

Abstract
Enzyme-linked immunosorbent assay (ELISA), flow cytometry, and Western blot are common bioanalytical techniques. Successful execution traditionally requires the use of one or more commercially available antibody-small-molecule dye, or antibody-reporter protein conjugates that recognize relatively short peptide tags (<15 amino acids). However, the size of antibodies, and their molecular complexity (by virtue of post-translational disulfide formation and glycosylation) typically requires either expression in mammalian cells or purification from immunized mammals. The preparation and purification of chemical dye- or reporter protein-antibody conjugates is often complicated and expensive, and not commonplace in academic laboratories. In response, researchers have developed comparatively simpler protein scaffolds for macromolecular recognition, which can be expressed with relative ease in E. coli and can be evolved to bind virtually any target. Nanobodies—a minimalist scaffold generated from camelid-derived heavy chain IgGs—is one such example. A multitude of nanobodies have been evolved to recognize a diverse array of targets, including a short peptide. Here, this peptide tag (termed BC2T), and BC2 nanobody-dye conjugates or reporter protein fusions are evaluated in ELISA, flow cytometry, and Western blot experiments, and compared to analogous experiments using commercially available antibody-conjugate/peptide tag pairs. Collectively, the utility and practicality of nanobody-based reagents in bioanalytical chemistry is demonstrated.
Graphical Abstract
A multitude of bioanalytical techniques and sensor platforms rely on monoclonal antibodies (principally immunoglobulins of isotype G, IgG, Figure 1a).1–5 Antibodies are large proteins (~150 kDa) that can be evolved in vitro—or generated by immunization—to recognize virtually any small-molecule or biopolymer target. Techniques such as Enzyme-linked immunosorbent assay (ELISA)6, flow cytometry5, and Western blot3 commonly rely on monoclonal antibodies that bind a small (<15 amino acid) peptide. When conjugated to a small-molecule dye or antibody-reporter protein conjugates, recognition of proteins containing the peptide ‘tag’ can occur, often in complex biological environments. Common peptide tags, for which excellent commercial antibodies and antibody-reporter conjugates exist, and include FLAG7, myelocytomatosis viral oncogene8 (myc), synthetic streptavidin binding Strep-tag9, and influenza hemmaglutinin10 (HA).
Figure 1.

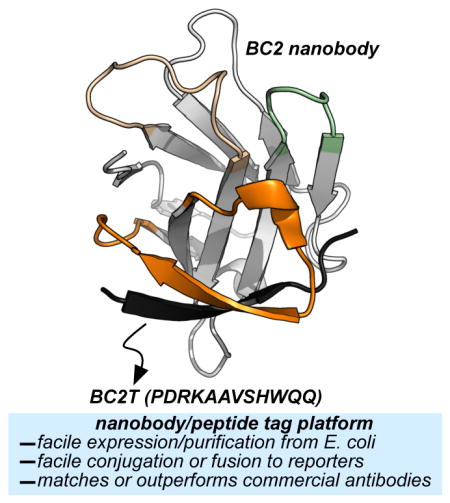
(a) Structure of IgG. Disulfide bonds are highlighted in red. Constant heavy-chain region 1 (CH1); constant light-chain (CL); variable light-chain (VL); variable heavy-chain (VH), and fragment antigen-binding (Fab) regions are highlighted with a blue background (PDB: 1IGY). (b) Architecture of a heavy-chain IgG (hcIgG), consisting of two heavy chains (CH3, CH2, VH) connected by disulfide bonds in the hinge region. The “nanobody” subunit is circled. (c) Structure of the recently reported nanobody BC2, bound to its peptide tag (BC2T, PDB: 5IVN). This complex was originally reported in Nature Scientific Reports 6, 19211 (2016).
While full-length IgG antibodies are very large (~150 kDa), target recognition is achieved within a relatively small region, termed the fragment antigen-binding (Fab) region (Figure 1a).11 Fab consists of a constant light-chain (CL) and a variable light-chain (VL) domain, linked to the constant (CH1) and the variable heavy-chain (VH) domains. When folded properly, six solvent-exposed loops from VL and VH are displayed, which participate in target recognition. Collectively, these loops are referred to as the complementary determining regions (CDRs).
Historically, antibodies have been a reagent of choice in bio-analytical techniques and sensor platforms (largely out of necessity), however their size and complexity requires isolation from mammalian cells or immunized mammals (principally goat, mouse, or rabbit).1 This relatively complicated production greatly adds to the cost of antibody-based reagents, which has negative consequences in basic research and commercial diagnostics development and application.1 Moreover, the inability of most academic labs to express and purify full-length antibodies, and chemically conjugate them to chemical dyes or reporter proteins, makes it challenging to prepare reagents ‘in house’.
