

Abstract
Background
Betulinic acid (BA) is a natural triterpenoid compound and exhibits a wide range of biological and medicinal properties including anti-inflammatory activity. Therefore, this theoretical investigation is performed to evaluate (a) physicochemical properties such as acid dissociation constant (pKa), distribution coefficient (logD), partition coefficient (logP), aqueous solubility (logS), solvation free energy, dipole moment, polarizability, hyperpolarizability and different reactivity descriptors, (b) pharmacokinetic properties like human intestinal absorption (HIA), cellular permeability, skin permeability (PSkin), plasma protein binding (PPB), penetration of the blood brain barrier (BBB), (c) toxicological properties including mutagenicity, carcinogenicity, risk of inhibition of hERG gene and (d) molecular mechanism of anti-inflammatory action which will aid the development of analytical method and the synthesis of BA derivatives.
Methods
The physicochemical properties were calculated using MarvinSketch 15.6.29 and Gaussian 09 software package. The pharmacokinetic and toxicological properties were calculated on online server PreADMET. Further, the molecular docking study was conducted on AutoDock vina in PyRx 0.8.
Results
The aqueous solubility increased with increasing pH due to the ionization of BA leading to decrease in distribution coefficient. The solvation energies in water, dimethyl sulfoxide (DMSO), acetonitrile, n-octanol, chloroform and carbon tetrachloride were − 41.74 kJ/mol, − 53.80 kJ/mol, − 66.27 kJ/mol, − 69.64 kJ/mol, − 65.96 kJ/mol and − 60.13 kJ/mol, respectively. From the results of polarizability and softness, it was clear that BA is less stable and hence, kinetically more reactive in water. BA demonstrated good human intestinal absorption (HIA) and moderate cellular permeability. Further, BA also exhibited positive CNS activity due to high permeability through BBB. The toxicological study revealed that BA was a mutagenic compound but noncarcinogenic in mice model. Moreover, molecular docking study of BA with PLA2 revealed that BA interacts with GLY22 & GLY29 through hydrogen bond formation and LEU2, PHE5, HIS6, ALA17, ALA18, HIS47 and TYR51 through different types of hydrophobic interactions. The binding affinity of BA was − 41.00 kJ/mol which is comparable to the binding affinity of potent inhibitor 6-Phenyl-4(R)-(7-Phenyl-heptanoylamino)-hexanoic acid (BR4) (− 33.89 kJ/mol).
Conclusions
Our computed properties may assist the development of analytical method to assay BA or to develop BA derivatives with better pharmacokinetic and toxicological profile.
Keywords: Physicochemical properties, Solvation free energy, Polarizability, Pharmacokinetic, Toxicology, Carcinogenic, Molecular docking
Background
Betulinic acid (BA), (3β-hydroxy-lup-20(29)-en-28-oic acid) (Fig. 1) is a natural pentacyclic lupane type triterpenoid compound and exhibits a wide range of biological and medicinal properties such as antivenom [1], anti-HIV [2, 3], antibacterial [2], antimalarial [4], anti-inflammatory [5–7] anthelmintic [8], antinociceptive [9], anti-HSV-1 [10, 11] and anticancer activities [12–14]. It is abundantly distributed throughout the plant kingdom [15]. The birch tree (Betula spp., Betulaceae) is one of the most widely reported sources of BA which can be obtained in considerable quantities [16, 17]. BA can also be isolated from various sources including Corypha taliera [18], Ziziphus spp. (Rhamnaceae) [19, 20], Syzygium spp. (Myrtaceae) [21], Diospyros spp. (Ebenaceae) [22, 23] and Paeonia spp. (Paeoniaceae) [24]. Betulin, the reduced form of BA, was one of the first natural products to be isolated from the bark of the white birch, Betula alba [25].
Fig. 1.
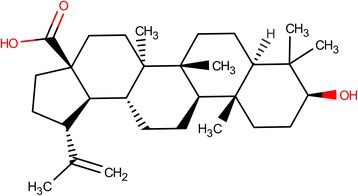
Structure of betulinic acid
BA was shown to exert its diverse pharmacological activities with variable median inhibitory concentrations (IC50) such as anticancer activity by inhibiting DNA Topoisomerases (Topos) II at IC50 of 56.12 μM [26], anti-HIV activity at IC50 of 23.65 μM [27], anti-malarial activity at IC50 of 56.71 μM [28], anti-fungal property at IC50 of 14.23 μM [29], anti-protozoal activity at IC50 of 50 μM [30].
BA also inhibited DNA polymerase beta at IC50 of 30.65 μM [31], protein tyrosine phosphatase 1B (PTP1B) at IC50 value of 1.5 μM [32], inhibited T. brucei GAPDH COX-1, COX-2 and LT formation in vitro with IC50 values of 240 μM [33], > 125 μM, > 125 μM and 102.2 μM [34], respectively. BA displayed potent anti-inflammatory activity by inhibiting Phospholipase A2 (PLA2) and showed 30% and 40% inhibition of PLA2 at concentrations of approximately 2.5 and 5 μM, respectively [5].
Earlier studies showed that BA exhibited anti-inflammatory activity by inhibiting TNF- alpha and increasing the production of anti-inflammatory cytokine IL-10 [35]. Kim and colleagues demonstrated that betulinic acid exerted its anti-inflammatory activity by inhibiting the nuclear factor-kappa beta pathway, production of nitric oxide (NO), prostaglandin E2 (PGE2), tumor necrosis factor- alpha, interleukin-6 (IL-6), and interleukin-1 beta levels [36]. Anti-inflammatory activity was also seen in encephalitogenic T cells where betulinic acid inhibited IL-17 and IFN-γ production [37]. Arachidonoyl trifluoromethyl ketone, bromoenol lactone, varespladib, varespladib methyl, ecopladib, efipladib, giripladib, pyrrophenone, pyrroxyphene, FPL67047XX, inhibitor 22, amide 23 (GK115), 2-oxoamides, 1,3-disubstituted propan-2-ones are all synthetic phospholipase A2 inhibitors that have been used for clinical cases [38]. Among natural compounds, extracts of curcumin, Ginkgo biloba and Centella asiatica have demonstrated to be phospholipase A2 inhibitors and have even been used to treat neurological disorders characterized by neuroinflammation [38]. Bernard and colleagues experimentally proved that betulin and betulinic acid were potent phospholipase A2 inhibitors [5].
Phospholipase A2 (PLA2) hydrolyzes the membrane glycerophospholipids and releases arachidonic acid eventually leading to the production of pro-inflammatory mediators such as leukotrienes, prostaglandins, platelet activating factors (PAF) [39]. Thus, inhibiting PLA2 activity and hence regulating the production of pro-inflammatory mediators for the development of therapeutics against inflammatory diseases [40, 41] is a plausible approach. However, the PLA2 exists in two isoforms [40, 42], which includes the low molecular weight (14 kDa) Ca+ 2 dependent extracellular PLA2 found in mammalian pancreases, several snake venoms, human platelets, human placentas, rheumatoid synovial fluids [43] and the high molecular weight (85 kDa) cytosolic PLA2 [44]. Convincing evidence suggests that the14 kDa PLA2 might be a potential target for the modulation of inflammatory diseases [40], here we have reported the molecular docking study of BA against human secretory (14 kDa) PLA2 (1KQU) to explore the molecular basis of anti-inflammatory action of BA.
Few computational and theoretical studies of BA have been reported. BA associates with human serum albumin via hydrogen bond with PHE206 & GLU354 and hydrophobic interactions with PHE206, ARG209, ALA210, ALA213, LEU327, GLY328, LEU331, ALA350 and LYS351, in the sub-domain IIA and IIB of the large hydrophobic cavity [45].
In this investigation, computational studies have been carried out to evaluate (a) physical and chemical properties such as acid dissociation constant (pKa), distribution coefficient (logD), partition coefficient (logP), aqueous solubility (logS), solvation free energy, dipole moment, polarizability, hyperpolarizability and different reactivity descriptors (chemical hardness, softness, chemical potential, electronegativity, electrophilicity index), (b) pharmacokinetic properties like human intestinal absorption (HIA), cellular permeability using Caco-2 cell model, skin permeability (PSkin), plasma protein binding (PPB), penetration of the blood brain barrier (BBB), (c) toxicological properties including mutagenicity, carcinogenicity, risk of inhibition of human ether-a-go-go-related (hERG) gene and (d) molecular mechanism of anti-inflammatory action of BA. The purpose of this study was to investigate the physicochemical, pharmacokinetic and toxicological properties and to correlate the calculated physicochemical properties with the absorption and distribution profile of BA. These in silico investigations will provide an insight for the development of analytical method to assay BA [46] or to develop BA derivatives with better pharmacokinetic & toxicological profile and having more potent anti-inflammatory activity.
Methods
Computational methods
The acid dissociation constant (pKa), distribution coefficient (logD), partition coefficient (logP) and aqueous solubility (logS) of betulinic acid (BA) over the pH range of 0.0 to 14.0at 298 K were calculated using MarvinSketch 15.6.29 (ChemAxon, Hungary) (http://www.chemaxon.com). The consensus logP method was applied to calculate distribution coefficient of the molecule.
