

Abstract
In the present study, SGC-7901/DDP cells were treated with different concentrations of parthenolide (PN) (2.5–15 µmol/l), cisplatin (DDP) (1.25–15 µg/ml) and PN+DDP. The proliferation inhibition rates were measured using an MTT assay, and the synergies of PN and DDP were analyzed. The effect of PN and DDP on SGC-7901/DDP cell proliferation demonstrated a time- and concentration-dependent association, and a synergy between PN and DDP was identified. DAPI staining and flow cytometry results indicated that 15 µmol/l PN significantly induced SGC-7901/DDP apoptosis and G1 phase arrest compared with the untreated control group. Western blotting analysis results indicated that among the apoptosis-associated proteins, there were dose-dependent increases in the protein expression of apoptosis regulator BAX, cellular tumor antigen p53, cleaved caspase-3 and cleaved capase-9, and decreases in apoptosis regulator Bcl-2 and Bcl-xL protein expression levels. Among the cell cycle-associated proteins, cyclin D1 expression was significantly decreased, cyclin-dependent kinase inhibitor 1 expression was significantly increased, and signal transducer and activator of transcription 3 (STAT3) activation was inhibited. Scratch and Transwell assay results revealed that PN significantly inhibited SGC-7901/DDP cell migration, and invasion. The present study demonstrated that PN induces SGC-7901/DDP apoptosis, inhibits SGC-7901/DDP proliferation, migration and invasion, and enhances the drug sensitivity of the cells to DDP. The underlying mechanisms may be associated with inhibition of the STAT3 signaling pathway and regulation of the downstream apoptotic protein and cyclin expression levels.
Keywords: gastric cancer, drug resistance, signal transducer and activator of transcription 3, parthenolide, proliferation, apoptosis
Introduction
Chemotherapy is the main treatment approach for advanced gastric cancer and adjuvant post-operative gastric cancer; however, multidrug resistance (MDR) often leads to the failure of chemotherapy (1). MDR is a distinct phenomenon enabling tumor cells to simultaneously develop cross-resistance to multiple structurally dissimilar anticancer drugs. Gastric cancer exhibits a strong MDR phenotype, resulting in a response rate to chemotherapy of <50% (2). The 5-year survival rate in advanced gastric cancer is 30%, which is possibly associated with the development of MDR. Therefore, approaches to intervene and reverse MDR in gastric cancer are required to improve the efficacy of chemotherapy and the survival rate of patients with gastric cancer.
Parthenolide (PN) is a sesquiterpene lactone extracted from Feverfew (Tanacetum parthenium), and is primarily used for the treatment of fever, arthritis and migraine (3). Previous studies have demonstrated that PN inhibits the growth and induces the apoptosis of various tumor cell types in vitro (4–6). Evidence suggests that PN induces anti-tumor effects primarily by targeting nuclear factor-κB (NF-κB) (7), producing reactive oxygen species (8) and activating c-Jun N-terminal kinase (9). Furthermore, previous studies have revealed that PN can enhance gastric, non-small cell lung and liver cancer cell sensitivity to chemotherapy (10); however, the underlying mechanisms remain unclear. In the present study, the effects and underlying mechanisms of PN treatment on the sensitivity of drug-resistant gastric cancer SGC-7901/DDP cells to cisplatin (DDP) were investigated, in order to provide a theoretical basis for the clinical application of PN.
Materials and methods
Reagents
The reagents and kits used in the present study were purchased as follows: PN (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany); DDP (Shandong Dezhou Taikang Pharmaceutical Co., Ltd., Dezhou, China); RPMI-1640 (Sigma-Aldrich; Merck KGaA); fetal bovine serum (Hangzhou Evergreen Biological Engineering Materials Co., Ltd., Hangzhou, China); Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) detection kit (Nanjing KGI Biological Technology Development Co., Ltd., Nanjing, China); MTT (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China); enhanced chemiluminescence (ECL; GE Healthcare, Chicago, IL, USA); and DAPI (Beyotime Institute of Biotechnology, Haimen, China). The SGC-7901/DDP drug-resistant gastric cancer cell line was purchased from Shanghai Bogoo Biotechnology Co., Ltd. (Shanghai, China). The antibodies were obtained from the following: Goat anti-rabbit and anti-mouse immunoglobulin G secondary antibodies (GE Healthcare Life Sciences, Chalfont, UK); primary antibodies directed against phosphorylated-signal transducer and activator of transcription 3 (p-STAT3), apoptosis regulator Bcl-xL and Bcl-2 (Santa Cruz Biotechnology, Inc., Dallas, TX, USA); β-actin (Sigma-Aldrich; Merck KGaA); and apoptosis regulator BAX (Bax), caspase-3, cleaved caspase-3, cellular tumor antigen p53, caspase-9, cleaved caspase-9, cyclin-dependent kinase inhibitor 1 (p21), cyclin D1 and STAT3 (Wanleibio Biotechnology Co., Ltd., Shanghai, China). Antibodies are detailed in Table I.
Table I.
Antibodies.
| Antibody target | Supplier | Catalog no. | Dilution |
|---|---|---|---|
| p-STAT3 | Santa Cruz Biotechnology, Inc., Dallas, TX, USA | Sc-293059 | 1:100 |
| Bcl-xL, | Santa Cruz Biotechnology, Inc. | Sc-7195 | 1:300 |
| Bcl-2 | Santa Cruz Biotechnology, Inc. | Sc-509 | 1:300 |
| β-actin | Sigma-Aldrich; Merck KGaA, Darmstadt, Germany | SAB2100037 | 1:300 |
Bcl-xL, B cell lymphoma extra-large; p-STAT, phosphorylated signal transducer and activator of transcription.
