

Saxena et al. find that, in diverse cell types, certain genome regions far from promoters accumulate H3K27me3, and optimal gene expression depends on enhancers clearing this repressive mark. These findings reveal new “anti-repressive” function for hundreds of tissue-specific enhancers.
Keywords: enhancers, H3K27me3, PRC2, CDX2, KDM6A
Abstract
Compacted chromatin and nucleosomes are known barriers to gene expression; the nature and relative importance of other transcriptional constraints remain unclear, especially at distant enhancers. Polycomb repressor complex 2 (PRC2) places the histone mark H3K27me3 predominantly at promoters, where its silencing activity is well documented. In adult tissues, enhancers lack H3K27me3, and it is unknown whether intergenic H3K27me3 deposits affect nearby genes. In primary intestinal villus cells, we identified hundreds of tissue-restricted enhancers that require the transcription factor (TF) CDX2 to prevent the incursion of H3K27me3 from adjoining areas of elevated basal marking into large well-demarcated genome domains. Similarly, GATA1-dependent enhancers exclude H3K27me3 from extended regions in erythroid blood cells. Excess intergenic H3K27me3 in both TF-deficient tissues is associated with extreme mRNA deficits, which are significantly rescued in intestinal cells lacking PRC2. Explaining these observations, enhancers show TF-dependent binding of the H3K27 demethylase KDM6A. Thus, in diverse cell types, certain genome regions far from promoters accumulate H3K27me3, and optimal gene expression depends on enhancers clearing this repressive mark. These findings reveal new “anti-repressive” function for hundreds of tissue-specific enhancers.
Genes are controlled through dispersed enhancers that enable access to DNA and bear active histone marks such as H3K4me1/2 and H3K27ac (Heinz et al. 2015; Long et al. 2016). Many transcription factors (TFs) bind thousands of such sites, but only a fraction of adjoining genes responds overtly to TF binding; other occupied sites may reflect minor activity, cis-element redundancy, or nontranscriptional functions (MacQuarrie et al. 2011; Cusanovich et al. 2014). Deletion of individual mammalian enhancers reveals dominant activity of single cis elements in some cases and redundant or context-selective functions in others (Montavon et al. 2011; Hnisz et al. 2015; Hay et al. 2016; Huang et al. 2016). Recognized enhancer classes include “shadow” enhancers, which are mutually redundant (Hong et al. 2008; Cannavo et al. 2016), and “super” or “stretch” enhancers, which bind TFs at high density and govern cell identity (Parker et al. 2013; Whyte et al. 2013). Other enhancer types with distinctive properties are not known in adult tissues.
H3K4me1/2 and H3K27ac are found at active promoters and enhancers (Barski et al. 2007; Heintzman et al. 2009). In contrast, the repressive mark H3K27me3, which appears at promoters and poised embryonic stem cell (ESC) enhancers (Creyghton et al. 2010; Rada-Iglesias et al. 2011), is generally absent from enhancers in adult cells. It is unclear whether this is because the methyltransferases in Polycomb repressor complex 2 (PRC2) are excluded from distal cis elements or whether H3K27me3 controls promoters only. Insulator or boundary elements occupied by the CCCTC-binding factor CTCF separate some H3K27me3 deposits from areas of active transcription (Cuddapah et al. 2009). Nevertheless, distant H3K27me3-rich regions have been identified computationally in cell lines (Pinello et al. 2014), and, during ESC differentiation into spinal neurons, the TF CDX2 facilitates elimination of H3K27me3 from a large region in the HoxA cluster (Mazzoni et al. 2013). It is unknown whether this is a locus-specific effect restricted to differentiating ESCs or a general activity of enhancers.
After mid-gestation, CDX2 expression is confined to the intestine, where it is essential for proper epithelial development and digestive functions (Gao et al. 2009; Verzi et al. 2010; Hryniuk et al. 2012; Stringer et al. 2012). In adult mouse intestinal villus cells, CDX2 binds nearly every active promoter and thousands of enhancers, frequently together with other tissue-restricted TFs such as HNF4A (Verzi et al. 2010, 2013). Although this scope of cis-element occupancy is commensurate with the enterocyte defects in Cdx2-null intestines, the fraction of essential occupancy and requirements with respect to adjoining histone modifications is unknown. Like other TFs, CDX2 binds a tiny fraction of sites that bear its preferred sequence, ATAAA; conversely, only ∼23% of CDX2-occupied sites carry this consensus motif. Thus, in accordance with the “TF collective” model (Calo and Wysocka 2013; Heinz et al. 2015; Long et al. 2016), its presence at other enhancers may not reflect direct DNA contacts but may occur through other TFs’ independent binding to their respective sequence motifs.
Here we report that about one-quarter of all CDX2-bound enhancers in wild-type intestines are compromised in Cdx2−/− cells for every enhancer feature that we examined. These sites control genes most affected by CDX2 loss and are not enriched for superenhancers. Hundreds of CDX2-dependent loci lie in H3K27me3-depleted zones near areas of high basal tissue-specific H3K27me3. Depletion of CDX2 causes broad, selective, and well-demarcated gains of H3K27me3, with marked attenuation of nearby genes. Blood cells reveal a similar exclusion of H3K27me3 from tissue-restricted enhancers that depend on the TF GATA1. Intestinal enhancers bind the H3K27 demethylase KDM6A, which is displaced from CDX2-dependent sites, and loss of PRC2 rescues the genes most affected in Cdx2−/− intestines, many of them to wild-type levels. Thus, beyond activating transcription, TF-dependent enhancers in the intestine, blood, and probably all adult cells actively clear H3K27me3 from regions where the mark is otherwise inclined to accumulate and restrict gene activity.
Results
Distinct enhancer activities underlie mRNA disturbances in Cdx2-null intestinal villi
Absence of CDX2 in differentiated mouse intestinal villus cells compromises organ function (Verzi et al. 2013). RNA sequencing (RNA-seq) on replicate samples of Cdx2−/− villus cells (Supplemental Fig. S1A) revealed reduced expression (q < 0.01) of 1525 genes (Supplemental Fig. S1B), and RT-qPCR on 10 representative genes across the spectrum of RNA deficiency confirmed the magnitude of losses detected by RNA-seq (Supplemental Fig. S1C). Among the down-regulated genes, 350 transcripts (23%) declined more than fourfold, and the levels of these transcripts in wild-type cells were no higher or lower than those of less affected genes (Supplemental Fig. S1D); thus, extreme dependence on CDX2 does not reflect basal expression levels per se. The low fraction of highly affected genes suggests that that absence of CDX2 perturbs most cis elements modestly, while others are more sensitive.
To determine CDX2 requirements at villus cell enhancers, we used ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) to identify its binding sites (Supplemental Table S1); these regions showed no binding in Cdx2−/− villus cells (Supplemental Fig. S1E). To examine TF dependencies, we defined enhancers (>2 kb upstream of or >1 kb downstream from transcription start sites [TSSs]) stringently as the 21,502 regions that gave strong ChIP-seq peaks with CDX2 as well as H3K4me2 and H3K27ac antibodies. H3K4me2 marks both enhancers and promoters and overlaps greatly with the enhancer mark H3K4me1 (Barski et al. 2007; Heintzman et al. 2009); Supplemental Figure S1F shows our validation of selected sites by qPCR. Highly concordant replicates of ChIP-seq for H3K27ac (Supplemental Fig. S1G) revealed that the average density of H3K27ac across these enhancers was lower in Cdx2−/− than in wild-type villus cells (Fig. 1A), reflecting the nearly complete absence of H3K27ac from 4951 vulnerable sites (type 1; 23%), while the remaining sites (type 2) retained wild-type levels or showed small gains (Fig. 1A; Supplemental Fig. S2A,B). In addition to reduced H3K27ac, vulnerable (type 1) enhancers showed lower levels of H3K4me2, loss of RNA polymerase II (RNAPII) and the SWI/SNF-B remodeling complex subunit PBRM1, and markedly lower signals in the assay for transposase-accessible chromatin (ATAC) (Buenrostro et al. 2013). In contrast, resistant (type 2) sites preserved every feature (Fig. 1B). Thus, in the absence of CDX2, most intestinal enhancers maintain integrity, but approximately one-quarter of them are wholly compromised.
