

Abstract
Patients with end‐stage liver disease develop acute decompensation (AD) episodes, which become more frequent and might develop into acute‐on‐chronic liver failure (ACLF). However, it remains unknown how AD induces acceleration of liver disease. We hypothesized that remodeling of collagen type III plays a role in the acceleration of liver cirrhosis after AD and analyzed its formation (Pro‐C3) and degradation (matrix metalloproteinase‐degraded type III collagen [C3M]) markers in animal models and human disease. Bile duct ligation induced different stages of liver fibrosis in rats. Fibrosis development (hydroxyprolin content, sirius red staining, α‐smooth muscle actin immunohistochemistry, messenger RNA of profibrotic cytokines), necroinflammation (aminotransferases levels), fibrolysis (matrix metalloproteinase 2 expression and activity, C1M, C4M), and Pro‐C3 and C3M were analyzed 2, 3, 4, 5, and 6 weeks after bile duct ligation (n = 5 each group). In 110 patients with decompensated liver cirrhosis who underwent a transjugular intrahepatic portosystemic shunt procedure for AD, clinical and laboratory parameters as well as Pro‐C3 and C3M were measured in blood samples from portal and hepatic veins and were collected just before the transjugular intrahepatic portosystemic shunt placement and 1‐3 weeks later. Animal studies showed increased markers of collagen type III deposition with fibrosis, necroinflammation, and decompensation of liver cirrhosis, defined as ascites development. Higher Pro‐C3 levels were associated with injury, disease severity scores (Model for End‐Stage Liver Disease, Child‐Pugh, chronic liver failure‐C AD), ACLF development, and mortality. C3M decreased with AD and the chronic liver failure‐C AD score. Collagen type III deposition ratio increased with the risk of ACLF development and mortality. Conclusion: We show for the first time that AD boosts collagen type III deposition in experimental and human cirrhosis, possibly contributing to the worsened outcome in patients with decompensated cirrhosis. (Hepatology Communications 2018;2:211–222)
Abbreviations
- α‐SMA
α‐smooth muscle actin
- ACLF
acute‐on‐chronic liver failure
- AD
acute decompensation
- BDL
bile duct ligation
- C3M
metalloproteinase‐degraded type III collagen
- CLIF‐C
chronic liver failure‐C
- ECM
extracellular matrix
- MELD
Model for End‐Stage Liver Disease
- MMP
matrix metalloproteinase
- Pro‐C3
formation of type III collagen
- TIPS
transjugular intrahepatic portosystemic shunt
Introduction
Chronic liver diseases are a growing health care burden. Liver cirrhosis is the common end stage of any chronic liver disease and can decompensate with its progression.1 Progressive fibrosis, increased hepatic vascular tone, and ongoing inflammation due to permanent liver injury are major causes of decompensation, morbidity, and mortality. These processes incite continuous extracellular matrix (ECM) protein remodeling, which reflects the progression of liver disease.2, 3, 4, 5, 6 During ECM remodeling, matrix metalloproteinases (MMPs) generate small peptide fragments, known as neoepitopes, that are released into the circulation. They can be measured with protein fingerprint technology using specific antibodies.7 The levels of these ECM markers depend on the degree of fibrosis4, 8, 9, 10 and also reflect antifibrotic therapy.5, 11 Degradation markers and collagen type III formation, in particular, mirror progression of fibrosis and diagnosis of portal hypertension4, 10, 12, 13; however, the roles of these markers in decompensation of liver cirrhosis have not been assessed.
As outlined above, severe liver fibrosis with portal hypertension induces acute decompensation (AD), such as variceal bleeding, refractory ascites, or hepatorenal syndrome, which can be treated with transjugular intrahepatic portosystemic shunt implantation (TIPS).14 In selected patients, TIPS decompresses the portal venous system, successfully treats ascites and variceal bleeding, and improves survival.14, 15, 16, 17 However, AD of liver cirrhosis can progress and develop into acute‐on‐chronic liver failure (ACLF) with poor survival.18 While deposition of ECM in the course of liver fibrosis has been known for some time, the role of collagen in patients with advanced liver cirrhosis and AD19 remains unclear.
The present work demonstrates for the first time that collagen III turnover markers show a specific pattern in the course of progression of liver fibrosis and can predict development of ACLF and mortality in patients with decompensated liver cirrhosis.
Materials and Methods
HUMAN STUDY DESIGN
We studied 110 patients with decompensated liver cirrhosis who underwent a TIPS procedure between 1996 and 2003. Inclusion criteria were age between 20 and 80 years, cirrhosis due to alcohol or viral hepatitis, and decompensated liver cirrhosis with indication for TIPS (bleeding in n = 41, refractory ascites in n = 51, or both in n = 18). Exclusion criteria were contraindications for TIPS placement,20, 21 i.e., serum levels of bilirubin >5 mg/dL, spontaneous bacterial peritonitis and manifest hepatic encephalopathy, pulmonary arterial hypertension, and cardiac insufficiency. An invasive control of the TIPS was performed as part of the routine in 74 patients 1‐3 weeks after TIPS insertion.10 Biochemical blood analyses were performed using standard tests. The local ethics committee of the University of Bonn approved the study (029/13), and all patients agreed and signed informed consent for the procedures in accordance with the Helsinki Declaration.