Instruments
General electrophoresis, horizontal and vertical electrophoresis tank, and semi-dry transfer blotter were purchased from Bio-Rad Laboratories, Inc., (Hercules, CA, USA). The Ti-U manual fluorescence/inverted phase contrast microscope was obtained from Nikon Corporation (Tokyo, Japan); BioSpectrum® Imaging system from UVP Inc., (Upland, CA, USA); microplate reader from Tecan Group Ltd. (Männedorf, Switzerland); and Alpha Automatic Image Analysis system from Bio-Techne (ProteinSimple; Minneapolis, MN, USA).
Cell culture
The human gastric cancer drug-resistant SGC-7901/DDP cell line was cultured in RPMI-1640 supplemented with 10% fetal bovine serum at 37°C with 5% CO2.
MTT assay
MTT was used to measure the effect of PN treatment on SGC-7901/DDP cell proliferation and sensitivity to DDP. SGC-7901/DDP cells in the exponential growth phase were harvested and made into single cell suspensions at a concentration of 5×104 cells/ml. The cells were seeded in 96-well plates (100 µl/well) and incubated for 24 h at 37°C. The SGC-7901/DDP cells were treated with various concentrations of PN (0, 2.5, 5, 10, 12.5 and 15 µmol/l) and DDP (0, 1.25, 2.5, 5, 10 and 15 µg/ml) with five-replicates/concentration. A total of 20 µl of 5 g/l MTT solution was added to each well at 24, 48 and 72 h, and then incubated for 4 h at 37°C. The supernatant was discarded and 150 µl dimethyl sulfoxide was added to each well. The plates were oscillated at a low speed for 2 min at room temperature (23–25°C) until all crystals were fully dissolved. The absorbance of each well was measured at a wavelength of 490 nm using a spectrophotometer. The growth inhibitory rate (%) was calculated using the following formula: (1-(absorbance of experimental well/absorbance of blank)) ×100%.
In order to prevent excessive cell death, based on the results from the above experiments, the combinations of less toxic concentrations of PN (5 and 10 µmol/l) and DDP (1.25 and 2.5 µg/ml) were selected. Therefore, the SGC-7901/DDP cells were treated with the following combinations: 5 µmol/l PN+1.25 µg/ml DDP; 5 µmol/l PN+2.5 µg/ml DDP; 10 µmol/l PN+1.25 µg/ml DDP; or 10 µmol/l PN+2.5 µg/ml DDP. The MTT assay was performed at 24 h following the treatments. The Zhenjun Jin method (11) was used to determine the synergy q=E(AB)/(EA+EB-EA × EB) EAB refers to growth inhibitory rate of combined treatment; and EA and EB refer to the growth inhibitory rate with either A or B treatment. q<0.85 indicates mutual antagonism of two drugs; q=0.85–1.15 indicates a simple additive effect of the two drugs; q>1.15 indicates that the two drugs have a synergistic effect.
Cell cycle distribution analysis via flow cytometry
SGC-7901/DDP cells in the exponential growth phase were seeded in 6-cm petri dishes at a density of 5×105 cells/dish. The medium was changed 12 h following seeding and the cells were treated with 0, 7.5 or 15 µmol/l of PN for 48 h at 37°C. The supernatant was discarded and the cells were digested with EDTA-free trypsin, washed twice with PBS, and fixed with 70% ethanol at −20°C overnight. Cell were washed twice with PBS, treated with RNaseA (a final concentration of 250 µg/ml) and incubated in a 37°C water bath for 30 min. The cells were stained with 5 µl PI in a dark room for 50 min at room temperature, and then the cell cycle distribution was measured using a FACSCalibur flow cytometer and FlowJo 7.6.1 (BD Biosciences, Franklin Lakes, NJ, USA).
Cell apoptosis detection via flow cytometry
SGC-7901/DDP cells in the exponential growth phase were seeded in 6-cm petri dishes at a density of 5×105 cells/dish. After culturing for 12 h, cells were treated with 0, 7.5, or 15 µmol/l PN for 48 h at 37°C. Cells were digested with EDTA-free trypsin, washed twice with PBS, stained for 15 min at room temperature in the dark using the Annexin V-FITC Apoptosis Detection kit and then the apoptosis rate was detected.
Cell apoptosis was detected by DAPI staining
SGC-7901/DDP cells in the exponential growth phase were seeded in 24-well culture plates at a density of 5×104 cells/well, and cultured for 24 h at 37°C. Cells were treated with 0, 7.5, or 15 µmol/l of PN for 24 h at 37°C. Cells were fixed with 0.1% paraformaldehyde for 30 min, washed with PBS for 5 min and stained with DAPI for 10 min at room temperature (23–25°C). The cells were observed using a fluorescence microscope (magnification, ×100) at an excitation wavelength of 359 nm.
Western blotting
SGC-7901/DDP cells in the exponential growth phase were seeded in 10 cm dishes at a density of 5×105 cells/ml and cultured for 24 h at 37°C. Cells were treated with different concentrations of PN (final concentration of 0, 7.5 or 15 µmol/l) and collected 48 h after treatment. The total protein was extracted using radioimmunoprecipitation assay buffer (P0013J; Beyotime Institute of Biotechnology) and phenylmethanesulfonyl fluoride (ST506; Beyotime Institute of Biotechnology). The protein quantity was determined using a bicinchoninic acid assay kit (P0011; Beyotime Institute of Biotechnology). Protein samples were boiled at 100°C for 5 min. A total of 30 µg protein/lane was loaded and subjected to SDS-PAGE (10% gel). The proteins were transferred to polyvinylidene difluoride membranes, blocked with 0.5% skim milk for 1 h (23–25°C) and incubated with the appropriate primary antibody at 4°C overnight. The membranes were washed with TBS-Tween 20 three times (5 min/wash) and then incubated with the corresponding secondary antibody at room temperature for 2 h. The membranes were washed with TBST and detected using an ECL luminescent liquid with cytometry. The quantitative analysis was performed with ImageJ software (version 1.45; National Institutes of Health, Bethesda, MD, USA. The relative expression of the target protein was calculated as the ratio of grey values of the target protein to the β-actin internal control.
