

Fang et al. show that Dbf4 is enriched at early origins through its interaction with forkhead transcription factors Fkh1 and Fkh2. Dbf4 interacts directly with Sld3 and promotes the recruitment of downstream limiting factors.
Keywords: DNA replication timing, limiting factors, Dbf4, forkhead transcription factor
Abstract
Initiation of eukaryotic chromosome replication follows a spatiotemporal program. The current model suggests that replication origins compete for a limited pool of initiation factors. However, it remains to be answered how these limiting factors are preferentially recruited to early origins. Here, we report that Dbf4 is enriched at early origins through its interaction with forkhead transcription factors Fkh1 and Fkh2. This interaction is mediated by the Dbf4 C terminus and was successfully reconstituted in vitro. An interaction-defective mutant, dbf4ΔC, phenocopies fkh alleles in terms of origin firing. Remarkably, genome-wide replication profiles reveal that the direct fusion of the DNA-binding domain (DBD) of Fkh1 to Dbf4 restores the Fkh-dependent origin firing but interferes specifically with the pericentromeric origin activation. Furthermore, Dbf4 interacts directly with Sld3 and promotes the recruitment of downstream limiting factors. These data suggest that Fkh1 targets Dbf4 to a subset of noncentromeric origins to promote early replication in a manner that is reminiscent of the recruitment of Dbf4 to pericentromeric origins by Ctf19.
In all eukaryotic cells, chromosome replication initiates at a large number of origins ranging from ∼500 in yeast to 50,000 in human. Interestingly, origins do not fire simultaneously but follow a well-defined timing program (Raghuraman et al. 2001; Rhind and Gilbert 2013; Yoshida et al. 2013; Fragkos et al. 2015). Despite its significance for gene expression, chromatin structure, genome stability, cell development, differentiation, and gene evolution (Gondor and Ohlsson 2009; Gilbert et al. 2010; Dileep et al. 2015), our knowledge of replication timing establishment still remains limited.
Initiation of eukaryotic DNA replication consists of at least two steps (Remus and Diffley 2009; Masai et al. 2010; Bell and Kaguni 2013; Li and Araki 2013). First, from late M to G1 phase, two hexameric DNA helicase Mcm2–7 rings are assembled into the prereplication complex (pre-RC) at each origin (Duzdevich et al. 2015; Ticau et al. 2015). Second, Sld3, Sld7, and Cdc45 are recruited to the pre-RC and assembled into the Cdc45–MCM–Sld3 (CMS) complex relying on MCM phosphorylation by Dbf4-dependent kinase (DDK) (Labib 2010; Heller et al. 2011; Tanaka et al. 2011; Deegan et al. 2016; Fang et al. 2016). Subsequently, the S-phase cyclin-dependent kinases (S-CDKs) phosphorylate Sld3 and Sld2 to stimulate their interactions with Dbp11, which leads to assembly of the preinitiative complex (pre-IC) containing GINS and DNA polymerase ε (Tanaka et al. 2007; Zegerman and Diffley 2007; Muramatsu et al. 2010).
A key step in determining origin firing is the origin association of Sld3 and Cdc45, which is a prerequisite of the pre-RC-to-pre-IC transition (Kamimura et al. 2001; Kanemaki and Labib 2006). Interestingly, Sld3 and Cdc45 are available in limiting amounts relative to the total number of origins in budding yeast. Overexpression of these limiting factors often results in advanced firing of some late origins (Mantiero et al. 2011; Tanaka et al. 2011; Douglas and Diffley 2012). Sld3 and Cdc45 are also found to be enriched at early origins in a DDK-dependent manner (Heller et al. 2011; Tanaka et al. 2011). The direct target of DDK is Mcm2–7, which alleviates its self-inhibition (Sheu and Stillman 2010; Sheu et al. 2016). DDK-catalyzed Mcm2–7 phosphorylation is a prerequisite of Cdc45 and Sld3 loading both in vitro and in vivo (Heller et al. 2011; Tanaka et al. 2011; Deegan et al. 2016; Fang et al. 2016).
In this study, we identified direct interactions between the regulatory DDK subunit Dbf4 and two forkhead transcription factors, Fkh1 and Fkh2. Both chromatin immunoprecipitation (ChIP) and in vitro reconstitution demonstrate that the preferential recruitment of Dbf4 to a subset of early origins depends on Fkh. A dbf4 mutant devoid of the Fkh interaction domain (dbf4ΔC) delays early origin firing, mimicking fkh mutations or deletion of the Fkh-binding sites (FBSs). Importantly, the role of Fkh1 in replication timing regulation can be bypassed by directly fusing Dbf4 with the DNA-binding domain (DBD; also called forkhead) of Fkh1. However, such a fusion selectively abolishes the origin firing at centromeric (CEN) regions due to the interference of Ctf19-mediated Dbf4 recruitment. Moreover, origin-associated Dbf4 promotes the recruitment of downstream Sld3–Cdc45 limiting factors through direct interaction with Sld3. These findings elucidate the molecular underpinnings of selective recruitment of limiting factors via forkhead transcription factors in determining the replication timing program at non-CEN regions.
Results
Fkh1/2 are necessary and sufficient to selectively recruit Dbf4 to early origins
To gain insight into the mechanism of selective association of limiting initiation factors at early origins, we performed a yeast two-hybrid screen to identify the putative interacting partners of Dbf4 given its crucial role in helicase activation and origin firing (Jackson et al. 1993; Dowell et al. 1994; Bousset and Diffley 1998; Labib 2010; Tanaka et al. 2011; Ramer et al. 2013; Deegan et al. 2016; Fang et al. 2016). Besides the DDK kinase subunit Cdc7, two transcription factors, Fkh1 and Fkh2, displayed robust interaction with Dbf4 (Fig. 1A). It is noteworthy that Fkh did not interact with other limiting factors such as Sld3, Sld7, and Cdc45. Fkh1 and Fkh2 are highly conserved forkhead box transcription factors that were reported recently to be important for determining early initiation of a subset of origins containing FBSs (Knott et al. 2012; Peace et al. 2016). However, their exact role in DNA replication has not yet been determined.
Figure 1.