TRANSJUGULAR INTRAHEPATIC PORTOSYSTEMIC SHUNT INSERTION AND HEMODYNAMIC MEASUREMENTS
TIPS (8‐10 mm Wallstent; Boston Scientific) placement was performed as described.20, 21 A single shot of antibiotic prophylaxis of cefuroxime (1.5 g) was administered at TIPS placement. Portal and hepatic venous pressures were measured invasively using a pressure transducer system (Combitrans; B. Braun Melsungen, Melsungen, Germany) and a multichannel monitor (Sirecust; Siemens). The difference between portal and hepatic venous pressures was defined as the portal hepatic pressure gradient. Arterial pressure and heart rate were monitored noninvasively. Biochemical parameters as well as portal and systemic hemodynamics were recorded. Blood from the portal and hepatic veins was collected as described.22 The blood sample from the portal vein was taken immediately after puncture of the vein. The hepatic venous sample was taken from the hepatic vein, which was used for the creation of TIPS, right before puncturing the portal vein. Portal venous samples were taken immediately after entering the portal vein but before dilation of the tract or insertion of the TIPS stent. At invasive TIPS control after a median of 10 days (range, 1‐3 weeks), a catheter was advanced into the portal vein. Blood from the portal and hepatic veins was collected. Protein fingerprint markers were assessed in the portal and the hepatic vein plasma samples.
ANIMAL STUDY DESIGN
Sprague‐Dawley rats with an initial body weight of 180‐200 g were used. Thirty rats underwent bile duct ligation (BDL) to induce liver fibrosis as described.23, 24, 25 A mortality of 20% led to a total of 24 rats for assessment. This study was authorized by the local committee for animal studies (Bezirksregierung Köln, 50.203.2‐BN22, 46/05). All rats received chow and water ad libitum and were weighed and fed daily. BDL rats were killed 2, 3, 4, 5, and 6 weeks after BDL (n = 5 per group, except for 4 weeks after BDL, n = 4). Blood was withdrawn and liver samples were cut into fragments and either snap frozen in liquid nitrogen and stored at –80°C or preserved in formaldehyde as described.11, 23, 24
PROTEIN FINGERPRINT MARKER ASSESSMENT
MMP‐degraded type III collagen (C3M)9 and formation of type III collagen (Pro‐C326) were examined. Additionally, MMP‐degraded type I (C1M) and type IV (C4M) collagen were measured.10 Briefly, each assay was run on a 96‐well streptavidin plate coated with the appropriate biotinylated synthetic peptide dissolved in an optimized assay and incubated for 30 minutes at 20°C. Next, 20 μL of calibrator peptide or sample in appropriate dilution was added to the wells, including 100 μL of horseradish peroxidase‐conjugated monoclonal antibody raised against the specific sequence of interest and incubated for 1 hour at 20°C or overnight at 4°C. Following incubation, 100 μL tetramethylbenzinidine (Kem‐En‐Tec cat. 4380H) was added, and the plate was incubated for 15 minutes at 20°C in the dark. In order to stop the reaction, 100 μL of stopping solution (1% H2SO4) was added and measured at 450 nm with 650 nm as reference. All incubation steps included shaking at 300 revolutions per minute. After coating and antibody incubation, the plate was washed 5 times in washing buffer (20 mM Tris, 50 mM NaCl, pH 7.2). A calibration curve was plotted using a four‐parametric fit model. Samples were measured within the detection range.
HEPATIC HYDROXYPROLINE CONTENT
Hepatic hydroxyproline was photometrically determined in liver hydrolysates.23, 24 Segments (200 mg) of snap‐frozen livers were hydrolyzed in HCl (6 N) at 110°C for 16 hours and filtered. Aliquots (50 μL) from each sample were incubated with chloramine T (2.5 mM) for 5 minutes and Ehrlich's reagent (410 mM) for 30 minutes at 60°C. Absorption was measured 3 times at 558 nm. For determination of hydroxyproline concentration, a standard curve for hydroxyproline was used. Results are expressed as micrograms per gram of wet liver tissue.
SIRIUS RED STAINING, α‐SMOOTH MUSCLE ACTIN IMMUNOHISTOCHEMISTRY, AND MORPHOMETRY
Liver specimens were fixed in 10% formalin. As described,23 paraffin‐embedded sections (2‐3 μm) were stained in 0.1% sirius red F3B in saturated picric acid (Chroma, Münster, Germany) to detect collagen fibers. For immunohistochemical staining of α‐smooth muscle actin (α‐SMA), sections were incubated with a mouse‐anti‐α‐SMA antibody (clone 1A4; Sigma‐Aldrich, St. Louis, MO) diluted 1:600 in Tris‐buffered saline for 60 minutes. A biotinylated rabbit‐anti‐mouse antibody absorbed with rat serum (Dako, Glostrup, Denmark) was used as secondary antibody and subsequently applied, diluted 1:200, for 45 minutes. Next, streptavidin‐conjugated alkaline phosphatase (1:200, 45 minutes; Dako) was attached to the complex. Finally, sections were developed with new fuchsin‐naphtol AS‐BI (Sigma‐Aldrich) and counterstained with hematoxylin. Stereomicroscopic analysis was performed as described.23 On sections with a surface of 1 cm2, 500 points were counted at 400× magnification. On sirius red staining, test points were classified as connective tissue (positive staining), hepatocytes, bile ducts, or other structures (not stained). α‐SMA‐positive cells were counted on α‐SMA‐immunohistologic sections and characterized by their location as sinusoidal, periductular, portal/septal, and others (e.g., vascular smooth muscle cells). The volume fraction of each structure was calculated as percentage of points overlaying the structure in relation to the total number of counted points.
QUANTITATIVE REVERSE‐TRANSCRIPTION POLYMERASE CHAIN REACTION OF CONNECTIVE TISSUE GROWTH FACTOR, PLATELET‐DERIVED GROWTH FACTOR, AND TRANSFORMING GROWTH FACTOR BETA 1
RNA was isolated from 30‐mg liver segments, using the RNeasy kit (Qiagen, Basel, Switzerland) with deoxyribonuclease treatment (Promega, Wallisellen, Switzerland), followed by reverse transcription with Moloney murine leukemia virus reverse transcriptase (Invitrogen, Basel, Switzerland) according to the manufacturer's instructions. Quantitative polymerase chain reaction was carried out on an ABI 7700 sequence detector (Applied Biosystems, Rotkreuz, Switzerland) with described sequences and accession numbers.23 Gene expression values were calculated based on the ΔCt method23, 25 and normalized to expression of glyceraldehyde 3‐phosphate dehydrogenase. Results were calculated as 2–ΔCt to express the fold increase of gene expression compared to sham‐operated rats.