The rapid progress in computational methods such as Gaussian family of methods (G1, G2, G2MP2, G3) and complete basis set extrapolation (CBS) method enable researchers to perform highly sophisticated calculations of enthalpies and thermochemical properties with minor errors in comparison to experimental data [47–51]. However, these methods are computationally very expensive. An alternative to these high cost calculations is the use of density functional theory (DFT) methods. Previous report revealed that [52] calculation of geometries (bond length, bond angle and dihedral angle) and thermochemical properties using DFT/B3LYP level of theory showed better agreement with the experiments. So, in the current investigation, the calculation of solvation free energy, dipole moment, polarizability, hyperpolarizability and global reactivity descriptor properties such as the chemical hardness, softness, chemical potential, electronegativity, electrophilicity index were conducted in gas phase and in different solvents namely water, dimethyl sulfoxide (DMSO), acetonitrile, n-octanol, chloroform and carbon tetrachloride with the B3LYP/6-31G(d) level of theory implemented in Gaussian 09 software package [53]. All calculations were conducted using the optimized geometry which was confirmed by the absence of imaginary frequency in the lowest energy state of the molecule. The Solvation Model on Density (SMD) [54] was used for all calculations involving the solvents. All calculations involving solvation were performed using the optimized solution-phase structures.
The pharmacokinetic and toxicological properties were calculated using online server PreADMET (https://preadmet.bmdrc.kr/). The pharmacokinetic properties such as human intestinal absorption (HIA), in vitro cellular permeability using Caco-2 cell model, skin permeability (PSkin), plasma protein binding (PPB) and penetration of the blood-brain barrier (BBB), interaction with P-glycoprotein (Pgp) and metabolism (both phase I and phase II) were calculated and predicted. In addition, BA was virtually screened to evaluate toxicological properties such as mutagenicity, carcinogenicity and risk of inhibition of human ether-a-go-go-related (hERG) gene.
Molecular docking study
Preparation of target protein X-ray structure
The first step of docking study is to select an appropriate X-ray crystal structure of the target protein that is already bound to its known ligand. This is because during docking, software searches complementary binding site/(s) for ligand within the search space of the target protein. That’s why ligand bound conformation of protein structure is the prerequisite to perform molecular docking. Moreover, the structure should be solved with a reasonable accuracy which is reflected in the statistics for data collection and processing of X-ray crystal structure. In addition, refinement statistics such as Rwork/Rfree, RMS deviation from ideality (bonds and angles) and validation parameters including Ramachandran outliers, rotamer outliers, bad bond count or bad angle count etc. also indicate the quality of the model structure. In our current investigation, we have selected the crystal structure of secretory PLA2 complexed with 6-Phenyl-4(R)-(7-Phenyl-heptanoylamino)-hexanoic acid (BR4) (PDB code: 1KQU) [55] since the structure was solved at 2.1 Å resolution with Rwork/Rfree of 0.209/0.240 and the validation parameters indicates good quality of the model structure. Water molecules and hetero atoms were then removed from the protein using PyMOL (Version 1.7.4.4, Schrödinger). Energy minimization was performed by applying YASARA force field level of theory in YASARA Energy Minimization Server (http://www.yasara.org/minimizationserver.htm).
Preparation of ligands
The structure of the BR4 and BA were drawn and optimized in Gaussian 09 [53] with density functional theory (DFT) at the B3LYP/6-31G(d) level of theory. The optimized structures of ligands (BR4 and BA) were saved in PDB format for docking study.
Protein-ligand docking
Several virtual screening tools are available to perform molecular docking such as BINDSURF, METADOCK, Lead Finder, FlexScreen, AutoDock vina etc. Scoring functions implemented in these softwares calculate the protein-ligand interactions energy by computing electrostatic, Van der Waals and hydrogen bonding terms [56–60]. In addition to these scoring functions AutoDock vina also calculates the hydrophobic interactions [60] which is crucial for calculating the interaction energies of protein and hydrophobic ligand. Since BA is hydrophobic in nature, we used AutoDock vina to explore not only the electrostatic, Van der Waals and hydrogen bonding but also the hydrophobic interactions.
The docking of target protein with the ligands was conducted using AutoDock vina [60] in PyRx 0.8 (https://pyrx.sourceforge.io/). Docking study was performed to get a set of possible conformations and orientations for the ligand at the binding site. Using PyRx software, the PLA2 and ligands were prepared after which docking was conducted using a grid whose center was (56.5961, 34.0180, 42.4808) and the dimensions were (25.00, 25.00, 25.00) Å. During the docking analysis, the drug molecules were flexible and the macromolecule was kept rigid. Ligand displaying the lowest binding affinity and ability to bind in the binding pocket of protein was chosen as the best conformation. The interactions of different residues of protein with ligands such as hydrogen bonds, electrostatic interactions, hydrophobic interactions and bond distances were analyzed by PyMOL and Discovery Studio visualizer v4.0.100.13345 (Figs. 2 and 3).
Fig. 2.
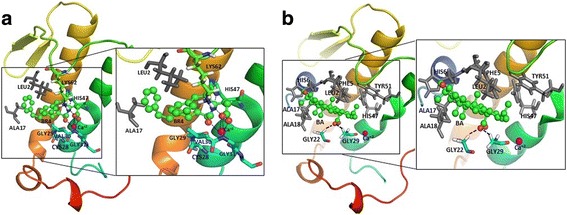
Interaction of (a). BR4 (green) and (b). BA (green) with PLA2 visualized in pymol. The calcium ion (Ca+ 2) is presented as magenta color sphere. Black dash indicates H bonding. Residues involve in hydrophobic interactions are shown in gray color
Fig. 3.
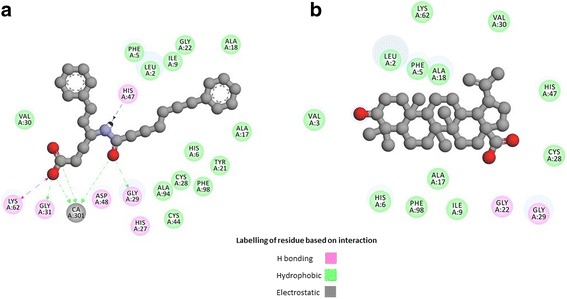
Interaction of (a). BR4 (gray) and (b). BA (gray) with PLA2 visualized in Drug Discovery studio
Before docking of BA, the protocol was validated by re-docking BR4 into the binding pocket of 1KQU to get the docked pose and root mean square deviation (RMSD). The result revealed that the first pose of BR4 almost superimposed (RMSD of heavy atoms constituting the backbone of molecule is 0.5271) with the experimental crystal structure of BR4 (Fig. 4). Thus, the docking method has reasonable accuracy and reproducibility and can be used for further docking experiments.
Fig. 4.
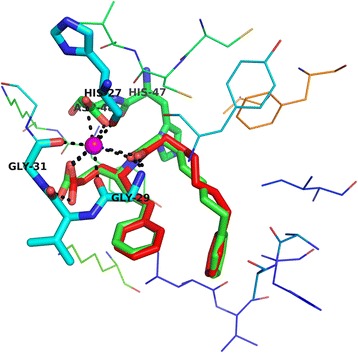
The superimposition of the best docking structure with the X-ray structure visualized in PyMOL; ( ) experimental ligand, (
) experimental ligand, ( ) docked ligand
) docked ligand
Results and discussion
Calculation of pKa
The calculation of pKa revealed that BA has two ionized species along with the unionized form (Fig. 5). It is clear from the figure that non-ionized form (Betulinic acid_1) predominates over the pH range of 0.0 to 4.6 whereas the betulinate form (Betulinic acid_3) dominates from pH 4.8 to 14.0. However, small amount of Betulinic acid_2 was present over the pH of 0.0 to 2.4.
Fig. 5.
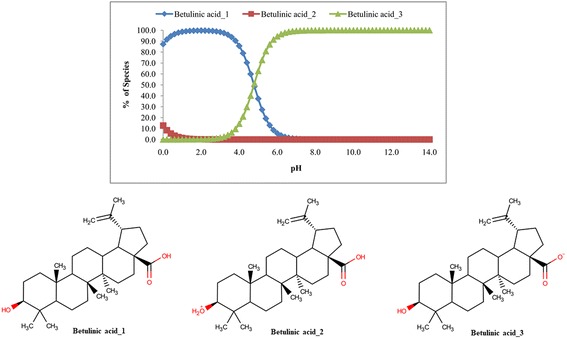
Microspecies distribution (%) of betulinic acid
Distribution (logD) coefficient and partition (logP) coefficient
The logD was calculated using consensus method implemented in MarvinSketch 15.6.29. The logD vs pH of BA is presented in Fig. 6. The logD value decreases with increasing pH of the solution suggesting that the prevalence of the unionized form of BA decreases and the ionized form (betulinate) increases with increasing pH of the solution. This result is in accordance with the calculation of pKa.
Fig. 6.