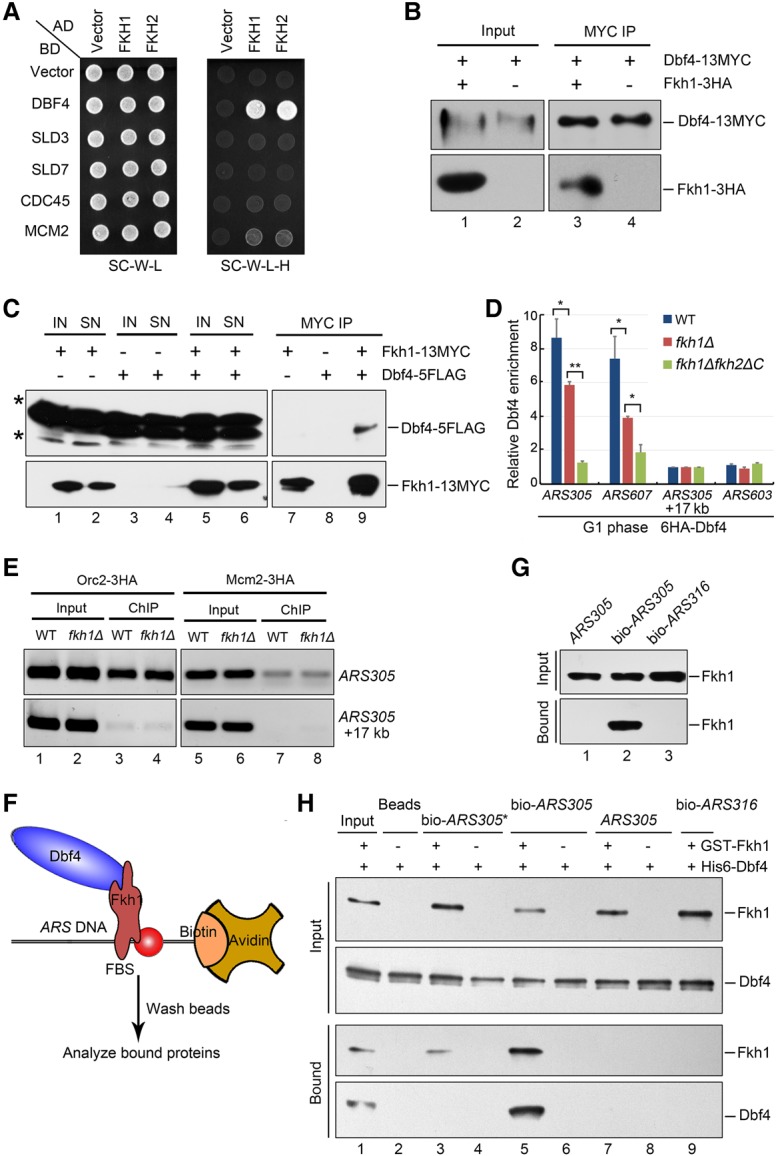
Fkh1/2 help to recruit Dbf4 onto early origins. (A) Both Fkh1 and Fkh2 forkhead transcription factors interact with Dbf4, but not other limiting firing factors, in a yeast two-hybrid assay. The yeast cells expressing the indicated proteins were grown at 30°C on either SC-W-L or SC-W-L-H plates. (B) Confirmation of Fkh1–Dbf4 interaction through immunoprecipitation. Dbf4 and Fkh1 were tagged with 13MYC and 3HA, respectively, at their genomic loci. Cell lysates were precipitated with an anti-MYC antibody. (C) Dbf4 coprecipitates with Fkh1 at their endogenous levels. Fkh1 was precipitated through its 13MYC tag, whereas the existence of Dbf4-5Flag in the precipitates was blotted against anti-Flag. Protein bands marked by an asterisk are cross-reactions. (IN) Input; (SN) supernatant. (D) Dbf4 is enriched at early origins in an Fkh1- and Fkh2-dependent manner. Cells with endogenous Dbf4 carrying a 6HA tag were arrested in G1 phase with α factor. Whole-cell extracts were prepared and subjected to ChIP as described in the Materials and Methods. (E) Fkh1 is dispensable for pre-RC assembly. ChIP of Orc2-3HA or Mcm2-3HA was conducted as described in D. (F) Schematic representation of the in vitro Dbf4 recruitment assay. Biotin-labeled ARS (bio-ARS) DNA fragments were immobilized to streptavidin beads. Purified recombinant Fkh1 and Dbf4 proteins were incubated with immobilized bio-ARS. After being washed three times, specifically bound fractions were detected by Western blotting. (G) Fkh1 binds to ARS305 (which contains FBSs) but not ARS605 (which does not). (H) In vitro reconstitution of Dbf4 recruitment to early origin ARS305. ARS305* (an ARS305 devoid of FBS), ARS316 (a late origin), and ARS305 with or without biotin label were used as controls.
To investigate whether the Dbf4–Fkh interactions identified here contribute to the establishment of replication timing, we carried out three sets of experiments. First, we corroborated the Dbf4–Fkh1 interaction with immunoprecipitation. For this purpose, DBF4 and FKH1 genes were tagged at their endogenous loci. Dbf4-13MYC was immunoprecipitated with an anti-MYC antibody and detected by Western blotting following SDS-PAGE. As shown in Figure 1B, a significant amount of Fkh1-3HA was detected in the precipitates of Dbf4-13MYC. Reciprocally, Dbf4 coprecipitated with Fkh1 in an independent immunoprecipitation experiment (Fig. 1C, lane 9). These results verified the physical association between Dbf4 and Fkh1 in vivo.
Second, we investigated the impact of Fkh–Dbf4 interaction on the enrichment of Dbf4 at origins during G1 phase using ChIP. Yeast cells with the endogenous Dbf4 carrying a 6HA epitope tag were grown and arrested with α factor in G1 phase, which is the stage of the cell cycle when the timing of origin activation has been determined (Wu and Gilbert 1996; Raghuraman et al. 1997; Gilbert et al. 2010). In agreement with earlier studies (Pasero et al. 1999; Katou et al. 2006; Natsume et al. 2013), Dbf4 was significantly enriched at early origins as FBS+ ARS305 and ARS607 compared with late origins as FBS− ARS603 (Fig. 1D). Strikingly, such enrichment of Dbf4 was reduced in fkh1Δ mutants and almost disappeared when combined with the loss of the Fkh2 C terminus (amino acids 584–862), which mediates a partially redundant role for Fkh1 in replication timing (Knott et al. 2012). In contrast, there was no apparent change in the enrichment of Orc2 and Mcm2 at ARS305 in the absence of Fkh1 (Fig. 1E), indicating that Fkh1 is unlikely to be involved in the pre-RC assembly stage. These results suggest that after pre-RC assembly, Fkh–Dbf4 interactions might be important for preferentially targeting Dbf4 to early origins in G1 phase.
Third, we asked whether Fkh1 is sufficient to recruit Dbf4 to the FBS+ early origins. To this end, we labeled an ARS DNA fragment with biotin (bio-ARS), which was efficiently immobilized by streptavidin beads (Supplemental Fig. S1A). Purified recombinant Fkh1 protein (Supplemental Fig. S1B) was incubated with immobilized ARS DNA (Fig. 1F). After three washes, bound fractions were analyzed by Western blotting. As shown in Figure 1G, Fkh1 was able to bind to FBS+ ARS305 but not to FBS− ARS316. When Dbf4 protein (Supplemental Fig. S1C) was added to this in vitro recruitment system, no bound fraction was detectable in either ARS305 or ARS316 (Fig. 1H, lanes 6,8). This result indicates that Dbf4 is not able to bind to ARS DNA per se. Therefore, the association between Dbf4 and ARS identified previously by yeast one-hybrid assay and other methods likely reflects an indirect one mediated by other proteins (Dowell et al. 1994; Katou et al. 2006; Jones et al. 2010). This turned out to be true; as shown in Figure 1H, Dbf4 prominently coprecipitated with bio-ARS305 beads only when Fkh1 was present (Fig. 1H, cf. lanes 5 and 6). To further confirm the Fkh1-dependent recruitment, we constructed an ARS305* with the FBS mutated. This mutation resulted in greatly decreased Fkh1 and barely detectable Dbf4 binding (Fig. 1H, lane 3). Association with late origins as ARS316 without FBS was not detectable either (Fig. 1H, lane 9). Taken together, these data demonstrate that Fkh1 is necessary and sufficient for selective Dbf4 loading onto FBS+ early origins.