MMP‐2 ZYMOGRAPHY
Proteolytic activity of tissue homogenates was examined by gelatin zymography as published.23 The supernatants of liver homogenates (20 μg of protein) were subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis using gels containing 0.3% gelatin. Proteolytic bands of 62 and 65 kDa, corresponding to the active and latent form of MMP‐2, respectively, were quantified by densitometry using AIDA software (Raytest, Urdorf, Switzerland).
STATISTICAL ANALYSIS
Plasma levels of the four protein fingerprint markers (Pro‐C3 and C3M; C1M and C4M) were logarithmically transformed to obtain normality and symmetry of variance. All correlations were calculated using Spearman's correlation. Comparison of the levels of protein fingerprints assessed in portal and hepatic venous blood at baseline and at the control visit were analyzed using the paired‐samples t test. Patient survival was calculated by Kaplan‐Meier analysis, while survival curves were compared using the log‐rank test. Univariate and multivariate analyses of patients' characteristics as predictors of mortality were calculated by Cox regression analysis. All data are shown as median with 95% confidence interval (CI) or as mean ± SEM. Differences between curves were calculated using two‐way analysis of variance. P < 0.05 was considered statistically significant. Statistical analyses were performed using SPSS 23 (IBM, Armonk, NY). Data were plotted using GraphPad Prism v.5 (Graph Pad Software, La Jolla, CA).
Results
PATIENT CHARACTERISTICS
The study population included 110 patients with cirrhosis, of whom 71 (64.5 %) were male individuals. The median age was 59 (36‐80) years. Etiology of cirrhosis was alcoholic liver cirrhosis in 81 (73.6%) patients, chronic viral hepatitis C in 13 (11.8%) patients, while 16 (14.6%) patients had other causes, including 11 (10%) patients with cryptogenic cirrhosis, 3 patients with primary biliary cirrhosis, and 2 patients suffering from autoimmune hepatitis. Median Model for End‐Stage Liver Disease (MELD) score was 8 (6‐18) and Child‐Turcotte‐Pugh score was 8 (5‐12). Before TIPS implantation, most patients showed esophageal varices (I‐II°, 65.6%; III‐IV°, 20.9%) and ascites (mild, 26.4%; severe, 46.4%). Hepatorenal syndrome was present in 25.5% of patients. The general characteristics and laboratory values of the study population are summarized in Tables 1 and 2.
Table 1.
CLINICAL PARAMETERS OF ALL PATIENTS (n = 110) AT ADMISSION
| Parameters | Value |
|---|---|
| Sex (male/female) | 71/39 |
| Age (years) | 59 (36‐80) |
| Etiology of cirrhosis (alcoholic/chronic viral hepatitis C/other) | 81/13/16 |
| Indication for TIPS implementation (bleeding/ascites/both) | 41/51/18 |
| Child‐Pugh score | 8 (5‐12) |
| Child‐Pugh category (A/B/C) | 17/67/26 |
| MELD score | 8 (6‐18) |
| Varices (esophageal: absent/°I‐II/°III‐IV/gastric varices) | 12/72/23/3 |
| Variceal bleeding (absent/present) | 51/59 |
| Ascites (absent/mild/severe) | 20/39/51 |
| Hepatorenal syndrome (absent/type I/type II) | 82/22/6 |
| Hepatic encephalopathy (absent/history of) | 98/12 |
Data are shown as median and ranges for continuous variables or total number for categorical variables.
Table 2.
PRESSURE GRADIENT AND LABORATORY PARAMETERS OF PATIENTS AT ADMISSION AND OUTCOME
| Parameters | At TIPS (n = 110) | Control Visit (n = 74) |
|---|---|---|
| PSPG (mm Hg) | 20 (11‐35) | 9 (2‐21)‡ |
| Bilirubin (mg/dL) | 1.2 (0.3‐14.8) | 1.4 (0.55‐10.4)‡ |
| gGT (IU/L) | 52 (8‐527) | 96 (12‐515)‡ |
| AST (IU/L) | 19 (8‐73) | 22 (12‐275)* |
| ALT (IU/L) | 18 (4‐94) | 21 (4‐357)‡ |
| INR | 1.2 (0.93‐2.23) | 1.2 (0.96‐1.92)‡ |
| Sodium (mEq/L) | 135 (119‐145) | 136 (118‐144)* |
| Platelet count (109/L) | 105 (27‐385) | 122 (46‐326) |
| Creatinine (mg/dL) | 1.1 (0.5‐8.2) | 1.0 (0.5‐7.1)† |
| BUN (mg/dL) | 44 (9‐225) | 32 (1.14‐118)‡ |
Data are shown as median and ranges. *P < 0.05, † P < 0.01, ‡ P < 0.001.
Abbreviations: ALT, alanine transaminase; AST, aspartate transaminase; BUN, blood urea nitrogen; gGT, gamma glutamyl transpeptidase; INR, international normalized ratio; PSPG, portal systemic pressure gradient.
COLLAGEN TYPE III REMODELING MARKERS BEFORE AND AFTER TIPS
The levels of formation marker Pro‐C3 in the hepatic vein were significantly higher than in the portal vein before and at a median of 10 days after TIPS placement (Fig. 1A,B), suggesting the liver to be the main origin of these markers. Interestingly, a comparison between the levels of the degradation marker C3M from the portal and the hepatic vein showed a similar difference but without reaching statistical significance after TIPS (Fig. 1A,B). Of note, when comparing levels of Pro‐C3 and C3M from the same compartment (hepatic or portal vein, respectively), both increased significantly after TIPS placement (Fig. 1C,D). These data raise the question as to whether this increase is due to mechanical liver parenchyma damage through TIPS placement or whether it reflects a true remodeling of collagen type III. Therefore, the time dependency of the levels of these markers after TIPS was analyzed (Fig. 1E,F). Importantly, after TIPS, levels of Pro‐C3 (Fig. 1F) and C3M (Fig. 1E) did not change significantly with time (≤8, 9‐11, >11 days), suggesting that mechanical liver injury is not involved in this increase.
