

Abstract
In heart failure (HF), dysregulated cardiac ryanodine receptors (RyR2) contribute to the generation of diastolic Ca2+ waves (DCWs), thereby predisposing adrenergically stressed failing hearts to life-threatening arrhythmias. However, the specific cellular, subcellular, and molecular defects that account for cardiac arrhythmia in HF remain to be elucidated. Patch-clamp techniques and confocal Ca2+ imaging were applied to study spatially defined Ca2+ handling in ventricular myocytes isolated from normal (control) and failing canine hearts. Based on their activation time upon electrical stimulation, Ca2+ release sites were categorized as coupled, located in close proximity to the sarcolemmal Ca2+ channels, and uncoupled, the Ca2+ channel-free non-junctional Ca2+ release units. In control myocytes, stimulation of β-adrenergic receptors with isoproterenol (Iso) resulted in a preferential increase in Ca2+ spark rate at uncoupled sites. This site-specific effect of Iso was eliminated by the phosphatase inhibitor okadaic acid, which caused similar facilitation of Ca2+ sparks at coupled and uncoupled sites. Iso-challenged HF myocytes exhibited increased predisposition to DCWs compared to control myocytes. In addition, the overall frequency of Ca2+ sparks was increased in HF cells due to preferential stimulation of coupled sites. Furthermore, coupled sites exhibited accelerated recovery from functional refractoriness in HF myocytes compared to control myocytes. Spatially resolved subcellular Ca2+ mapping revealed that DCWs predominantly originated from coupled sites. Inhibition of CaMK∏ suppressed DCWs and prevented preferential stimulation of coupled sites in Iso-challenged HF myocytes. These results suggest that CaMK∏-(and phosphatase)-dependent dysregulation of junctional Ca2+ release sites contributes to Ca2+-dependent arrhythmogenesis in HF.
Keywords: Ryanodine receptor, Ca2+ waves, Arrhythmia, Microdomains, Refractoriness, Ventricular myocyte
Introduction
In ventricular myocytes, most of the ryanodine receptors (RyR2s), sarcoplasmic reticulum (SR) Ca2+ release channels, are organized in clusters located in immediate proximity to sarcolemmal Ca2+ channels. These junctional RyR2 clusters comprise coupled Ca2+ release sites that initiate Ca2+-induced Ca2+ release during the systolic action potential (AP) [21, 40, 43]. Structural and functional data indicate that there is a fraction of extra-junctional RyR2s that do not co-localize (couple) with the sarcolemmal Ca2+ channels [4, 14, 19, 40, 44, 46]. The non-junctional RyR2 clusters form (uncoupled/non-coupled) Ca2+ release sites and appear to play a secondary role by amplifying primary Ca2+ release. These populations of distinct Ca2+ release sites are exposed to different local ionic and signaling environments, including the Na+/Ca2+ “fuzzy space”, domain-specific CaMK∏ and reactive oxygen species signaling, and potential gradients in [Ca2+]SR [1, 13, 18, 23, 47, 50]. As a consequence, the intrinsic properties and physiological modulation of coupled and uncoupled sites might be different. Indeed, recent studies have suggested that coupled sites are selectively influenced by CaMK∏ localized in the dyadic cleft [19, 20, 33].
Heart failure (HF) is a leading cause of death that occurs either as a result of pump failure or malignant arrhythmia [29, 49]. Although various mechanisms can underlie disrupted Ca2+ homeostasis in HF [39, 51, 52], dysfunction of RyR2s has consistently been reported in both animal models of HF and in human HF [8, 52], It was shown that in HF, RyR2s become abnormally active, or ‘leaky’, owing to an accumulation of posttranslational modifications due to phosphorylation and oxidation [8, 10, 11, 45, 48, 52]. This dysregulated RyR2 activity has the potential to decrease systolic contraction while giving rise to diastolic Ca2+ waves (DCWs) and delayed after-depolarizations, precursors of triggered arrhythmias. Mechanistically, DCWs in HF myocytes were linked to accelerated recovery from a state of functional quiescence, i.e., refractoriness, that RyR2s normally enter following each systolic Ca2+ release [10, 17, 35]. Furthermore, shortened refractoriness in HF was attributed to abnormal CaMK∏ phosphorylation and oxidation of RyR2 [8, 10]. However, presently, very little is known about the contribution of anatomically and functionally distinct release sites in myocyte Ca2+ handling and the role of subdomain-specific Ca2+ signaling in arrhythmogenesis.
The objective of the present study was to define the subcellular determinants of arrhythmogenic DCWs by quantifying functional differences in Ca2+ signaling from anatomically distinct sites (coupled vs. non-coupled) and their relative roles in the genesis of DCWs. Using a canine model of tachypacing-induced chronic HF, we show that excessive activation of coupled SR Ca2+ release sites underlies increased susceptibility of failing myocytes to arrhythmogenic DCWs.
Materials and methods
An expanded Methods section can be found in the Electronic Supplementary Material.
Canine model of heart failure
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). All animal procedures were approved by Institutional Animal Care and Use Committee of the Ohio State University. Ventricular dysfunction was induced by right ventricular tachypacing, as described previously [10, 30]. Briefly, adult hound dogs (17–31 kg) of either sex were chronically instrumented with modified pacemakers (St. Jude Medical, MN) with the pacing lead (St. Jude Medical, MN) placed in the right ventricle apex. Tachypacing was performed at: 180 bpm for 2 weeks, 200 bpm for 6 weeks, and 180 bpm thereafter. This tachypacing protocol invariably induced HF evidenced by elevated concentration of brain natriuretic peptide, LV dysfunction, and functional impairment [10, 30] (electronic supplementary material, Table 1). Cellular studies were performed following 16 weeks of tachypacing unless otherwise stated.
Table 1.