Dbf4 C terminus-mediated interaction with Fkh1/2 is important for early origin firing
Next, we mapped the interaction domains of Fkh1/2 with Dbf4. Fkh1/2 bear a conserved FHA domain at their N termini and a forkhead domain (also known as the DBD; amino acids 231–374) within their C termini (Supplemental Fig. S2A). We first mutated a highly conserved arginine (R80) residue to alanine within the FHA domain, which often mediates protein–protein interactions (Li et al. 2012). Unexpectedly, R80A substitution did not interrupt the Fkh1–Dbf4 interaction in yeast two-hybrid assays (Fig. 2A, top panel). When we split Fkh1 into two halves, both the N and C halves showed positive interactions with Dbf4, indicating that more than one site of Fkh1 are able to bind Dbf4 (Fig. 2A, middle panel). If R80A mutation was introduced in the N half, it abolished FkhN–Dbf4 interaction (Fig. 2A, bottom panel), indicating that the FHA domain is one of the interaction sites of Fkh1. FHA-mediated association often shows bias to phosphorylated substrates. Coincidently, Dbf4 has been reported to be phosphorylated during the cell cycle in budding yeast (Weinreich and Stillman 1999). To address the possibility that phosphorylation of Dbf4 promotes interaction with Fkh proteins, endogenous Dbf4 protein was purified from yeast cells and treated with λ-phosphatase prior to incubation with recombinant GST-Fkh1 protein. Fkh1 displayed a lower affinity to phosphatase-treated Dbf4 than to native Dbf4 (Fig. 2B, cf. lanes 6 and 5). This result implicates that the Fkh–Dbf4 interaction might be regulated in vivo.
Figure 2.

The Dbf4–Fkh1 interaction is important for early origin firing. (A) Both the N-terminal FHA domain and the C terminus of Fkh1 interact with Dbf4. Fkh1 fragments and mutants were constructed and used in yeast two-hybrid assays as described in Figure 1A. Fivefold serial dilutions of yeast cells expressing the indicated proteins were grown at 30°C on either SC-W-L or SC-W-L-H plates. (B) Fkh1–Dbf4 association is likely enhanced by Dbf4 phosphorylation in vivo. Dbf4 was purified from yeast cells and treated with phosphatase prior to incubation with recombinant GST-Fkh1 protein. (C) Schematic of the motifs in Dbf4 interacting with other proteins. (HBRCT) A noncanonical BRCT domain with an additional helix. (D) Mapping the domain of Dbf4 interacting with Fkh1. Dbf4 truncations were constructed and used in yeast two-hybrid assays as described in A. (E) The Dbf4 C terminus is sufficient to bind directly to Fkh1 in vitro. Purified recombinant GST-Fkh1 and 6His-Dbf4 (656–704) proteins were incubated with glutathione-Sepharose beads in the binding buffer containing 1 µg/µL BSA. The protein bands were revealed by immunoblot against anti-His and anti-GST antibodies, respectively. The band marked by an asterisk is a cross-reaction. (F) ARS305 fails to fire in an interaction-defective dbf4ΔC mutant as analyzed by changes in DNA copy number. An average was calculated from three independent experiments. Error bars represent standard deviations (SDs). (*) P-value < 0.05.
Meanwhile, we used a similar strategy to map which regions in Dbf4 are involved in Fkh interaction (Fig. 2C). Among a series of truncations, loss of the very C-terminal 50 amino acids in Dbf4 (Dbf4C; 656–704) resulted in abolished interaction with Fkh1 (Fig. 2D) but did not affect interaction with Cdc7. Moreover, Dbf4C alone was able to support interaction with both Fkh1 and Fkh2 (Fig. 2D; Supplemental Fig. S2B). Finally, pull-down assays confirmed the direct association between Dbf4C and Fkh1 in vitro (Fig. 2E). Together, these results indicate that Dbf4C is necessary and sufficient to bind Fkh.
To directly examine the possible role of Fkh–Dbf4 interaction in the replication program, we next compared dbf4ΔC with wild type for origin duplication time and efficiency. Yeast cells were collected after release from G1 arrest for the indicated time points. Genomic DNA was prepared for each sample, and the copy number of ARS305 relative to a late origin ARS1412 was measured by quantitative PCR (qPCR) (Hayano et al. 2012). The ARS305/ARS1412 ratio was normalized to 1 for samples harvested in G1. In wild type, the early origin ARS305 was duplicated before 40 min (Fig. 2F). In fkh1Δ and fkh1Δfkh2ΔC alleles, the ARS305/ARS1412 ratio was reduced to ∼1.2 at 40 min and during the entire S phase, indicating that ARS305 loses priority over late origin ARS1412 in terms of timing and/or efficiency. These results are consistent with previous BrdU immunoprecipitation (BrdU-IP) profiles of these origins and support the view that Fkh is a critical determinant of the replication program (Knott et al. 2012; Peace et al. 2016). Intriguingly, an interaction-defective mutant, dbf4ΔC, phenocopied the replication defects observed in fkh alleles (Fig. 2F). Moreover, in dbf4ΔC cells, Fkh1 persisted in binding to these origins, as shown by ChIP assays (Supplemental Fig. S2C). This result indicates that loss of Dbf4–Fkh interaction does not affect the origin recruitment of Fkh1, in good agreement with the direct binding of Fkh1 and the forkhead domain to early origins in vitro, as shown in Figures 1G and 3A. Thus, the early origin firing defects observed in dbf4ΔC are very likely caused by reduced origin recruitment of Dbf4 due to a lack of the Fkh–Dbf4 interaction.
Directly targeting Dbf4 to origin DNA can bypass the requirement of Fkh1
An alternative possibility is that deletion of the Dbf4 C terminus could reduce DDK activity or impair interaction with replication factors other than Fkh (Harkins et al. 2009; Jones et al. 2010). To test these possibilities, we adopted a strategy to tether the Dbf4ΔC protein directly to FBS+ ARS DNA. DNA binding of Fkh1 is mediated by an evolutionarily conserved forkhead domain (amino acids 231–374) (Supplemental Fig. S3A; Stroud et al. 2006). We then deleted an NxxRHxxS motif within the forkhead domain (referred to here as forkhead*). By using the in vitro binding assay described in Figure 1G, we showed that the Fkh1 protein with a mutated forkhead* completely lost its ability to bind FBS+ ARS305 (Fig. 3A), reinforcing the notion that the forkhead domain is necessary for DNA binding. Next, we fused Dbf4ΔC with forkhead to directly target Dbf4 to FBS+ early origins (Fig. 3B). Indeed, the Dbf4ΔC–forkhead fusion successfully restored firing of ARS607 (Fig. 3C) and ARS305, albeit with a moderate delay for the latter (Supplemental Fig. S3B,C). In contrast, in both cases, no effects were observed in a fusion construct with inactivated forkhead*. These results demonstrate that the firing problem of Fkh-activated origins can be overcome through directly targeting Dbf4ΔC to origins. Therefore the replication timing phenotype observed in dbf4ΔC is not due to interfering with the DDK kinase activity but is directly caused by the compromised Fkh–Dbf4 interaction. Altogether, these data allow us to conclude that the Fkh-dependent origin association of Dbf4 is critical in promoting early origin firing.
Figure 3.