Figure 1.

Differential enhancer response to CDX2 loss associates strongly with reduced gene expression. (A) Average H3K27ac ChIP signals in wild-type (black) and Cdx2−/− (blue) villus cells at 21,504 enhancers. Differential analysis (Supplemental Fig. S2A) revealed two enhancer classes, type 1 (red) and type 2 (blue) enhancers, which showed heavy losses or preservation, respectively, of H3K27ac in Cdx2−/− cells. Representative Integrative Genomics Viewer (IGV) tracks from the Slc26a3 locus show CDX2 at wild-type enhancers and loss (red arrows) or preservation (blue arrows) of H3K27ac in Cdx2−/− epithelium. Scales refer to the range of signals in individual genome tracks. (B) Heat maps of modified histones; TF, SWI/SNF, and RNAPII binding; and ATAC combined with sequencing (ATAC-seq) in wild-type and Cdx2−/− epithelium, demonstrating attenuation of all signals at type 1—but not type 2—enhancers. Rows are ordered by decreasing H3K27ac signal in wild type and represent a distance of ±1.5 kb from the enhancer summits. (C) Density of the consensus CDX2 and HNF4A motifs ±200 base pairs (bp) around type 1 and type 2 enhancer summits; curves depict average motif densities. (D) Distributions of type 1 (red) and type 2 (blue) enhancers located ±50 kb from the 1525 genes significantly reduced (q < 0.01) in Cdx2−/− intestines. Pie charts show the fraction of genes with nearby type 1, type 2, and superenhancers among genes reduced less than twofold, twofold to fourfold, or fourfold or greater. P-values refer to the observed versus expected distributions of each enhancer type. (n.s.) Not significant. Type 1 enhancers are significantly overrepresented and underrepresented among genes reduced fourfold or greater and less than twofold, respectively. (E) Predictions of enhancer regulatory function by BETA (binding and expression target analysis) reveal a strong association of type 1 and a weaker association of type 2 enhancers with genes down-regulated (green) in Cdx2−/− intestines; neither enhancer type is associated with up-regulated genes (red). Plots depict the cumulative score of regulatory potential for every gene based on enhancer distances from the TSS, dashed lines represent the background of unaltered genes, and P-values denote the significance of up or down associations relative to the background.
In wild-type cells, CDX2 bound type 2 and type 1 enhancers equally well (Fig. 1B), indicating that this vulnerability does not reflect either weak or exceptional basal occupancy. Among the many possible explanations, we found only that CDX2-binding motifs were significantly more abundant in type 1 regions (Fig. 1C). In contrast, the motif for HNF4A, an intestinal TF that co-occupies most CDX2-binding sites (Verzi et al. 2010, 2013), was equally enriched at type 1 and type 2 sites. This marked difference suggests that CDX2 may contact DNA directly at type 1 enhancers (which depend on CDX2) and, in line with the “TF collective” model for DNA binding (Calo and Wysocka 2013; Heinz et al. 2015; Long et al. 2016), occupy type 2 enhancers indirectly through other TFs.
To impute the function of CDX2-dependent enhancers, we mapped their distributions within ±50 kb of all genes down-regulated (q < 0.01) in Cdx2−/− villus cells. The fraction of type 1 sites increased in proportion to the magnitude of RNA deficiency, deviating most from the expected distribution near genes that decline fourfold or greater (P < 0.0001, Fig. 1D). Genes with large mRNA losses often had multiple type 1 enhancers within 100 kb (range 0–15 enhancers, mean 1.97), and, affirming the activating property of intestinal CDX2 (Verzi et al. 2010, 2013), we observed no relation of binding sites to genes that increase in Cdx2−/− cells (Supplemental Fig. S2C). In BETA (binding and expression target analysis) (Wang et al. 2013), type 1 sites associated far more significantly (P < 10−78) than type 2 sites (P < 10−9) with genes that decline in the Cdx2−/− epithelium (Fig. 1E), and neither group was associated with up-regulated genes (P = 1). Thus, CDX2-dependent type 1 enhancers nicely explain the transcriptional deficits that occur in the absence of this TF.
Superenhancers identify tissue-specific loci and gene activity (Parker et al. 2013; Whyte et al. 2013). To inquire whether type 1 enhancers may be a proxy for these composite cis elements, we identified superenhancers (Whyte et al. 2013; Hnisz et al. 2015) in wild-type villus cells using established parameters: extreme density of Mediator1 (MED1) binding (which identified 345 regions) and H3K27ac marks (which identified 913 superenhancers) (Supplemental Fig. S2D). These regions included highly active intestinal loci such as solute carrier and cytoskeletal genes and bound CDX2 as strongly as other sites, and their constituent enhancers were depleted for type 1 and enriched for type 2 sites (P < 0.0001, Supplemental Fig. S2E). Superenhancers were distributed equally near genes showing small and large changes in the Cdx2−/− epithelium (Fig. 1D) and, unlike type 1 sites, had largely intact histone marks in the mutant cells (Supplemental Fig. S2F). Thus, transcriptional deficits in Cdx2-null cells correlate better with type 1 enhancers than with superenhancers.
Unexpected regional features of exceptionally vulnerable loci
Although genes are controlled mainly through enhancers (Heintzman et al. 2009; Shen et al. 2012), we asked whether the promoters of the genes most sensitive to CDX2 loss (ΔRNA ≥ 4×) might differ from those of less vulnerable genes (ΔRNA < 4×) in the wild-type villus epithelium. Indeed, promoters associated with large RNA deficits showed lower average signals for H3K4me3, H3K27ac, RNAPII, PBRM1, and ATAC compared with promoters with <4× reduced mRNA even though CDX2 bound both groups at comparable strength (Fig. 2A; Supplemental Fig. S3A). As noted above, this is not because severely affected genes had lower basal activity; rather, RNA levels of genes in the ΔRNA ≥ 4× and ΔRNA < 4× groups were similar in wild-type villi (Fig. 2A). Moreover, histone marking and other features at 9732 active promoters showed little variation over a >10 log2 range of RNA levels, and genes most vulnerable to CDX2 loss were scattered across this spectrum (Supplemental Fig. S3B).
Figure 2.

Genes reduced in Cdx2−/− intestines show basal characteristics that suggest locus vulnerability. (A) Histone modifications, chromatin access (ATAC), factor binding, and basal mRNA expression in wild-type villus cells at the promoters (TSS ±2 kb) of genes down-regulated in the Cdx2−/− epithelium. Genes are ordered by the degree of reduced expression and separated into groups that changed fourfold or greater or less than fourfold. (B) H3K27me3 ChIP signals at genes reduced in Cdx2−/− cells, ordered as in the adjoining heat map. Signals are shown for gene bodies, all scaled to 10 kb, as well as 7 kb upstream (including promoters) and 4 kb downstream. The corresponding aggregate plots show average H3K27me3 signals at genes reduced fourfold or greater (orange) or less than fourfold (green) in Cdx2−/− cells and genes not expressed (reads per kilobase transcript per million mapped reads [RPKM] = 0; gray curve) in wild-type intestines. The violin plot represents the log transformed ratio of signal over gene bodies and 5 kb upstream of and 4 kb downstream from genes reduced fourfold or greater and less than fourfold in Cdx2−/− intestines. (C) IGV tracks of ChIP-seq and RNA-seq data from a representative locus, Enpp7 (log2 8.9-fold reduced mRNA level). Red arrowheads denote type 1 enhancers. Scales refer to the signal range in individual genome tracks. (D) Average H3K27me3 ChIP signals in the extended vicinity (±10 kb) of type 1 (red) and type 2 (blue) enhancers in wild-type (left) and Cdx2−/− (right) villus epithelia.
