reichert CM, Mercer JA. 1999. Absence of the tight junctional protein AF-6 disrupts epithelial cell–cell junctions and cell polarity during mouse development. Curr Biol 9: 880–888. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
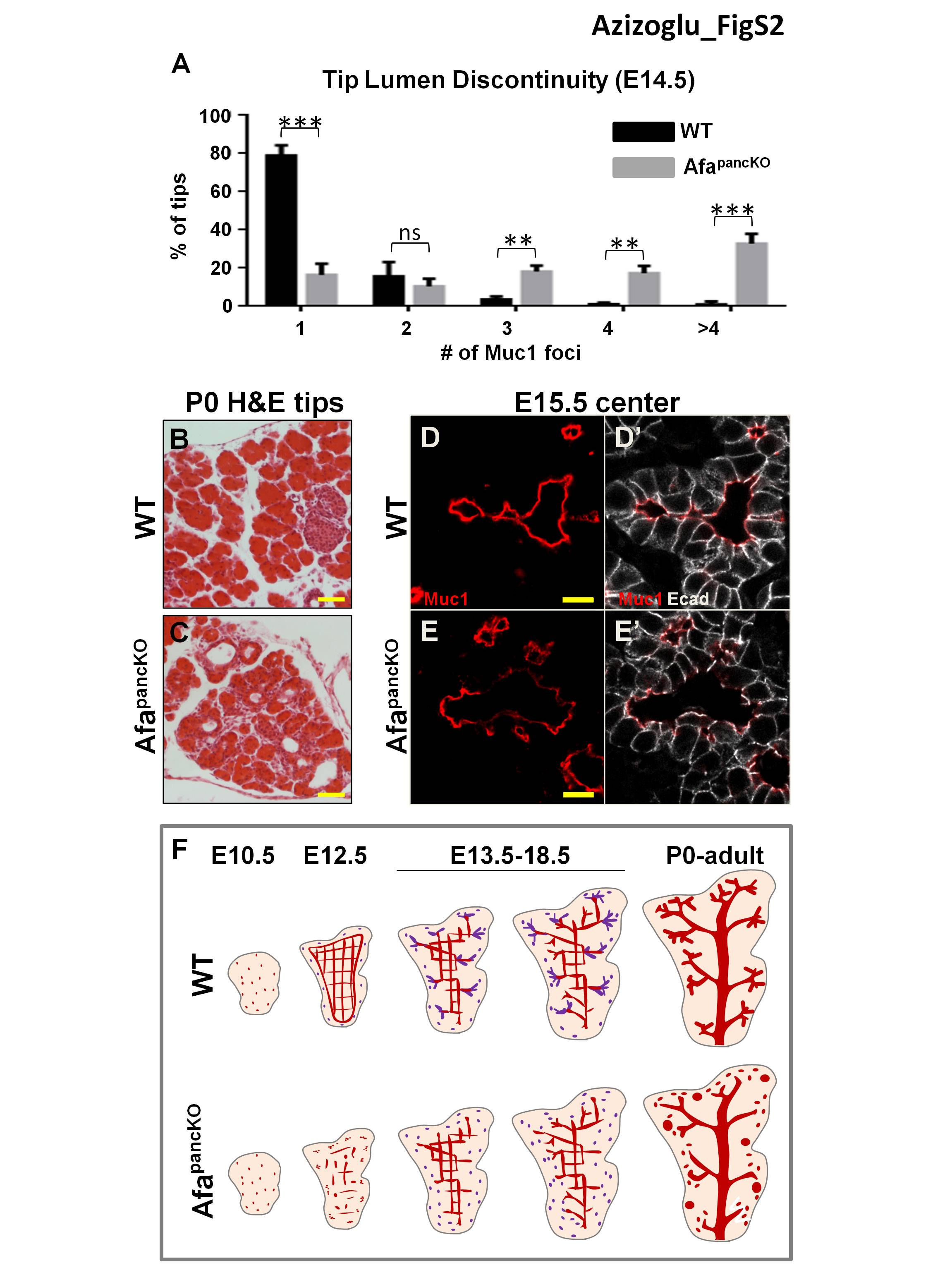{kind=link}