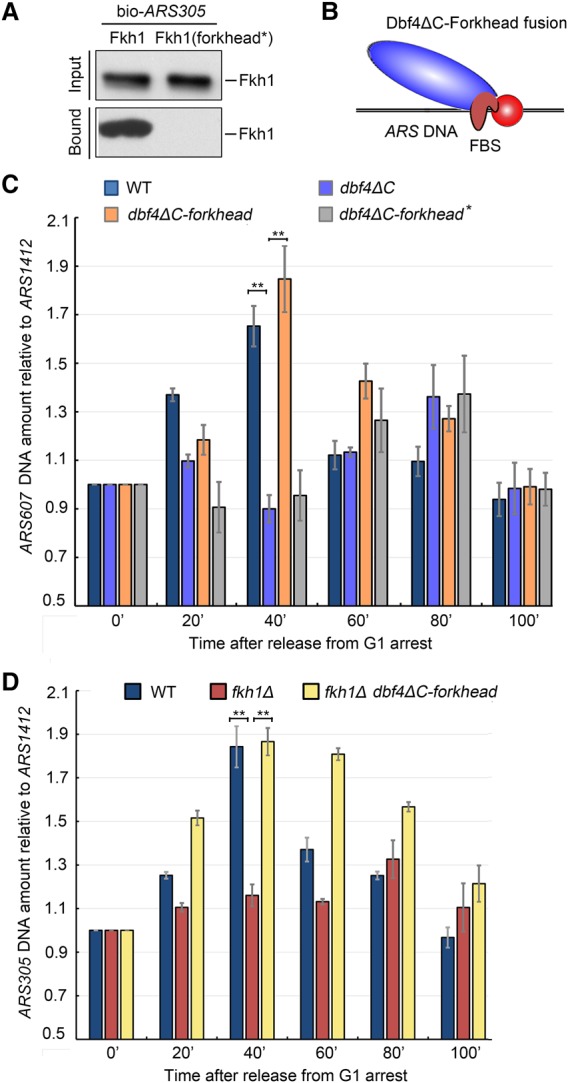
Fusing Fkh1-DBD to Dbf4ΔC restores the early origin replication. (A) The forkhead domain (DBD; amino acids 231–374) of Fkh1 is responsible for DNA binding. (Forkhead*) A forkhead mutant with deletion of a conserved eight-amino-acid patch (NxxRHxxS) within the DBD (see Supplemental Fig. S3A for sequence alignment). In vitro recruitment assays were performed as described in Figure 1G. (B) Schematic representation of the Dbf4ΔC–forkhead fusion. (C) Replication of ARS607 can be rescued by fusing Dbf4ΔC with the forkhead domain but not with the forkhead* mutant. An average was calculated from three independent experiments, as described in Figure 2F. Error bars represent SDs. (**) P-value < 0.01. See also Supplemental Figure S3B for the results of ARS305, and Supplemental Figure S3C for the flow cytometry profiles of these samples. (D) The role of Fkh1 in ARS305 replication can be bypassed via Dbf4ΔC–forkhead fusion. An average was calculated from three independent experiments. Error bars represent SDs. (**) P-value < 0.01.
Because the forkhead domain contains only DNA-binding activity and is unlikely to retain transcription factor function, the above results also suggest that the role of Fkh in regulating replication timing is very likely independent of its transcription role. Following this notion, we then examined whether Dbf4–forkhead fusion can completely bypass the requirement of Fkh1 for early origin firing. This turned out to be true. If we fused forkhead with Dbf4 in a fkh1Δ background, it rescued ARS305 firing to 40 min—equivalent to wild-type timing (Fig. 3D; Supplemental Fig. S3C). Interestingly, the ARS305/ARS1412 ratio dropped significantly more slowly in Dbf4–forkhead fusion than wild type, suggesting that the firing of FBS− late origins might be inhibited because the recycle usage of Dbf4 might be blocked under fusion conditions. These results strengthen the functional importance of fine-tuning origin usage through Fkh1-dependent targeting of Dbf4 to specific origins. These data also allow us to conclude that the role of Fkh1 in DNA replication is independent of its transcription function and lies solely in promoting origin loading of Dbf4.
Genome-wide replication profiles of fkh1Δ and the Dbf4–forkhead fusion
To confirm these results and extend the analysis to the entire yeast genome, we next monitored DNA replication in wild-type and fkh1Δ cells by deep sequencing (Müller et al. 2014). Briefly, cells were arrested in G1 with α factor and released synchronously into S phase in the presence of 200 mM hydroxyurea (HU) to allow initiation but restrict elongation to a region of ∼5 kb around active origins (Poli et al. 2012). Cells were collected in G1, and, 60 min after release into S phase, genomic DNA was extracted and sequenced with an Illumina HiSeq 4000 platform. To generate replication profiles, the ratio of uniquely mapped reads in HU-arrested cells to G1 samples was calculated. Unreplicated regions of the genome in HU presented the same copy number as in the G1 sample and showed a ratio close to 1. In contrast, origin firing resulted in an increased relative copy number around active origins (Fig. 4A). To quantify origin activity, the sum of unique reads in G1 and HU samples calculated for 5-kb regions centered on 504 annotated origins (Crabbe et al. 2010) was normalized and expressed as a ratio of HU to G1. In wild-type cells, this relative copy number was proportional to time of replication (Trep) of 230 individual origins (Fig. 4B) for which a Trep was determined (Yabuki et al. 2002). This is consistent with earlier studies (Poli et al. 2012; Yoshida et al. 2014) and indicates that relative copy number represents a good proxy for origin activity in HU-arrested cells. A comparison of biological replicates confirmed the robustness of the method (R2 = 0.96) (Supplemental Fig. S4A).
Figure 4.

Genome-wide analysis of origin usage in wild-type and fkh1Δ cells. (A) Replication profiles of wild-type, fkh1Δ, fkh1Δ DBF4–forkhead, fkh1Δ DBF4–forkhead*, and DBF4-GST cells for a representative region on chromosome XII. Cells were arrested in G1 for 170 min and released synchronously into S phase for 60 min in the presence of 200 mM HU. Relative copy number was determined by deep sequencing as the ratio of normalized reads in HU and G1 cells. Peaks corresponding to Fkh-dependent origins are labeled in orange, and pericentromeric origins are labeled in green. (B) Scatter plots of relative DNA copy number in wild-type, fkh1Δ, fkh1Δ DBF4–forkhead, fkh1Δ DBF4–forkhead*, and DBF4-GST cells versus the Trep (Yabuki et al. 2002) for 230 origins. Fkh1-dependent origins are labeled in orange, and pericentromeric origins are labeled in green. (C) Relative copy number at Fkh origins (n = 54) in wild-type, fkh1Δ, fkh1Δ DBF4–forkhead, fkh1Δ DBF4–forkhead*, and DBF4-GST cells. (D) Relative copy number at CEN origins (n = 31) in wild-type, fkh1Δ, fkh1Δ DBF4–forkhead, fkh1Δ DBF4–forkhead*, and DBF4-GST cells. (****) P < 0.0001; (**) P < 0.01; (ns) nonsignificant, Wilcoxon matched-pairs signed rank test.
In fkh1Δ cells, we observed a reduction of the activity of ARS305 relative to wild-type cells (Supplemental Fig. S4B), which is consistent with the results obtained by qPCR (Figs. 2F, 3D). However, the activity of the origins ARS306 to ARS309, which are not regulated by Fkh1 (Knott et al. 2012), was unchanged. Other examples of Fkh1-dependent and Fkh1-independent origins are shown on representative regions on chromosomes XII (Fig. 4A) and XIII (Supplemental Fig. S4G). Overall, we identified 54 origins showing a reduced activity in the fkh1Δ mutant in two independent sets of experiments (Supplemental Fig. S4C). These origins overlapped with 83% of the origins previously identified by BrdU-IP-seq (BrdU-IP combined with sequencing) as down-regulated in fkh1Δ cells (Supplemental Fig. S4D) and 52% of the origins down-regulated in the fkh1Δ fkh2ΔC double mutant (Knott et al. 2012). We also identified 60 origins showing an increased activity in the absence of Fkh1 (Supplemental Fig. S4C), presumably because these origins have access to larger pools of nucleotide and initiation factors upon inactivation of Fkh1-dependent origins. Together, these data indicate that deep sequencing of genomic DNA in G1- and HU-arrested cells represents an accurate method to monitor DNA replication in Fkh1-deficient cells.