Notably, basal H3K27me3 levels at these promoters were higher than those at genes with ΔRNA < 4× in Cdx2−/− intestines; this elevation was evident in replicate wild-type samples and extended for variable distances from vulnerable promoters, often >5 kb in either direction (Fig. 2B). ChIP for H3K27me3 was validated by qPCR at selected regions (Supplemental Fig. S3C). Active histone marks, chromatin access, RNAPII, and PBRM1 were further reduced at highly vulnerable promoters—but not at modestly affected genes—in Cdx2−/− villi (Supplemental Fig. S3A), and H3K27me3 was further increased at those TSSs and gene bodies and beyond (Fig. 2B). Thus, in addition to a high fraction of nearby type 1 enhancers, the genes most sensitive to CDX2 deficiency show signs of basal promoter vulnerability in wild-type cells; this convergence of cis-element features likely reflects locus-wide effects. Although various mechanisms are proposed for H3K27me3 spread (Margueron and Reinberg 2011; Blackledge et al. 2015), there is little precedent for long-range effects emanating from promoters. We therefore asked whether domains of elevated H3K27me3 might encompass both promoters and type 1 enhancers. Indeed, whereas H3K27me3 signals were comparably low at type 2 and type 1 enhancers per se, average signals in the extended vicinity of type 1 enhancers were higher in wild-type cells and further elevated in Cdx2−/− cells (Fig. 2C,D).
Significant and broad increase of H3K27me3 at select loci in Cdx2−/− intestines
Away from promoters or gene bodies, H3K27me3 has been detected mainly at poised ESC enhancers, where an acetyl group (H3K27ac) replaces trimethylataion when cells differentiate (Creyghton et al. 2010; Rada-Iglesias et al. 2011; Zentner et al. 2011). However, type 1 enhancers in adult wild-type villus cells are not “poised” but are highly active, with abundant H3K27ac and other features of transcriptional activity (Fig. 1). Although intergenic H3K27me3+ domains have not been studied as a group (Calo and Wysocka 2013; Heinz et al. 2015; Long et al. 2016), CDX2 is reported to facilitate removal of H3K27me3 from the Hoxa1-a9 region in differentiating ESCs to specify cervical versus thoracic neuron identity (Mazzoni et al. 2013). We postulated that this is a general function and that in the adult intestine, type 1 enhancers at non-Hox loci rely on CDX2 to prevent regional H3K27me3 accumulation.
Using the MEDIPS package in R (Lienhard et al. 2014), we identified significant changes in H3K27me3 (P < 0.05) at 630 enhancers in the Cdx2−/− villus epithelium; nearly all of these changes were gains occurring at 12.7% (designated type 1A) of all type 1 sites, with mean H3K27me3 counts >50 in Cdx2−/− cells (Fig. 3A). Type 1A sites accounted for nearly all of the average gain in Cdx2−/− cells, while H3K27me3 levels near the remaining sites (type 1B) were comparable with those near type 2 enhancers (Fig. 3B). Type 1A enhancers predominate near genes that decline substantially in Cdx2−/− villi (P = 0.0002) (Fig. 3C), underscoring their likely functional role, and abut regions where basal H3K27me3 levels exceed the genome background (Fig. 3D). These extended regions may have eluded recognition previously because intergenic H3K27me3 typically spans multiple adjacent nucleosomes and therefore does not give conventional peaks in ChIP analysis. Moreover, H3K27me3 signals lower than the high levels present at certain promoters are not readily distinguished from the “background.” The signals near type 1A enhancers in wild-type intestinal cells approximate those near expressed genes with low mRNA levels, are notably higher than the empiric genome background (Jadhav et al. 2016), and drew our attention because they are substantially elevated in Cdx2−/− cells (Supplemental Fig. S4A). One possibility is that this elevation trivially reflects altered cell fractions in Cdx2−/− villi; however, the ratio of enterocytes to goblet cells was intact (Supplemental Fig. S4B).
Figure 3.

The absence of CDX2 leads to H3K27me3 accumulation in extended regions surrounding type 1 enhancers. (A) Scatter plots of average H3K27me3 signals (RPKM values, n = 2) at all type 1 (left) and type 2 (right) regions ±2.5 kb from enhancer summits in wild-type (X-axis) versus Cdx2−/− (Y-axis) villi. Dot colors represent log2 fold changes in H3K27me3, and dot diameters represent the statistical significance of the difference (−log10 P-value). Slopes of linear regression (gray lines) differ significantly (P < 0.0001) for type 1 and type 2 enhancers. (B) H3K27me3 ChIP signals ±10 kb from the summits of enhancers of types 1A, 1B, and 2, represented as average profiles in wild-type and Cdx2−/− villi, revealing high basal H3K27me3 near type 1A sites, further increased in the Cdx2−/− epithelium. (Bottom) Violin plots for ratios of normalized ChIP-seq counts in Cdx2−/− and wild-type cells verify the selective increase at type 1A enhancers. (C) Type 1A enhancers are overrepresented near (±100 kb) genes reduced fourfold or greater in Cdx2−/− villi. P-values are indicated ([n.s.] not significant) for the fractions of genes where association with type 1A enhancers deviates from expected values. (D) A representative locus, Scin, showing significant H3K27me3 gain in Cdx2−/− cells. The red bar denotes a well-demarcated domain of H3K27me3 gain, with boundaries adjacent to CTCF (blue)-binding sites and TSSs of neighboring genes. Scales refer to the signal range in genome tracks. (E) Aggregate plots of the 322 regions depleted of H3K27me3 in wild-type intestines and gain of this mark, but not of H3K9me3, in Cdx2−/− intestines. All affected domains are scaled to 10 kb and depicted in relation to regions 10 kb (unscaled) upstream and downstream. Shaded areas represent the standard error of the mean (SEM).
Areas of local H3K27me3 depletion in wild-type intestines and significant elevation in Cdx2−/− villi ranged from 1.3 to 177 kb (mean 17.2 kb, median 8.1 kb) and showed sharp boundaries of H3K27me3 gain at one or both ends (Fig. 3D,E; Supplemental Fig. S4C). These boundaries occurred most often near type 1A enhancers (60.8%) or sites occupied by the boundary factor CTCF (26.7%) but rarely near type 1B (4.3%) or type 2 (6.5%) sites Supplemental Fig. S4D). They demarcated 322 regions of clear and extensive H3K27me3 gain, which we refer to here as type 1A “domains” (Fig. 3E), each encompassing an average of two type 1A enhancers (range one to 17). Type 1A domains had gained H3K27me3 by 5 d of Cdx2 deletion, and H3K27me3 levels were further increased 3 and 7 d later (Supplemental Fig. S4E). H3K9me3, a marker of heterochromatin (Nakayama et al. 2001; Becker et al. 2016), was not depleted at the same regions in wild-type cells or increased in Cdx2-null cells (Fig. 3E). Thus, certain enhancers selectively prevent incursion of H3K27me3 from areas of high local density into extended genomic domains, especially near genes that are highly sensitive to CDX2 loss.