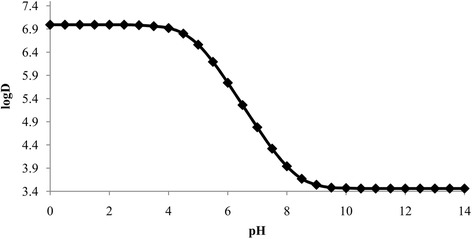
Distribution coefficient (logD) of betulinic acid over the pH range of 0.0 to 14.0
The partition coefficient (logP) of BA was also computed and presented in Table 1. The logP calculated in MarvinSketch 15.6.29 was 6.64 whereas the experimental logP of BA were reported as 6.61 [61] and 6.85 [62].
Table 1.
Comparison of partition coefficient of betulinic acid
| logP | |
|---|---|
| MarvinSketch 15.6.29 | 6.64 |
| Experimental (40) | 6.61 |
| Experimental (41) | 6.85 |
Aqueous solubility (logS)
The aqueous solubility of BA in terms of logS is presented in Fig. 7. The figure indicates that the solubility increases with increasing pH of the solution. However, the intrinsic solubility of BA was found − 7.34 (0.000000046 mol/L) and the aqueous solubility at pH 7.4 was − 4.79 (0.000016 mol/L) indicating that BA is practically insoluble in water.
Fig. 7.
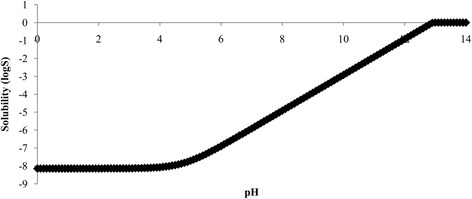
Solubility (logS) of betulinic acid over the pH range of 0.0 to 14.0
Solvation free energy
The solvation free energies of BA were calculated with the SMD model [54] The values are summarized in Table 2. The solvation energies of BA in water, DMSO, acetonitrile, n-octanol, chloroform and carbontetrachloride were − 41.74 kJ/mol, − 53.80 kJ/mol, − 66.27 kJ/mol, − 69.64 kJ/mol, − 65.96 kJ/mol and − 60.13 kJ/mol, respectively. It could therefore be concluded that solvation free energy increases with decreasing polarity of polar nonprotic solvent (DMSO and acetonitrile). The highest value of solvation energy was in non-polar protic solvent (n-octanol). The free energy decreases with decreasing polarity of non-polar aprotic solvent (chloroform and carbon tetrachloride). Thus, it could be stated that BA associates with non-polar portion (hydrocarbon chain of n-octanol) via hydrophobic interactions. Electrostatic interactions might arise due to the association of the carboxyl group of BA with the polar portion (hydroxyl group of n-octanol) (Table 5).
Table 2.
Solvation free energy (kJ/mol) of betulinic acid in different solvents with SMD
| Medium (dielectric constant) | Solvation free energy (kJ/mol) |
|---|---|
| Water (78.3) | −41.74 |
| DMSO (46.8) | −53.80 |
| Acetonitrile (35.7) | −66.27 |
| n-Octanol (9.9) | − 69.64 |
| Chloroform (4.7) | −65.96 |
| Carbon tetrachloride (2.2) | −60.13 |
Table 5.
Molecular Orbital Energy (eV) (HOMO and LUMO) of betulinic acid in different solvents with SMD
| Medium (dielectric constant) | Molecular Orbital Energy (eV) | ||
|---|---|---|---|
| HOMO | LUMO | ∆E | |
| Gas Phase | −6.305 | 0.133 | 6.438 |
| Water (78.3) | −6.262 | 0.116 | 6.378 |
| DMSO (46.8) | −6.243 | 0.321 | 6.564 |
| Acetonitrile (35.7) | −6.244 | 0.321 | 6.565 |
| n-Octanol (9.9) | −6.241 | 0.201 | 6.441 |
| Chloroform (4.7) | −6.222 | 0.286 | 6.508 |
| Carbon tetrachloride (2.2) | −6.229 | 0.251 | 6.480 |
Dipole moment
The dipole moment of BA is found to be higher in different solvents than that of the gas phase. Table 3 presents the dipole moments computed in the gas phase and different solvents (water, DMSO, acetonitrile, n-octanol, chloroform and carbon tetrachloride) at the B3LYP level of theory with 6-31G(d) basis set using SMD solvation model. The dipole moments were 2.98D, 5.13D, 4.50D, 4.53D, 4.54D, 3.96D and 3.46D in the gas phase, water, DMSO, acetonitrile, n-octanol, chloroform and carbon tetrachloride, respectively. Therefore, increasing dielectric constant of the solvent is accompanied by a gradual increase in the dipole moment. In other words, the dipole moment increases with the increasing polarity of the solvent (Fig. 8).
Table 3.
Dipole moment (Debye, (D)) of betulinic acid in gas phase and in different solvents using SMD
| Medium (dielectric constant) |
Dipole Moment (D) |
|---|---|
| Gas | 2.98 |
| Water (78.3) | 5.13 |
| DMSO (46.8) | 4.50 |
| Acetonitrile (35.7) | 4.53 |
| n-Octanol (9.9) | 4.54 |
| Chloroform (4.7) | 3.96 |
| Carbon tetrachloride (2.2) | 3.46 |
Fig. 8.
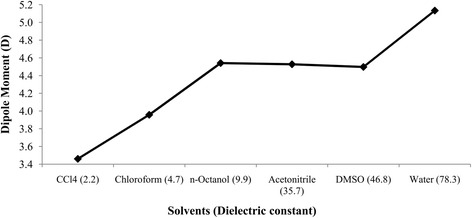
Effect of solvent polarity on dipole moment (D) of betulinic acid
Polarizability and first order hyperpolarizability
The polarizability(α) of BA was calculated using the following equation:
The quantities αxx, αyy and αzz are known as principal values of polarizability tensor.
The calculated polarizability of BA in different solvents is presented in Table 4, which showed that polarizability (αtot) ranged from 351.90 to 448.67 a.u. The plot of polarizability vs. solvent is shown in Fig. 9. It is clear from the figure that the polarizability gradually increases when going from lower to higher dielectric constant. This suggests that the kinetic reactivity of BA increases with increasing polarity of the solvent [63].
Table 4.
Effect of solvent polarity on polarizability (a.u.) and first order hyperpolarizability (a,u)
| Medium (dielectric constant) | αxx | αyy | αzz | αtot | βx | βy | βz | βtot |
|---|---|---|---|---|---|---|---|---|
| Gas Phase | 355.73 | 300.52 | 260.89 | 305.72 | 103.92 | −121.26 | −37.58 | 164.06 |
| Water (78.3) | 447.54 | 459.46 | 439.01 | 448.67 | 154.68 | − 170.92 | −87.52 | 246.57 |
| DMSO (46.8) | 441.82 | 450.17 | 428.25 | 440.08 | 132.92 | − 155.98 | −68.48 | 216.07 |
| Acetonitrile (35.7) | 442.00 | 448.66 | 425.71 | 438.79 | 130.61 | −155.78 | −70.89 | 215.30 |
| n-Octanol (9.9) | 434.46 | 426.93 | 394.48 | 418.62 | 139.39 | − 157.93 | −74.51 | 223.43 |
| Chloroform (4.7) | 418.61 | 395.58 | 356.17 | 390.12 | 124.74 | − 146.22 | −58.57 | 200.93 |
| Carbon tetrachloride (2.2) | 393.70 | 352.38 | 309.63 | 351.90 | 117.66 | − 134.29 | −46.43 | 184.48 |
Fig. 9.
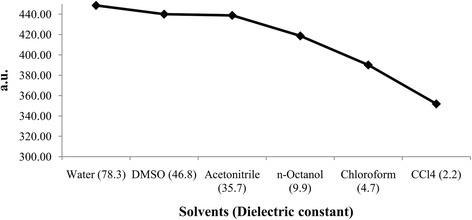
Effect of solvent polarity on polarizability of betulinic acid
The first order hyperpolarizability (βtot) is the measure of the nonlinear optical activity and can be calculated using the following equation:
Where,
The βtot for different solvents was listed in Table 4, which displayed that the hyperpolarizability in different solvents ranged from 184.48 to 246.57 a.u. Moreover, the hyperpolarizability increases as the dielectric constant increases except for non-polar protic n-octanol where the hyperpolarizability is higher than that of acetonitrile and DMSO (Fig. 10).
Fig. 10.
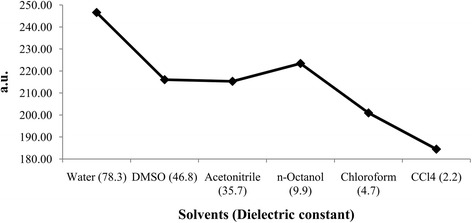
Effect of solvent polarity on first order hyperpolarizability of betulinic acid
Global reactivity descriptors
The highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO-LUMO) energy gap represents the stability or reactivity of molecules. Higher energy gap indicates greater stability and vice versa. The values of HOMO-LUMO energy gap in various solvents are presented in Table 5 and their trend is shown in Fig. 11. From Fig. 11, it is clear that the HOMO-LUMO energy gap is the highest in DMSO and acetonitrile, indicating greater stability of BA in these solvents, however lowest HOMO-LUMO energy gap was found in water meaning the titled molecule is less stable and hence, more reactive in water which is in agreement with calculated polarizability, chemical hardness and softness. The Table 5 and Fig. 11 suggest that the molecule is stabilized with decreasing polarity of the solvent i.e. the molecule is less likely to be kinetically reactive.