Figure 1.
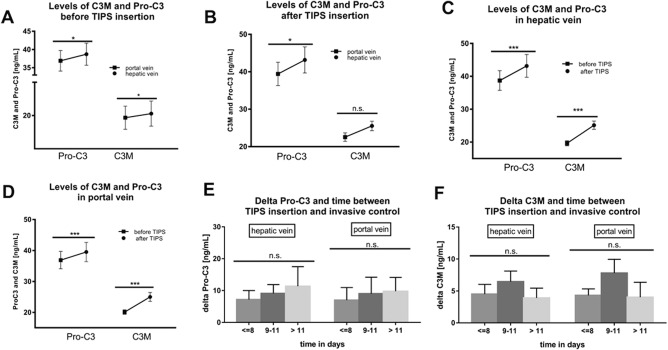
Evolution of levels of Pro‐C3 and C3M before and after TIPS. (A) Before the TIPS procedure, levels of Pro‐C3 and C3M were significantly higher in the hepatic vein compared to the portal vein (P < 0.05). (B) After TIPS, Pro‐C3 levels were significantly higher in the hepatic vein compared to the portal vein (P < 0.05), while the difference for C3M did not reach statistical significance. A significant increase in Pro‐C3 and C3M levels after the TIPS procedure was demonstrated in (C) for the hepatic vein and (D) for the portal vein (P < 0.001). (E) C3M and (F) Pro‐C3 levels over time after TIPS (<8, 9‐11, and >11 days) as they did not change significantly (P > 0.05). *P < 0.05, **P < 0.01, ***P < 0.001; data represent mean ± SEM. Abbreviation: n.s., not significant.
COLLAGEN TYPE III FORMATION MARKER Pro‐C3 AND SURVIVAL
As with previous observations4, 10, 12, 13 in patients with liver fibrosis and/or compensated liver cirrhosis, patients with high Pro‐C3 levels had a poorer outcome than patients with lower levels (Fig. 2A). The cutoff to discriminate between these two groups was set at 20 ng/mL in the hepatic vein (30 out of 110 patients), which represents the thirty‐third percentile.
Figure 2.
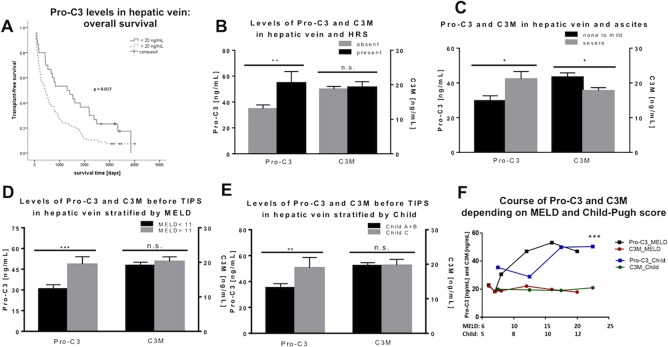
Pro‐C3 and C3M levels in the hepatic vein and their relationship with severity of human cirrhosis. (A) Kaplan‐Meier stratified for Pro‐C3 levels at a threshold of 20 ng/mL in the hepatic vein. (B) Pro‐C3 and C3M levels in the hepatic vein in patients with and without previous episodes of HRS. (C‐E) Higher Pro‐C3 levels in the hepatic vein and lower levels of C3M in patients with (C) severe ascites. Higher Pro‐C3 levels in patients with (D) higher MELD score (<11) and (E) Child‐Pugh C score, while C3M levels did not change significantly. (F) Distribution of Pro‐C3 and C3M levels according to MELD and Child‐Pugh scores. A significant divergence of Pro‐C3 and C3M curves is demonstrated. *P < 0.05, **P < 0.01, ***P < 0.001; data represent mean ± SEM. Abbreviation: n.s., not significant.
COLLAGEN TYPE III FORMATION MARKER Pro‐C3 LEVELS ARE INCREASED IN HEPATORENAL SYNDROME
Patients with previous episodes of hepatorenal syndrome (HRS), which indicates severely impaired renal function, had significantly higher Pro‐C3 levels in the hepatic vein (Fig. 2B). Interestingly, collagen type III degradation marker C3M levels did not differ between the groups with and without previous HRS.
COLLAGEN TYPE III FORMATION MARKER Pro‐C3 LEVELS ARE INCREASED IN PATIENTS WITH ASCITES, WHILE COLLAGEN TYPE III DEGRADATION MARKER C3M LEVELS ARE DECREASED
In patients with no or mild (i.e., treatable with diuretics) ascites, Pro‐C3 levels were significantly lower compared to patients with severe (refractory) ascites, indicating increased collagen type III formation. Inversely, we found C3M levels to be significantly decreased in patients with severe ascites compared to those with mild or no ascites, indicating decreased collagen type III degradation at the same time (Fig. 2C).
COLLAGEN TYPE III TURNOVER MARKERS AND PROGNOSTIC SCORES
Patients with a MELD score of 11 or higher had significantly higher levels of Pro‐C3 before TIPS compared to those with lower MELD scores. Levels of C3M, however, did not differ between the high and low MELD groups (Fig. 2D). A similar picture was observed in patients with Child‐Pugh stage C, who also had significantly higher Pro‐C3 levels before TIPS compared to patients with Child‐Pugh stages A or B, while C3M levels were not statistically different (Fig. 2E). As this possibly suggests promoted collagen type III formation in patients with more advanced liver cirrhosis, we calculated Pro‐C3 and C3M levels in the hepatic vein according to MELD and Child‐Pugh scores. As shown in Fig. 2F, levels of Pro‐C3 continually increased with increasing MELD and Child‐Pugh scores, reflecting decompensated states of liver cirrhosis, while levels of C3M did not congruently increase but instead did not change significantly.