In response to challenges encountered with antibody-based reagents, numerous researchers have developed non-immunoglobulin proteins, or minimalist forms of structurally simpler immunoglobulins, as scaffolds for tailored recognition.11 Many of these scaffolds mimic the structure of IgG Fab VH, but are comparatively robust and simple to express as recombinant proteins in E. coli. One such scaffold is derived from heavy-chain IgGs (hcIgGs, Figure 1b). hcIgGs are produced in camelids, and in contrast to IgGs produced in other mammals, lack a light chain. Thus, recognition is achieved through a single VH domain, as opposed to the combination of VH and VL domains in IgG. In isolation, the VH domain of hcIgG is referred to as a ‘nanobody’, a small (~15 kDa) protein that can be expressed in E. coli with relative ease, and evolved in the laboratory to recognize a diverse array of targets, through interactions involving one or more CDR loops (CDR 1–3, Figure 1c).11,12
In contrast to full-length antibodies, and their fragments, nanobodies often express well (>20 mg/L), as a folded and soluble recombinant protein, from E. coli. By virtue of their stability and ready expression, purification, and manipulation, researchers have used nanobodies in numerous applications, including therapeutic discovery13, bioimaging14, and sensors15,16. Rothbauer and coworkers recently reported a nanobody referred to as BC2 that binds a short peptide (PDRKAAVSHWQQ, referred to herein as BC2 tag, BC2T) with excellent affinity (KD ~1.4 nM) and selectivity, principally through interactions involving CDR 3 (Figure 1c).17 Here, the utility of the BC2/BC2T recognition platform is evaluated in common bioanalytical techniques (ELISA, flow cytometry, and Western blot). Throughout, outcomes from the BC2 nanobody/BC2T platform are compared to those from analogous experiments using commercially available antibody-reporter conjugates and their peptide binding partners.
ELISA typically requires (1) immobilization of a protein (‘protein a’) onto a surface (2) incubation with a binding partner (‘protein b’) equipped with a small peptide tag; (3) treatment with an antibody-reporter protein conjugate, which recognizes the peptide tag, and generates a signal following addition of a small-molecule substrate (Figure 2a). HorseRadish Peroxidase (HRP) is commonly used as a reporter protein.4 Here, a direct comparison between the BC2/BC2T platform and commercially available antibodies that bind the myc tag (EQKLISEEDL), or His6 (HHHHHH) is provided.
Figure 2.
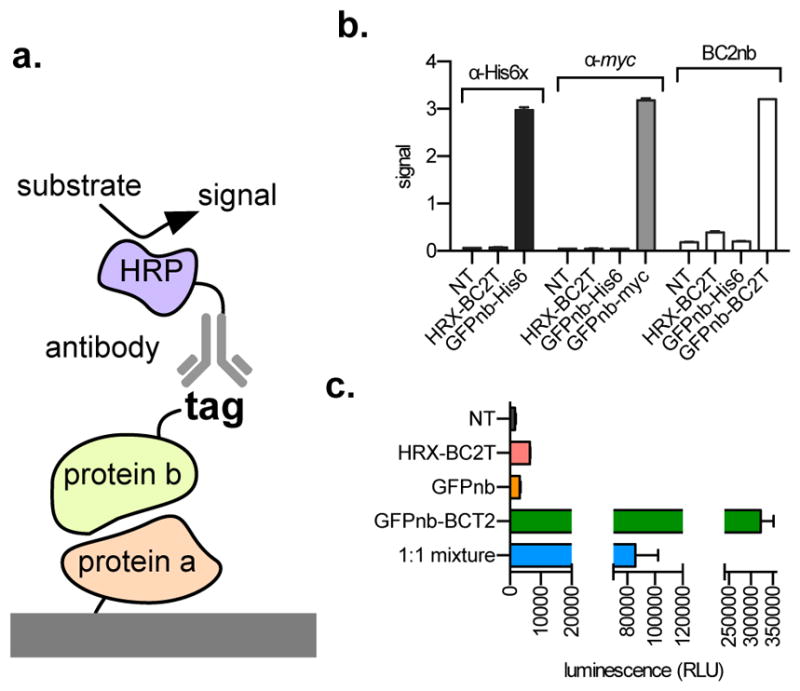
(a) Scheme of an Enzyme-Linked Immunosorbent Assay (ELISA). (b) ELISA data: immobilized GFP is treated with buffer (NT), and either HRX-BCT2, GFPnb-His6, GFPnb-myc, or GFPnb-BC2T, then either anti-His6-HRP, anti-myc-HRP, or the BC2nb-HRP conjugate, and HRP substrate. Signal is the observed absorbance at 655 nm. (c) ELISA data: GFP was immobilized onto streptavidin coated plates, then treated with buffer (NT, black), HRX-BC2T (red), GFP nanobody (GFPnb, orange), GFPnb-BC2T (green), or a 1:1 mixture of GFP nanobody and GFPnb-BC2T (blue), followed by nLuc substrate. All experiments were performed in triplicate. Error bars represent standard deviation of three experiments. α = anti; NT = no treatment. RLU = relative luminescence units.