Fig. 11.
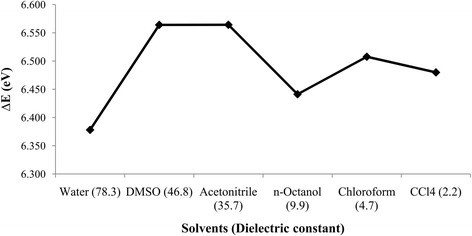
Effect of solvent polarity on HOMO-LUMO energy gap of betulinic acid
The global chemical reactivity descriptors such as softness, hardness, chemical potential and electrophilicity index can be calculated from the HOMO-LUMO energy gap of a molecule [64–68]. Using Koopman’s theorem for closed-shell molecules the hardness (η), chemical potential(μ), electronegativity (χ) and softness (S) are calculated according to the following equation:
Where
Ionization potential, I = -EHOMO
Electron affinity, A = -ELUMO
The electrophilicity index (ω) is calculated according to equation derived (by Parr et al., [68]) [58] as follows:
The molecular properties of BA in different solvents are presented in Table 6. No systematic trend was found in the case of chemical hardness & softness, chemical potential, electronegativity and electrophilicity index. However, the highest chemical softness, electronegativity and electrophilicity index was found in water (polar protic) followed by n-octanol (non-polar aprotic) and carbon tetrachloride (non-polar aprotic).
Table 6.
Effect of solvent polarity on molecular properties of betulinic acid
| Medium (dielectric constant) | Chemical hardness (η) | Softness (S) | Chemical potential (μ) | Electronegativity (χ) | Electrophilicity index (ω) |
|---|---|---|---|---|---|
| Gas Phase | 3.219 | 0.311 | −3.086 | 3.086 | 1.479 |
| Carbon tetrachloride (2.2) | 3.240 | 0.309 | −2.989 | 2.989 | 1.379 |
| Chloroform (4.7) | 3.254 | 0.307 | −2.968 | 2.968 | 1.354 |
| n-Octanol (9.9) | 3.221 | 0.310 | −3.020 | 3.020 | 1.416 |
| Acetonitrile (35.7) | 3.282 | 0.305 | −2.962 | 2.962 | 1.336 |
| DMSO (46.8) | 3.282 | 0.305 | −2.961 | 2.961 | 1.336 |
| Water (78.3) | 3.189 | 0.314 | −3.073 | 3.073 | 1.480 |
Pharmacokinetic study
Pharmacokinetic studies such as absorption, distribution and metabolism of BA was done by using the web based application PreADMET (https://preadmet.bmdrc.kr/). The calculated absorption, distribution and metabolism parameters are presented in Table 7.
Table 7.
Absorption, distribution and metabolism of betulinic acid
| Absorption | |||
| HIA (%) | PCaco-2 (nm/s) | Pskin | |
| 96.00 | 21.86 | −2.11 | |
| Distribution | |||
| PPB (%) | Cbrain/Cblood | P-Glycoprotein (Inhibition) | P-Glycoprotein (Substrate) |
| 100.00 | 8.20 | Inhibitor | Substrate |
| Metabolism | |||
| Phase I | Phase II | ||
| Enzyme | Inhibitor/Substrate | Enzyme | Substrate/Non-substrate |
| Cytochrome P450 2C19 (Inhibitor) | Non | UDP-glucuronosyltransferase (UGT) | Non-substrate |
| Cytochrome P450 2C9 (Inhibitor) | Inhibitor | Sulfotransferase (SULT) | Non-substrate |
| Cytochrome P450 2D6 (Inhibitor) | Non | ||
| Cytochrome P450 2D6 (substrate) | Non | ||
| Cytochrome P450 3A4 ((Inhibitor) | Inhibitor | ||
| Cytochrome P450 3A4 (substrate) | Substrate | ||
The calculated human intestinal absorption HIA of BA (Table 7) was found to be 95.996% which suggests that BA is well absorbed through the intestinal cell [69]. BA was moderately permeable since values for absorption through Caco-2 cell (PCaco-2) was 21.86 [70]. The skin permeability (PSkin) is a vital parameter for assessment of drugs and chemical that might require transdermal administration [71]. BA was found to be impermeable through skin since a calculated PSkin was − 2.11 encountered which proves BA is not permeable through the skin.
The distribution properties were assessed by measuring the brain to blood partitioning (Cbrain/Cblood) and plasma protein binding (PPB). Generally, compounds with more than 90% of PPB are classified as strongly bound chemicals whereas less than 90% are weakly bound chemicals (https://preadmet.bmdrc.kr/adme-prediction/). The calculated value of PPB for BA was 100.00% indicating BA is a strongly bound chemical and hence, the free form of BA will be less available in the systematic circulation. Based on Cbrain/Cblood ratio all chemicals fall under three categories namely high absorption to CNS (Cbrain/Cblood value more than 2.0), moderate absorption to CNS (Cbrain/Cblood value within 2.0–0.1) and low absorption to CNS (Cbrain/Cblood value less than 0.1) [72]. The ratio of Cbrain/Cblood (8.20) suggests high absorption of BA to CNS indicating higher ability to cross blood brain barrier (BBB).
The exchangeable fraction (sum of unbound fraction and fraction dissociated from the protein) of drug determines the amount of drug that will penetrate the BBB [73]. Besides, Pardridge and colleagues have reported the permeation of protein-bound compounds through the BBB [74–76]. Since BA is a strongly protein bound chemical (100%) so it could be assumed that BA will penetrate the BBB either due to the fraction dissociated from the plasma protein or as a protein-bound form or both.
P-glycoprotein (Pgp) is the product of the multi drug resistance (MDR) gene and an ATP dependent efflux transporter that affects the absorption, distribution and excretion of clinically important drugs [77]. Over-expression of this protein, which may result in MDR is a major cause of the failure of cancer chemotherapy, and decreased efficacy of antibiotics [78, 79]. The prediction of Pgp substrates, which facilitates early identification and elimination of drug candidates of low efficacy or high potential of MDR [80, 81]. Identifying molecules that interact with Pgp transporters is important for drug discovery, but it is commonly determined through laborious in vitro and in vivo studies [82]. Computational classification model can be used to screen molecules and predict the likeliness to be substrate for Pgp [82]. The in silico screening revealed that BA is a dual inhibitor and substrate for Pgp like quinidine [83]. This is because due to complex modulatory interactions with the Pgp which make BA to function as combination of substrate and inhibitor [83].
The computed metabolism of BA (Table 7) displayed that it is an inhibitor of CYP2C9 and a dual inhibitor and substrate for CYP3A4 due to complex modulation of CYP3A4 in phase I reaction. In phase II reaction BA is neither a substrate for UDP-glucuronosyltransferase (UGT) nor sulfotransferase (SULT).
Toxicological study
The PreADMET server (https://preadmet.bmdrc.kr/) was used to evaluate the carcinogenicity of BA. It was found that although BA is mutagenic but it demonstrated noncarcinogenicity in mice. On the other hand, BA is unlikely to be an inhibitor of human ether-a-go-go-related (hERG) gene (low risk). Inhibition of the hERG gene has been linked to long QT syndrome [84]. The results have been in summarized in Table 8.
Table 8.
Toxicological properties of betulinic acid
| Mutagenicity (Ames test) |
Carcinogenicity in Mouse |
hERG (Inhibition) |
|---|---|---|
| Mutagenic | Negative | Low risk |
Protein optimization
YASARA Energy Minimization Server (http://www.yasara.org/minimizationserver.htm) [85] was used to optimize the structure of PLA2. This server utilizes a new partly knowledge-based all atom force field derived from Amber, whose parameters have been optimized to attain the protein structure as close as to its native structure with maximum accuracy [85]. The simulation was performed in a water sphere containing ions.
The structure validation Z-scores and force field energies before and after the minimization are displayed in Table 9. The energies of the structure before and after optimization were − 68,549.6 and − 83,729.2 kJ/mol, respectively. This indicates that the protein structure was stabilized by an amount of − 15,179.6 kJ/mol. In addition, the structure validation Z-scores of before and after optimization was − 0.1 and 1.15 indicating good optimization of the target protein. The structure of PLA2 after energy minimization in a water sphere is presented in Fig. 11. PyMOL (Version 1.7.4.4, Schrödinger) was used to evaluate the root mean square deviation (RMSD) between the initial and final structure of PLA2 which was found to be 0.363.
Table 9.