ANIMAL STUDIES
To further investigate whether there are any mechanistic implications from the findings from our patient samples, we conducted animal studies as described above. These are summarized in Fig. 3A. In this model, we observed histologically increasing stages of fibrosis with increasing time after BDL, as shown by sirius red and α‐SMA staining (Supporting Fig. S1).
Figure 3.
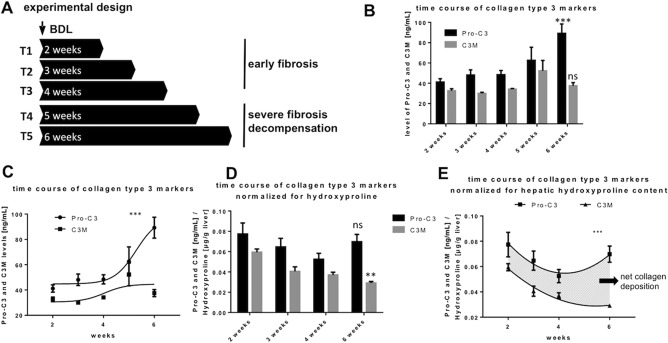
Levels of Pro‐C3 and C3M with severity of experimental fibrosis in BDL‐rats. (A) Experimental design of the rodent studies with sacrifice at 5 different time points, T1‐3 representing early stages of fibrosis, T4‐5 representing decompensated fibrosis of the liver. (B) Levels of Pro‐C3 and C3M at different time points of sacrifice. Pro‐C3 increased significantly in severe stages of fibrosis. C3M did not change significantly. (C) Significant divergence of levels of Pro‐C3 and C3M with increasing severity of fibrosis. (D) Levels of Pro‐C3 and C3M at different time points of sacrifice normalized for hydroxyproline content per gram of liver tissue. C3M decreased significantly in severe stages of fibrosis. Pro‐C3 did not change significantly. (E) Significant divergence of levels of Pro‐C3 and C3M with increasing severity of fibrosis. The area between the curves represents net collagen type III deposition in the liver (shaded area). *P < 0.05, **P < 0.01, ***P < 0.001; data represent mean ± SEM. Abbreviation: ns, not significant.
CORRELATION OF PROFIBROTIC CYTOKINES AND NECROINFLAMMATION
Along with progression of liver fibrosis, we could also observe increasing necroinflammation with increasing levels of aspartate aminotransferase and alanine aminotransferase. Profibrotic cytokines connective tissue growth factor, platelet‐derived growth factor, and transforming growth factor β1 showed a positive correlation with alanine aminotransferase and aspartate aminotransferase levels as markers of necroinflammation (Supporting Fig. S2A).
DYNAMICS OF COLLAGEN TYPE III TURNOVER MARKERS
Pro‐C3 levels significantly increased continuously with increasing time after BDL, which equates to more advanced fibrosis (Fig. 3B). Interestingly, the accompanying levels of C3M remained unchanged with advanced fibrosis, even in decompensated states, which began 4 and 5 weeks after BDL when the animals developed ascites (Fig. 3B). Statistical analysis revealed the two curves (Pro‐C3 and C3M) to diverge significantly (Fig. 3C). Because liver fibrosis is an accumulation of ECM, we normalized Pro‐C3 and C3M levels for hydroxyproline content (Fig. 3D). This revealed that levels of C3M per each microgram of hydroxyproline decrease with increasing severity of liver fibrosis. In contrast, Pro‐C3 levels per each microgram of hydroxyproline remained stable with increasing severity of liver fibrosis (Fig. 3D). Importantly, the curves of Pro‐C3 and C3M again diverged significantly, suggesting a progressive net collagen type III deposition due to increased formation and decreased degradation with advanced and decompensated liver fibrosis (Fig. 3E).
CORRELATION OF FIBROSIS, MMP2 ACTIVITY, AND COLLAGEN TYPES I, III, AND IV DEGRADATION
As expected, with increasing stages of fibrosis, we could show increasing ECM deposition in sirius red staining with a strong correlation with α‐SMA‐positive staining as a marker of hepatic stellate cell activation. Overall, MMP2 increased with progression of fibrosis. More importantly, MMP2 activity increased. However, there was a negative correlation of C1M, C3M, and C4M (normalized for hydroxyproline content of the liver) with sirius red staining, suggesting decreased collagen degradation in advanced fibrosis (Supporting Fig. S2B). Of note, C3M showed the strongest correlation with sirius red staining, suggesting that degradation of collagen type III, in particular, is impacted in advanced liver fibrosis (Supporting Fig. S2B). Interestingly, C1M only weakly correlated with C3M and C4M, while C3M and C4M strongly correlated with each other and with MMP2 activity.
COLLAGEN TYPE III DEPOSITION RATIO (Pro‐C3/C3M) IS INDEPENDENT OF THE TIPS PROCEDURE
Because the absolute values of Pro‐C3 and C3M failed to estimate collagen type III deposition in the liver, we calculated the ratio of collagen type III formation and degradation markers Pro‐C3 and C3M (Pro‐C3/C3M), assuming that this ratio reflects net production and therefore deposition of collagen type III. Importantly, we observed a higher net deposition ratio in the hepatic vein than in the portal vein before and after TIPS (data not shown). There was no significant change in the deposition ratio during the observed time after TIPS (≤8, 9‐11, and >11 days; data not shown), which suggests that while TIPS placement itself does not impact deposition ratio, in some patients an increased deposition ratio after TIPS can be found. This would also suggest that changes in the rate of liver perfusion after TIPS have a mild effect on serum levels of collagen type III markers.