{kind=link}
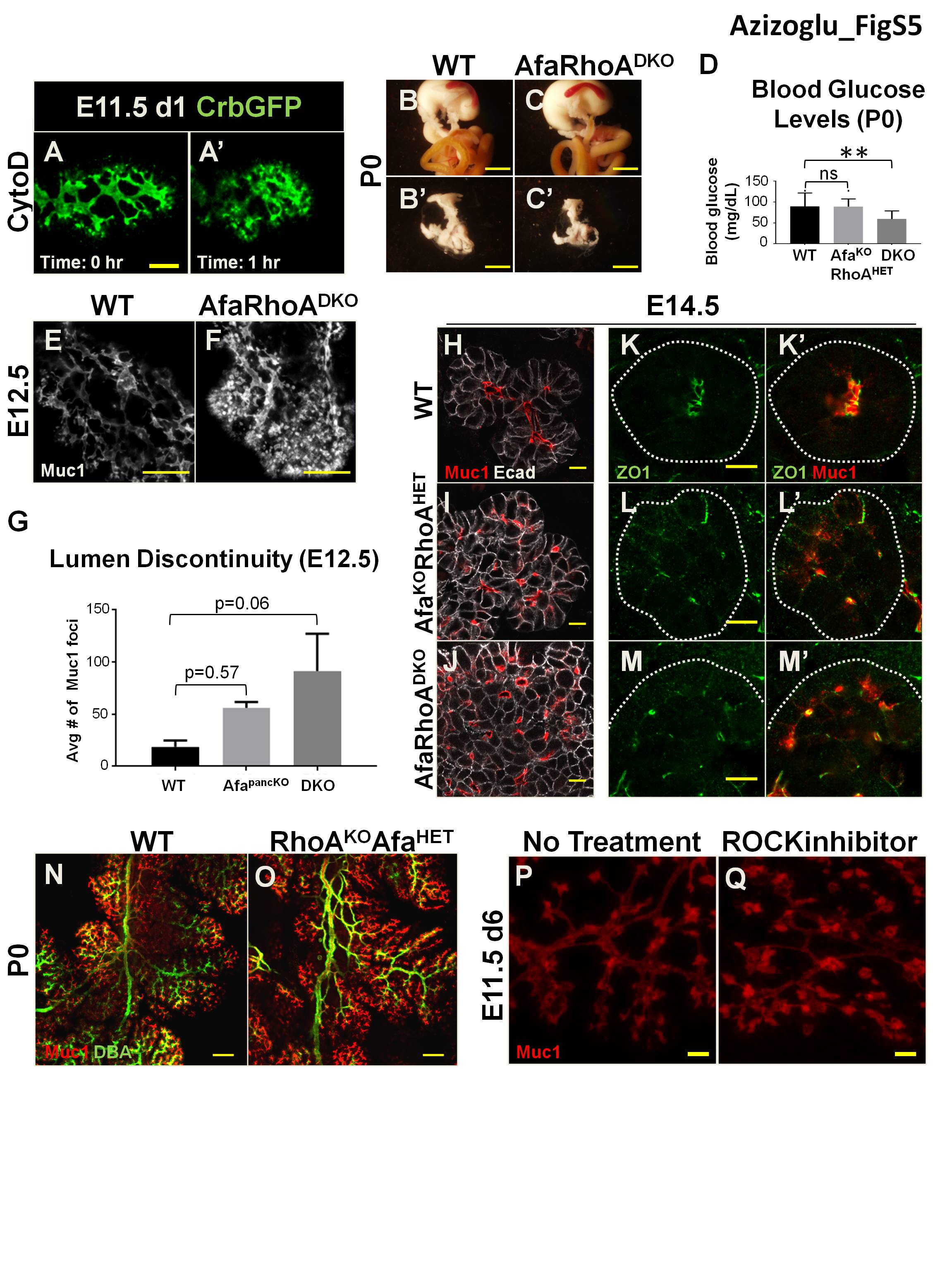{kind=link}
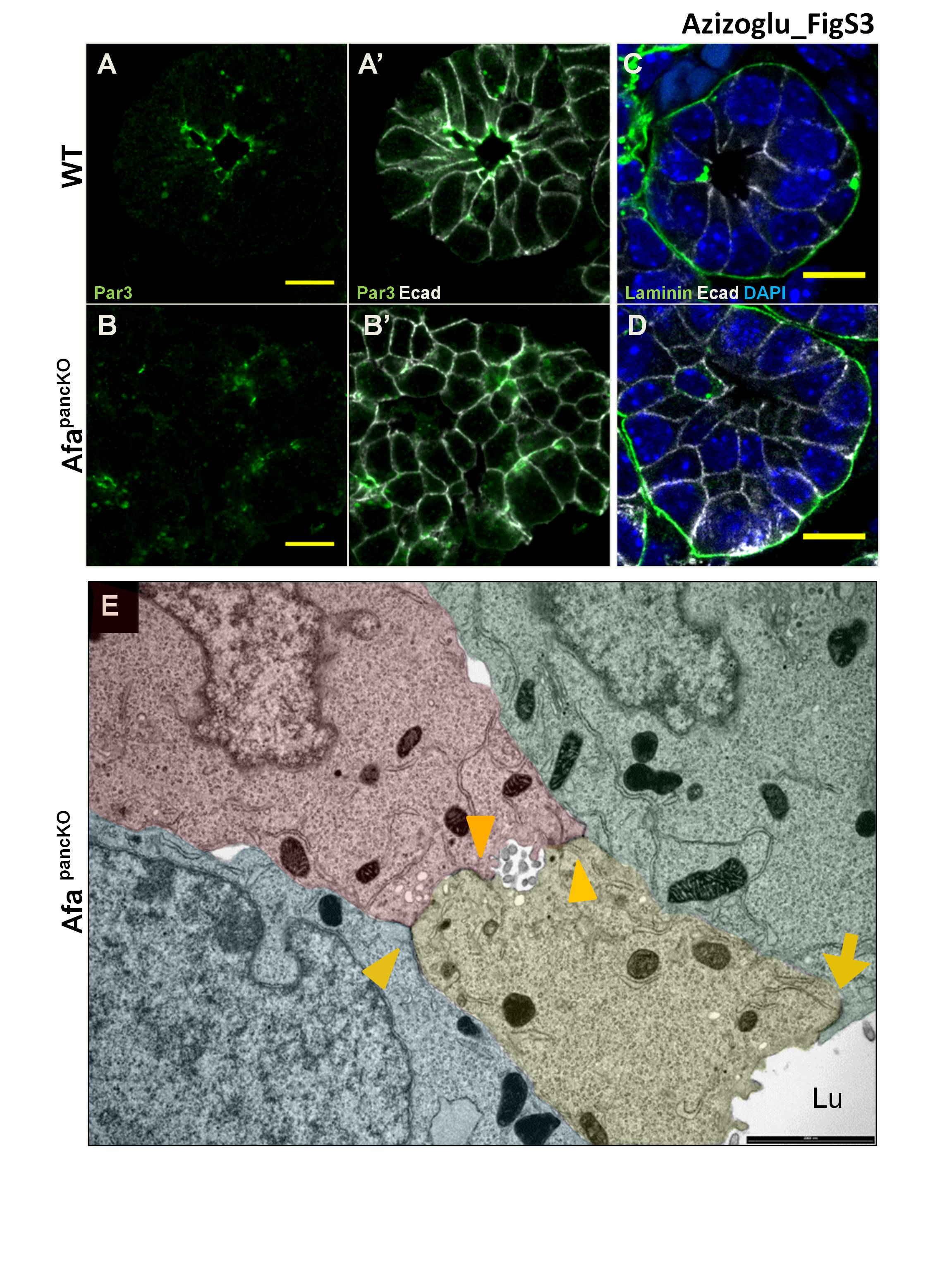{kind=link}