Scratch migration assay
SGC-7901/DDP cells in the exponential growth phase were seeded in 6-well plates at a density of 5×105 cells/well and cultured until 70% confluence was achieved. A scratch was made in each well using a 100-µl pipette tip. The cells were treated with various concentrations of serum-free PN (0, 2.5 or 5 µmol/l). Images were acquired at 24, 48 and 72 h after treatment. The scratch width was measured and the mobility was calculated as follows: (Distance/scratch width) ×100%.
Transwell cell invasion assay
In order to assess the invasive ability of cells, 120 µg Matrigel was added to a Transwell chamber. The gel was diluted with two volumes of serum-free RPMI-1640 and incubated at 37°C for 30 min until the gel solidified. RPMI-1640 containing 10% FBS was added to the lower chamber as a chemoattractant. SGC-7901/DDP cells in the exponential growth phase were digested with EDTA-free trypsin and viable cell counts were performed by eye. A 200-µl suspension containing 5×104 cells was added to each Transwell chamber and cultured for 48 h at 37°C. The experiment was performed in triplicate. The liquid in the upper chamber was discarded. The non-invasive cells and Matrigel on the membrane surface were removed using a wet cotton swab. The membrane was rinsed with saline, slightly dried, fixed in formalin (4%) for 15 min and stained with crystal violet for 12 min (23–25°C). The penetrating cell count was performed in three different fields at a magnification of ×100 using a light microscope.
Statistical analysis
Data are presented as the mean ± standard deviation. Statistical analysis was performed using SPSS (version 10.0; SPSS, Inc., Chicago, IL, USA) using one-way analysis of variance. Comparisons between groups were measured using the post-hoc test. Multiple comparisons amongst groups were performed using the Student-Newman-Keuls test. P<0.05 was considered to indicate a statistically significant difference.
Results
Cell proliferation inhibitory effects of PN and the changes in the sensitivity of SGC-7901/DDP to DDP
All concentrations of PN, except 2.5 µmol/l, significantly inhibited SGC-7901/DDP cell proliferation in a time- and concentration-dependent manner compared with untreated cells (Fig. 1). DDP inhibited SGC-7901/DDP cell proliferation in a similar time- and concentration-dependent manner (Fig. 2). To investigate the changes in the sensitivity of SGC-7901/DDP cells to the DDP, SGC-7901/DDP cells were treated with combinations of PN and DDP as described above. The rate of proliferation inhibition in each group is presented in Fig. 3. The q value for each group was calculated using the Zhenjun Jin method. With the exception of the minimal concentration group (5 µmol/l PN+1.25 µg/ml DDP), the q values for all other groups were >1.15, indicating that the two drugs have a synergistic effect.
Figure 1.
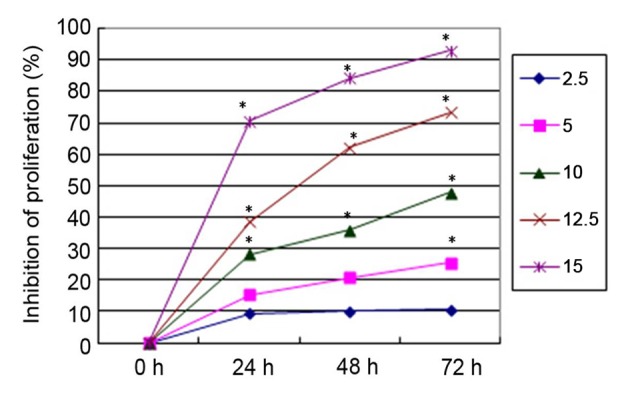
Inhibitory effects of different concentrations of parthenolide (2.5–15 µmol/l) on SGC-7901/DDP cell proliferation. *P<0.05 vs. untreated control.
Figure 2.
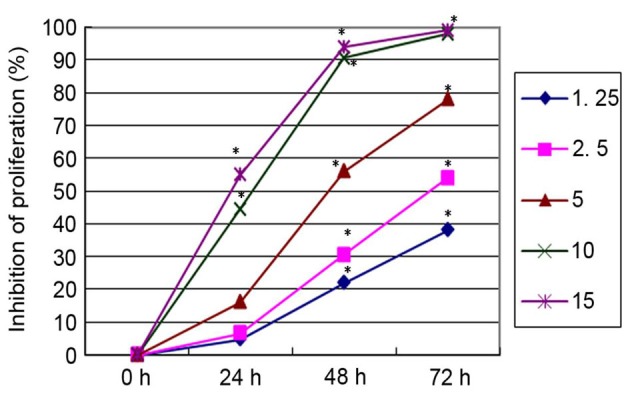
Inhibitory effects of different concentrations of cisplatin (1.25–15 µg/ml) on the SGC-7901/DDP cell proliferation. *P<0.05 vs. untreated control.
Figure 3.