COLLAGEN TYPE III DEPOSITION RATIO (Pro‐C3/C3M) IS INCREASED IN PATIENTS WITH HRS AND SEVERE ASCITES
We observed a significantly higher deposition ratio in patients with HRS (Fig. 4A). Accordingly, we also found a significantly higher deposition ratio in patients with severe ascites compared to patients with or without mild ascites (Fig. 4B).
Figure 4.

Pro‐C3/C3M ratio and disease severity in TIPS patients. (A) Significantly higher Pro‐C3/C3M ratio in patients with HRS. (B) Significantly higher Pro‐C3/C3M ratio in patients with severe ascites. (C) Patients with MELD score >11 and Child‐Pugh C show a significantly higher Pro‐C3/C3M ratio. (D) Significantly higher Pro‐C3/C3M ratio in patients with CLIF‐C AD score >49.5, and patients with Pro‐C3‐/C3M ratio >1.5 show significantly higher CLIF‐C AD scores. (E) Significant correlation of Pro‐C3/C3M ratio with ACLF. (F) Kaplan‐Meier stratified for Pro‐C3/C3M ratio at a threshold of 1.5 in the hepatic vein. *P < 0.05, **P < 0.01, ***P < 0.001; data represent mean ± SEM.
COLLAGEN TYPE III DEPOSITION RATIO (Pro‐C3/C3M) INCREASED WITH ACUTE DECOMPENSATION AND MORTALITY
After stratifying the deposition ratio for prognostic scores, we found a significantly higher ratio for patients with higher MELD and Child‐Pugh scores (Fig. 4C). Indeed, patients with higher chronic liver failure‐C (CLIF‐C) AD score, reflecting a high risk of ACLF,27 also had a higher deposition ratio (Fig. 4D). Likewise, a higher deposition ratio was significantly associated with a higher CLIF‐C AD score (Fig. 4D) and with survival as shown in a Kaplan‐Meier plot using a ratio cutoff of 1.5 (fiftieth percentile) (Fig. 4F). However, multivariate analysis did not reveal a collagen type III deposition ratio as an independent predictor for survival.
Discussion
This study demonstrates for the first time that acute decompensation boosts collagen type III net deposition in the liver and that it possibly induces aggravation of liver disease leading to ACLF with increased mortality. Continuous liver injury leads to ECM remodeling in response to tissue injury and to development of progressive fibrosis in affected patients.28, 29, 30, 31 Development of ascites and acute decompensation are decisive events in patients with liver cirrhosis. From that moment onward, patients are at a much higher risk of developing further decompensation and face a higher mortality. To date, little is known about the hepatic pathophysiologic processes after the first decompensation. One could speculate that fibrosis is already present and that the main pathophysiologic processes are systemic inflammation and extrahepatic complications. However, our study in animal models of biliary cirrhosis clearly shows that the development of ascites boosts collagen type III deposition in the liver. This was not only associated with increased formation, similar to the general ECM proteins, but also associated with decreased degradation of this specific collagen type. This was also confirmed in our TIPS patients.
Besides the animal data directly comparing the soluble and indirect but noninvasive markers of liver fibrosis, we have previously shown that patients with compensated chronic liver disease show increased collagen type III turnover, i.e., formation and degradation.4, 26 Interestingly, not only the formation marker Pro‐C3 but also the degradation marker C3M is correlated with increased liver fibrosis in human immunodeficiency virus /hepatitis C virus coinfected patients with early liver disease as well as in another cohort with compensated advanced liver disease.4, 10 The present work confirms these findings in the animal model, demonstrating the robustness of these markers. However, patients receiving TIPS have already experienced AD. While Pro‐C3 correlates with the progression of disease and decompensating events, such as HRS, the degradation marker C3M levels decreased with a higher risk of ACLF development as indicated by the CLIF‐C AD score.30 This underlines what the animal model data had suggested, namely that degradation of collagen type III is attenuated in patients with acute decompensation.
Interestingly, the increase of Pro‐C3 serum levels after TIPS and higher levels of Pro‐C3 in more advanced fibrosis were consistent. This was also the case in patients who experienced decompensation. At first glance, this seems to be contradictory. Our study reveals that the increase in Pro‐C3 in all patients after TIPS insertion might be due to an increase in collagen type III synthesis associated with deterioration of liver function with changes in liver perfusion after TIPS. This has been shown several times by us and other studies.4, 22, 32, 33 However, the cutoff for survival benefit is much lower than the cutoff for the discrimination of developing acute decompensation in patients receiving TIPS. Moreover, acute decompensation is more strongly associated with the ratio between formation and degradation of collagen type III than just with Pro‐C3 levels.
We could also clearly demonstrate a correlation between net collagen type III deposition with advancing chronic liver disease and acute decompensation in rodent animal studies. Because we initially showed a significant increase in Pro‐C3 levels without significant changes in C3M levels, whether the net collagen deposition results predominantly from increased collagen type III formation or decreased degradation remains to be studied in future projects. However, when normalized for hepatic hydroxyproline content, which represents the entire ECM deposition and therefore general fibrosis, we found a significant decrease in C3M levels.
Our data possibly explain why in about half of the patients in the CLIF Acute‐on‐Chronic Liver Failure in Cirrhosis (CANONIC) study, no precipitating event was determined for development of ACLF. One hypothesis is that acute decompensation itself leads to enhanced and uncontrolled collagen type III deposition, deterioration of liver injury, and thereby induction of ACLF (Fig. 4F). One pathophysiologic explanation, suggested by this study, is that a massive deposition of ECM in the liver diminishes the last reserve of liver function, resulting in organ failure and ACLF with a poor outcome. Further studies with quantification of collagen type III deposition are warranted to confirm this hypothesis.