TF-dependent exclusion of H3K27me3 from enhancers in another cell type
To determine whether TF-dependent exclusion of H3K27me3 occurs in other tissues, we examined the role in blood cells of a TF that mirrors intestinal CDX2 function: GATA1, which binds numerous erythroid enhancers (Yu et al. 2009) and is required for red blood cell differentiation (Pevny et al. 1991). As GATA1 deficiency is lethal in embryos, its activity has been studied in Gata1-null ESCs induced to differentiate in the presence (G1EER4) or absence (G1E) of Gata1 rescue (Fig. 4A; Weiss et al. 1997; Rylski et al. 2003); G1E and G1EER4 epigenomes have been mapped extensively (Jain et al. 2015). We defined erythroid enhancers based on both GATA1 binding and H3K27ac marks in G1ER4 cells. Similar to differences between Cdx2−/− and wild-type intestinal villus enhancers, Gata1-null G1E cells showed lower average H3K27ac at 7174 enhancers, and this average reflected total H3K27ac loss at 2828 sites (type 1; 39%) in G1E cells; H3K27ac was preserved at the remaining (type 2) enhancers (Supplemental Fig. S5A). Compared with genes with ΔRNA < 4× in G1E cells, those with ΔRNA ≥ 4× (q < 0.01) also showed lower promoter H3K4me3 and ATAC signals in Gata1-proficient G1EER4 cells (Supplemental Fig. S5B). Moreover, even in the presence of GATA1, the promoters and bodies of these highly GATA1-dependent genes showed higher H3K27me3 levels than less dependent genes, revealing basal locus-wide differences, and those levels increased further in G1E cells (Supplemental Fig. S5B). Thus, patterns of differential enhancer and gene susceptibility and different basal levels of H3K27me3 in GATA1-dependent erythroid cells parallel our findings on intestinal CDX2 dependency.
Figure 4.

H3K27me3 accumulates around type 1A enhancers in GATA1-null erythroid cells. (A) Schema for GATA1-null (G1E) ESCs and the requirement for GATA1 in erythroid lineage differentiation (GATA1+ G1EER4 cells). (B) Comparison of H3K27me3 signals (log2 RPKM) at the 2828 type 1 enhancers in G1E (Y-axis) and G1EER4 (X-axis) cells. The gray line represents linear regression, and the colors of the dots represent log2 fold changes in H3K27me3; this approach identified 772 differential sites. (C) Representative IGV tracks for H3K27me3 and H3K27ac in G1E and G1EER4 cells at the Btg2 locus. The black bar denotes the domain of H3K27me3 gain in G1E. GATA1-occupied sites in G1EER4 show the H3K27ac-marked promoter and type 1A enhancers within this domain. (D) Aggregate plots of the 300 regions that are depleted of H3K27me3 in G1EER4 cells and gain the mark in G1E cells. The black bar denotes the 10-kb distance to which all regions are fitted with 10-kb (unscaled) regions on both sides. Shaded areas = SEM. (E) Distinct genomic regions acquire H3K27me3 in TF-deficient blood and intestinal cells. (F) Aggregate plots of the 322 and 300 regions depleted for H3K27me3 in wild-type intestinal and erythroid cells, respectively, showing the basal marking at the same regions in the other wild-type tissue. Data are displayed as in D and show average elevations over the genome H3K27me3 background. (G) Representative IGV tracks for H3K27me3. Bars denotes the domain of H3K27me3 gain in the Cdx2−/− intestine (blue) or G1E cells (red), bound by dotted lines. Examples illustrate areas of tissue-specific gain flanked by elevated H3K27me3 in both tissues (top; Tle3 and Plekho2) or only one tissue (bottom; Enpp7 [high only in the intestine] and Mgat4e [only in blood]).
Analysis of H3K27me3 in 5-kb regions surrounding erythroid type 1 enhancers identified 772 sites that bind GATA1 in G1EER4 cells and have significantly more H3K27me3 in G1E than in G1EER4 cells, fitting the definition of type 1A enhancers (Fig. 4B,C). These sites lie in 300 well-demarcated domains (Fig. 4C) that show strong aggregate increases of H3K27me3 in G1E cells (Fig. 4D) and concentrate near genes reduced fourfold or greater in G1E cells (Supplemental Fig. S5C) but barely overlap with their CDX2-dependent counterparts in intestinal cells (Fig. 4E). Although peri-enhancer domains lacking H3K27me3 thus appear tissue-specific, the average signals in their neighborhoods were higher than the genome background in both tissues (Fig. 4F); these averages reflect extended regions of high H3K27me3 in both tissues in some cases and tissue-selective elevations in others (Fig. 4G). Thus, CDX2 and GATA1, crucial TFs in distinct cell types, exclude H3K27me3 from extended domains that are enriched for this mark, some common to blood and intestinal cells and others tissue-restricted. Type 1A domains in both cell types (Supplemental Table S4) are enriched near the genes most responsive to TF loss.
Cis elements recruit the H3K27 demethylase KDM6A/UTX
Our findings collectively suggest that type 1A domains are intrinsically predisposed to acquiring H3K27me3, with deposition and erasure occurring dynamically. One possible way for enhancers to offset H3K27me3 spread is to recruit a demethylase such as KDM6A/UTX, which limits H3K27me3 accumulation at promoters (Agger et al. 2007; Arcipowski et al. 2016) and is reported to bind certain heart-specific and T-cell-specific enhancers (Lee et al. 2012; Beyaz et al. 2017). To test whether intestinal CDX2-dependent enhancers recruit KDM6A to avoid H3K27me3 accumulation, we first confirmed that the KDM6A antibody recognizes a single protein of the expected mass by immunoblotting (Supplemental Fig. S6A); ChIP-seq in wild-type intestinal villus cells revealed KDM6A binding at nearly all active enhancers and promoters, well correlated with CDX2 occupancy (Fig. 5A; Supplemental Fig. S6B), and qPCR of selected sites verified ChIP-seq results (Supplemental Fig. S6B–D). KDM6A binding in wild-type villi was similar at type 1A, 1B, and 2 enhancers and was selectively displaced in Cdx2−/− cells from type 1, but not type 2, enhancers (Fig. 5B,C). Notably, 78% of type 1A domain edges showed nearby KDM6A binding—more than any other factor that we interrogated (Supplemental Figs. S4D, S6E). Thus, active intestinal cis elements bind KDM6A, and its displacement from type 1A—among other type 1—enhancers can explain H3K27me3 accumulation in areas of high adjacent H3K27me3.
Figure 5.

KDM6A is present at CDX2-bound intestinal cis elements and preferentially lost from type 1 enhancers in the Cdx2−/− epithelium. (A) Comparisons of log2 RPKM for CDX2 (Y-axis) and KDM6A (X-axis) ChIP-seq signals at intestinal enhancers. Heat represents density of points. (Yellow) Most; (gray) single points. r = Pearson correlation coefficient. (B) KDM6A binding at type 1 and type 2 enhancers in wild-type and Cdx2−/− villi. ChIP-seq data are plotted ±1.5 kb from enhancer summits. The adjoining aggregate (top) and violin (bottom) plots separate average signals at the different enhancer types, showing preferential KDM6A displacement from type 1, but not type 2, enhancers in Cdx2−/− cells. (C) IGV tracks showing KDM6A loss at type 1 (A and B) enhancers in the Car4 locus in Cdx2−/− villi. By definition, H3K27me3 is increased near type 1A, but not type 1B, enhancers.