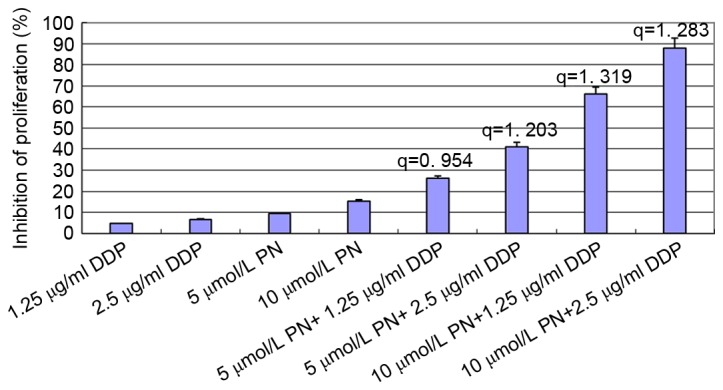
Proliferation inhibition rate of SGC-7901/DDP cell 24 h following combined treatments of PN and DDP. The Zhenjun Jin method was used to determine the synergy (q). With the exception of the minimal concentration group (5 µmol/l PN+1.25 µg/ml DDP), the q values for all other groups were >1.15, indicating that the two drugs have a synergistic effect. DDP, cisplatin; PN, parthenolide.
PN induces cell cycle arrest in SGC-7901/DDP cells
SGC-7901/DDP cells were treated with different concentrations of PN (0, 7.5 or 15 µmol/l) and the cell cycle distribution was analyzed 48 h following treatment (Fig. 4). The ratio of G0/G1 in the untreated and 7.5, and 15 µmol/l PN groups was 27.2±1.05, 32.7±1.50 and 35.8±1.39%, respectively; the percentage of S-phase cells was 63.98±2.74, 60.49±2.11, and 58.94±3.10%, respectively. Furthermore, the percentage of G2/M phase cells was 8.82±0.57, 6.78±0.73 and 4.72±0.46%, respectively. Compared with the untreated control group, the population of S and G2/M phase cells in the treatment groups significantly decreased but not significantly, and G1 phase cells significantly increased in a concentration-dependent manner (P<0.05; Fig. 4).
Figure 4.

Effect of parthenolide (7.5 or 15 µmol/l) on SGC-7901/DDP cell cycle arrest. *P<0.05 compared with untreated control group.
PN induces SGC-7901/DDP cell apoptosis
SGC-7901/DDP cells were treated with 7.5 and 15 µmol/l of PN and the cell apoptosis rates were detected 48 h following treatment (Fig. 5). The early apoptosis rates in the 7.5 and 15 µmol/l treated groups were 6.87±0.63 and 17.7±1.15%, respectively. Compared with the control group (5.30±1.31%), the early apoptosis rate was markedly increased in the 7.5 µmol/l group and significantly increased in the 15 µmol/l group (P<0.05) compared with that in the untreated control group. The late apoptosis rates were 2.88±0.54, 3.4±0.61 and 4.18±0.78% following no treatment, and treatment with 7.5 and 15 µmol/l PN, respectively. No statistically significant differences were identified between these groups (data not shown).
Figure 5.

Effect of parthenolide (7.5 or 15 µmol/l) on SGC-7901/DDP cell apoptosis using flow cytometry. *P<0.05 compared with untreated control group.
PN induces SGC-7901/DDP cell apoptosis as determined using DAPI staining
SGC-7901/DDP cells were treated with 7.5 or 15 µmol/l PN and observed using DAPI staining after 24 h (Fig. 6). The cells in the untreated control group possessed normally shaped nuclei (round or oval) with uniform staining and evenly distributed nuclear chromatin. The shrinkage, cohesion and fragmentation (spherical or particulate matter) of nuclei were observed 48 h after PN treatment. The number of apoptotic cells increased in a PN concentration-dependent manner, as indicated by the number of dense fluorescent particles (Fig. 6). The SGC-7901/DDP cell apoptosis rates were 0.88±0.05, 10.56±0.82 and 18.85±1.13% in the untreated, 7.5, and 15 µmol/l PN groups, respectively. The increases in apoptosis rates in the 7.5 and 15 µmol/l groups were significant compared with the untreated control group (P<0.05; Fig. 6).
Figure 6.
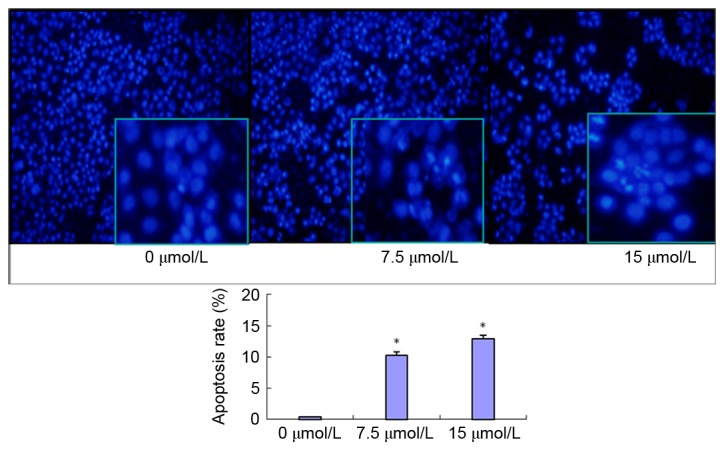
Effect of PN (7.5 or 15 µmol/l) on SGC-7901/DDP cell apoptosis via DAPI detection with fluorescence microscopy (magnification, ×100). *P<0.05 compared with untreated control group.
Expression of apoptosis- and cell cycle-associated proteins
Among the apoptosis-associated proteins, following PN treatment (7.5 or 15 µmol/l) Bax and p53 expression was significantly upregulated, Bcl-xL and Bcl-2 was significantly downregulated, and cleaved caspase-3 and −9 was significantly increased in a dose-dependent manner as compared with the expression in the untreated control groups (all P<0.05; Fig. 7). Among the cell cycle-associated proteins, cyclin D1 expression was significantly decreased, p21 was significantly increased, and P-STAT3 activation was significantly inhibited (all P<0.05; Fig. 7).