Properties of end-diastolic Ca2+ sparks recorded in control and HF myocytes in the presence of 100 nM Iso
| Parameter | All sparks
|
Coupled
|
Non-coupled
|
|||
|---|---|---|---|---|---|---|
| Control | HF | Control | HF | Control | HF | |
| Frequency (100 µm−1 s−1) | 7.7 ± 1.2 | 11.9 ± 1.6* | 5.6 ± 1.1 | 9.7 ± 1.8* | 9.2 ± 1.2& | 13.2 ± 1.7 |
| Peak (ΔF/F0) | 1.2 ± 0.1 | 1.3 ± 0.1 | 1.2 ± 0.1 | 1.1 ± 0.2 | 1.2 ± 0.1 | 1.4 ± 0.1& |
| FWHM (µm) | 3.3 ± 0.1 | 3.5 ± 0.1 | 3.4 ± 0.1 | 3.2 ± 0.2 | 3.2 ± 0.1 | 3.8 ± 0.2*& |
| FDHM (ms) | 23 ± 1 | 23 ± 1 | 24 ± 1 | 20 ± 1 | 23 ± 1 | 25 ± 1& |
| TTP (ms) | 20 ± 1 | 19 ± 1 | 20 ± 1 | 20 ± 3 | 22 ± 2 | 19 ± 1 |
| Max Rate (ΔF/F0/ms) | 0.08 ± 0.01 | 0.08 ± 0.01 | 0.08 ± 0.01 | 0.06 ± 0.01 | 0.09 ± 0.01 | 0.09 ± 0.01 |
Data presented as mean ± sem. Data were collected from 18 control and 10 HF myocytes, respectively
Peak Ca2+ spark amplitude, FWHM half-width of Ca2+ spark, FDHM duration of Ca2+ spark at ½ of amplitude, TTP time to peak of Ca2+ spark
p < 0.05 vs. control,
p < 0.05 vs. coupled
Ventricular myocyte isolation
The dogs were anesthetized with pentobarbital sodium (50 mg/kg intravenously; Nembutal, Abbott Laboratories, IL), and the heart was rapidly removed and perfused with ice-cold cardioplegic solution containing the following (mM): NaCl 110, CaCl2 1.2, KCl 16, MgCl2 16, and NaHCO3 10. Left circumflex artery was cannulated and used to perfuse both the left atria and left ventricle. Right atria and right ventricle were removed from the preparation. The heart was perfused for 10 min with a perfusion buffer containing (mM) NaCl 130, KCl 5.4, MgCl2 3.5, NaH2PO4 0.5, Glucose 10, HEPES 5, and taurine 20 supplemented with 0.1 mM EGTA; this was followed by heart perfusion with the perfusion buffer containing 0.3 mM Ca2+, 0.12 mg/ml of soybean trypsin inhibitor (Thermo Fisher Scientific, MA), and 1.33 mg/ml of type ∏ collagenase (lot number 44C14804B, activity 265 U/mg, Worthington Biochemical Corp, NJ) for 30 min. Following enzymatic digestion, mid-myocardial section of the left ventricle was dissected from the heart and placed in shaking water bath at 37° C for additional 10 min. Single myocytes were obtained by filtering through nylon mesh followed 2–3 cycles of gravity sedimentation (1× g for 10 min). Cells were resuspended in the low Ca2+ (0.4 mM CaCl2) perfusion buffer supplemented with 1% bovine serum albumin and plated on 12 mm diameter glass coverslips (CS-12R, Warner Instruments, CT) coated with mouse laminin. Myocytes were stored either at room temperature or at +4° C.
Ca2+ imaging
Cellular experiments were performed using an RC-25 open bath imaging chamber (Warner Instruments, CT) mounted on PM-6 magnetic platform (Warner Instruments, CT). The chamber was continuously perfused with an external solution containing (in mM): 140 NaCl, 5.4 KCl, 2.0 CaCl2, 0.5 MgCl2, 5.6 glucose, and 10 HEPES (pH 7.4). In intact myocytes, Ca2+ transients were elicited by field stimulation through the pair of platinum electrodes using 4 ms square voltage pulses generated by SD9 stimulator (Grass Technologies/Astro-Med Inc, RI). Ca2+ dyes were loaded by incubating myocytes in a low Ca2+ (0.4 mM CaCl2) external solution containing either 9 µM Fluo-3 AM (Molecular Probes/Thermo Fisher Scientific, MA) or 8 μM Rhod-4 AM (AAT Bioquest, CA) at room temperature for 20–30 min; following dye washout, 20–40 min was allowed for de-esterification of AM esters. In patch-clamped myocytes, Ca2+ transients were induced by a series (10–25) of voltage stimuli shaped as a typical control AP. Whole-cell patch-clamp configuration was established using an Axopatch 200B amplifier coupled to Digi-datal322A data acquisition system (Axon Instruments Inc./Molecular Devices, CA). Patch pipettes were filled with the following solution (mM): 90 K-aspartate, 50 KCl, 5 MgATP, 5 NaCl, 1 MgCl2, 0.1 Tris GTP, 10 HEPES, and 0.1 Rhod-2 tri-potassium salt (Molecular Probes/Thermo Fisher Scientific, MA); pH 7.2. To inhibit CaMK∏ activity, myocytes were treated with the low Ca2+ external solution containing 1 μM KN-93 at room temperature for 30–50 min. Such treatment did not significantly affected Ca2+ current density recorded in control myocytes (Figure S5, electronic supplementary material). Intracellular Ca2+ imaging was performed using Olympus FluoView FV 1000 (Olympus America Inc., PA) and Nikon AIR (Nikon Instruments Inc., NY) confocal microscope systems, each equipped with 60x oil-immersion objective lens (NA 1.4). Rhod dyes were excited with 561 nm laser and fluorescence was collected at 570–620 nm wavelengths. Fluo-3 was excited with 488 nm line of argon laser and signal was collected at 500–600 nm wavelengths. In the line-scanning mode, images were acquired along the central axis of the myocytes at a speed of ~ 2 ms per line with pixel size ranging from 0.08 to 0.41 µm. In resonant scanning mode, images were recorded at the myocyte midsection at ~110 frames per second with pixel size of 0.27–0.41 µm. Fluorescence signals were normalized to the baseline cellular fluorescence (F0).
Data analysis
To classify image pixels into coupled/non-coupled Ca2+ release groups, median time to 30% of amplitude of spatially averaged Ca2+ transients (T30) was determined and pixels with the response time less than T30 were assigned into early response (coupled) group (e.g. Figure 1, blue rectangles); image regions with response time longer than T30 were categorized into the delayed-response (uncoupled) group (Fig. 1, red rectangles). If line-scan image region categorized into coupled or uncoupled group had widths of less than half of the sarcomere length (<0.8 µm), it was removed from the corresponding group and marked with the gray rectangles, as illustrated in Fig. 1. Image pixels with a response time equal to T30 were considered as “intermediate” and also marked with the gray rectangles (Fig. 1).
Fig. 1.

Site-specific regulation of Ca2+ release by β-adrenergic receptor (β-AR) stimulation. a and b: a Representative traces of ‘typical’ control action potentials (APs) used as a voltage command and line-scan images with corresponding profiles of Ca2+ transients recorded in control myocyte under baseline (a, n = 14) and in the presence of 100 nM isoproterenol (Iso, b, n = 18), a β-AR agonist. b Line-scan images corresponding to the part of the image marked with yellow box in panel a illustrate work-flow of data analysis. Time to 30% of the spatially averaged amplitude of Ca2+ transient was detected (white line) and pixels with the response time less than median time to 30% of the amplitude (T30, red dashed line) were assigned into early (coupled) group (blue rectangles). Image regions reaching 30% of the amplitude with the response time more than T30 were categorized into delayed (uncoupled) group (red rectangles). The remaining pixels marked with gray rectangles (see “Materials and methods” for detailed description). This pixel classification was used to group end-diastolic Ca2+ sparks, c Average frequency of Ca2+ sparks observed in coupled and uncoupled regions
Diastolic Ca2+ sparks were detected using the variance stabilization and local baseline subtraction approach described by Bankhead et al. [6]. Briefly, the square root transform was applied to the images for variance stabilization followed by a sequence of image smoothing performed using cubic B-spline filter with 2(n−1)—1 zeros inserted between filter coefficients at each smoothing iteration (n is a number of smoothing iterations). Baseline (Ca2+ transient) signal was approximated using five smoothing iterations; two smoothing iterations were used for the Ca2+ spark images. Following baseline signal subtraction, the Ca2+ spark images were thresholded using threshold of five standard deviations. End-diastolic Ca2+ spark properties were analyzed using Ca2+ sparks detected during last 600 ms of diastolic interval (cycle length 2000 ms).