Function of H3K27me3 at intestinal enhancers
H3K27me3 accumulation in type 1A domains in TF-null cells could represent a repressive force or reflect reduced transcription. Although the absence of H3K27me3 at many highly repressed loci argues against the latter possibility, perhaps only type 1A loci, which abut regions of high basal marking, are susceptible. The combined absence of CDX2 and PRC2 could indicate whether the H3K27me3 deposits are functional and reveal CDX2's relative contributions toward classical gene activation and prevention of gene repression. In the first few days after intestinal loss of PRC2, only selected fetal genes are derepressed, with no effect on genes expressed at high levels in wild-type villus cells, such as Cdx2 or the genes down-regulated in Cdx2−/− intestines (Jadhav et al. 2016). To determine the importance of H3K27me3 accumulation in the absence of CDX2, we crossed Cdx2Flox (Verzi et al. 2010), EedFlox (Xie et al. 2014), and Villin-CreER−T2 (el Marjou et al. 2004) mice to enable inducible intestine-restricted depletion of both H3K27 methylation and CDX2 (Fig. 6A). Following Cre activation, Villin-CreER−T2;Cdx2Fl/Fl;EedFl/Fl mice showed epithelial loss of CDX2 and H3K27me3 (Fig. 6A), with the latter reflected, for example, in ∼103-fold elevation of Hoxb13 mRNA (Supplemental Fig. S6F). Lone depletion of H3K27me3 in Eed−/− intestines did not significantly perturb expression of the genes affected in the absence of CDX2 alone (Fig. 6B). When superimposed on CDX2 loss, however, EED deficiency partially rescued these mRNA deficits (Fig. 6B). Small changes in large numbers of genes conferred statistical significance to every group, but the extent of rescue was highest among genes reduced fourfold or greater in Cdx2−/− intestines (Fig. 6B), where 51.3% of genes near type 1A enhancers were restored at least twofold, and some (e.g., Scin and Slc26a6) were rescued fully (Fig. 6C; Supplemental Fig. S6G). However, gene expression in the Cdx2−/−;Eed−/− epithelium was not globally restored. Some genes down-regulated up to 30-fold in Cdx2−/− cells (e.g., Enpp7) showed minimal rescue; with a paucity of intact type 2 enhancers, such genes depend crucially on CDX2 for activation. Moreover, rare genes located >100 kb from type 1A sites were also rescued, likely reflecting imperfect distance-based enhancer assignments. Despite these limitations, the mRNA profile of Cdx2−/−;Eed−/− cells demonstrates that in addition to a conventional role in gene activation, CDX2-dependent prevention of regional H3K27me3 deposits is important for gene activity, especially near type 1A enhancers.
Figure 6.
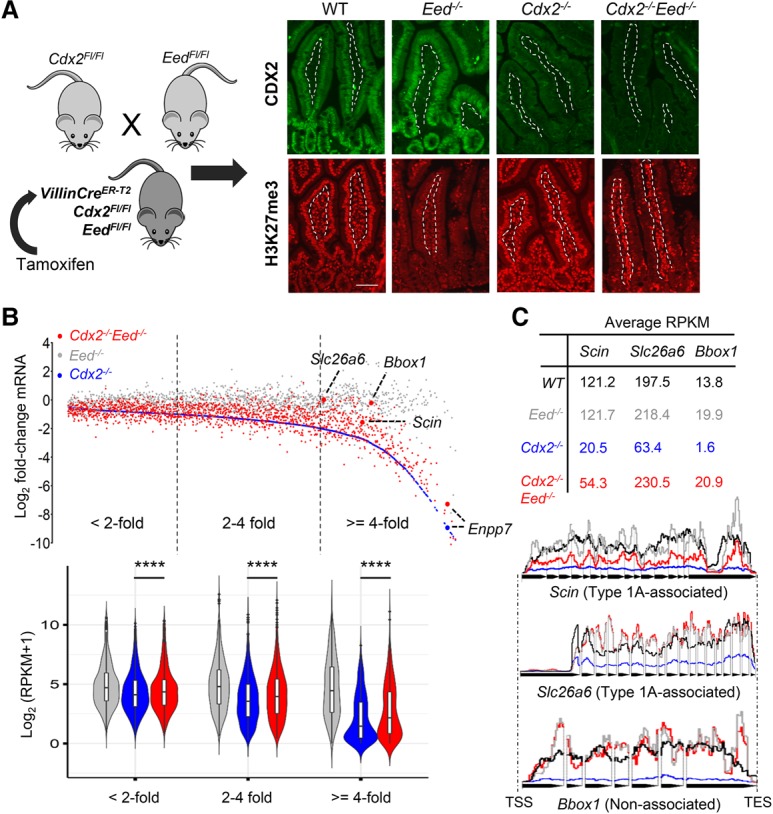
Deletion of the PRC2 component gene Eed eliminates H3K27me3 and partially rescues CDX2-dependent gene expression. (A) Mouse breeding and treatment scheme to eliminate both CDX2 and PRC2 activity, verified by CDX2 (top) and H3K27me3 (bottom) immunostains on the third day after 5 d of tamoxifen treatment to activate Cre. Dashed lines demarcate the epithelium, selectively targeted by Villin-Cre, from the intestinal lamina propria. Bar, 10 µm. (B) Scatter plot of all genes reduced in Cdx2−/− (blue; arranged in order of increasing effect) and their levels in Eed−/− (gray) and Cdx2−/−;Eed−/− (red) intestinal cells. Dots represent the mean from two replicates of each genotype, and the violin plots below represent RPKM values of bins of genes that decrease less than twofold, twofold to fourfold, or fourfold or greater in Cdx2−/− villi. (****) P < 0.0001. Boxes within the violins show median values at the 75th and 25th percentiles, and the whiskers show 1.5 times the interquartile range. Significance was determined by the Friedman test. EED loss substantially rescued many transcripts, especially among those most affected in Cdx2−/− cells, with some genes restored to wild-type levels. (C) Slc26a6 and Scin (two type 1A-associated genes) and Bbox1 (an unlinked gene) are highlighted by RPKM values and collated tracks (data were averaged from duplicates, introns excluded). In contrast, Enpp7 in B showed minimal rescue.
Discussion
Nucleosomes and compacted chromatin are known barriers to gene activity (Wang et al. 2011); less clear are the extent of other transcriptional constraints, including repressive histone modifications or how TFs overcome diverse impediments. We examined the collective function of nearly 5000 TF-dependent (type 1) villus enhancers. Although CDX2 binds thousands of additional enhancers and Cdx2-null intestines gradually fail (Verzi et al. 2010), only 23% of affected genes suffer fourfold or greater in Cdx2−/− cells. Other intestinal genes are likely spared by the persistence of intact type 2 sites, which qualify as adult “shadow” enhancers (Hong et al. 2008; Cannavo et al. 2016) and highlight the extent of enhancer redundancy in vivo. Notably, CDX2-dependent enhancers were significantly more enriched for the CDX2 motif than type 2 sites, suggesting that enhancer integrity might depend crucially on the principal TF that contacts DNA. Although intestinal superenhancers contain discrete type 1 sites, as a group, they are not enriched at highly CDX2-dependent loci, again probably because type 2 enhancers insensitive to CDX2 loss provide adequate activity. Our studies reveal the exclusion of H3K27me3 from large intergenic domains as a crucial activity of certain TF-dependent enhancers characterized by this “anti-repressive” function. Thus, although TF-dependent enhancers function primarily to activate genes, a unique fraction—type 1A enhancers—has an additional and potent role in overcoming regional repressive forces (Fig. 7).
Figure 7.

Model depicting chromatin at anti-repressive and classic enhancers in wild-type and TF-deficient cells. (Top left) Whereas all enhancers have some activating function, ∼700 “anti-repressive” enhancers within ∼300 domains (pink) in two mouse tissues showed an additional role in preventing local H2K27me3 accumulation. These enhancers associate most with genes highly affected by TF deficiency, which induced regional H3K27me3 deposits (top right; pink) not seen at most enhancers (bottom right; blue). Inferred effects on transcription of target genes are represented by the thickness of the green arrows.