Both collagen type III turnover markers (Pro‐C3 and C3M) have been found in the urine of patients with kidney fibrosis.34 Nevertheless, the exact route of elimination remains unknown. Of note, the net collagen type III deposition in our cohort was especially pronounced with significant renal dysfunction, a hallmark of ACLF development in Western countries as well as the CANONIC cohort. One might expect accumulation of these soluble fragments in renal dysfunction (as they are cleared through the kidney),34 but in fact C3M levels were lower in patients with severe renal dysfunction, as determined by very low sodium urinary excretion. Based on these considerations, interpretation of our experimental and human data suggests that AD boosts collagen type III deposition through decreased degradation, possibly representing one of the mechanisms by which the first acute decompensation deteriorates outcome.
Although this study has a well‐characterized patient cohort backed by rodent experimental data, it has several limitations. While all samples were obtained prospectively, data collection and measurements were carried out retrospectively. However, we could rule out change over time of these markers in previous experiments. Also, the patients received a bare metal stent to create TIPS, which is no longer the current standard of care. Yet, the latter might be an advantage because at the time this routine included an invasive control of TIPS 1‐3 weeks later, thus enabling the collection of follow‐up samples. Also, high pro‐C3 levels possibly reflect a higher degree of fibrosis rather than an independent factor as our multivariate analysis did not reveal Pro‐C3 to be an independent predictor for survival. However, we observed increasing formation markers whereas degradation markers did not increase in the same way. This suggests collagen deposition in acute decompensation. The investigation of the exact mechanism as to why collagen type III degradation seems to be strongly associated with acute decompensation compared to collagen type I and IV degradation goes beyond the scope of this study. This hypothesis and any related mechanisms require further confirmation in the future. Finally, the patients did not receive liver biopsies in order to assess the deposition of collagen type III.
In summary, this study shows for the first time that markers for collagen type III formation increase while markers of degradation decrease with acute decompensation and development of ACLF, reflecting mortality in decompensated cirrhosis. Moreover, our study might offer a novel precipitating event in the development of ACLF, namely the boost of deposition of ECM in the liver due to acute decompensation and systemic inflammation, which eventually diminishes the last reserve of liver function, resulting in organ failure and ACLF with a poor outcome.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1135/full.
Supporting Information 1
Acknowledgment
We thank Gudrun Hack, Lise Larsen, and Silke Bellinghausen for their excellent technical assistance.
Potential conflict of interest: Dr. Bendtsen advises Ferring and received grants from Norgine. The other authors have nothing to report.
Supported by grants from the Deutsche Forschungsgemeinschaft (SFB TRR57) to J. T., European Union's Horizon 2020 Research and Innovation Programme (No. 668031) to J. T. and A. K., and the Cellex Foundation to J. T.
REFERENCES
- 1. Chirapongsathorn S, Talwalkar JA, Kamath PS. Readmission in cirrhosis: a growing problem. Curr Treat Options Gastroenterol 2016;14:236‐246. [DOI] [PubMed] [Google Scholar]
- 2. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008;134:1655‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jansen C, Leeming DJ, Mandorfer M, Byrjalsen I, Schierwagen R, Schwabl P, et al. PRO‐C3‐levels in patients with HIV/HCV‐Co‐infection reflect fibrosis stage and degree of portal hypertension. PloS One 2014;9:e108544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leeming DJ, Anadol E, Schierwagen R, Karsdal MA, Byrjalsen I, Nielsen MJ, et al. Combined antiretroviral therapy attenuates hepatic extracellular matrix remodeling in HIV patients assessed by novel protein fingerprint markers. AIDS 2014. 10;28:2081‐2090. [DOI] [PubMed] [Google Scholar]
- 6. Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest 2007;117:539‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karsdal MA, Nielsen MJ, Sand JM, Henriksen K, Genovese F, Bay‐Jensen A‐C, et al. Extracellular matrix remodeling: the common denominator in connective tissue diseases. Possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev Technol 2013;11:70‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leeming DJ, Nielsen MJ, Dai Y, Veidal SS, Vassiliadis E, Zhang C, et al. Enzyme‐linked immunosorbent serum assay specific for the 7S domain of collagen type IV (P4NP 7S): a marker related to the extracellular matrix remodeling during liver fibrogenesis. Hepatol Res 2012;42:482‐493. [DOI] [PubMed] [Google Scholar]
- 9. Veidal SS, Karsdal MA, Nawrocki A, Larsen MR, Dai Y, Zheng Q, et al. Assessment of proteolytic degradation of the basement membrane: a fragment of type IV collagen as a biochemical marker for liver fibrosis. Fibrogenesis Tissue Repair 2011;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leeming DJ, Karsdal MA, Byrjalsen I, Bendtsen F, Trebicka J, Nielsen MJ, et al. Novel serological neo‐epitope markers of extracellular matrix proteins for the detection of portal hypertension. Aliment Pharmacol Ther 2013;38:1086‐1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schierwagen R, Leeming DJ, Klein S, Granzow M, Nielsen MJ, Sauerbruch T, et al. Serum markers of the extracellular matrix remodeling reflect antifibrotic therapy in bile‐duct ligated rats. Front Physiol 2013;4:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karsdal MA, Henriksen K, Nielsen MJ, Byrjalsen I, Leeming DJ, Gardner S, et al. Fibrogenesis assessed by serological type III collagen formation identifies patients with progressive liver fibrosis and responders to a potential antifibrotic therapy. Am J Physiol Gastrointest Liver Physiol 2016;311:G1009‐G1017. [DOI] [PubMed] [Google Scholar]