Figure 7.

Effect of PN (7.5 or 15 µmol/l) on the protein expression levels of apoptosis- and cell cycle-associated proteins in SGC-7901/DDP cells using western blotting. *P<0.05; #P<0.01 compared with untreated control group. p-STAT3, phosphorylated-signal transducer and activator of transcription 3; Bcl-xL, apoptosis regulator Bcl-xL; Bcl-2, apoptosis regulator Bcl-2; Bax, apoptosis regulator BAX; p53, cellular tumor antigen p53; p21, cyclin-dependent kinase inhibitor 1.
PN decreases SGC-7901/DDP cell migration
The effect of PN on SGC-7901/DDP cell migration is presented in Fig. 8. The mobility rate of the untreated control group at 24, 48 and 72 h were 11.85±2.55, 25.57±2.75, and 43.32±2.52%, respectively. The mobility rate of the 2.5 µmol/l PN group at 24, 48 and 72 h were 8.09±0.83, 22.70±2.92, and 31.11±3.85%, respectively. The mobility rate of the 5 µmol/l PN group at 24, 48 and 72h were 3.13±0.76, 7.62±1.17, and 16.54±3.02%, respectively. The differences between the 2.5 and 5 µmol/l PN groups, and the untreated control group at each time point were statistically significant (P<0.05).
Figure 8.
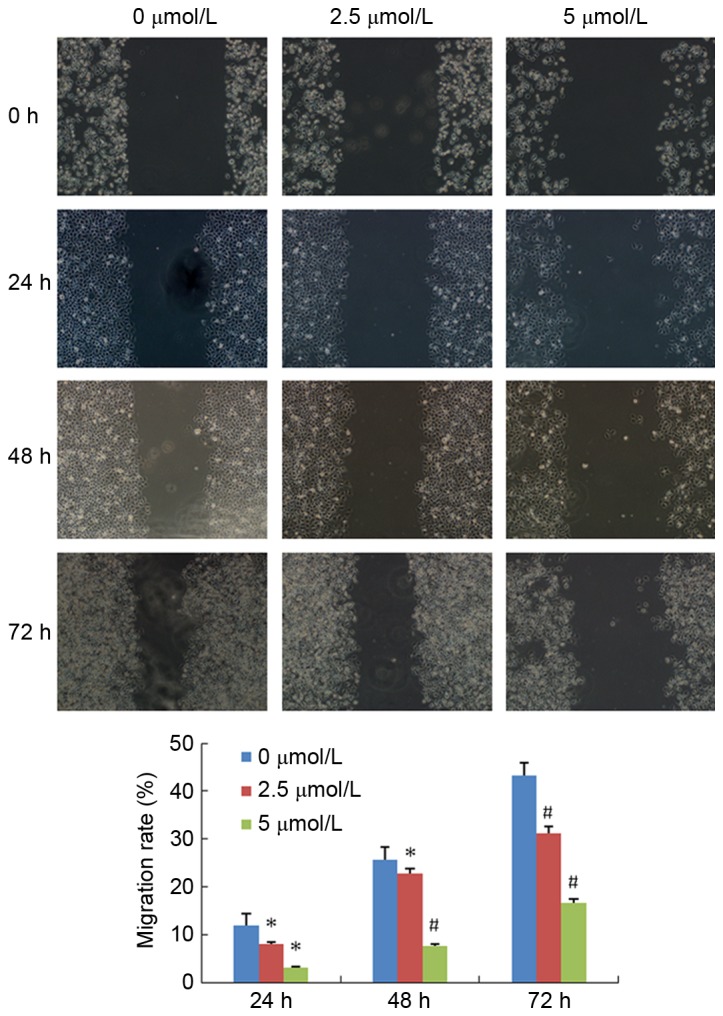
Effect of PN (2.5 or 5 µmol/l) on SGC-7901/DDP cell migration using a Scratch migration assay (magnification, ×100). *P<0.05; #P<0.01 compared with untreated control group.
PN decreases SGC-7901/DDP cell invasion ability
The effect of PN on invasion ability is presented in Fig. 9. The number of transmembrane cells in the untreated control, and 2.5 and 5 µmol/l PN groups were 737±33.57, 344±12.81, and 127±8.02, respectively. The differences between the treated groups and the untreated control group were statistically significant (P<0.05).
Figure 9.
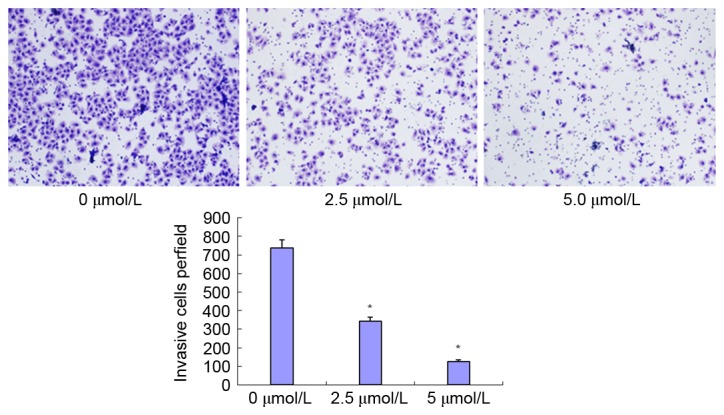
Effect of PN (2.5 or 5 µmol/l) on SGC-7901/DDP cell invasion ability using a Transwell invasion assay (magnification, ×100). *P<0.05 compared with untreated control group.