H3K27me3 was implicated previously in silencing promoters and developmental enhancers, including reports of one enhancer's role in clearing the mark from α-globin promoter CpG island in humanized mouse erythroid cells (Vernimmen et al. 2011) and the CDX2-mediated clearance of H3K27me3 from the Hoxa locus during ESC differentiation (Mazzoni et al. 2013). We found that hundreds of tissue-restricted enhancers in adult cells prevent H3K27me3 from marking genomic domains that range in size from 1300 base pairs (bp) to 177 kb, abut regions of high basal H3K27me3, and have sharp boundaries that coincide with CTCF, type 1A enhancers, and especially KDM6A binding. Genes highly dependent on CDX2 are enriched near these type 1A domains. Although CDX2 and GATA1, in principle, could prevent H3K27me3 deposition as a peripheral consequence of activating local transcription, this is unlikely to be the principal basis because excess H3K27me3 appears at only a fraction of genes highly affected in the respective TF-null tissues. Moreover, half of the genes highly repressed in Cdx2−/− intestines are rescued between twofold and fully when PRC2 activity is absent. Therefore, type 1A domains appear to be at intrinsic risk for spread of H3K27me3, a mark known to propagate linearly until it encounters a block (Calo and Wysocka 2013), and “anti-repressive” enhancers counter that risk. Our data indicate that different regions in each tissue qualify as type 1A domains, a likely sequela of embryonic gene expression, with some areas bearing elevated H3K27me3 in multiple tissues. These levels, albeit lower than those found at promoters, impose a barrier to transcription that “anti-repressive” enhancers help breach. The interspersing of active mammalian enhancers among developmentally repressed regions has an evolutionary parallel in Drosophila Polycomb response elements (Erceg et al. 2017).
Some CDX2-dependent genes, such as Scin and Slc26a6, were fully rescued in the combined absence of CDX2 and PRC2, indicating the primacy of preventing H3K27me3 deposits in these loci. Other highly CDX2-dependent genes, such as Enpp7, remained inactive in the absence of PRC2, implying that they need CDX2 for other activities. Together, these findings reflect type 1A enhancers’ dual roles in H3K27me3 removal and conventional gene activation. Indeed, beyond their role in constraining H3K27me3, we identified no attribute unique to type 1A enhancers; even KDM6A demethylase binds “anti-repressive” enhancers similar to other cis elements. In these respects, “anti-repressive” sites are analogous to “superenhancers” or “stretch enhancers”—dense clusters of discrete cis elements that individually resemble other dispersed enhancer units (Parker et al. 2013; Whyte et al. 2013). The sum of the data thus implies that individual enhancers recruit TFs, ancillary factors, and active histone marks, but their roles in “shadow,” “super,” or “anti-repressive” capacities may be dictated less by intrinsic properties than by their relation to other cis elements and the local chromatin context. The few hundred “anti-repressive” enhancers and type 1A domains that we identified in intestinal or blood cells approximate the number of superenhancers in any tissue (Parker et al. 2013; Whyte et al. 2013), and our findings reveal their crucial role in overcoming transcriptional barriers imposed by regional H3K27me3 deposition.
Materials and methods
Experimental mice and treatments
Mice were maintained on a C57Bl/6 background with minimal or no 129/Sv contribution. Animals were handled with procedures approved by the Animal Care and Use Committee of the Dana-Farber Cancer Institute. Mice of both sexes were at least 7 wk old at the time of treatments and cell isolations, and littermates served as controls. Villin-CreER(T2) mice carrying one or more floxed alleles received five daily intraperitoneal injections of tamoxifen (Sigma, T5648) prepared in sunflower oil at doses of 1 mg per 25 g of body weight, adjusted according to daily measures of weight. Control mice were injected with the same volume of sunflower oil. These animals were euthanized on the first, third, or seventh day after the last tamoxifen injection.
Epithelial cell and RNA isolation and mRNA-seq
All molecular analyses were performed on purified villus epithelium, which was harvested by incubating fresh jejuna (middle approximately one-third of the small intestine) in 5 mM EDTA in phosphate-buffered saline (PBS; pH 8) for 45 min at 4°C. Villi were collected in cold PBS by retention on a 70-µm strainer, washed in PBS, and either fixed for ChIP-seq or used to prepare single-cell suspensions for ATAC combined with sequencing (ATAC-seq). RNA was isolated using TRIzol (Thermo Fisher Scientific, 15596026), RNeasy kit (Qiagen, 74104), and RNase-free DNase (Qiagen, 79254). Total RNA was used to make RNA-seq libraries using the TruSeq RNA sample preparation kit version 2 (Illumina, RS-122-2001), following the manufacturer's instructions. Libraries were sequenced on Illumina NextSeq 500.
Seventy-five-base-pair single-end sequencing reads were aligned to the mouse reference genome Mm9 (NCBI build 37) using TopHat version 2.0.6 (Trapnell et al. 2009). Raw read counts were obtained using HTSeq (Anders et al. 2015) to estimate transcript levels. HTSeq counts were used in DESeq2 (Love et al. 2014) to normalize the data and identify significant (q < 0.01) differential expression between control (wild-type, n = 2) and mutant (Cdx2−/−, n = 3) samples. RPKM (reads per kilobase transcript per million mapped reads) values were determined using Cufflinks version 2.2.1 (Trapnell et al. 2010), and Pearson correlation coefficients of RPKM values for all genes were calculated and plotted using the Corrgram package in R (http://www.R-project.org). To visualize tracks in the Integrative Genomics Viewer (IGV) version 2.3 (Robinson et al. 2011), we used the bamCoverage module in deepTools2 (Ramirez et al. 2016). Differential expression profiles for erythroid cell RNA-seq data (Supplemental Table S2) were likewise obtained using DESeq2. Genes significantly reduced in Cdx2−/− villi were assessed for changes (Eed−/−) or rescued expression (Cdx2−/−Eed−/−) by comparing RPKM values. Significant differences within groups of genes reduced less than twofold, twofold to fourfold, or fourfold or greater in Cdx2−/− cells were determined by Friedman's test, and violin plots of log transformed RPKM values were generated using the ggplot2 package in R. Log2 (fold change) values determined using DESeq2 were used to fit and plot smoothing splines in R, and we determined the significance of different distributions using the Kolmogorov-Smirnov test. Average signals over exons were obtained by merging individual BedGraph files from replicate wild-type and experimental samples and plotted in R as traces over all exons (excluding introns) between TSSs and transcription end sites.
Histology and immunohistochemistry
Tissues from the same mice used for molecular studies were fixed overnight in 4% paraformaldehyde, washed in PBS, dehydrated in ethanol, and embedded in paraffin. Five-micrometer tissue sections were deparaffinized, rehydrated, and stained with Alcian blue or processed for immunohistochemistry. Antigens were retrieved in 10 mM sodium citrate (pH 6) followed by overnight incubations in antibodies against CDX2 or H3K27me3 (Supplemental Table S3) at 4°C and 1-h incubations with anti-rabbit or anti-goat IgG conjugated to Cy3 or Alexa fluor 488 (Thermo Fisher Scientific). Images were captured as described previously (Jadhav et al. 2016).