When DNA copy number in wild-type and fkh1Δ cells was plotted relative to the Trep at the 230 origins for which a Trep is available, we noticed that the activity of the 54 Fkh1-dependent origins (Fig. 4B, orange) was reduced in fkh1Δ cells, whereas other origins (Fig. 4B, gray) were not affected. Importantly, we observed a significant rescue of Fkh origins when fkh1Δ cells were complemented with the Dbf4–forkhead fusion protein corresponding to the full-length Dbf4 protein fused to Fkh1-DBD but not with the Dbf4–forkhead* fusion protein that is unable to bind DNA (Fig. 4A–C). Together, these data validate that the Fkh-mediated recruitment of Dbf4 defines a rate-limiting step in establishing the temporal program of FBS+ origins distributed throughout the genome.
Dbf4–forkhead fusion specifically interferes with the firing of CEN origins
Intriguingly, we also detected early origins whose activity was completely lost in cells expressing the Dbf4–forkhead fusion proteins but not in wild-type and fkh1Δ cells (Fig. 4A; Supplemental Fig. S4G; green peaks). A visual inspection of chromosome maps revealed that all of these origins are located in the vicinity of centromeres. Interestingly, we also found that Dbf4 enrichment in CEN origins (ARS902, ARS1209, and ARS1513) are not significantly affected in the absence of Fkh1 (Supplemental Fig. S4H, cf. Fig. 1D). A previous study demonstrated that early replication of CEN origins depends on the recruitment of Dbf4 by Ctf19, a component of the kinetochore. Moreover, C-terminal tagging of Dbf4 with either MYC or FRB abolishes its localization at centromeres but not at non-CEN origins (Natsume et al. 2013). Consistently, we were able to detect a physical interaction between Dbf4 and Ctf19 in yeast two-hybrid assays when Dbf4 was fused at the N terminus but not at the C terminus (Supplemental Fig. S4I). Therefore, we reasoned that the fusion of Fkh1-DBD to Dbf4 could interfere with the recruitment of DDK at centromeres. To further confirm this hypothesis, we replaced the forkhead domain with a GST tag and integrated the DBF4-GST at the endogenous DBF4 locus. Remarkably, this construct interfered with the replication of CEN origins to the same extent as the Dbf4–forkhead fusion proteins (Figs. 4A,B,D). Altogether, these results indicate that Fkh1 targets Dbf4 to a subset of replication origins to promote early replication in a manner that is reminiscent of the recruitment of Dbf4 to pericentromeric origins by Ctf19 (Natsume et al. 2013).
Dbf4 recruits downstream limiting factors Sld3–Cdc45
The above results also imply that origin association of Dbf4 is sufficient to trigger downstream events required for origin firing. We thus hypothesized that other factors essential for initiating replication can be subsequently recruited to origins via Dbf4. To test this idea, we investigated whether Dbf4 interacts with downstream limiting factors such as Sld2, Sld3, or Cdc45. Among them, only Sld3 exhibited interaction with Dbf4 in yeast two-hybrid assays (Fig. 5A). The direct interaction between Dbf4 and Sld3 was validated with an in vitro pull-down assay using purified recombinant proteins (Fig. 5B). We further proved that the Dbf4 N terminus (1–390) is sufficient for Sld3 binding in vitro (Fig. 5B, lane 6). These results raise the possibility that the loading of Sld3 might be facilitated by origin-associated Fkh–Dbf4. Supporting this idea, similar to the Dbf4 ChIP results shown in Figure 1D, Sld3 enrichment at early origins ARS305/607 was lost in fkh and Fkh interaction-defective dbf4ΔC mutants (Fig. 5C). Since all of these mutants can be rescued by tethering Dbf4 to origins, these results indicate that the preferential association of Sld3 to early origins largely depends on the interaction between Dbf4 and Fkh. In addition, Sld3 also interacted with another downstream limiting factor, Cdc45 (Fig. 5A), which is consistent with the fact that Sld3–Cdc45 are recruited to origins in a mutually dependent fashion (Kamimura et al. 2001). Together, these data suggest that the Fkh-dependent origin association of Dbf4 promotes the subsequent recruitment of the limiting factors Sld3–Cdc45 and defines a critical upstream step during origin firing (Fig. 5D).
Figure 5.
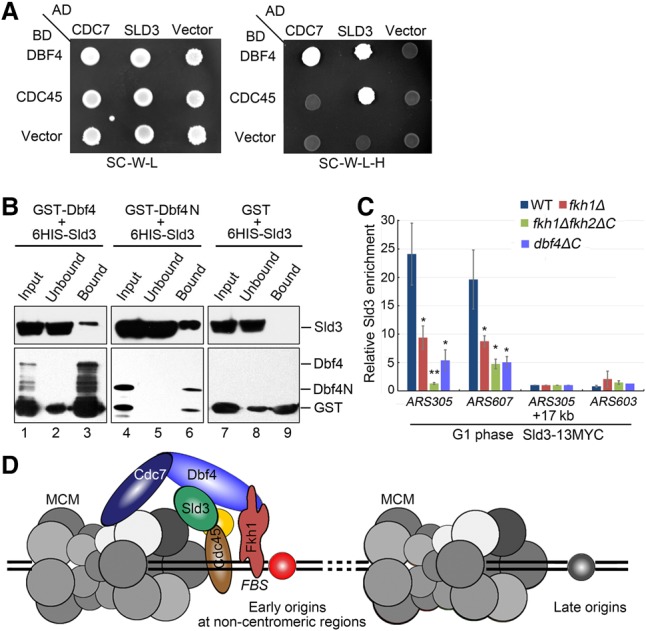
The preferential association of limiting factors Sld3–Cdc45 with early origins depends on Fkh and Dbf4. (A) Yeast two-hybrid assays of Dbf4–Sld3 and Sld3–Cdc45 pairs. (B) Direct association between Dbf4 and Sld3. Purified recombinant His6-Sld3 and GST-Dbf4 or GST-Dbf4 (1–390) were mixed with glutathione beads in the binding buffer containing 1 µg/µL BSA. The protein bands were revealed by immunoblot against anti-His and anti-GST antibodies, respectively. (C) Enrichment of Sld3 at early origins is abrogated in fkh1Δfkh2ΔC. Sld3-13MYC ChIP was conducted as described in the Materials and Methods. (D) A Fkh-dependent recruitment model of Dbf4 and downstream limiting factors. Dbf4 is recruited to a subset of FBS+ early origins at chromosome arms through interaction with Fkh1 or Fkh2. Origin-associated Dbf4 promotes the recruitment of the initiation factors such as Sld3 and Cdc45, which are essential for CMS assembly.
Discussion
Despite tremendous progress made in the analysis of replication initiation and timing during the last decade, the mechanisms by which limiting initiation factors are assembled at thousands of origins in a temporally coordinated manner remain to be determined. Here, we found that among these limiting factors, origin association of Dbf4 in G1 phase defines a rate-limiting step in determining the time of origin activation.