Discussion
Previous studies have demonstrated that PN can inhibit the proliferation of tumor cells and enhance the sensitivity of certain cancer cell lines to anti-cancer drugs (7,12). Gao et al (13) suggested that PN enhances the sensitivity of non-small cell lung cancer cells to chemotherapy drugs via modulation of the NF-κB/I-κB kinase (IKK) signal cascade through IKKβ. Furthermore, Liu et al (14) and another study (15) revealed that PN is able to enhance the sensitivity of drug-resistant hepatocellular carcinoma cells to chemotherapy drugs through inhibition of NF-κB activity, the downregulation of P-glycoprotein, multidrug resistance-associated protein, Bcl-2 and proto-oncogene Wnt family member 1 expression levels, and the upregulation of p53 expression. The present study demonstrated that PN significantly inhibited the proliferation of the drug-resistant gastric cancer cell line SGC7901/DDP in a time- and concentration-dependent manner. Additionally, PN and DDP co-treatment demonstrated a synergistic effect. The inhibitory effect of PN on SGC7901/DDP cell proliferation may be due to cycle arrest and the induction of apoptosis. PN treatment was demonstrated to induce early phase apoptosis and G1 phase cycle arrest. Furthermore, the results in the present study revealed that the anticancer effects of PN are associated with the upregulation of p53 and Bax, the downregulation of Bcl-2 and Bcl-xL, and the activation of capase-9 and caspase-3. Previously, these proteins have all been demonstrated to be involved in the regulation of apoptosis (12,16), thus these results suggest that PN treatment activates the apoptotic signaling pathway. In addition, the results of the current study revealed that PN significantly decreased cyclin D1, increased p21 expression and inhibited STAT3 activation.
STAT3 in the cytoplasm is a bifunctional protein. Previous studies (17,18) have revealed that the STAT3 downstream target genes, including Bcl-xL, Bcl-2, induced myeloid leukemia cell differentiation protein Mcl-1, cyclin D1, proto-oncogene c-Myc, Jun proto-oncogene AP-1 transcription factor subunit, Fas cell surface death receptor and vascular endothelial growth factor, which are closely associated with cell proliferation, apoptosis, and angiogenesis. Under normal physiological conditions, the activation of STATs is rapid and transient. Sustained activation of this signaling pathway in tumor cells can induce the upregulation of cell cycle regulatory factors and anti-apoptotic proteins, consequently promoting abnormal proliferation, and migration of malignant tumors (19). Therefore, STAT3 is considered an oncogene (19). Previous studies demonstrated that STAT3 signaling pathway may be associated with tumor cell drug-resistance to chemotherapy (20,21). The aberrant activation of STAT3 can aid in the evasion of cancer cell death caused by drugs, thus inducing drug resistance. The inhibition of the STAT3 signaling pathway may become a potential cancer therapeutic target in the future. For example, Yue et al (22) demonstrated that the overactivation of STAT3 promotes DDP-resistant ovarian cancer progression, and can be used as a gene marker for drug resistance and cancer recurrence. Ovarian cancer cells with high expression levels of STAT3 and Bcl-xL exhibit DDP, and Taxol resistance; whereas, low STAT3 expression cell lines are sensitive to the drug treatment (23). Inhibiting the STAT3 signaling pathway can promote apoptosis of drug-resistance cancer cells and increase the sensitivity of cancer cells to various drugs (23). In addition, a previous study demonstrated that STAT3 activation-induced expression of Bcl-xL can enhance the survival and drug resistance capability of human tumor cells; and that silencing of the STAT3 gene downregulates the expression of Bcl-xL (24). In addition, inhibition of Bcl-xL protein expression through siRNA knockdown in the gastric cancer MGC-803 cell line promotes programmed cell death (24).
STAT3 overactivation has been identified in various types of tumor cells, including breast and ovarian cancer, and head and neck squamous cell carcinoma (25,26). In addition, Yu et al (27) demonstrated that the activity of STAT3 in gastric cancer cell lines is higher compared with that in normal gastric epithelial cells. STAT3-DNA binding activity is higher in poorly differentiated gastric adenocarcinoma cell lines (SGC7901, MKN45 and AGS) compared with in highly differentiated gastric cancer cell lines (MKN28 and NCI-SNU-1) (27). Furthermore, p-STAT3 expression in poorly differentiated gastric cancer tissue is higher compared with in the adjacent normal mucosa, suggesting that the overactivation of STAT3 serves a role in gastric cancer (27).
The results of the present study revealed that PN significantly inhibited STAT3 phosphorylation, modulated the expression levels of the downstream cell cycle-associated proteins, including cyclin D1 and p21, and downregulated the apoptosis-associated protein expression, including Bcl-2 and Bcl-XL. Cyclin D1 is an essential protein in the regulation of the cell cycle G1 phase. p21 is an important member of the cell cycle protein kinase inhibitor gene family. Decreased cyclin D1 and increased p21 expression results in increased cell cycle arrest at the G1 phase. Bcl-2 and Bcl-xL are important members of the anti-apoptotic protein family. Bcl-2 and Bcl-XL downregulation can enhance tumor cell susceptibility to drug-induced apoptosis. Song et al (28) revealed that PN inhibits the activation of STAT3 signaling pathway; Carlisi et al (29) also demonstrated that PN significantly reduces STAT3 and STAT5 phosphorylation, and increases the sensitivity of liver cells to apoptosis. Cheng et al and Ghantous et al (30–32) also confirmed that the anti-tumor effects of PN are not only due to the inhibition of NF-κB, but also associated with p21 upregulation, and the downregulation of cyclin D1 and Bcl-xL. The results of the current study are consistent with these previous studies. However, STAT3 converges various carcinogenic kinase signaling pathways, including the epidermal growth factor, interleukin-6/Janus kinase and Src signaling pathways. The mechanisms underlying the interactions of PN with the upstream and downstream factors in these complex signaling pathways warrants further investigation. In addition, the present study revealed that PN can inhibit SGC7901/DDP cell migration and invasion. These results suggest that PN is a promising anticancer drug for the treatment of patients with gastric cancer.