Optimization parameter of phospholipase A2 (1KQU)
| Force field energy (kJ/mol) | Structure validation Z-score | RMSD of initial and final structure | |
|---|---|---|---|
| Before optimization | −68,549.6 | −0.1 | 0.363 |
| After optimization | −83,729.2 | 1.15 | |
| Difference (∆) | −15,179.6 | 1.25 |
Molecular docking
Molecular docking of BA was carried out with human PLA2 using AutoDock Vina, to identify and understand the binding mode of BA and the intermolecular interactions with the target protein. All reported PLA2 inhibitors possess three key enzyme binding components such as Ca+ 2 binding oxygen atom, a HIS47 binding H-bond donor and a hydrophobic component that bind the active site of the enzyme [55]. So, PLA2 inhibitors should form electrostatic interactions with Ca+ 2 ion, H bond formation with the catalytic HIS47 and different types of hydrophobic interactions with hydrophobic residues which line the active site cavity of the enzyme. In addition, the inhibitors should interact and displace the HIS6; a unique residue to human PLA2 enzyme [55]. From our docking study, it was found that BA interacts with PLA2 at a binding affinity of − 41.00 kJ/mol whereas for BR4 it was − 33.89 kJ/mol. BA lies deep within the active site activity and makes numerous close contacts (< 5.4 Å) with the enzyme through hydrogen bond formation with GLY22 & GLY29 and hydrophobic interactions with the LEU2, PHE5, HIS6, ALA17, ALA18, TYR51 and the catalytic residue HIS47 (Figs. 2 and 3 and Table 10). However, no interactions of BA with calcium were observed as it was found in case of bovine PLA2 [5]. Therefore, BA has the ability to interact with the catalytic residue HIS47 and the base forming residue HIS6 of human PLA2.
Table 10.
Binding affinity (kJ/mol) and interactions of BR4 and BA with phospholipase A2 (1KQU)
| Compound Name | Binding affinity (kJ/mol) | Amino acid and ligand (BR4/BA) interactions | Bond Distances (Å) | Category of interaction | Types of interaction |
|---|---|---|---|---|---|
| 6-Phenyl-4(R)-(7-Phenyl-heptanoylamino)-hexanoic acid (BR4) | −33.89 | LYS62[H…O]BR4 | 2.870 | Hydrogen Bond;Electrostatic | Salt Bridge;Attractive Charge |
| [Ca+ 2…O]BR4 | 4.024 | Electrostatic | Attractive Charge | ||
| BR4[NH…N]HIS47 | 1.829 | Hydrogen Bond | Conventional Hydrogen Bond | ||
| GLY29[NH…O]BR4 | 1.796 | Hydrogen Bond | Conventional Hydrogen Bond | ||
| GLY31[NH…O]BR4 | 2.374 | Hydrogen Bond | Conventional Hydrogen Bond | ||
| CYS28[H…O]BR4 | 2.601 | Hydrogen Bond | Carbon Hydrogen Bond | ||
| VAL30[H…O]BR4 | 2.388 | Hydrogen Bond | Carbon Hydrogen Bond | ||
| LYS62[H…O]BR4 | 2.941 | Hydrogen Bond | Carbon Hydrogen Bond | ||
| [Ca+ 2…O]BR4 | 2.767 | Other | Metal-Acceptor | ||
| [Ca+ 2…O]BR4 | 2.489 | Other | Metal-Acceptor | ||
| BR4[Pi…Pi]BR4 | 4.726 | Hydrophobic | Pi-Pi T-shaped | ||
| GLY29-VAL30[CON…BR4] | 4.326 | Hydrophobic | Amide-Pi Stacked | ||
| BR4[Pi…Alkyl]LEU2 | 4.891 | Hydrophobic | Pi-Alkyl | ||
| BR4[Pi…Alkyl]ALA17 | 4.480 | Hydrophobic | Pi-Alkyl | ||
| BR4[Pi…Alkyl]ALA18 | 5.058 | Hydrophobic | Pi-Alkyl | ||
| BR4[Pi…Alkyl]LEU2 | 4.483 | Hydrophobic | Pi-Alkyl | ||
| Betulinic acid (BA) | −41.00 | GLY22[H…O]BA | 2.493 | Hydrogen Bond | Carbon Hydrogen Bond |
| GLY29[H…O]BA | 2.714 | Hydrogen Bond | Carbon Hydrogen Bond | ||
| BA[Pi…Sigma]PHE5 | 3.783 | Hydrophobic | Pi-Sigma | ||
| BA[Alkyl…Alkyl]LEU2 | 5.406 | Hydrophobic | Alkyl | ||
| BA[Alkyl…Alkyl]LEU2 | 4.322 | Hydrophobic | Alkyl | ||
| BA[Alkyl…Alkyl]ALA18 | 3.715 | Hydrophobic | Alkyl | ||
| BA[Alkyl…Alkyl]LEU2 | 3.813 | Hydrophobic | Alkyl | ||
| BA[Alkyl…Alkyl]ALA18 | 3.870 | Hydrophobic | Alkyl | ||
| BA[Alkyl…Alkyl]ALA18 | 3.892 | Hydrophobic | Alkyl | ||
| BA[Alkyl…Alkyl]LEU2 | 4.924 | Hydrophobic | Alkyl | ||
| LEU2[Alkyl…Alkyl]BA | 3.962 | Hydrophobic | Alkyl | ||
| ALA17[Alkyl…Alkyl]BA | 5.402 | Hydrophobic | Alkyl | ||
| ALA17[Alkyl…Alkyl]BA | 5.356 | Hydrophobic | Alkyl | ||
| ALA18[Alkyl…Alkyl]BA | 3.737 | Hydrophobic | Alkyl | ||
| PHE5[Pi…Alkyl]BA | 5.290 | Hydrophobic | Pi-Alkyl | ||
| PHE5[Pi…Alkyl]BA | 4.239 | Hydrophobic | Pi-Alkyl | ||
| HIS6[Pi…Alkyl]BA | 5.133 | Hydrophobic | Pi-Alkyl | ||
| HIS6[Pi…Alkyl]BA | 4.162 | Hydrophobic | Pi-Alkyl | ||
| HIS47[Pi…Alkyl]BA | 5.074 | Hydrophobic | Pi-Alkyl | ||
| HIS47[Pil…Alkyl]BA | 5.073 | Hydrophobic | Pi-Alkyl | ||
| TYR51[Pi…Alkyl]BA | 4.321 | Hydrophobic | Pi-Alkyl |
On the other hand, the standard compound BR4 forms hydrogen bonding with LYS62, HIS47, GLY29, GLY31, CYS28, VAL30 and LYS62. Different types of hydrophobic interactions were also displayed by BR4 with GLY29-VAL30, LEU2 and ALA1. Moreover, BR4 exhibited attractive electrostatic interactions with Ca+ 2 ions.
Binding affinity and in vitro inhibitory activity
A comparison was made between the binding affinities and in vitro inhibitory activities (IC50) of LY 311727, Manoalide, Indomethacin, Manoalogue, Aristolochic Acid and 1,1,1-Trifluoro-2-heptadecanone. The data has been presented in Table 11. Similar protocol described earlier was followed during performing docking of these compounds with PLA2 (1KQU). From the table it was clear that generally compounds with higher binding affinities also possess higher inhibitory activities. The binding affinities and reported PLA2 inhibitory concentration of BA are − 41.00 kJ/mol and ~ 2.5 μM for 30% & ~ 5 μM for 40% [5] inhibition which is consistent in accordance to our findings of binding affinity and in vitro inhibitory activity.
Table 11.
Binding affinities (kJ/mol) and in vitro inhibitory activity (IC50) of different PLA2 inhibitors
Conclusions
The acid dissociation constant (pKa), distribution coefficient (logD), partition coefficient (logP), aqueous solubility (logS), solvation free energy, dipole moment, polarizability, hyperpolarizability and different reactivity descriptors such as the chemical potential, electrophilicity, chemical hardness and chemical softness of betulinic acid have been calculated using MarvinSketch 15.6.29 and Gaussian 09 software. The calculated properties showed that betulinic acid has three species (one unionized and two ionized) over the pH range of 0.0 to 14.0 suggesting the unionized form is likely to exist in the acidic pH of the stomach however the betulinate form (ionized form) would be the predominant form in the alkaline pH of the intestine.
The logP, logD and logS values suggest that betulinic acid is non-polar and hydrophobic. This property would enable betulinic acid to pass through the lipid bilayer membrane of the cells. This hypothesis was corroborated by the results of HIA and permeability of betulinic acid through Caco-2 cell. It was found that BA has low skin permeability hence topical application BA as an anti-inflammatory agent can be achieved. Further, being lipophilic in nature betulinic acid would also exhibit positive CNS activity due to high permeability through BBB as evident from our in silico data.
The computational investigation of solvation free energy in different solvents explains the relative stability or reactivity of betulinic acid. Our theoretical calculation suggests that betulinic acid stabilized more in non-polar protic solvent due to hydrophobic interaction between the steroidal nucleus with the non-polar region of solvent and the electrostatic interaction between carboxyl group of betulinic acid with the polar portion of the solvent. Due to greater interaction with non-polar solvent the HOMO-LUMO gap becomes shorter as compared to the polar solvent (Table 5 and Fig. 11). It could also be inferred that it is this property which facilitates the hydrophobic interactions and formation of hydrogen bond of BA at the binding site cavity of PLA2.