A similar approach was used to detect Ca2+ waves: six and two smoothing iterations were used for baseline and Ca2+ wave images, respectively. The Ca2+ wave images were thresholded using a 3.8 standard deviation threshold and reconstructed to include image pixels with signal >mean plus two standard deviations. K-mean clustering of the areas of detected objects was used to separate the Ca2+ waves from the Ca2+ sparks.
Images were analyzed using MATLAB (2014b, The MathWorks, Inc., MA). An original code for Ca2+ sparks detection was obtained from Bankhead et al. [6]. Aggregated data were analyzed using R software environment (R Foundation for Statistical Computing, http://www.R-project.org). Results are expressed as the mean ± SEM. Statistical significance between two groups was defined by Student’s t test p values of <0.05.
All materials were obtained from Sigma-Aldrich (St. Lois, MI, USA) unless specified otherwise.
Results
Differential regulation of coupled and uncoupled Ca2+ release sites by β-adrenergic receptor stimulation in control myocytes
Based on activation time following electrical stimulation, Ca2+ release sites in cardiac myocytes can be categorized as early and delayed, corresponding to L-type Ca2+ channel-coupled and uncoupled sites, respectively [19, 20, 41] (see Materials and Methods, and Figure S1, electronic supplementary material). We examined the properties of diastolic Ca2+ sparks originated at coupled and uncoupled sites in AP-stimulated cardiac myocytes (at 0.5 Hz) under baseline conditions and in the presence of the β-adrenergic receptor (β-AR) agonist, isoproterenol (Iso). As shown in Fig. 1a, the frequency of Ca2+ sparks originating from coupled sites was not different from the frequency of sparks originated from uncoupled sites in the absence of Iso. Treatment of myocytes with Iso (100 nM) resulted in a disproportional increase in the frequency of uncoupled site compared to coupled sites (Fig. 1b). Of note, uncoupled site spark frequency also had a markedly broadened Iso dose-dependency compared to that of coupled sites (Fig. 2). For example, Iso at 10 nM produced no further stimulation of coupled site sparks but increased uncoupled site frequency by approximately threefold (Fig. 2b). At the same time, coupled and uncoupled site sparks exhibited similar amplitudes and spatio-temporal properties (Table 1).
Fig. 2.

Sensitivity of Ca2+ sparks and myocyte SR Ca2+ content to Iso. The dose-dependent effects of Iso on the frequency of Ca2+ sparks recorded throughout the myocyte (a, all sparks) and recorded at coupled and uncoupled Ca2+ release sites (b). c, Iso effects on the SR Ca2+ content. Each data point represents data collected from 4 to 18 control myocytes
SR Ca2+ content was measured in AP-stimulated myocytes at different Iso concentrations by rapid application of 15 mM caffeine (Fig. 2c). Notably, the SR Ca2+ content reached maximum at 3 nM Iso and did not further increase at higher Iso concentrations. Taken together, these data suggest that the preferential stimulation of uncoupled sites by Iso is attributable to site-specific modulation of RyR2s, independent of SR Ca2+ content.
To assess the role of protein phosphatases in site-specific regulation of Ca2+ release by β-AR stimulation, we examined the effect of Iso in the presence of the protein phosphatase inhibitor okadaic acid (OA, 1.2 µM). OA increased the frequency of sparks compared to the effects of Iso alone (Fig. 3). Interestingly, OA exerted more pronounced effects on coupled Ca2+ release sites (108 vs. 52% increase in the activity uncoupled sites), indicating that the activity of coupled Ca2+ release sites is tightly regulated by protein phosphatases in control myocytes.
Fig. 3.

Phosphatase inhibitor eliminates site-specific effects of Iso. a: a representative traces of ‘typical’ control APs used as a voltage command and line-scan images with corresponding profiles of Ca2+ transients recorded in control myocyte in the presence of 100 nM Iso plus 1.2 µM okadaic acid (OA), a protein phosphatase inhibitor, a: b Line-scan images corresponding to the part marked with yellow box of the image shown in panel a; the images illustrate the onset of Ca2+ transient, classification of the line-scan image regions into early and delayed, and end-diastolic Ca2+ activity. b Average frequency of Ca2+ sparks recorded throughout the myocytes, at the coupled and uncoupled regions in control myocytes in the presence of 100 nM Iso alone (data presented in Figs. 1 and 2) and in the presence of Iso plus OA (n = 11)
Increased Ca2+ spark activity at coupled Ca2+ release sites in HF myocytes
HF is associated with increased frequency of Ca2+ sparks and Ca2+ waves that become particularly pronounced and promote arrhythmogenesis during β-AR stimulation of failing myocytes [8, 32]. [8, 32]. Consistent with the previous reports [7, 10, 45], Ca2+ spark frequency was significantly increased in HF myocytes compared to control at baseline (i.e., the absence of Iso; Figure S4 electronic supplementary material). To probe the subcellular mechanism of arrhythmogenesis in adrenergically stressed failing hearts, we examined the contributions of coupled (early) vs. uncoupled (delayed) sites to HF-de-pendent alterations in Ca2+ handling in Iso-challenged HF myocytes with the same protocol that we used for control myocytes above. In agreement with the previous reports [10, 26], the propensity for Ca2+ waves and the overall Ca2+ spark frequency were significantly higher in HF myocytes than in control cells (Fig. 4a). This increase in overall Ca2+ spark frequency in HF myocytes was attributable to an increased frequency of coupled sites (Fig. 4b). Whereas the frequency of uncoupled sites was not different between HF and control myocytes, coupled site spark frequency increased ~ twofold to a level similar to that for uncoupled sites in both HF and control cells. These differences in frequency were associated with no significant differences in most of the spatio-temporal properties of Ca2+ sparks (Table 1). Of note, in the absence of Iso end-diastolic Ca2+ spark frequencies under AP-clamp pacing were not different between control and HF myocytes (data not shown).
Fig. 4.