Acknowledgements
The present study was supported by the Natural Science Foundation of Liaoning Province Funded Project (grant no. 2013022070) and the Liaoning Medical Youth Science and Technology Start-up Funding Project (grant no. Y2012Z004).
References
- 1.Fodale V, Pierobon M, Liotta L, Petricoin E. Mechanism of cell adaptation: When and how do cancer cells develop chemoresistance? Cancer J. 2011;17:89–95. doi: 10.1097/PPO.0b013e318212dd3d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi H, Lu D, Shu Y, Shi W, Lu S, Wang K. Expression of multidrug resistance-related proteins p-glycoprotein, glutathione-s-transferases, topoisomerase-II and lung resistance protein in primary gastric cardiac adenocarcinoma. Hepatogastroenterology. 2008;55:1530–1536. [PubMed] [Google Scholar]
- 3.Kishida Y, Yoshikawa H, Myoui A. Parthenolide, a natural inhibitor of nuclear factor-kappaB, inhibits lung colonization of murine osteosarcoma cells. Clin Cancer Res. 2007;13:59–67. doi: 10.1158/1078-0432.CCR-06-1559. [DOI] [PubMed] [Google Scholar]
- 4.Kawasaki BT, Hurt EM, Kalathur M, Duhagon MA, Milner JA, Kim YS, Farrar WL. Effects of the sesquiterpene lactone parthenolide on prostate tumor-initiating cells: An integrated molecular profiling approach. Prostate. 2009;69:827–837. doi: 10.1002/pros.20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu JW, Cai MX, Xin Y, Wu QS, Ma J, Yang P, Xie HY, Huang DS. Parthenolide induces proliferation inhibition and apoptosis of pancreatic cancer cells in vitro. J Exp Clin Cancer Res. 2010;29:108. doi: 10.1186/1756-9966-29-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W, Adachi M, Kawamura R, Sakamoto H, Hayashi T, Ishida T, Imai K, Shinomura Y. Parthenolide-induced apoptosis in multiple myeloma cells involves reactive oxygen species generation and cell sensitivity depends on catalase activity. Apoptosis. 2006;11:2225–2235. doi: 10.1007/s10495-006-0287-2. [DOI] [PubMed] [Google Scholar]
- 7.Dai Y, Guzman ML, Chen S, Wang L, Yeung SK, Pei XY, Dent P, Jordan CT, Grant S. The NF (Nuclear factor)-κB inhibitor parthenolide interacts with histone deacetylase inhibitors to induce MKK7/JNK1-dependent apoptosis in human acute myeloid leukaemia cells. Br J Haematol. 2010;151:70–83. doi: 10.1111/j.1365-2141.2010.08319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shanmugam R, Kusumanchi P, Cheng L, Crooks P, Neelakantan S, Matthews W, Nakshatri H, Sweeney CJ. A water-soluble parthenolide analogue suppresses in vivo prostate cancer growth by targeting NFkappaB and generating reactive oxygen species. Prostate. 2010;70:1074–1086. doi: 10.1002/pros.21141. [DOI] [PubMed] [Google Scholar]
- 9.Holcomb BK, Yip-Schneider MT, Waters JA, Beane JD, Crooks PA, Schmidt CM. Dimethylamino parthenolide enhances the inhibitory effects of gemcitabine in human pancreatic cancer cells. J Gastrointest Surg. 2012;16:1333–1340. doi: 10.1007/s11605-012-1913-7. [DOI] [PubMed] [Google Scholar]
- 10.Yip-Schneider MT, Nakshatri H, Sweeney CJ, Marshall MS, Wiebke EA, Schmidt CM. Parthenolide and sulindac cooperate to mediate growth suppression and inhibit the nuclear factor-kappa B pathway in pancreatic carcinoma cells. Mol Cancer Ther. 2005;4:587–594. doi: 10.1158/1535-7163.MCT-04-0215. [DOI] [PubMed] [Google Scholar]
- 11.Zhang SQ, Zhang SH, Xue XH, Wang XJ, Jiang JT. Synergism between a siRNA targeted to survivin and 5-FU in inhibiting MCF-7 cell proliferation in vitro. Nan Fang Yi Ke Da Xue Xue Bao. 2006;26:251–254. [PubMed] [Google Scholar]
- 12.Gunn EJ, Williams JT, Huynh DT, Iannotti MJ, Han C, Barrios FJ, Kendall S, Glackin CA, Colby DA, Kirshner J. The natural products parthenolide and andrographolide exhibit anti-cancer stem cell activity in multiple myeloma. Leuk Lymphoma. 2011;52:1085–1097. doi: 10.3109/10428194.2011.555891. [DOI] [PubMed] [Google Scholar]
- 13.Gao ZW, Zhang DL, Guo CB. Paclitaxel efficacy is increased by parthenolide via nuclear factor-kappaB pathways in in vitro and in vivo human non-small cell lung cancer models. Curr Cancer Drug Targets. 2010;10:705–715. doi: 10.2174/156800910793605776. [DOI] [PubMed] [Google Scholar]
- 14.Liu D, Liu Y, Liu M, Ran L, Li Y. Reversing resistance of multidrug-resistant hepatic carcinoma cells with parthenolide. Future Oncol. 2013;9:595–604. doi: 10.2217/fon.13.15. [DOI] [PubMed] [Google Scholar]
- 15.Sohma I, Fujiwara Y, Sugita Y, Yoshioka A, Shirakawa M, Moon JH, Takiguchi S, Miyata H, Yamasaki M, Mori M, Doki Y. Parthenolide, an NF-κB inhibitor, suppresses tumor growth and enhances response to chemotherapy in gastric cancer. Cancer Genomics Proteomics. 2011;8:39–47. [PubMed] [Google Scholar]