ChIP-seq and ATAC-seq
ChIP for TFs and modified histones (Supplemental Table S1) was performed on cross-linked or native chromatin as described previously (Verzi et al. 2013; Jadhav et al. 2016). Briefly, jejunal villus epithelial cells were cross-linked at room temperature with 1% formaldehyde (Sigma, F8775) or 2 mM disuccinimidyl glutarate (DSG; Thermo Fisher Scientific, 20593) and 1% formaldehyde or treated with micrococcal nuclease (MNase) (Sigma, N3755). Cross-linked cells were resuspended in RIPA lysis buffer and sonicated to obtain 200- to 800-bp chromatin fragments. For native ChIP, cells were resuspended in a digestion buffer (40 mM Tris-HCl at pH 7.6, 1 mM CaCl2, 0.2% Triton-X 100, EDTA-free protease inhibitor cocktail [Roche], 0.5 mM phenyl methyl sulfonyl fluoride [PMSF], 5 mM Na butyrate) and treated with MNase for 5–6 min at 37°C. Reactions were terminated by adding 5 mM EDTA followed by dialysis (Slide-A-Lyzer, Thermo Fisher Scientific, 66380) at 4°C in RIPA buffer. Lysates were incubated overnight at 4°C with antibodies (Supplemental Table S3) followed by Protein A and G Dynabeads (Thermo Fisher Scientific, 10002D and 10004D) at 1:1 ratio for 3 h. Beads were washed twice in the sonication buffer, once in high-salt buffer (20 mM Tris-HCl at pH 8, 1 mM EDTA, 0.5 M NaCl, 0.1% SDS, 1% Triton X-100), once in LiCl buffer (10 mM Tris-HCl at pH 8, 1mM EDTA, 250 mM LiCl, 1% NP-40), and once in TE buffer. Cross-links were reversed overnight by incubation at 65°C in 1% SDS and 0.1 M NaHCO3 followed by treatment with Proteinase K (Thermo Fisher Scientific, 25530049) for 1 h at 55°C. DNA was purified with QIAQuick PCR purification kits (Qiagen, 28106). Libraries were prepared using ThruPLEX DNA-seq kits (Rubicon Genomics, R400427) and purified using Ampure XP beads (Beckman Coulter, A63881).
Jejunal villus epithelium was digested for 30–40 min at 37°C in 4× TrypLE (Thermo Fisher Scientific, A1217702) to obtain single-cell suspensions, and 40,000–50,000 cells were used for transposition, following established protocol (Buenrostro et al. 2013). Briefly, cells were resuspended in cold lysis buffer (10 mM Tris-Cl at pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Igepal CA-360), and crude nuclear pellets were incubated in transposition mix (25 µL of 2× reaction buffer from Nextera kit [Illumina, FC-121-1030], 2.5 µL of Nextera Tn5 transposase, 22.5 µL of nuclease-free water) for 30 min at 37°C. Transposed DNA was purified using MinElute PCR purification kit (Qiagen, 28004); PCR-amplified for optimal number of cycles using NEBNext High-Fidelity master mix (New England Biolabs, M0541L), a common forward primer, and uniquely barcoded reverse primers for each sample; and purified using MinElute PCR purification kits (Qiagen), and primer dimers were removed using Ampure XP beads (Beckman Coulter).
Primers for ChIP-qPCR are in Supplemental Table S3. Libraries were sequenced on an Illumina NextSeq 500 instrument to obtain 75-bp single-end reads, which were aligned to the mouse reference genome mm9 (NCBI Build 37) using Bowtie2 (Langmead and Salzberg 2012). Densities of CDX2 and HNF4A motif instances were calculated against the genome background and represented within ±200 bp of type 1 or type 2 enhancer summits as scatter and aggregate plots using R. We used the bamCoverage module in deepTools2 (Ramirez et al. 2016) to create .bigwig files. For MNase-ChIP, we used NPS (Zhang et al. 2008b) to generate .wig files. H3K27ac or MED1 peaks called (q < 0.01) by MACS2 (Zhang et al. 2008a) were used in ROSE (Whyte et al. 2013) to call superenhancers, excluding 2-kb regions around TSSs and with the default stitching distance of 12.5 kb. The LSD package in R was used to represent ChIP-seq counts as density plots for KDM6A and CDX2.
Replicate ATAC-seq .bam files were merged using Samtools (Li et al. 2009). For comparative visualizations, data sets from experimental and control samples were quantile-normalized using Haystack (Pinello et al. 2014) followed by deepTools2 or SitePro (Liu et al. 2011) and RStudio to create heat maps and aggregate plots. Normalized ChIP or ATAC counts for promoter marks were calculated at ±1 kb from the TSS over 10-bp nonoverlapping bins (1-kb bins for H3K27me3), and the corresponding bins in wild-type and Cdx2−/− were used to calculate log transformed ratios to create violin plots in ggplot2. For enhancer violin plots, H3K27me3 or KDM6A ChIP signals were calculated for distances ±10 kb or ±0.5 kb from enhancer summits, respectively. Log transformed ratios of Cdx2−/− over wild type were calculated and plotted for each enhancer.
Delineation of the intestinal villus enhancer set and differential H3K27ac and H3K27me3 analyses
CDX2 ChIP-seq peaks were filtered for promoters and intersected with nonpromoter sites bearing H3K27ac to define a working set of 21,502 enhancers. Two highly concordant experimental replicates each from wild-type and Cdx2−/− epithelia were assessed for input-corrected differential H3K27ac signal ±750 bp from each enhancer summit using the MEDIPS package (Lienhard et al. 2014) in R for edgeR-based analysis. Similarly, enhancers were assessed for differential H3K27me3 signals ±2.5 kb from the summit in wild-type and Cdx2−/− epithelia (n = 2 each) using the MEDIPS package in R. Results were displayed in a scatter plot prepared using the Plotly package (Plotly Technologies, Inc.). Among intestinal enhancers, 2290 showed higher H3K27ac in the Cdx2−/− epithelium (false discovery rate [FDR]-adjusted P < 0.05), and the other 19,212 sites were designated as type 1 or type 2.
Log2 transformed RPKM values for H3K27me3 in wild-type and Cdx2−/− or Gata1-null cells were used to create a regression model, and GraphPad Prism was used to compare the lines of regression for enhancers of types 1 and 2. Enhancers with significantly increased H3K27me3 in mutant cells (P < 0.05) were designated as type 1A and examined in IGV. Regions of surrounding elevated H3K27me3 were manually curated to delineate H3K27me3-high domains, with edges (boundaries) defined by the position where H3K27me3 levels were the same as in wild-type cells. In Figures 3E and 4, D and F, and Supplemental Figure S4E, type 1A regions are represented by scaling to the same length.
Enhancer–gene associations
TSSs of the 1525 genes showing reduced expression (q < 0.01) in the Cdx2−/− epithelium were extended by 50 kb upstream and downstream, and enhancer summits within this 100-kb distance were assigned to each gene. We used R to array genes in order of fold attenuation or fold increase in Cdx2−/− cells and then plotted all type 1 and type 2 enhancers assigned to that gene. Down-regulated genes were likewise associated with superenhancers within 100 kb. To determine the significance of enhancer distributions, we considered differences between the observed and expected fractions of the total number of type 1, type 2, or type 1A enhancers mapping to each gene group using a binomial test in GraphPad Prism. We used BETA (Wang et al. 2013) to associate genes with enhancers and quantify these associations using 100-kb distance limits, a significance threshold of FDR-adjusted P < 0.01 for differential gene expression in wild-type and mutant cells, and other default parameters.
Data availability
All data from this study (Supplemental Table S1) were deposited in the Gene Expression Omnibus (GSE98724). Data for analysis of GATA1-deficient and rescued erythroid blood cells (Supplemental Table S2) are from the ENCODE consortium and Jain et al. (2015).
Supplementary Material
Acknowledgments
We thank S. Robine [Villin-CreER(T2)] and S.H. Orkin (EedFl) for sharing mice; K. Nalapareddy, S. Madha, and Z. Herbert for technical support; and M. Verzi, P. Cejas, H. Long, and M. Brown for valuable discussions. This work was supported by National Institutes of Health awards R01DK082889 and R01DK081113 to R.A.S., F32DK115080 to M.S., F32DK103453 to U.J., and P50CA127003. M.S., A.K.S.R., and R.A.S. conceived and designed the study; M.S., A.K.S.R., R.S., and U.J. acquired data; M.S., A.K.S.R., and N.K.O. performed computational analyses; R.A.S. supervised the study; and M.S. and R.A.S. drafted the manuscript with input from all authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.308536.117.