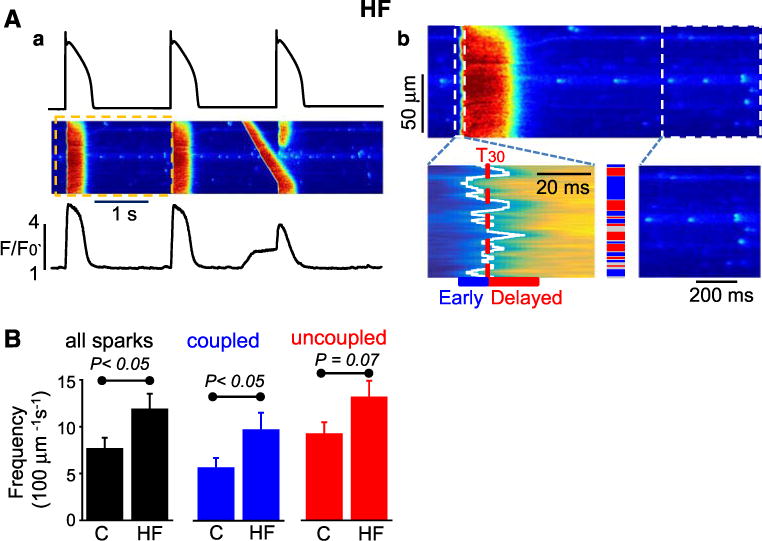
HF preferentially increases activity of the coupled Ca2+ release sites. a: a, Representative traces of ‘typical’ control APs used as a voltage command and line-scan images with corresponding profiles of Ca2+ transients recorded in HF myocyte in the presence of 100 nM Iso. a: b, Line-scan images corresponding to the part, marked with yellow box, of the image shown in panel a; the images illustrate the onset of Ca2+ transient, classification of the line-scan image regions into early and delayed, and end-diastolic Ca2+ activity. b Average frequency of Ca2+ sparks recorded throughout the myocytes, and at the coupled and uncoupled regions in control (c, n = 18) and heart failure (HF, n = 10) myocytes
Accelerated recovery from refractoriness of Ca2+ release in HF myocytes
We previously demonstrated that shortened RyR2 refractoriness facilitates synchronization of aberrant diastolic Ca2+ release and promotes arrhythmogenesis in failing cardiomyocytes [8, 10, 15]. To evaluate the contributions of coupled and uncoupled sites to this aberration, we measured site-specific restitution of SR Ca2+ release using a standard two-pulse protocol. Consistent with our previous findings [8], restitution of globally measured SR Ca2+ release (dF/dt max) occurred significantly faster in HF myocytes than in control cells (Fig. 5). Functional recovery of coupled sites occurred manifestly faster in HF myocytes than in control cells (Fig. 5b). At uncoupled sites, Ca2+ release showed no signs of refractoriness even at the shortest time intervals examined in HF or control cells and no significant differences in the functional recovery were revealed at these sites between the groups. Thus, accelerated recovery of global SR Ca2+ release in HF myocytes could be attributed predominantly to shortened refractoriness of coupled Ca2+ release sites. Considered with the results on site-specific alterations in Ca2+ spark frequency (Fig. 5), these findings suggest that HF is associated with preferential facilitation of coupled sites as compared to uncoupled sites.
Fig. 5.

HF speeds up recovery of coupled Ca2+ release sites. a Restitution of Ca2+ transient was measured in control and HF myocytes using two-pulse protocol. Upper panels show line-scan images of Rhod-2 fluorescence during Ca2+ release activation in response to the first depolarizing pulse (control pulse) and in response to the second pulse that occurred with 0.5, 0.8, and 1.2 s delay, respectively. Ca2+ release sites were classified as early (blue rectangles) and delayed (red rectangles) as described in Fig. 1. Black traces show restitution of spatially averaged Ca2+ transients, while blue and red traces illustrate restitution of Ca2+ release at coupled and uncoupled sites. b Time-course of Ca2+ release recovery recorded in control and HF myocytes was analyzed for all, coupled and uncoupled Ca2+ release sites and expressed as time-dependent restitution of gain function. Gain function for all and coupled release sites was calculated as the ratio of maximum rate of changes of fluorescence (maximum Ca2+ release flux) to the peak density of Ca2+ current; the gain for the uncoupled sites was calculated as the ratio of maximum Ca2+ release fluxes recorded at uncoupled and coupled sites, respectively. Data were obtained from 4 control and 5 HF myocytes, respectively. *p < 0.05
Preferential regulation of coupled Ca2+ release sites by CaMKΠ
Enhanced functional activity of Ca2+ release in HF settings, including this model of canine HF, has previously been attributed to excessive phosphorylation of RyR2 by CaMK∏ [2, 10, 31, 38]. To assess the role of CaMK∏ in site-specific regulation of Ca2+ activity, we utilized the CaMK∏ inhibitor KN-93 (1 µM). Consistent with our previous results [10], this treatment significantly reduced the propensity to DCWs in field-stimulated HF myocytes in the presence of Iso (Fig. 6a, b). Moreover, inhibition of CaMK∏ also resulted, on average, in a twofold decrease in the frequency of Ca2+ spark recorded in HF myocytes (Fig. 6c). As illustrated in Fig. 6d, inhibition of CaMK∏ in HF myocytes preferentially suppressed Ca2+ sparks at the coupled release sites. In agreement with the results of field stimulation experiments described above, KN-93 also preferentially suppressed the activity of coupled Ca2+ release sites in HF myocytes stimulated by a standard AP waveform under AP-clamp (Figure S6 electronic supplementary material). Further support for the critical role of CaMK∏ in site-specific regulation of Ca2+ release was obtained in experiments utilizing the selective auto-camtide-3 derived inhibitory peptide (AC3) [3]. In these experiments, the addition of AC3 to the pipette solution significantly suppressed the frequency of DCWs and preferentially inhibited the activity of coupled Ca2+ release sites in AP-clamped HF myocytes (Figure S7 electronic supplementary material).
Fig. 6.