- 16.Al-Fatlawi AA, Al-Fatlawi AA, Irshad M, Rahisuddin, Ahmad A. Effect of parthenolide on growth and apoptosis regulatory genes of human cancer cell lines. Pharm Biol. 2015;53:104–109. doi: 10.3109/13880209.2014.911919. [DOI] [PubMed] [Google Scholar]
- 17.Caldera V, Mellai M, Annovazzi L, Valente G, Tessitore L, Schiffer D. Stat3 expression and its correlation with proliferation and apoptosis/autophagy in gliomas. J Oncol. 2008;2008:219241. doi: 10.1155/2008/219241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunigal S, Lakka SS, Sodadasu PK, Estes N, Rao JS. Stat3-siRNA induces Fas-mediated apoptosis in vitro and in vivo in breast cancer. Int J Oncol. 2009;34:1209–1220. [PMC free article] [PubMed] [Google Scholar]
- 19.Barré B, Vigneron A, Perkins N, Roninson IB, Gamelin E, Coqueret O. The STAT3 oncogene as a predictive marker of drug resistance. Trends Mol Med. 2007;13:4–11. doi: 10.1016/j.molmed.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Huang S, Chen M, Shen Y, Shen W, Guo H, Gao Q, Zou X. Inhibition of activated Stat3 reverses drug resistance to chemotherapeutic agents in gastric cancer cells. Cancer Lett. 2012;315:198–205. doi: 10.1016/j.canlet.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Kohsaka S, Wang L, Yachi K, Mahabir R, Narita T, Itoh T, Tanino M, Kimura T, Nishihara H, Tanaka S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol Cancer Ther. 2012;11:1289–1299. doi: 10.1158/1535-7163.MCT-11-0801. [DOI] [PubMed] [Google Scholar]
- 22.Yue P, Zhang X, Paladino D, Sengupta B, Ahmad S, Holloway RW, Ingersoll SB, Turkson J. Hyperactive EGF receptor, Jaks and Stat3 signaling promote enhanced colony-forming ability, motility and migration of cisplatin-resistant ovarian cancer cells. Oncogene. 2012;31:2309–2322. doi: 10.1038/onc.2011.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duan Z, Ames RY, Ryan M, Hornicek FJ, Mankin H, Seiden MV. CDDO-Me, a synthetic triterpenoid, inhibits expression of IL-6 and Stat3 phosphorylation in multi-drug resistant ovarian cancer cells. Cancer Chemother Pharmacol. 2009;63:681–689. doi: 10.1007/s00280-008-0785-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei XY, Zhong M, Feng LF, Yan CY, Zhu BY, Tang SS, Liao DF. Silencing of Bcl-XL expression in human MGC-803 gastric cancer cells by siRNA. Acta Biochim Biophys Sin (Shanghai) 2005;37:555–560. doi: 10.1111/j.1745-7270.2005.00077.x. [DOI] [PubMed] [Google Scholar]
- 25.Min H, Wei-hong Z. Constitutive activation of signal transducer and activator of transcription 3 in epithelial ovarian carcinoma. J Obstet Gynaecol Res. 2009;35:918–925. doi: 10.1111/j.1447-0756.2009.01045.x. [DOI] [PubMed] [Google Scholar]
- 26.Peyser ND, Freilino M, Wang L, Zeng Y, Li H, Johnson DE, Grandis JR. Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene. 2016;35:1163–1169. doi: 10.1038/onc.2015.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu LF, Zhu YB, Qiao MM, Zhong J, Tu SP, Wu YL. Constitutive activation and clinical significance of Stat3 in human gastric cancer tissues and cell lines. Zhonghua Yi Xue Za Zhi. 2004;84:2064–2069. (In Chinese) [PubMed] [Google Scholar]
- 28.Song JM, Qian X, Upadhyayya P, Hong KH, Kassie F. Dimethylaminoparthenolide, a water soluble parthenolide, suppresses lung tumorigenesis through down-regulating the STAT3 signaling pathway. Curr Cancer Drug Targets. 2014;14:59–69. doi: 10.2174/15680096113136660108. [DOI] [PubMed] [Google Scholar]
- 29.Carlisi D, D'Anneo A, Angileri L, Lauricella M, Emanuele S, Santulli A, Vento R, Tesoriere G. Parthenolide sensitizes hepatocellular carcinoma cells to TRAIL by inducing the expression of death receptors through inhibition of STAT3 activation. J Cell Physiol. 2011;226:1632–1641. doi: 10.1002/jcp.22494. [DOI] [PubMed] [Google Scholar]
- 30.Czyz M, Lesiak-Mieczkowska K, Koprowska K, Szulawska-Mroczek A, Wozniak M. Cell context-dependent activities of parthenolide in primary and metastatic melanoma cells. Br J Pharmacol. 2010;160:1144–1157. doi: 10.1111/j.1476-5381.2010.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng G, Xie L. Parthenolide induces apoptosis and cell cycle arrest of human 5637 bladder cancer cells in vitro. Molecules. 2011;16:6758–6768. doi: 10.3390/molecules16086758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghantous A, Saikali M, Rau T, Gali-Muhtasib H, Schneider-Stock R, Darwiche N. Inhibition of tumor promotion by parthenolide: Epigenetic modulation of p21. Cancer Prev Res (Phila) 2012;5:1298–1309. doi: 10.1158/1940-6207.CAPR-12-0230. [DOI] [PubMed] [Google Scholar]