First, Dbf4 is preferentially recruited to a series of early origins in G1 phase by forkhead transcription factors Fkh1 and Fkh2. We demonstrated a direct physical interaction between Dbf4 and Fkh, which is necessary and sufficient for origin association of Dbf4 in vitro and in vivo. In the absence of Fkh1, 54 origins are repeatedly down-regulated, which can be rescued by fusing Dbf4 with Fkh1-DBD. Meanwhile, ∼60 non-Fkh1-dependent origins show a mild increase in activity in fkh1Δ cells. Dbf4 and other limiting initiation factors that are no longer tethered to Fkh origins are now available to activate these origins earlier or more efficiently. These origins may also benefit from the availability of more dNTPs following the loss of Fkh1-dependent origins. The patterns of down-regulated and up-regulated origins are basically consistent with the BrdU-IP-seq profiles reported by Aparicio's group (Knott et al. 2012). Based on the dimerization of Fkh1/2 (Ostrow et al. 2017), they propose an origin clustering model, which may further contribute to the local enrichment of Dbf4 at the replication factories (Saner et al. 2013).
Second, Dbf4 is targeted to different classes of origins with different chromatin contexts through distinct mechanisms. For a subset of non-CEN origins, Fkh1/2 mediate the recruitment of Dbf4. In contrast, for all CEN origins, Dbf4 recruitment would depend on interaction with a kinetochore scaffold protein, Cft19, as reported previously by Tanaka's group (Natsume et al. 2013). Although the very C terminus of Dbf4 seems to be required for both Fkh- and Ctf19-mediated recruitment pathways, tagging Dbf4 in the C terminus interferes with the latter, but not the former, pathway.
Third, Rif1 is a global marker of late origins from yeast to humans (Cornacchia et al. 2012; Hayano et al. 2012; Yamazaki et al. 2012). Intriguingly, Rif1 represses the origin firing through recruiting protein phosphatase 1 to counteract DDK-dependent MCM phosphorylation (Dave et al. 2014; Hiraga et al. 2014; Mattarocci et al. 2014). All of these studies point to the notion that DDK (recruitment and activity) is the bona fide determinant in establishing the global replication timing program across evolution.
It is also noteworthy that Dbf4 may have functions other than the regulation of the Cdc7 kinase. The essential role of DDK has been demonstrated to lie in the phosphorylation of MCM, which mediates the recruitment of Sld3 (Randell et al. 2010; Deegan et al. 2016). Besides the essential role as a DDK entity, this study indicates an additional scaffold role of Dbf4 in choreographing origin firing through direct interactions with upstream Fkh1/2 and downstream Sld3 proteins.
Transcription and chromatin structure have been shown to influence origin timing and/or efficiency, which is thought to regulate chromatin accessibility of origins to limiting factors (Gondor and Ohlsson 2009; Knott et al. 2009; Gilbert et al. 2010; Yoshida et al. 2013, 2014). It is noteworthy that Fkh transcription factors are able to directly recruit Dbf4 and downstream limiting factors. Combined with previous transcriptomic data (Knott et al. 2012; Peace et al. 2016), our Dbf4–forkhead fusion experiments argue strongly that the role of Fkh proteins in replication timing establishment is completely separable from their transcription function. As far as we know, this may represent the first direct link between transcription factors and the regulation of replication timing and efficacy.
Materials and methods
Yeast two-hybrid assay
Yeast two-hybrid assays were performed as described previously (Quan et al. 2015). Gal4-AD (pGADT7) and Gal4-BD (pGBKT7) vectors were used for the construction of plasmids expressing prey and bait proteins. AH109 was transformed with pGBKT7-derived bait and pGADT7-derived prey plasmids. To quantify two-hybrid interactions, cotransformed cells were spotted at fivefold serial dilutions on selective media and grown for 2–3 d.
Immunoprecipitation
Immunoprecipitation was carried out basically as described previously (Lou et al. 2008). Briefly, yeast protein extracts (input) were prepared for immunoprecipitation by bead-beating in lysis buffer. MYC-tagged proteins were immunoprecipitated using an anti-MYC monoclonal antibody (9E10) conjugated to protein G-Sepharose. Two percent of input or supernatant was run together with immunoprecipitation (30% of the precipitates). Blots were probed in phosphate-buffered saline containing 0.1% Tween and 2% dried milk. 9E10 (1:1000) was used to detect MYC-tagged proteins, M2 (1:1000) was used to detect Flag-tagged proteins, and polyclonal sera against Fkh1 (1:10000) and Mcm2 (1:10000) raised in this study were used to detect those proteins.
GST pull-down
GST or GST-Fkh1 was induced in BL21 cells for 3 h at 37°C using 0.1 mM IPTG. Cells were sonicated in lysis buffer containing 1% Triton X-100. Proteins with a GST tag were purified from soluble extracts by binding to glutathione-agarose (GE) and eluted in 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, and 10% glycerol containing 10 mM glutathione followed by dialysis against the same buffer. For GST pull-downs, 6His-Dbf4p, GST-Fkh1, or GST was incubated with glutathione-agarose in the presence of binding buffer for 1 h at 4°C. The glutathione agarose beads were washed extensively, and bound proteins were separated on 8% PAGE gels containing SDS. Blots were probed with monoclonal antibody against GST (1:1000) and His (1:1000).
ChIP
We performed ChIP from extracts of cross-linked cells with specific and nonspecific antibodies (Tanaka et al. 2011). One microgram of purified anti-MYC or anti-HA was used. After purification of associated DNA fragments, we used a “real-time” PCR machine (ABI, ViiA7) to calculate the enrichment of particular sequences in the specific immunoprecipitation. The ratio of immunoprecipitated DNA to total DNA in whole-cell extract was normalized as described in each figure legend and is shown as fold enrichment (Natsume et al. 2013).
In vitro origin-binding assay
Biotin-labeled origin DNA conjugated to streptavidin magnetic beads was incubated with different concentrations of purified proteins in a solution containing 20 mM Tris-HCl (pH 7.5), 100 µM EDTA, 10% glycerol, 40 µg/mL BSA, 10 mM magnesium acetate, and 200 µM DTT in a final reaction volume of 50 µL (Heller et al. 2011; Bruck and Kaplan 2015). The reactions were incubated for 1 h at 25°C. After the incubation, the magnetic beads were collected at room temperature using a magnet (Dynal). The supernatant was removed, and the beads were washed three times with the binding buffer. The beads were resuspended in 20 µL of sample buffer and boiled, and 50% of the binding reaction was resolved by 8% SDS-PAGE. Origin-binding proteins were analyzed by immunoblots.
Cell synchronization and flow cytometry analysis
α Factor (5 µg/mL) was added for cell synchronization in G1 phase. G1-arrested cells were released by filter-washing twice in fresh medium and continued growth for the indicated time. Samples were collected and fixed with 70% ethanol and then processed for flow cytometry using a BD Biosciences FACS Verse machine.
Protein expression and purification
Full-length and truncated forms of pGEX4T-1-Fkh1, pGEX-4T-1-Fkh1*, pGEX-4T-1-Fkh1(1–230), pGEX-4T-1-Fkh1(303–584), pET28a-Dbf4, pET-28a-Dbf4(656–704), pET28a-Cdc7, and pET28a-Fkh1 constructs used in the pull-down experiments were expressed in Escherichia coli BL21 (DE3) RIL codon-plus (Stratagene) and purified by affinity tags and conventional column chromatography.
Replication timing analysis
Log phase cultures were arrested in G1 phase at 25°C using α factor and then released into fresh medium without α factor for 100 min, and time-lapse culture were collected every 20 min. Genomic DNA was prepared for the G1-phase (0-min time point) and S-phase (20/40/60/80/100-min) cells and quantified by qPCR. The ratio of the amount of genomic DNA in S phase to that in G1 was calculated for each locus. The ARS1412 locus was used for normalization (Hayano et al. 2012). Two-tailed t-tests for each mutant against wild type were performed from at least three replicates. A P-value of <0.05 (*) was deemed significant and is indicated by one asterisk, whereas a P-value of <0.01 is indicated by two asterisks. Standard deviations are indicated in all panels.