CaMK∏ inhibition preferentially suppresses activity of coupled Ca2+ release sites in HF myocytes. a Line-scan images of HF myocyte periodically stimulated at 0.5 Hz in the presence of 1 µM Iso alone, and in the presence of 1 µM Iso plus 1 µM KN-93, a CaMK∏ inhibitor. Upper trace illustrates timing of field stimulation. b Average data for the effect of KN-93 on frequency of diastolic Ca2+ waves per diastolic interval (DI), and on the fraction of cells with the Ca2+ waves. Data were obtained from 19 (Iso 1 µM alone) and 20 (Iso + KN93) HF myocytes. c Line-scan images of HF myocyte periodically stimulated at 0.5 Hz in the presence of 10 nM Iso alone, and in the presence of 10 nM Iso plus 1 µM KN-93. Left panels illustrate classification of Ca2+ release sites into early (coupled, blue rectangles) and delayed (uncoupled, red rectangles). d Summary of the effects of KN-93 on frequency of Ca2+ spark recorded from all, coupled and uncoupled Ca release sites. Data were obtained from 20 (Iso 10 nM alone) and 17 (Iso + KN93) HF myocytes
If increased coupled site activity in HF cells is, indeed, due to preferential modulation by CaMK∏, then it could be possible to mimic the HF phenotype by local stimulation of CaMK∏ at coupled sites in control cells. Therefore, we used the L-type Ca channel agonist Bay K8644 to stimulate CaMK∏ at coupled sites [22]. As demonstrated in Figure S8 (electronic supplementary material), BayK8644 applied in the presence of Iso caused a significant increase in Ca2+ influx associated with a noticeable reduction in Ca2+ current inactivation in AP-clamped control myocytes. At the same time, BayK8644 significantly increased the frequency of DCWs in paced control myocytes (Figure S9 A–C electronic supplementary material). Analysis of diastolic Ca2+ sparks showed that Bay K8644 predominantly increased the activity of coupled Ca2+ release sites (Figure S9 D electronic supplementary material). Taken together, these data support the idea that CaMK∏ mediates its pro-arrhythmic effects in HF myocytes by selectively affecting RyR2 properties at the early Ca2+ release sites.
L-type Ca2+ channels as potential targets for CaMK∏
L-type Ca2+ channels are known as potential targets for CaMK∏ phosphorylation which, in general, has been shown to increase channel activity [12]. Increased Ca2+ entry into junctional domains could stimulate coupled Ca2+ release sites, thus contributing to the observed CaMK∏-dependent alterations in Ca2+ release in HF myocytes. To assess whether this potential mechanism contributed to the remodeling of intracellular Ca2+ signaling in HF, we examined the functional characteristics and phosphorylation status of the L-type Ca2+ channels in HF vs. control myocytes. As summarized in Table S2 (electronic supplementary material), peak Ca2+ current in HF myocyte was either decreased (at the baseline) or unchanged (in the presence of 100 nM Iso) when compared to the currents in control myocytes. In addition, inactivation of the Ca2+ currents was not significantly different between control and HF myocytes. These functional effects are not consistent with increased CaMK∏-dependent phosphorylation of the L-type Ca2+ channels in HF myocytes. We also performed Western blot analysis of phosphorylation statuses of Cav1.2 α1C and β2 subunits extracted from the membrane fractions of control and HF samples. Phosphorylation levels of these subunits of L-type Ca2+ channels were not significantly different in control and HF (Figure S10, electronic supplementary material). Thus, our data do not support a critical role for CaMK∏-dependent phosphorylation of the L-type Ca2+ channels in the generation of arrhythmogenic Ca2+ waves in HF myocytes.
The role of coupled sites in initiation of DCWs in HF myocytes
To directly examine the role of coupled and uncoupled sites in the generation of arrhythmogenic Ca2+ waves in HF myocytes, we mapped Ca2+ wave initiation using fast 2D confocal Ca2+ imaging. First, we categorized Ca2+ release sites as coupled and uncoupled according to their activation time on AP stimulation (Fig. 7a). Then, we visualized initiation and spread of DCWs. Having assigned recorded Ca2+ wave initiation sites into ‘in focus’ and ‘out-of-focus’ groups (see electronic supplementary materials, Methods and Fig. 2), we overlaid ‘in focus’ DCW initiation sites with the ‘onset map’ (Fig. 7b, f). As illustrated in Fig. 7c DCW initiation sites for the Ca2+ wave shown in panel 7B coincided with the early Ca2+ release sites (onset time of 20–40 ms). On average, 45% (10 out of 22) of recorded initiation sites of Ca2+ waves were assigned to out-of focus group. Analysis of the ‘in focus’ initiation sites showed that the majority of pixels comprising the initial area Ca2+ wave corresponded to coupled Ca2+ release sites(Fig. 7d). These data suggest that coupled Ca2+ release sites underlie the initiation of DCWs in HF myocytes.
Fig. 7.

Ca2+ waves in HF myocytes originate preferentially from coupled Ca2+ release sites. a: a-c, XY images of HF myocyte during onset of Ca2+ transient in response to electrical stimulation in the presence of 100 nM Iso. a: d Time-course of changes in spatially averaged Rhod-4 fluorescence. a: e Activation map of systolic Ca2+ release illustrates time to 30% of the amplitude of spatially averaged Ca2+ transient expressed for each pixel. Nuclear area was excluded from the analysis. b: a–c XY images of HF myocyte exemplify time-dependent evolution of a diastolic Ca2+ wave. b: d Myocyte map shows initiation and propagation of the Ca2+ wave. Asterisk indicates site of Ca2+ wave initiation. b: e Image illustrates initial stages of Ca2+ wave propagation. White area indicates initiation site of the Ca2+ wave. The image corresponds to the white square (15 × 15 µm) shown in panel b: d. Scale bars in a and b correspond to 10 µm. b: f Juxtaposition of the initial site of the Ca2+ wave with the Ca2+ release activation map (white square in panel a: e). c Distribution of cell image pixels during onset of Ca2+ transient (for the cell shown in a and b). d Summary data for localization of the initiation sites of Ca2+ waves calculated for 9 HF myocytes
Discussion
In the present study, we investigated the role of subcellular compartmentalization of Ca2+ signaling in catecholamine-dependent arrhythmogenesis in failing canine cardiac myocytes. In particular, we used a well-characterized canine model of tachypacing-induced HF to determine how chronic HF affects diastolic activities of sarcolemmal Ca2+ channel-coupled and -uncoupled Ca2+ release sites in adrenergically challenged cardiomyocytes prone to arrhythmogenic DCWs. Our main findings are as follows: (1) upon β-AR stimulation of control cells with Iso uncoupled sites exhibited higher diastolic activity (measured as frequency of topographically mapped Ca2+ sparks) than coupled sites, and this site-specific modulation was lost in HF cells that presented indiscriminate facilitation of both coupled and uncoupled sites. (2) The shortened overall Ca2+ release refractoriness underlying the increased arrhythmogenic potential of HF cells was specifically attributable to accelerated functional restitution of coupled sites; (3) initiation of DCWs in HF cells was mapped preferentially to coupled sites; and (4) the site-specific effects of Iso in control and HF myocytes depended on protein phosphatase and CaMK∏ activities. These findings provide new insights into subcellular and molecular mechanisms of cardiac arrhythmias in the failing heart.