BA was found to have 100% plasma protein binding and hence it could be deduced that it is unlikely to be eliminated promptly from the systemic circulation upon administration. Since BA acts as both inhibitor and substrate for P glycoprotein due to complex modulatory interactions with the Pgp [83], it is unlikely to be pumped out of the cells completely hence reducing the possibility of resistance mediated by efflux pump.
BA is likely to be metabolized by Cytochrome P450 3A4. However, it must be noted that it is an inhibitor for the same enzyme which could be overcome with the administration of Cytochrome P450 3A4 inducer. Since BA was also found to be an inhibitor of Cytochrome P450 2C9, it should not be used when a drug (which acts as a substrate for this enzyme) needs to be metabolized.
The toxicological study revealed that although betulinic acid is a mutagenic compound, it was noncarcinogenic in mice model. Moreover, molecular docking study revealed that betulinic acid interacts with GLY22 & GLY29 through hydrogen bond formation and LEU2, PHE5, HIS6, ALA17, ALA18, HIS47 and TYR51 through different types of hydrophobic interactions with a binding affinity of − 41.00 kJ/mol.
These computed molecular properties may assist to develop analytical method [46] to assay BA and the pharmacokinetic, toxicology and molecular docking study may provide a guide to synthesize betulinic acid derivatives with better pharmacokinetic & toxicological properties with potent phospholipase A2 inhibitory activity.
Acknowledgements
We would like to thank Dr. Shahidul M. Islam, Department of Chemistry, College of Science, Sultan Qaboos University for his help to conduct the research.
Funding
One of us (MAR) is thankful to the Ministry of Education, Government of the People’s Republic of Bangladesh for a research grant (No. 37.01.0000.078.02.018.13–125/10/2/2015) for the period 2014–15 to 2016–17 to carry out the work.
Availability of data and materials
Data and material are included in the article.
Abbreviations
- BA
Betulinic acid
- BR4
6-Phenyl-4(R)-(7-Phenyl-heptanoylamino)-hexanoic acid
- DMSO
Dimethyl sulfoxide
- HOMO
Highest occupied molecular orbital
- LUMO
Lowest unoccupied molecular orbital
- PDB
Protein Data Bank
- PLA2
phospholipase A2
- RMSD
Root mean square deviation
- SMD
Solvation Model on Density
Authors’ contributions
Conceived and designed the experiments: MFK. Performed theoretical investigations: MFK, RBR, and AC. Analyzed the data: MFK, NN. Wrote the manuscript: MFK, RBR and MAR. All authors read and approved the final manuscript for publication.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there is no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mohammad Firoz Khan, Email: firoz@sub.edu.bd.
Nusrat Nahar, Email: nusratnahar17@gmail.com.
Ridwan Bin Rashid, Email: ridwan@sub.edu.bd.
Akhtaruzzaman Chowdhury, Email: akhtarcy@yahoo.com.
Mohammad A. Rashid, Email: rashidma@du.ac.bd
References
- 1.Carvalho BM, Santos JD, Xavier BM, Almeida JR, Resende LM, Martins W, Marcussi S, Marangoni S, Stábeli RG, Calderon LA, Soares AM. Snake venom PLA2s inhibitors isolated from Brazilian plants: synthetic and natural molecules. Biomed Res Int. 2013;2013:1–8. doi: 10.1155/2013/153045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fujoka T, Kashiwada Y, Robert EK. Anti-AIDS agents. Betulinic acid and platanic acid as anti-HIV principles from Syzygium claviflorum and the anti-HIV activity of structurally related triterpenoids. J Nat Prod. 1994;57:243–247. doi: 10.1021/np50104a008. [DOI] [PubMed] [Google Scholar]
- 3.Kashiwada Y, Wang HK, Nagao T, Kitanaka S, Yasuda I, Fujioka T, Yamagishi T, Cosentino LM, Kozuka M, Okabe H, Ikeshiro Y. Anti-AIDS agents. 30. Anti-HIV activity of oleanolic acid, pomolic acid, and structurally related triterpenoids 1. J Nat Prod. 1998;61(9):1090–1095. doi: 10.1021/np9800710. [DOI] [PubMed] [Google Scholar]
- 4.Bringmann G, Saeb W, Assi LA, Francois G, Narayanan AS, Peters K, Peters EM. Betulinic acid: isolation from Triphyophyllum peltatum and Ancistrocladus heyneanus, antimalarial activity, and crystal structure of the benzyl ester. Planta Med. 1997;63(03):255–257. doi: 10.1055/s-2006-957666. [DOI] [PubMed] [Google Scholar]
- 5.Bernard P, Scior T, Didier B, Hibert M, Berthon JY. Ethnopharmacology and bioinformatic combination for leads discovery: application to phospholipase a 2 inhibitors. Phytochemistry. 2001;58(6):865–874. doi: 10.1016/S0031-9422(01)00312-0. [DOI] [PubMed] [Google Scholar]
- 6.Huguet AI, del Carmen RM, Máñez S, Giner RM. Ríos JL. Effect of triterpenoids on the inflammation induced by protein kinase C activators, neuronally acting irritants and other agents. Eur J Pharmacol. 2000;410(1):69–81. doi: 10.1016/S0014-2999(00)00860-8. [DOI] [PubMed] [Google Scholar]
- 7.Mukherjee PK, Saha K, Das J, Pal M, Saha BP. Studies on the anti-inflammatory activity of rhizomes of Nelumbo Nucifera. Planta Med. 1997;63(04):367–369. doi: 10.1055/s-2006-957705. [DOI] [PubMed] [Google Scholar]
- 8.Enwerem NM, Okogun JI, Wambebe CO, Okorie DA, Akah PA. Anthelmintic activity of the stem bark extracts of Berlina grandiflora and one of its active principles, Betulinic acid. Phytomedicine. 2001;8(2):112–114. doi: 10.1078/0944-7113-00023. [DOI] [PubMed] [Google Scholar]
- 9.Kinoshita K, Akiba M, Saitoh M, Ye Y, Koyama K, Takahashi K, Kondo N, Yuasa H. Antinociceptive effect of triterpenes from cacti. Pharm Biol. 1998;36(1):50–55. doi: 10.1076/phbi.36.1.50.4614. [DOI] [Google Scholar]
- 10.Ryu SY, Lee CK, Lee CO, Kim HS, Zee OP. Antiviral triterpenes from Prunella vulgaris. Arch Pharm Res. 1992;15(3):242–245. doi: 10.1007/BF02974063. [DOI] [Google Scholar]
- 11.Ryu SY, Lee CK, Ahn JW, Lee SH, Zee OP. Antiviral activity of triterpenoid derivatives. Arch Pharm Res. 1993;16(4):339–342. doi: 10.1007/BF02977528. [DOI] [Google Scholar]
- 12.Fulda S, Jeremias I, Steiner HH, Pietsch T, Debatin KM. Betulinic acid: a new cytotoxic agent against malignant brain-tumor cells. Int J Cancer. 1999;82(3):435–441. doi: 10.1002/(SICI)1097-0215(19990730)82:3<435::AID-IJC18>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 13.Fulda S, Debatin KM. Betulinic acid induces apoptosis through a direct effect on mitochondria in neuroectodermal tumors. Med Pediatr Oncol. 2000;35(6):616–618. doi: 10.1002/1096-911X(20001201)35:6<616::AID-MPO27>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 14.Zuco V, Supino R, Righetti SC, Cleris L, Marchesi E, Gambacorti-Passerini C, Formelli F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002;175(1):17–25. doi: 10.1016/S0304-3835(01)00718-2. [DOI] [PubMed] [Google Scholar]
- 15.Cichewicz RH, Kouzi SA. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med Res Rev. 2004;24(1):90–114. doi: 10.1002/med.10053. [DOI] [PubMed] [Google Scholar]
- 16.O'Connell MM, Bentley MD, Campbell CS, Cole BJ. Betulin and lupeol in bark from four white-barked birches. Phytochemistry. 1988;27(7):2175–2176. doi: 10.1016/0031-9422(88)80120-1. [DOI] [Google Scholar]
- 17.Cole BJ, Bentley MD, Hua Y. Triterpenoid extractives in the outer bark of Betula lenta (black birch) Holzforschung. 1991;45(4):265–268. doi: 10.1515/hfsg.1991.45.4.265. [DOI] [Google Scholar]
- 18.Chowdhury A, Alam MA, Rashid RB, Al-Mansur MA, Rahman MS, Rashid MA. Steroids and triterpenoids from Corypha taliera Roxb: a critically endangered palm species of Bangladesh. Res J Med Plant. 2013;7:125–129. doi: 10.3923/rjmp.2013.125.129. [DOI] [Google Scholar]
- 19.Jagadeesh SG, Krupadanam GD, Srimannarayana G, Murthy SS, Kaur A, Raja SS. Tobacco caterpillar antifeedent from the gotti stem wood triterpene betulinic acid. J Agric Food Chem. 1998;46(7):2797–2799. doi: 10.1021/jf970768b. [DOI] [Google Scholar]