- 13. Nielsen MJ, Veidal SS, Karsdal MA, Ørsnes‐Leeming DJ, Vainer B, Gardner SD, et al. Plasma Pro‐C3 (N‐terminal type III collagen propeptide) predicts fibrosis progression in patients with chronic hepatitis C. Liver Int 2015;35:429‐437. [DOI] [PubMed] [Google Scholar]
- 14. Trebicka J. Emergency TIPS in a Child‐Pugh B patient: when does the window of opportunity open and close? J Hepatol 2017;66:442‐450. [DOI] [PubMed] [Google Scholar]
- 15. Salerno F, Cammà C, Enea M, Rössle M, Wong F. Transjugular intrahepatic portosystemic shunt for refractory ascites: a meta‐analysis of individual patient data. Gastroenterology 2007;133:825‐834. [DOI] [PubMed] [Google Scholar]
- 16. García‐Pagán JC, Caca K, Bureau C, Laleman W, Appenrodt B, Luca A, et al.; Early TIPS (Transjugular Intrahepatic Portosystemic Shunt) Cooperative Study Group . Early use of TIPS in patients with cirrhosis and variceal bleeding. N Engl J Med 2010;362:2370‐2379. [DOI] [PubMed] [Google Scholar]
- 17. Bureau C, Thabut D, Oberti F, Dharancy S, Carbonell N, Bouvier A, et al. Transjugular intrahepatic portosystemic shunts with covered stents increase transplant‐free survival of patients with cirrhosis and recurrent ascites. Gastroenterology 2017;152:157‐163. [DOI] [PubMed] [Google Scholar]
- 18. Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al.; CANONIC Study Investigators of the EASL–CLIF Consortium . Acute‐on‐chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 2013;144:1426‐1437, 1437.e1‐9. [DOI] [PubMed] [Google Scholar]
- 19. Trebicka J. Predisposing factors in acute‐on‐chronic liver failure. Semin Liver Dis 2016;36:167‐173. [DOI] [PubMed] [Google Scholar]
- 20. Brensing KA, Hörsch M, Textor J, Schiedermaier P, Raab P, Schepke M, et al. Hemodynamic effects of propranolol and nitrates in cirrhotics with transjugular intrahepatic portosystemic stent‐shunt. Scand J Gastroenterol 2002;37:1070‐1076. [DOI] [PubMed] [Google Scholar]
- 21. Brensing KA, Textor J, Perz J, Schiedermaier P, Raab P, Strunk H, et al. Long term outcome after transjugular intrahepatic portosystemic stent‐shunt in non‐transplant cirrhotics with hepatorenal syndrome: a phase II study. Gut 2000;47:288‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trebicka J, Krag A, Gansweid S, Appenrodt B, Schiedermaier P, Sauerbruch T, et al. Endotoxin and tumor necrosis factor‐receptor levels in portal and hepatic vein of patients with alcoholic liver cirrhosis receiving elective transjugular intrahepatic portosystemic shunt. Eur J Gastroenterol Hepatol 2011;23:1218‐1225. [DOI] [PubMed] [Google Scholar]
- 23. Trebicka J, Hennenberg M, Odenthal M, Shir K, Klein S, Granzow M, et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J Hepatol 2010;53:702‐712. [DOI] [PubMed] [Google Scholar]
- 24. Trebicka J, Hennenberg M, Laleman W, Shelest N, Biecker E, Schepke M, et al. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho‐kinase and activation of endothelial nitric oxide synthase. Hepatology 2007;46:242‐253. [DOI] [PubMed] [Google Scholar]
- 25. Klein S, Klösel J, Schierwagen R, Körner C, Granzow M, Huss S, et al. Atorvastatin inhibits proliferation and apoptosis, but induces senescence in hepatic myofibroblasts and thereby attenuates hepatic fibrosis in rats. Lab Invest 2012;92:1440‐1450. [DOI] [PubMed] [Google Scholar]
- 26. Nielsen MJ, Nedergaard AF, Sun S, Veidal SS, Larsen L, Zheng Q, et al. The neo‐epitope specific PRO‐C3 ELISA measures true formation of type III collagen associated with liver and muscle parameters. Am J Transl Res 2013;5:303‐315. [PMC free article] [PubMed] [Google Scholar]
- 27. Jalan R, Pavesi M, Saliba F, Amorós A, Fernandez J, Holland‐Fischer P, et al.; CANONIC Study Investigators ; EASL‐CLIF Consortium . The CLIF Consortium Acute Decompensation score (CLIF‐C ADs) for prognosis of hospitalised cirrhotic patients without acute‐on‐chronic liver failure. J Hepatol 2015;62:831‐840. [DOI] [PubMed] [Google Scholar]
- 28. Liedtke C, Luedde T, Sauerbruch T, Scholten D, Streetz K, Tacke F, et al. Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair 2013;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsochatzis EA, Senzolo M, Germani G, Gatt A, Burroughs AK. Systematic review: portal vein thrombosis in cirrhosis. Aliment Pharmacol Ther 2010;31:366‐374. [DOI] [PubMed] [Google Scholar]
- 30. Mehal W, To U. New approaches for fibrosis regression in alcoholic cirrhosis. Hepatol Int 2016;10:773‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buck M, Garcia‐Tsao G, Groszmann RJ, Stalling C, Grace ND, Burroughs AK, et al. Novel inflammatory biomarkers of portal pressure in compensated cirrhosis patients. Hepatology 2014;59:1052‐1059. [DOI] [PubMed] [Google Scholar]
- 32. Berres M‐L, Asmacher S, Lehmann J, Jansen C, Görtzen J, Klein S, et al. CXCL9 is a prognostic marker in patients with liver cirrhosis receiving transjugular intrahepatic portosystemic shunt. J Hepatol 2015;62:332‐339. [DOI] [PubMed] [Google Scholar]
- 33. Berres M‐L, Lehmann J, Jansen C, Görtzen J, Meyer C, Thomas D, et al. Chemokine (C‐X‐C motif) ligand 11 levels predict survival in cirrhotic patients with transjugular intrahepatic portosystemic shunt. Liver Int 2016;36:386‐394. [DOI] [PubMed] [Google Scholar]
- 34. Genovese F, Boor P, Papasotiriou M, Leeming DJ, Karsdal MA, Floege J. Turnover of type III collagen reflects disease severity and is associated with progression and microinflammation in patients with IgA nephropathy. Nephrol Dial Transplant 2016;31:472‐479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1135/full.
Supporting Information 1