Coupled and uncoupled RyR2 dusters
In cardiac myocytes, Ca2+ handling shows substantial spatial heterogeneity [37,42]. RyR2s are typically grouped in release units composed on average of 14–63 channels [5, 25, 40]. While most of SR Ca2+ release units face sarcolemmal Ca2+ channels in the juxtaposed t-tubule membrane, thus forming couplons [21, 40, 43], a certain fraction of release units is not associated with the Ca2+ channels [4, 14, 24, 27, 28, 44, 46]. Coupled and uncoupled RyR2s can functionally be distinguished by different response times (i.e., fast and delayed, respectively) during AP activation [19, 20, 24, 27, 28]. They can be further quantified by correlating Ca2+ release rise times with distances to the nearest t-tubule membrane stained with a membrane dye [19, 24]. Consistent with previous reports [16, 19, 24], we found a significant fraction of uncoupled sites in canine myocytes examined in this study, thus revealing a substantial contribution of non-junctional sites to the generation of the Ca2+ transient. Notably, in HF myocytes, this fraction was not significantly altered (Figure S2, electronic supplementary material).
The role of release site heterogeneity in the generation of arrhythmogenic Ca2+ release in HF
HF is associated with increased diastolic activity of RyR2s (i.e., “leak”) secondary to their increased phosphorylation and oxidation [8, 11]. Using the same canine model of HF that was employed here, we previously demonstrated that changes in RyR2 function translate into impaired Ca2+ signaling refractoriness that accounts for increased predisposition to arrhythmogenic DCWs in paced HF myocytes [10]. Normally, refractoriness prevents CICR from spontaneous activation during the diastolic period. When refractoriness is impaired (shortened), Ca2+ sparks that arise synchronously throughout the cell upon reloading of the SR give rise to DCWs. Here, we specifically examined the role of coupled and uncoupled RyR2s in generation of DCWs. We found that aberrant Ca2+ release arises preferentially from fast, i.e., coupled, Ca2+ release sites. This conclusion is based on the following evidence: first, the increase in overall release site activity (sparks) in HF cells was entirely attributable to increased spark frequency at fast sites (Fig. 4); two-pulse restitution experiments demonstrated that the shortened overall release refractoriness associated with increased arrhythmogenic potential of HF cells can be ascribed to shortened refractoriness of coupled sites (Fig. 5); and finally, mapping of subcellular Ca2+ directly demonstrated the predominant role of fast sites in the genesis of DCWs (Fig. 7). Thus, these experiments show for the first time that arrhythmogenic diastolic Ca2+ release in failing myocytes arises from RyR2s localized in couplons adjacent to sarcolemmal Ca2+ channels. In line with these results, Dries et al. [20] recently showed that in normal pig myocytes challenged by Iso, Ca2+ waves tend to arise at coupled sites.
Given the similar functional activities of coupled and uncoupled sites in HF myocytes in this study (Fig. 4), it is not obvious why DCWs arise preferentially at coupled sites. The observed site specificity of Ca2+ wave generation could be ascribed to particulars of subcellular architecture as well as the distribution of Ca2+ buffers and Ca2+ transport systems including SERCA at the coupled vs. uncoupled sites. For example, the localization of coupled sites in close proximity to the T-tubule membrane, but farther away from sites of re-uptake, could facilitate lateral spread of Ca2+ and the cross activation of neighboring sites required for the transition of localized release to a propagating Ca2+ wave. Further studies including modeling with accurate representation of the spatial organization of Ca2+ release are needed to delineate the factors accounting for the specific role of coupled site in DCW generation.
Mechanism of site-specific modulation of RyR2s in normal and HF myocytes
β-AR stimulation is a critical factor in the initiation of Ca2+-dependent arrhythmias. In general, it is thought that β-AR stimulation causes arrhythmia by increasing the SR Ca2+ load (via PKA-dependent phosphorylation of phospholamban) and facilitating RyR2 phosphorylation by PKA and CaMK∏. However, the specific subcellular and molecular mechanisms remain to be clarified. A key finding of this study is that while in control myocytes, Iso resulted in preferential stimulation of uncoupled sites, in HF cells, Iso increased spark frequency at both coupled and uncoupled sites. Importantly, inhibiting protein phosphatase activity altered the preferential Iso-dependent stimulation of uncoupled sites in control myocytes leading to an indiscriminate facilitation of coupled and uncoupled sites (Fig. 3). Thus, the site-specific modulation of RyR2 observed in normal (but not HF) cells could be attributed to elevated protein phosphatase activity at junctional sites. This site-specific modulation is likely to involve direct effects on RyR2 through changing its phosphorylation status rather than indirect effects via phospholamban/SERCA-dependent changes in SR cisternae Ca2+ content (Fig. 2c). This control mechanism is evidently lost in HF myocytes that exhibit Iso-dependent facilitation of activities of both coupled and uncoupled sites. Consistent with this notion, binding of protein phosphatases (PP1 and PP2A) to RyR2 is reportedly decreased in failing hearts [2, 9, 36]. In further support of the role of altered compartmentalization of RyR2 phosphorylation mechanisms in HF, the β-AR-mediated increase in coupled site activity in HF myocytes was sensitive to inhibition of CaMK∏ (Fig. 6). Previously, we and others showed that Ca2+-de-pendent arrhythmogenesis was associated with increased CaMK∏ phosphorylation of RyR2s [2, 10, 31, 38]. Our present results show for the first time that arrhythmogenesis in HF myocytes is specifically attributable to CaMK∏-dependent facilitation of coupled sites.
Besides phosphorylation of RyR2, the observed site-specific effects of CaMK∏ on SR Ca2+ release in HF myocytes could involve phosphorylation of the L-type Ca2+ channels [12]. However, we found no functional or biochemical evidence for enhanced CaMK∏-dependent phosphorylation of Ca2+ channels in HF (Table S2 and Figure S10 electronic supplementary material). Therefore, this pathway is unlikely to play a significant role in mediating local pro-arrhythmic remodeling of Ca2+ signaling in the HF model employed in the current study.
As another interesting possibility, we recently showed that local Na+/Ca2+ signaling involving neuronal Na+ channel plays an important role in the genesis of Ca2+-dependent arrhythmia [34]. Stimulation of neuronal Na+ channels by CaMK∏ results in increased Na+ influx and hence increased Ca2+ level in the junctional space. Thus, CaMK∏-dependent activation of neuronal Na+ channel could account for increased arrhythmogenic potential of coupled sites in the settings of leaky RyR2s in HF. Further studies are required to define the specific molecular factors involved in differential regulation of coupled and uncoupled sites in normal and diseased hearts.