Genome-wide replication profiling
Strains (W303-1a, DS1, DS6, DS8, and DS13 in Supplemental Table S1) were grown at 25°C in YEP medium supplemented with 2% glucose. Cells were arrested in G1 for 170 min with 2 µg/mL α factor (GenePep) and released into S phase by the addition of 75 µg/mL pronase into medium containing 0.2 M HU for 60 min. For genomic DNA extraction, 100 mL of yeast cells at 1 × 107 cells per milliliter was shaken five times for 2 min in NIB buffer (17% [v/v] glycerol, 50 mM MOPS buffer, 150 mM potassium acetate, 2 mM magnesium chloride, 500 µM spermidine, 150 µM spermine at pH 7.2) with zirconium beads on a Vibrax (VXR basic, Ika) at 4°C. DNA was isolated using Qiagen genomic DNA extraction kit according to the manufacturer's instructions. DNA was fragmented using sonication (∼200- to 500-base-pair [bp] size range). Sequencing libraries were prepared using a ThruPLEX DNA-seq kit (Rubicon Genomics). Next-generation sequencing was performed on a HiSeq 4000 (Illumina). Single-end reads of 50 bp were aligned to the Saccharomyces cerevisiae genome (2011) and rDNA sequence with Bowtie, allowing only perfect matches. Relative copy number was determined as the ratio of normalized reads in HU and G1 cells. Replication profiles were smoothed with a sliding window of 1000 bp and displayed using Integrated Genome Browser version 8.2 (Nicol et al. 2009). Statistical analyses were performed using GraphPad Prism 7.
Supplementary Material
Acknowledgments
We thank Dr. Qun He, Dr. Li-Lin Du, and members of the Lou laboratory for helpful discussion. We thank Ismael Padioleau for help with bioinformatic analyses. This work was supported by National Natural Science Foundation of China 31630005, 31770084, 31628011, and 31771382; Opening Project of the State Key Laboratory of Microbial Resources; Program for Extramural Scientists (2016SKLAB6-10) of the State Key Laboratory of Agrobiotechnology; and Chinese Universities Scientific Fund 2015TC039. Work in P.P.’s laboratory was supported by grants from Agence Nationale pour la Recherche (ANR), Institut National du Cancer (INCa), Ligue contre le Cancer (équipe labellisée), and the MSDAvenir fund. K.G. and M.S. were supported by the Foundation for Polish Science (TEAM). H.L. and P.P. conceived and designed the research; D.F., A.L., D.S., X.W., and Q.C. performed experiments; M.S. and K.G. prepared libraries for next-generation sequencing and carried out next-generation sequencing; P.P., A.L., R.F., and C.Y. did the bioinformatic analysis; and H.L., P.P., and A.L. analyzed the data and wrote the paper.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.306571.117.
References
- Bell SP, Kaguni JM. 2013. Helicase loading at chromosomal origins of replication. Cold Spring Harb Perspect Biol 5: a010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousset K, Diffley JFX. 1998. The Cdc7 protein kinase is required for origin firing during S phase. Genes Dev 12: 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck I, Kaplan DL. 2015. The replication initiation protein Sld3/Treslin orchestrates the assembly of the replication fork helicase during S phase. J Biol Chem 290: 27414–27424. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cornacchia D, Dileep V, Quivy JP, Foti R, Tili F, Santarella-Mellwig R, Antony C, Almouzni G, Gilbert DM, Buonomo SBC. 2012. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J 31: 3678–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe L, Thomas A, Pantesco V, De Vos J, Pasero P, Lengronne A. 2010. Analysis of replication profiles reveals key role of RFC–Ctf18 in yeast replication stress response. Nat Struct Mol Biol 17: 1391–1397. [DOI] [PubMed] [Google Scholar]
- Dave A, Cooley C, Garg M, Bianchi A. 2014. Protein phosphatase 1 recruitment by Rif1 regulates DNA replication origin firing by counteracting DDK activity. Cell Rep 7: 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deegan TD, Yeeles JT, Diffley JF. 2016. Phosphopeptide binding by Sld3 links Dbf4-dependent kinase to MCM replicative helicase activation. EMBO J 35: 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dileep V, Rivera-Mulia JC, Sima J, Gilbert DM. 2015. Large-scale chromatin structure–function relationships during the cell cycle and development: insights from replication timing. Cold Spring Harb Symp Quant Biol 80: 53–63. [DOI] [PubMed] [Google Scholar]
- Douglas ME, Diffley JF. 2012. Replication timing: the early bird catches the worm. Curr Biol 22: R81–R82. [DOI] [PubMed] [Google Scholar]
- Dowell SJ, Romanowski P, Diffley JF. 1994. Interaction of Dbf4, the Cdc7 protein kinase regulatory subunit, with yeast replication origins in vivo. Science 265: 1243–1246. [DOI] [PubMed] [Google Scholar]
- Duzdevich D, Warner Megan D, Ticau S, Ivica Nikola A, Bell Stephen P, Greene Eric C. 2015. The dynamics of eukaryotic replication initiation: origin specificity, licensing, and firing at the single-molecule level. Mol Cell 58: 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Cao Q, Lou H. 2016. Sld3–MCM interaction facilitated by Dbf4-dependent kinase defines an essential step in eukaryotic DNA replication initiation. Front Microbiol 7: 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkos M, Ganier O, Coulombe P, Mechali M. 2015. DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 16: 360–374. [DOI] [PubMed] [Google Scholar]
- Gilbert DM, Takebayashi S-I, Ryba T, Lu J, Pope BD, Wilson KA, Hiratani I. 2010. Space and time in the nucleus: developmental control of replication timing and chromosome architecture. Cold Spring Harb Symp Quant Biol 75: 143–153. [DOI] [PubMed] [Google Scholar]
- Gondor A, Ohlsson R. 2009. Replication timing and epigenetic reprogramming of gene expression: a two-way relationship? Nat Rev Genet 10: 269–276. [DOI] [PubMed] [Google Scholar]
- Harkins V, Gabrielse C, Haste L, Weinreich M. 2009. Budding yeast Dbf4 sequences required for Cdc7 kinase activation and identification of a functional relationship between the Dbf4 and Rev1 BRCT domains. Genetics 183: 1269–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayano M, Kanoh Y, Matsumoto S, Renard-Guillet C, Shirahige K, Masai H. 2012. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev 26: 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RC, Kang S, Lam WM, Chen S, Chan CS, Bell SP. 2011. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 146: 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga S, Alvino GM, Chang F, Lian HY, Sridhar A, Kubota T, Brewer BJ, Weinreich M, Raghuraman MK, Donaldson AD. 2014. Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev 28: 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Pahl PM, Harrison K, Rosamond J, Sclafani RA. 1993. Cell cycle regulation of the yeast Cdc7 protein kinase by association with the Dbf4 protein. Mol Cell Biol 13: 2899–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DR, Prasad AA, Chan PK, Duncker BP. 2010. The Dbf4 motif C zinc finger promotes DNA replication and mediates resistance to genotoxic stress. Cell Cycle 9: 2018–2026. [DOI] [PubMed] [Google Scholar]
- Kamimura Y, Tak YS, Sugino A, Araki H. 2001. Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae. EMBO J 20: 2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemaki M, Labib K. 2006. Distinct roles for Sld3 and GINS during establishment and progression of eukaryotic DNA replication forks. EMBO J 25: 1753–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katou Y, Kaneshiro K, Aburatani H, Shirahige K. 2006. Genomic approach for the understanding of dynamic aspect of chromosome behavior. Methods Enzymol 409: 389–410. [DOI] [PubMed] [Google Scholar]