- 20.Schühly W, Heilmann J, Calis I, Sticher O. New triterpenoids with antibacterial activity from Zizyphus joazeiro. Planta Med. 1999;65(08):740–743. doi: 10.1055/s-1999-14054. [DOI] [PubMed] [Google Scholar]
- 21.Chang CW, Wu TS, Hsieh YS, Kuo SC, Chao PD. Terpenoids of Syzygium Formosanum. J Nat Prod. 1999;62(2):327–328. doi: 10.1021/np980313w. [DOI] [PubMed] [Google Scholar]
- 22.del Carmen RM, Giner RM, Manez S, Gueho J, Julien HR, Hostettmann K, Rios JL. Investigations on the steroidal anti-inflammatory activity of triterpenoids from Diospyros Leucomelas. Planta Med. 1995;61(01):9–12. doi: 10.1055/s-2006-957988. [DOI] [PubMed] [Google Scholar]
- 23.Singh P, Sharma S. Triterpenoid constituents of the seeds of Diospyros melanoxylon, Tecomella undulata and Terminalia bellirica. J Indian Chem Soc. 1997;74(6):504–505. [Google Scholar]
- 24.Lin HC, Ding HY, Wu YC. Two novel compounds from Paeonia s uffruticosa. J Nat Prod. 1998;61(3):343–346. doi: 10.1021/np9704258. [DOI] [PubMed] [Google Scholar]
- 25.Krasutsky PA. Birch bark research and development. Nat Prod Rep. 2006;23(6):919–942. doi: 10.1039/b606816b. [DOI] [PubMed] [Google Scholar]
- 26.Wada SI, Tanaka R. Betulinic acid and its derivatives, potent DNA topoisomerase II inhibitors, from the bark of Bischofia Javanica. Chem Biodivers. 2005;2(5):689–694. doi: 10.1002/cbdv.200590045. [DOI] [PubMed] [Google Scholar]
- 27.Reutrakul V, Chanakul W, Pohmakotr M, Jaipetch T, Yoosook C, Kasisit J, Napaswat C, Santisuk T, Prabpai S, Kongsaeree P, Tuchinda P. Anti-HIV-1 constituents from leaves and twigs of Cratoxylum arborescens. Planta Med. 2006;72(15):1433–1435. doi: 10.1055/s-2006-951725. [DOI] [PubMed] [Google Scholar]
- 28.Steele JP, Warhurst DC, Kirby GC, Simmonds MJ. In vitro and in vivo evaluation of betulinic acid as an antimalarial. Phytother Res. 1999;13(2):115–119. doi: 10.1002/(SICI)1099-1573(199903)13:2<115::AID-PTR404>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z, ElSohly HN, Jacob MR, Pasco DS, Walker LA, Clark AM. Natural products inhibiting Candida Albicans secreted aspartic proteases from Tovomita krukovii. Planta Med. 2002;68(01):49–54. doi: 10.1055/s-2002-20049. [DOI] [PubMed] [Google Scholar]
- 30.Domínguez-Carmona DB, Escalante-Erosa F, García-Sosa K, Ruiz-Pinell G, Gutierrez-Yapu D, Chan-Bacab MJ, Giménez-Turba A, Peña-Rodríguez LM. Antiprotozoal activity of betulinic acid derivatives. Phytomedicine. 2010;17(5):379–382. doi: 10.1016/j.phymed.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Ma J, Starck SR, Hecht SM. DNA polymerase β inhibitors from Tetracera boiviniana. J Nat Prod. 1999;62(12):1660–1663. doi: 10.1021/np990326p. [DOI] [PubMed] [Google Scholar]
- 32.Choi JY, Na M, Hyun Hwang I, Ho Lee S, Young Bae E, Yeon Kim B, Seog AJ. Isolation of betulinic acid, its methyl ester and guaiane sesquiterpenoids with protein tyrosine phosphatase 1B inhibitory activity from the roots of Saussurea Lappa CB Clarke. Molecules. 2009;14(1):266–272. doi: 10.3390/molecules14010266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nyasse B, Nono JJ, Nganso Y, Ngantchou I, Schneider B. Uapaca genus (Euphorbiaceae), a good source of betulinic acid. Fitoterapia. 2009;80(1):32–34. doi: 10.1016/j.fitote.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Wenzig EM, Widowitz U, Kunert O, Chrubasik S, Bucar F, Knauder E, Bauer R. Phytochemical composition and in vitro pharmacological activity of two rose hip (Rosa Canina L.) preparations. Phytomedicine. 2008;15(10):826–835. doi: 10.1016/j.phymed.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 35.Costa JF, Barbosa-Filho JM, de Azevedo Maia GL, Guimarães ET, Meira CS, Ribeiro-dos-Santos R, de Carvalho LC, Soares MB. Potent anti-inflammatory activity of betulinic acid treatment in a model of lethal endotoxemia. Int Immunopharmacol. 2014;23(2):469–474. doi: 10.1016/j.intimp.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 36.Kim KS, Lee DS, Kim DC, Yoon CS, Ko W, Oh H, Kim YC. Anti-inflammatory effects and mechanisms of action of coussaric and betulinic acids isolated from Diospyros kaki in lipopolysaccharide-stimulated RAW 264.7 macrophages, 2016. Molecules. 21(9):1206. [DOI] [PMC free article] [PubMed]
- 37.Blaževski J, Petković F, Momčilović M, Paschke R, Kaluđerović GN, Stojković MM, Miljković D. Betulinic acid regulates generation of neuroinflammatory mediators responsible for tissue destruction in multiple sclerosis in vitro. Acta Pharmacol Sin. 2013;34(3):424–431. doi: 10.1038/aps.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ong WY, Farooqui T, Kokotos G, Farooqui AA. Synthetic and natural inhibitors of phospholipases A2: their importance for understanding and treatment of neurological disorders. ACS Chem Neurosci. 2015;6(6):814–831. doi: 10.1021/acschemneuro.5b00073. [DOI] [PubMed] [Google Scholar]
- 39.Irvine RF. How is the level of free arachidonic acid controlled in mammalian cells? Biochem J. 1982;204(1):3. doi: 10.1042/bj2040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayer RJ, Marshall LA. New insights on mammalian phospholipase A2 (s); comparison of arachidonoyl-selective and-nonselective enzymes. FASEB J. 1993;7(2):339–348. doi: 10.1096/fasebj.7.2.8440410. [DOI] [PubMed] [Google Scholar]
- 41.Posadas I, Terencio MC, De Rosa S, Payá M. Cavernolide: a new inhibitor of human sPLA2 sharing unusual chemical features. Life Sci. 2000;67(24):3007–3014. doi: 10.1016/S0024-3205(00)00875-4. [DOI] [PubMed] [Google Scholar]
- 42.Friess H, Shrikhande S, Riesle E, Kashiwagi M, Baczako K, Zimmermann A, Uhl W, Büchler MW. Phospholipase A2 isoforms in acute pancreatitis. Ann Surg. 2001;233(2):204–212. doi: 10.1097/00000658-200102000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dennis EA, Rhee SG, Billah MM, Hannun YA. Role of phospholipase in generating lipid second messengers in signal transduction. FASEB J. 1991;5(7):2068–2077. doi: 10.1096/fasebj.5.7.1901288. [DOI] [PubMed] [Google Scholar]
- 44.Clark JD, Milona N, Knopf JL. Purification of a 110-kilodalton cytosolic phospholipase A2 from the human monocytic cell line U937. Proc Natl Acad Sci U S A. 1990;87(19):7708–7712. doi: 10.1073/pnas.87.19.7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malleda C, Ahalawat N, Gokara M, Subramanyam R. Molecular dynamics simulation studies of betulinic acid with human serum albumin. J Mol Model. 2012;18(6):2589–2597. doi: 10.1007/s00894-011-1287-x. [DOI] [PubMed] [Google Scholar]
- 46.Khan MF, Rita SA, Kayser MS, Islam MS, Asad S, Rashid RB, Bari MA, Rahman MM, Al Aman DA, Setu NI, Banoo R. Theoretically guided analytical method development and validation for the estimation of rifampicin in a mixture of isoniazid and pyrazinamide by UV spectrophotometer. Front Chem. 2017;5(27):1–12. [DOI] [PMC free article] [PubMed]
- 47.Bach RD, Ayala PY, Schlegel HB. A reassessment of the bond dissociation energies of peroxides. An ab initio study. J Am Chem Soc. 1996;118(50):12758–12765. doi: 10.1021/ja961838i. [DOI] [Google Scholar]
- 48.Glukhovtsev MN, Bach RD. Ab initio study on the thermochemistry of vinyl radical and cation. Chem Phys Lett. 1998;286(1):51–55. doi: 10.1016/S0009-2614(97)01475-9. [DOI] [Google Scholar]
- 49.Abboud JL, Castano O, Dávalos JZ, Gomperts R. The standard enthalpies of formation of 1-and 2-Adamantyl cations and radicals. An ab initio study. Chem Phys Lett. 2001;337(4):327–330. doi: 10.1016/S0009-2614(01)00212-3. [DOI] [Google Scholar]
- 50.Li ZH, Wong MW. Scaling of correlation basis set extension energies. Chem Phys Lett. 2001;337(1):209–216. doi: 10.1016/S0009-2614(01)00188-9. [DOI] [Google Scholar]
- 51.Espinosa-García J. Theoretical C–H bond dissociation enthalpies for CH 3 OCl and CH 3 OBr. Chem Phys Lett 2000;316(5):563-568.