References
- Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. 2007. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449: 731–734. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcipowski KM, Martinez CA, Ntziachristos P. 2016. Histone demethylases in physiology and cancer: a tale of two enzymes, JMJD3 and UTX. Curr Opin Genet Dev 36: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837. [DOI] [PubMed] [Google Scholar]
- Becker JS, Nicetto D, Zaret KS. 2016. H3K9me3-dependent heterochromatin: barrier to cell fate changes. Trends Genet 32: 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyaz S, Kim JH, Pinello L, Xifaras ME, Hu Y, Huang J, Kerenyi MA, Das PP, Barnitz RA, Herault A, et al. 2017. The histone demethylase UTX regulates the lineage-specific epigenetic program of invariant natural killer T cells. Nat Immunol 18: 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackledge NP, Rose NR, Klose RJ. 2015. Targeting Polycomb systems to regulate gene expression: modifications to a complex story. Nat Rev Mol Cell Biol 16: 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. 2013. Modification of enhancer chromatin: what, how, and why? Mol Cell 49: 825–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannavo E, Khoueiry P, Garfield DA, Geeleher P, Zichner T, Gustafson EH, Ciglar L, Korbel JO, Furlong EE. 2016. Shadow enhancers are pervasive features of developmental regulatory networks. Curr Biol 26: 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. 2009. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res 19: 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Pavlovic B, Pritchard JK, Gilad Y. 2014. The functional consequences of variation in transcription factor binding. PLoS Genet 10: e1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. 2004. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39: 186–193. [DOI] [PubMed] [Google Scholar]
- Erceg J, Pakozdi T, Marco-Ferreres R, Ghavi-Helm Y, Girardot C, Bracken AP, Furlong EE. 2017. Dual functionality of cis-regulatory elements as developmental enhancers and Polycomb response elements. Genes Dev 31: 590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, White P, Kaestner KH. 2009. Establishment of intestinal identity and epithelial–mesenchymal signaling by Cdx2. Dev Cell 16: 588–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D, Hughes JR, Babbs C, Davies JO, Graham BJ, Hanssen LL, Kassouf MT, Oudelaar AM, Sharpe JA, Suciu MC, et al. 2016. Genetic dissection of the α-globin super-enhancer in vivo. Nat Genet 48: 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK. 2015. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 16: 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, Bradner JE, Young RA. 2015. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell 58: 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JW, Hendrix DA, Levine MS. 2008. Shadow enhancers as a source of evolutionary novelty. Science 321: 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryniuk A, Grainger S, Savory JG, Lohnes D. 2012. Cdx function is required for maintenance of intestinal identity in the adult. Dev Biol 363: 426–437. [DOI] [PubMed] [Google Scholar]
- Huang J, Liu X, Li D, Shao Z, Cao H, Zhang Y, Trompouki E, Bowman TV, Zon LI, Yuan GC, et al. 2016. Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev Cell 36: 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav U, Nalapareddy K, Saxena M, O'Neill NK, Pinello L, Yuan GC, Orkin SH, Shivdasani RA. 2016. Acquired tissue-specific promoter bivalency is a basis for PRC2 necessity in adult cells. Cell 165: 1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain D, Mishra T, Giardine BM, Keller CA, Morrissey CS, Magargee S, Dorman CM, Long M, Weiss MJ, Hardison RC. 2015. Dynamics of GATA1 binding and expression response in a GATA1-induced erythroid differentiation system. Genom Data 4: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee JW, Lee SK. 2012. UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev Cell 22: 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhard M, Grimm C, Morkel M, Herwig R, Chavez L. 2014. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics 30: 284–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. 2011. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol 12: R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Prescott SL, Wysocka J. 2016. Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167: 1170–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQuarrie KL, Fong AP, Morse RH, Tapscott SJ. 2011. Genome-wide transcription factor binding: beyond direct target regulation. Trends Genet 27: 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469: 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni EO, Mahony S, Peljto M, Patel T, Thornton SR, McCuine S, Reeder C, Boyer LA, Young RA, Gifford DK, et al. 2013. Saltatory remodeling of Hox chromatin in response to rostrocaudal patterning signals. Nat Neurosci 16: 1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montavon T, Soshnikova N, Mascrez B, Joye E, Thevenet L, Splinter E, de Laat W, Spitz F, Duboule D. 2011. A regulatory archipelago controls Hox genes transcription in digits. Cell 147: 1132–1145. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. 2001. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292: 110–113. [DOI] [PubMed] [Google Scholar]
- Parker SC, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, Program NCS, et al. 2013. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci 110: 17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevny L, Simon MC, Robertson E, Klein WH, Tsai SF, D'Agati V, Orkin SH, Costantini F. 1991. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 349: 257–260. [DOI] [PubMed] [Google Scholar]
- Pinello L, Xu J, Orkin SH, Yuan GC. 2014. Analysis of chromatin-state plasticity identifies cell-type-specific regulators of H3K27me3 patterns. Proc Natl Acad Sci 111: E344–E353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T. 2016. deepTools2: a next generation Web server for deep-sequencing data analysis. Nucleic Acids Res 44: W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rylski M, Welch JJ, Chen YY, Letting DL, Diehl JA, Chodosh LA, Blobel GA, Weiss MJ. 2003. GATA-1-mediated proliferation arrest during erythroid maturation. Mol Cell Biol 23: 5031–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. 2012. A map of the cis-regulatory sequences in the mouse genome. Nature 488: 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer EJ, Duluc I, Saandi T, Davidson I, Bialecka M, Sato T, Barker N, Clevers H, Pritchard CA, Winton DJ, et al. 2012. Cdx2 determines the fate of postnatal intestinal endoderm. Development 139: 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernimmen D, Lynch MD, De Gobbi M, Garrick D, Sharpe JA, Sloane-Stanley JA, Smith AJ, Higgs DR. 2011. Polycomb eviction as a new distant enhancer function. Genes Dev 25: 1583–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi MP, Shin H, He HH, Sulahian R, Meyer CA, Montgomery RK, Fleet JC, Brown M, Liu XS, Shivdasani RA. 2010. Differentiation-specific histone modifications reveal dynamic chromatin interactions and partners for the intestinal transcription factor CDX2. Dev Cell 19: 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi MP, Shin H, San Roman AK, Liu XS, Shivdasani RA. 2013. Intestinal master transcription factor CDX2 controls chromatin access for partner transcription factor binding. Mol Cell Biol 33: 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bai L, Bryant GO, Ptashne M. 2011. Nucleosomes and the accessibility problem. Trends Genet 27: 487–492. [DOI] [PubMed] [Google Scholar]
- Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, Liu XS. 2013. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc 8: 2502–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MJ, Yu C, Orkin SH. 1997. Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a gene-targeted cell line. Mol Cell Biol 17: 1642–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. 2013. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153: 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, Orkin SH. 2014. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14: 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Riva L, Xie H, Schindler Y, Moran TB, Cheng Y, Yu D, Hardison R, Weiss MJ, Orkin SH, et al. 2009. Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol Cell 36: 682–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Tesar PJ, Scacheri PC. 2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 21: 1273–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008a. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shin H, Song JS, Lei Y, Liu XS. 2008b. Identifying positioned nucleosomes with epigenetic marks in human from ChIP-seq. BMC Genomics 9: 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data from this study (Supplemental Table S1) were deposited in the Gene Expression Omnibus (GSE98724). Data for analysis of GATA1-deficient and rescued erythroid blood cells (Supplemental Table S2) are from the ENCODE consortium and Jain et al. (2015).