Comparison with previous results
Previously, Dries et al. [19] showed selective modulation (activation) of coupled sites in porcine myocytes, which was dependent on CaMK∏ and was lost following infarction. While being in apparent agreement about the role of coupled sites in arrhythmogenesis, our results and those of Dries et al. seem to have some important differences. In our study, (β-AR stimulation selectively increased the activity of uncoupled as opposed to coupled sites in control myocytes; HF led to increased activity of coupled sites. Thus, it appears that in our study, the normal regulation of coupled sites is negative rather than positive (as in Dries et al.), such that loss of this inhibitory influence in HF results in increased activity of coupled sites and enhanced arrhythmogenesis. The specific reasons behind these divergent results are not clear. They could involve species differences as well as differences in experimental conditions, including differences in pacing rate. Consistent with the latter possibility, in porcine myocytes, spark rate at coupled vs. uncoupled sites was not different at slow pacing rates [19]. Further experiments are needed to explain these differences and define the specific mechanisms of compartmentalized regulation of RyR2s in normal and diseased hearts.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (Grants HL074045 and HL063043 to S. Györke, HL089836 to C. A. Carnes, and HL121796 to D.Terentyev) and the Russian Science Foundation (N15-15-20008 to S. Györke).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00395-017-0633-2) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Ethical approval All animal procedures were approved and conducted in accordance with the guidelines issued by Institutional Animal Care and Use Committee of the Ohio State University, in compliance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. The manuscript does not contain human data.
References
- 1.Acsai K, Antoons G, Livshitz L, Rudy Y, Sipido KR. Microdomain [Ca2+] near ryanodine receptors as reported by L-type Ca2+ and Na +/Ca2+ exchange currents. J Physiol. 2011;589:2569–2583. doi: 10.1113/jphysiol.2010.202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 3.Anderson ME, Braun AP, Wu Y, Lu T, Wu Y, Schulman H, Sung RJ. Kn-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early after depolarizations in rabbit heart. J Pharmacol Exp Ther. 1998;287:996–1006. [PubMed] [Google Scholar]
- 4.Asghari P, Scriven DRL, Hoskins J, Fameli N, van Breemen C, Moore EDW. The structure and functioning of the couplon in the mammalian cardiomyocyte. Protoplasma. 2012;249(Suppl 1):S31–S38. doi: 10.1007/s00709-011-0347-5. [DOI] [PubMed] [Google Scholar]
- 5.Baddeley D, Jayasinghe ID, Lam L, Rossberger S, Cannell MB, Soeller C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc Natl Acad Sci USA. 2009;106:22275–22280. doi: 10.1073/pnas.0908971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bankhead P, Scholfield CN, Curtis TM, McGeown JG. Detecting Ca2+ sparks on stationary and varying baselines. Am J Physiol Cell Physiol. 2011;301:C717–C728. doi: 10.1152/ajpcell.00032.2011. [DOI] [PubMed] [Google Scholar]
- 7.Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Györke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. 2007;93:4083–4092. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belevych AE, Radwański PB, Carnes CA, Györke S. “Ryanopathy”: causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc Res. 2013;98:240–247. doi: 10.1093/cvr/cvt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belevych AE, Sansom SE, Terentyeva R, Ho H-T, Nishijima Y, Martin MM, Jindal HK, Rochira JA, Kunitomo Y, Abdellatif M, Carnes CA, Elton TS, Györke S, Terentyev D. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One. 2011;6:e28324. doi: 10.1371/joumal.pone.0028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, Carnes CA, Györke S. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res. 2011;90:493–502. doi: 10.1093/cvr/cvr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol. 2014;76:107–127. doi: 10.1146/annurev-physiol-020911-153308. [DOI] [PubMed] [Google Scholar]
- 12.Bers DM, Morotti S. Ca2+ current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Front Pharmacol. 2014 doi: 10.3389/fphar.2014.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bers DM, Shannon TR. Calcium movements inside the sarcoplasmic reticulum of cardiac myocytes. J Mol Cell Cardiol. 2013;58:59–66. doi: 10.1016/j.yjmcc.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biesmans L, Macquaide N, Heinzel FR, Bito V, Smith GL, Sipido KR. Subcellular heterogeneity of ryanodine receptor properties in ventricular myocytes with low T-tubule density. PLoS ONE. 2011;6:e25100. doi: 10.1371/joumal.pone.0025100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brunello L, Slabaugh JL, Radwanski PB, Ho H-T, Belevych AE, Lou Q, Chen H, Napolitano C, Lodola F, Priori SG, Fedorov VV, Volpe P, Fill M, Janssen PML, Györke S. Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc Natl Acad Sci USA. 2013;110:10312–10317. doi: 10.1073/pnas.1300052110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crossman DJ, Ruygrok PN, Ruygrok PR, Soeller C, Cannell MB. Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS ONE. 2011;6:e17901. doi: 10.1371/joumal.pone.0017901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DelPrincipe F, Egger M, Niggli E. Calcium signalling in cardiac muscle: refractoriness revealed by coherent activation. Nat Cell Biol. 1999;1:323–329. doi: 10.1038/14013. [DOI] [PubMed] [Google Scholar]
- 18.Despa S, Shui B, Bossuyt J, Lang D, Kotlikoff MI, Bers DM. Junctional cleft [Ca2+]i measurements using novel cleft-targeted Ca2+ sensors. Circ Res. 2014;115:339–347. doi: 10.1161/CIRCRESAHA.115.303582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dries E, Bito V, Lenaerts I, Antoons G, Sipido KR, Macquaide N. Selective modulation of coupled ryanodine receptors during microdomain activation of calcium/calmodulin-dependent kinase ∏ in the dyadic cleft. Circ Res. 2013;113:1242–1252. doi: 10.1161/CIRCRESAHA.113.301896. [DOI] [PubMed] [Google Scholar]
- 20.Dries E, Santiago DJ, Johnson DM, Gilbert G, Holemans P, Korte SM, Roderick HL, Sipido KR. Calcium/calmodulin-dependent kinase ∏ and nitric oxide synthase 1-dependent modulation of ryanodine receptors during β-adrenergic stimulation is restricted to the dyadic cleft. J Physiol. 2016;594:5923–5939. doi: 10.1113/JP271965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franzini-Armstrong C, Protasi F, Ramesh V. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys J. 1999;77:1528–1539. doi: 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao Z, Singh MV, Hall DD, Koval OM, Luczak ED, Joiner MA, Chen B, Wu Y, Chaudhary AK, Martins JB, Hund TJ, Mohler PJ, Song L-S, Anderson ME. Catecholamine-independent heart rate increases require CaMK∏. Circ Arrhythm Electrophysiol. 2011;4:379–387. doi: 10.1161/CIRCEP.110.961771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorelik J, Wright PT, Lyon AR, Harding SE. Spatial control of the βAR system in heart failure: the transverse tubule and beyond. Cardiovasc Res. 2013;98:216–224. doi: 10.1093/cvr/cvt005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinzel FR, Bito V, Volders PGA, Antoons G, Mubagwa K, Sipido KR. Spatial and temporal inhomogeneities during Ca2+ release from the sarcoplasmic reticulum in pig ventricular myocytes. Circ Res. 2002;91:1023–1030. doi: 10.1161/01.RES.0000045940.67060.DD. [DOI] [PubMed] [Google Scholar]