- 52.BecNe AD. Densityϋfunctional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 53.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H. Fox DJ Gaussian 09, revision a. 02. Wallingford, CT: Gaussian. Inc.; 2009. [Google Scholar]
- 54.Marenich AV, Cramer CJ, Truhlar DG. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B. 2009;113(18):6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 55.Hansford KA, Reid RC, Clark CI, Tyndall JD, Whitehouse MW, Guthrie T, McGeary RP, Schafer K, Martin JL, Fairlie DP. D-tyrosine as a Chiral Precusor to potent inhibitors of human nonpancreatic Secretory Phospholipase A2 (IIa) with Antiinflammatory activity. Chembiochem. 2003;4(2–3):181–185. doi: 10.1002/cbic.200390029. [DOI] [PubMed] [Google Scholar]
- 56.Sánchez-Linares I, Pérez-Sánchez H, Cecilia JM, García JM. High-throughput parallel blind virtual screening using BINDSURF. BMC Bioinf. 2012;13(14):S13. doi: 10.1186/1471-2105-13-S14-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Imbernón B, Cecilia JM, Pérez-Sánchez H, Giménez D. METADOCK: A parallel metaheuristic schema for virtual screening methods. Int J High Perform Comput Appl. 2017; p. 1–15.
- 58.Cerón-Carrasco JP, Cerezo J, Requena A, Zuñiga J, Contreras-García J, Chavan S, Manrubia-Cobo M, Pérez-Sánchez H. Labelling Herceptin with a novel oxaliplatin derivative: a computational approach towards the selective drug delivery. J Mol Model. 2014;20(9):2401. doi: 10.1007/s00894-014-2401-7. [DOI] [PubMed] [Google Scholar]
- 59.Navarro-Fernández J, Pérez-Sánchez H, Martínez-Martínez I, Meliciani I, Guerrero JA, Vicente V, Corral J, Wenzel W. In silico discovery of a compound with nanomolar affinity to antithrombin causing partial activation and increased heparin affinity. J Med Chem. 2012;55(14):6403–6412. doi: 10.1021/jm300621j. [DOI] [PubMed] [Google Scholar]
- 60.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elusiyan CA, Msagati TA, Shode FO, Mamba BB. Measurements of distribution coefficients and lipophilicity values for oleanolic acid and betulinic acid extracted from indigenous plants by hollow fibre supported liquid membrane. Bull Chem Soc Ethiop. 2011;25(3):321–32.
- 62.Claude B, Morin P, Lafosse M, Andre P. Evaluation of apparent formation constants of pentacyclic triterpene acids complexes with derivatized β-and γ-cyclodextrins by reversed phase liquid chromatography. J Chromatogr A. 2004;1049(1):37–42. doi: 10.1016/S0021-9673(04)01143-4. [DOI] [PubMed] [Google Scholar]
- 63.Suhasini M, Sailatha E, Gunasekaran S, Ramkumaar GR. Vibrational and electronic investigations, thermodynamic parameters, HOMO and LUMO analysis on Lornoxicam by density functional theory. J Mol Struct. 2015;1100:116–128. doi: 10.1016/j.molstruc.2015.07.003. [DOI] [Google Scholar]
- 64.Chattaraj PK, Maiti B, Sarkar U. Philicity: a unified treatment of chemical reactivity and selectivity. J Phys Chem A. 2003;107(25):4973–4975. doi: 10.1021/jp034707u. [DOI] [Google Scholar]
- 65.Parr RG, Donnelly RA, Levy M, Palke WE. Electronegativity: the density functional viewpoint. J Chem Phys. 1978;68(8):3801–3807. doi: 10.1063/1.436185. [DOI] [Google Scholar]
- 66.Parr RG, Pearson RG. Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc. 1983;105(26):7512–7516. doi: 10.1021/ja00364a005. [DOI] [Google Scholar]
- 67.Parr RG, Chattaraj PK. Principle of maximum hardness. J Am Chem Soc. 1991;113(5):1854–1855. doi: 10.1021/ja00005a072. [DOI] [Google Scholar]
- 68.Parr RG, Szentpaly LV, Liu S. Electrophilicity index. J Am Chem Soc. 1999;121(9):1922–1924. doi: 10.1021/ja983494x. [DOI] [Google Scholar]
- 69.Yee S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man—fact or myth. Pharm Res. 1997;14(6):763–766. doi: 10.1023/A:1012102522787. [DOI] [PubMed] [Google Scholar]
- 70.Yazdanian M, Glynn SL, Wright JL, Hawi A. Correlating partitioning and Caco-2 cell permeability of structurally diverse small molecular weight compounds. Pharm Res. 1998;15(9):1490–1494. doi: 10.1023/A:1011930411574. [DOI] [PubMed] [Google Scholar]
- 71.Singh S, Singh J. Transdermal drug delivery by passive diffusion and iontophoresis: a review. Med Res Rev. 1993;13(5):569–621. doi: 10.1002/med.2610130504. [DOI] [PubMed] [Google Scholar]
- 72.Chen C, Yang J. Predictive model of blood-brain barrier penetration of organic compounds. Acta Pharmacol Sin. 2005;26(4):500–512. doi: 10.1111/j.1745-7254.2005.00068.x. [DOI] [PubMed] [Google Scholar]
- 73.Tanaka H, Mizojiri K. Drug-protein binding and blood-brain barrier permeability. J Pharmacol Exp Ther. 1999;288(3):912–918. [PubMed] [Google Scholar]
- 74.Cornford EM, Pardridge WM, Braun LD, Oldendorf WH. Increased blood-brain barrier transport of protein-bound anticonvulsant drugs in the newborn. J Cereb Blood Flow Metab. 1983;3(3):280–286. doi: 10.1038/jcbfm.1983.42. [DOI] [PubMed] [Google Scholar]
- 75.Pardridge WM, Sakiyama R, Fierer G. Transport of propranolol and lidocaine through the rat blood-brain barrier. Primary role of globulin-bound drug. J Clin Invest. 1983;71(4):900. doi: 10.1172/JCI110844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Terasaki TE, Pardridge WM, Denson DD. Differential effect of plasma protein binding of bupivacaine on its in vivo transfer into the brain and salivary gland of rats. J Pharmacol Exp Ther. 1986;239(3):724–729. [PubMed] [Google Scholar]
- 77.Schinkel AH. P-glycoprotein, a gatekeeper in the blood–brain barrier. Adv Drug Deliv Rev. 1999;36(2):179–194. doi: 10.1016/S0169-409X(98)00085-4. [DOI] [PubMed] [Google Scholar]
- 78.Cabrera MA, González I, Fernández C, Navarro C, Bermejo M. A topological substructural approach for the prediction of P-glycoprotein substrates. J Pharm Sci. 2006;95(3):589–606. doi: 10.1002/jps.20449. [DOI] [PubMed] [Google Scholar]
- 79.Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest. 1998;101(2):289. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.de Cerqueira LP, Golbraikh A, Oloff S, Xiao Y, Tropsha A. Combinatorial QSAR modeling of P-glycoprotein substrates. J Chem Inf Model. 2006;46(3):1245–1254. doi: 10.1021/ci0504317. [DOI] [PubMed] [Google Scholar]
- 81.Wang YH, Li Y, Yang SL, Yang L. Classification of substrates and inhibitors of P-glycoprotein using unsupervised machine learning approach. J Chem Inf Model. 2005;45(3):750–757. doi: 10.1021/ci050041k. [DOI] [PubMed] [Google Scholar]
- 82.Joung JY, Kim HJ, Kim HM, Ahn SK, Nam KY, No KT. Prediction models of p-glycoprotein substrates using simple 2d and 3d descriptors by a recursive partitioning approach. Bull Kor Chem Soc. 2012;33(4):1123–1127. doi: 10.5012/bkcs.2012.33.4.1123. [DOI] [Google Scholar]
- 83.Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9(4):310–322. doi: 10.2174/138920008784220664. [DOI] [PubMed] [Google Scholar]
- 84.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440(7083):463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 85.Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins. 2009;77(S9):114–122. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lister MD, Glaser KB, Ulevitch RJ, Dennis EA. Inhibition studies on the membrane-associated phospholipase A2 in vitro and prostaglandin E2 production in vivo of the macrophage-like P388D1 cell. Effects of manoalide, 7, 7-dimethyl-5, 8-eicosadienoic acid, and p-bromophenacyl bromide. J Biol Chem. 1989;264(15):8520–8528. [PubMed] [Google Scholar]
- 87.Lobo IB, Hoult JR. Groups I, II and III extracellular phospholipases A2: selective inhibition of group II enzymes by indomethacin but not other NSAIDs. Agents Actions. 1994;41(1–2):111–113. doi: 10.1007/BF01986409. [DOI] [PubMed] [Google Scholar]
- 88.Santa Cruz Biotechnology. https://www.scbt.com/scbt/browse/chemicals-Protein-Interacting-Inhibitors-Activators-Substrates-protein-inhibitors-pla2-inhibitors/_/N-16mpppv. Access Date: 13 May 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data and material are included in the article.