- Knott SR, Viggiani CJ, Tavare S, Aparicio OM. 2009. Genome-wide replication profiles indicate an expansive role for Rpd3L in regulating replication initiation timing or efficiency, and reveal genomic loci of Rpd3 function in Saccharomyces cerevisiae. Genes Dev 23: 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott SR, Peace JM, Ostrow AZ, Gan Y, Rex AE, Viggiani CJ, Tavare S, Aparicio OM. 2012. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell 148: 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib K. 2010. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev 24: 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Araki H. 2013. Loading and activation of DNA replicative helicases: the key step of initiation of DNA replication. Genes Cells 18: 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Coic E, Lee K, Lee CS, Kim JA, Wu Q, Haber JE. 2012. Regulation of budding yeast mating-type switching donor preference by the FHA domain of Fkh1. PLoS Genet 8: e1002630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Komata M, Katou Y, Guan Z, Reis CC, Budd M, Shirahige K, Campbell JL. 2008. Mrc1 and DNA polymerase ɛ function together in linking DNA replication and the S phase checkpoint. Mol Cell 32: 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Mackenzie A, Donaldson A, Zegerman P. 2011. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J 30: 4805–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M. 2010. Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem 79: 89–130. [DOI] [PubMed] [Google Scholar]
- Mattarocci S, Shyian M, Lemmens L, Damay P, Altintas DM, Shi T, Bartholomew CR, Thoma NH, Hardy CF, Shore D. 2014. Rif1 controls DNA replication timing in yeast through the PP1 phosphatase Glc7. Cell Rep 7: 62–69. [DOI] [PubMed] [Google Scholar]
- Müller CA, Hawkins M, Retkute R, Malla S, Wilson R, Blythe MJ, Nakato R, Komata M, Shirahige K, de Moura APS, et al. 2014. The dynamics of genome replication using deep sequencing. Nucleic Acids Res 42: e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu S, Hirai K, Tak Y-S, Kamimura Y, Araki H. 2010. CDK-dependent complex formation between replication proteins Dpb11, Sld2, Pol ε, and GINS in budding yeast. Genes Dev 24: 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T, Muller CA, Katou Y, Retkute R, Gierlinski M, Araki H, Blow JJ, Shirahige K, Nieduszynski CA, Tanaka TU. 2013. Kinetochores coordinate pericentromeric cohesion and early DNA replication by Cdc7-Dbf4 kinase recruitment. Mol Cell 50: 661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol JW, Helt GA, Blanchard JSG, Raja A, Loraine AE. 2009. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25: 2730–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrow AZ, Kalhor R, Gan Y, Villwock SK, Linke C, Barberis M, Chen L, Aparicio OM. 2017. Conserved forkhead dimerization motif controls DNA replication timing and spatial organization of chromosomes in S. cerevisiae. Proc Natl Acad Sci 114: E2411–E2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero P, Duncker BP, Schwob E, Gasser SM. 1999. A role for the Cdc7 kinase regulatory subunit Dbf4p in the formation of initiation-competent origins of replication. Genes Dev 13: 2159–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peace JM, Villwock SK, Zeytounian JL, Gan Y, Aparicio OM. 2016. Quantitative BrdU immunoprecipitation method demonstrates that Fkh1 and Fkh2 are rate-limiting activators of replication origins that reprogram replication timing in G1 phase. Genome Res 26: 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J, Tsaponina O, Crabbé L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A, Pasero P. 2012. dNTP pools determine fork progression and origin usage under replication stress. EMBO J 31: 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan Y, Xia Y, Liu L, Cui J, Li Z, Cao Q, Chen Xiaojiang S, Campbell Judith L, Lou H. 2015. Cell-cycle-regulated interaction between Mcm10 and double hexameric Mcm2–7 is required for helicase splitting and activation during S phase. Cell Rep 13: 2576–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghuraman MK, Brewer BJ, Fangman WL. 1997. Cell cycle-dependent establishment of a late replication program. Science 276: 806–809. [DOI] [PubMed] [Google Scholar]
- Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL. 2001. Replication dynamics of the yeast genome. Science 294: 115–121. [DOI] [PubMed] [Google Scholar]
- Ramer MD, Suman ES, Richter H, Stanger K, Spranger M, Bieberstein N, Duncker BP. 2013. Dbf4 and Cdc7 promote DNA replication through interactions with distinct Mcm2–7 subunits. J Biol Chem 288: 14926–14935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randell JC, Fan A, Chan C, Francis LI, Heller RC, Galani K, Bell SP. 2010. Mec1 is one of multiple kinases that prime the Mcm2–7 helicase for phosphorylation by Cdc7. Mol Cell 40: 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus D, Diffley JF. 2009. Eukaryotic DNA replication control: lock and load, then fire. Curr Opin Cell Biol 21: 771–777. [DOI] [PubMed] [Google Scholar]
- Rhind N, Gilbert DM. 2013. DNA replication timing. Cold Spring Harb Perspect Biol 5: a010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saner N, Karschau J, Natsume T, Gierliński M, Retkute R, Hawkins M, Nieduszynski CA, Blow JJ, de Moura APS, Tanaka TU. 2013. Stochastic association of neighboring replicons creates replication factories in budding yeast. J Cell Biol 202: 1001–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu YJ, Stillman B. 2010. The Dbf4–Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 463: 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu Y-J, Kinney JB, Stillman B. 2016. Concerted activities of Mcm4, Sld3, and Dbf4 in control of origin activation and DNA replication fork progression. Genome Res 26: 315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud JC, Wu Y, Bates DL, Han A, Nowick K, Paabo S, Tong H, Chen L. 2006. Structure of the forkhead domain of FOXP2 bound to DNA. Structure 14: 159–166. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Umemori T, Hirai K, Muramatsu S, Kamimura Y, Araki H. 2007. CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 445: 328–332. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Nakato R, Katou Y, Shirahige K, Araki H. 2011. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr Biol 21: 2055–2063. [DOI] [PubMed] [Google Scholar]
- Ticau S, Friedman LJ, Ivica NA, Gelles J, Bell SP. 2015. Single-molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading. Cell 161: 513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich M, Stillman B. 1999. Cdc7p–Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J 18: 5334–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JR, Gilbert DM. 1996. A distinct G1 step required to specify the Chinese hamster DHFR replication origin. Science 271: 1270–1272. [DOI] [PubMed] [Google Scholar]
- Yabuki N, Terashima H, Kitada K. 2002. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells 7: 781–789. [DOI] [PubMed] [Google Scholar]
- Yamazaki S, Ishii A, Kanoh Y, Oda M, Nishito Y, Masai H. 2012. Rif1 regulates the replication timing domains on the human genome. EMBO J 31: 3667–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Poveda A, Pasero P. 2013. Time to be versatile: regulation of the replication timing program in budding yeast. J Mol Biol 425: 4696–4705. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Bacal J, Desmarais D, Padioleau I, Tsaponina O, Chabes A, Pantesco V, Dubois E, Parrinello H, Skrzypczak M, et al. 2014. The histone deacetylases sir2 and rpd3 act on ribosomal DNA to control the replication program in budding yeast. Mol Cell 54: 691–697. [DOI] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF. 2007. Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 445: 281–285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.