- 25.Hou Y, Jayasinghe I, Crossman DJ, Baddeley D, Soeller C. Nanoscale analysis of ryanodine receptor clusters in dyadic couplings of rat cardiac myocytes. J Mol Cell Cardiol. 2015;80:45–55. doi: 10.1016/j.yjmcc.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 26.Kubalova Z, Terentyev D, Viatchenko-Karpinski S, Nishijima Y, Gyorke I, Terentyeva R, da Cunha DN, Sridhar A, Feldman DS, Hamlin RL, Carnes CA, Gyorke S. Abnormal intrastore calcium signaling in chronic heart failure. Proc Natl Acad Sci USA. 2005;102:14104–14109. doi: 10.1073/pnas.0504298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, Sipido KR. Reduced synchrony of Ca2 + release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63–73. doi: 10.1016/j.cardiores.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 28.Louch WE, M0rk HK, Sexton J, Strømme TA, Laake P, Sjaastad I, Sejersted OM. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574:519–533. doi: 10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mozaffarian D, Anker SD, Anand I, Linker DT, Sullivan MD, Cleland JG, Carson PE, Maggioni AP, Mann DL, Pitt B, Poole-Wilson PA, Levy WC. Prediction of mode of death in heart failure: the Seattle Heart Failure Model. Circulation. 2007;116:392–398. doi: 10.1161/CIRCULATIONAHA.106.687103. [DOI] [PubMed] [Google Scholar]
- 30.Nishijima Y, Feldman DS, Bonagura JD, Ozkanlar Y, Jenkins PJ, Lacombe VA, Abraham WT, Hamlin RL, Carnes CA. Canine nonischemic left ventricular dysfunction: a model of chronic human cardiomyopathy. J Card Fail. 2005;11:638–644. doi: 10.1016/j.cardfail.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 31.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XHT. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase ∏ promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med. 2004;14:61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Popescu I, Galice S, Mohler PJ, Despa S. Elevated local [Ca2+] and CaMK∏ promote spontaneous Ca2+ release in ankyrin-B-deficient hearts. Cardiovasc Res. 2016;111:287–294. doi: 10.1093/cvr/cvw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Radwański PB, Ho H-T, Veeraraghavan R, Brunello L, Liu B, Belevych AE, Unudurthi SD, Makara MA, Priori SG, Volpe P, Armoundas AA, Dillmann WH, Knollmann BC, Mohler PJ, Hund TJ, Györke S. Neuronal Na+ Channels Are Integral Components of Pro-arrhythmic Na+/Ca2+ signaling nanodomain that promotes cardiac arrhythmias During β-adrenergic stimulation. JACC Basic Transl Sci. 2016;1:251–266. doi: 10.1016/j.jacbts.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramay HR, Liu OZ, Sobie EA. Recovery of cardiac calcium release is controlled by sarcoplasmic reticulum refilling and ryanodine receptor sensitivity. Cardiovasc Res. 2011;91:598–605. doi: 10.1093/cvr/cvrl43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reiken S, Gaburjakova M, Guatimosim S, Gomez AM, D’Ar-miento J, Burkhoff D, Wang J, Vassort G, Lederer WJ, Marks AR. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J Biol Chem. 2003;278:444–453. doi: 10.1074/jbc.M207028200. [DOI] [PubMed] [Google Scholar]
- 37.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 38.Sag CM, Wadsack DP, Khabbazzadeh S, Abesser M, Grefe C, Neumann K, Opiela MK, Backs J, Olson EN, Brown JH, Neef S, Maier SK, Maier LS. Calcium/calmodulin-dependent protein kinase ∏ contributes to cardiac arrhythmogenesis in heart failure. Circ Heart Fail. 2009;2:664–675. doi: 10.1161/CIRCHEARTFAILURE.109.865279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schulte JS, Fehrmann E, Tekook MA, Kranick D, Fels B, Li N, Wehrens XHT, Heinick A, Seidl MD, Schmitz W, Muller FU. Cardiac expression of the CREM repressor isoform CREM-IbΔC-X in mice leads to arrhythmogenic alterations in ventricular cardiomyocytes. Basic Res Cardiol. 2016;111:15. doi: 10.1007/s00395-016-0532-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scriven DRL, Asghari P, Moore EDW. Microarchitecture of the dyad. Cardiovasc Res. 2013;98:169–176. doi: 10.1093/cvr/cvt025. [DOI] [PubMed] [Google Scholar]
- 41.Shkryl VM, Blatter LA. Ca2+ release events in cardiac myocytes up close: insights from fast confocal imaging. PLoS ONE. 2013;8:e61525. doi: 10.1371/joumal.pone.0061525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sipido KR, Cheng H. T-tubules and ryanodine receptor microdomains: on the road to translation. Cardiovasc Res. 2013;98:159–161. doi: 10.1093/cvr/cvt077. [DOI] [PubMed] [Google Scholar]
- 43.Soeller C, Crossman D, Gilbert R, Cannell MB. Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proc Natl Acad Sci USA. 2007;104:14958–14963. doi: 10.1073/pnas.0703016104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci USA. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Terentyev D, Györke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Györke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torres NS, Sachse FB, Izu LT, Goldhaber JI, Spitzer KW, Bridge JH. A modified local control model for Ca2+ transients in cardiomyocytes: junctional flux is accompanied by release from adjacent non-junctional RyRs. J Mol Cell Cardiol. 2014;68:1–11. doi: 10.1016/j.yjmcc.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verdonck F, Mubagwa K, Sipido KR. [Na(+)] in the subsarcolemmal “fuzzy” space and modulation of [Ca2+](i) and contraction in cardiac myocytes. Cell Calcium. 2004;35:603–612. doi: 10.1016/j.ceca.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 48.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci USA. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Writing Group Members. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després J-P, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics Committee, Stroke Statistics Subcommittee Heart Disease and Stroke Statistics-2016 Update: a Report From the American Heart Association. Circulation. 2016;133:e38–e360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 50.Zhang H, Gomez AM, Wang X, Yan Y, Zheng M, Cheng H. ROS regulation of microdomain Ca2+ signalling at the dyads. Cardiovasc Res. 2013;98:248–258. doi: 10.1093/cvr/cvt050. [DOI] [PubMed] [Google Scholar]
- 51.Zhang X, Ai X, Nakayama H, Chen B, Harris DM, Tang M, Xie Y, Szeto C, Li Y, Li Y, Zhang H, Eckhart AD, Koch WJ, Molkentin JD, Chen X. Persistent increases in Ca2+ influx through Cavl.2 shortens action potential and causes Ca2+ overload-induced afterdepolarizations and arrhythmias. Basic Res Cardiol. 2016;111:4. doi: 10.1007/s00395-015-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W, Terentyev D. Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch. 2014;466:1129–1137. doi: 10.1007/s00424-014-1469-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.