First, Green Fluorescent Protein (GFP) was immobilized onto the surface of a multi-well plate. Following a washing step, GFP-coated wells were treated with either buffer (NT), HRX-BC2T (which has no appreciable affinity for GFP), or a GFP-binding nanobody-His6 fusion protein (GFPnb-His6), which tightly binds GFP (KD ~ 1nM14,18). After washing steps to remove unbound material, wells were incubated with a commercially available anti-His6 antibody-HRP conjugate, and HRP substrate. Unsurprisingly, no appreciable signal is observed in wells incubated with HRX-BC2T (indicating no interaction between HRX and GFP, Figure 2b, black). However, strong signal is generated in GFP immobilized wells following treatment with GFPnb-His6, and subsequent incubation with anti-His6-HRP and HRP substrate (Figure 2b, black). Similarly, when GFP immobilized wells are treated with buffer (NT), HRX-BC2T, or GFPnb-His6, no appreciable signal is observed after subsequent incubation with anti-myc-HRP and HRP substrate (Figure 2b, gray). However, signal that compares favorably to the analogous His6/anti-His6 experiment is observed in wells that contain immobilized GFP, follwing treatment with GFPnb-myc tag, and subsequent incubation with anti-myc-HRP and HRP substrate (Figure 2b, gray). Satisfyingly, signal that compares favorably to the analogous experiments described above is observed when wells containing immobilized GFP are treated with GFPnb-BC2T, and subsequent incubation with ‘in house’ prepared BC2nb-HRP conjugate and HRP substrate (Figure 2b, white, Supporting Information, Figure S-5). As expected, no appreciable signal was observed when GFP containing wells were treated with buffer (NT), HRX-BC2T, or GFPnb-His6, following subsequent treatment with BC2nb-HRP and HRP substrate (Figure 2b, white). Additionally, no appreciable signal was observed in wells lacking immobilized GFP (Supporting Information, Figure S-1)
Fundamentally, this paper aims to accentuate practical remedies afforded by the use of nanobody-based reagents in bioanalytical chemistry. It therefore went unnoticed that chemical conjugation of HRP to BC2 is likely an impediment to broad use of this reagent—including laboratories without experience in bioconjugation. A more practical solution is expression of the BC2 nanobody as a fusion to a reporter protein. Unfortunately, the BC2 nanobody-HRP fusion does not express as a soluble protein in E. coli. However, a recently reported bioluminescent ‘nanoluciferase’ protein (nLuc19)—developed by Promega—expresses as a fusion to BC2 nanobody (Supporting Information, Figure S-5). Satisfyingly, the BC2 nanobody-nLuc performed well in our ELISA analysis. First, biotinylated GFP was immobilized onto streptavidin-coated plates. Wells containing immobilized GFP were then incubated with either buffer (NT), HRX-BC2T, GFPnb, or GFPnb-BC2T. Following washing steps to remove unbound material, wells were treated with the BC2-nLuc fusion protein, washed again, then treated with the nLuc substrate (“NanoGlo™”). As expected, no appreciable signal was generated in wells containing immobilized GFP, but incubated with either HRX or GFP-binding nanobody lacking the BC2T peptide (Figure 2c, red and orange, respectively). In contrast, we observed robust signal in lanes containing immobilized GFP in complex with the GFP-binding nanobody genetically fused to the BC2T peptide (Figure 2c, green). When immobilized GFP was treated with a solution containing equal parts GFP-binding nanobody-BC2T peptide and GFP-binding nanobody (without the tag), a ~50% decrease in luminescence is observed, compared to wells treated with only the GFP-binding nanobody equipped with the BC2T peptide (Figure 2c, blue). In contrast, no appreciable signal was observed in wells that were treated identically, but lack immobilized GFP (Supporting Information, Figure S-2). Collectively, these data show that an ‘in house’ prepared BC2 nanobody-nLuc fusion protein, when paired with binding partners containing the BC2 tag, is an excellent reagent for ELISA.
The BC2 nanobody/BC2T platform was next evaluated in the context of flow cytometry, a commonly used technique to evaluate protein-protein and protein-nucleic acid interactions on the surface of yeast or bacteria, and enrichment of binders from a protein library by Fluorescence Activated Cell Sorting (FACS). In a typical flow cytometry experiment, bacteria20–22 or yeast23–25 display a peptide or protein that is flanked by a peptide tag recognized by a commercial antibody-fluorescent dye conjugate. Interaction between the tag and antibody-reporter conjugate allows researchers to quantitate display efficiency. Concomitantly, the peptide or protein displaying cells are treated with a binding target that is also fluorescently tagged.
Traditionally, yeast display efficiency has been measured using either a commercially available antibody-dye conjugate that binds to an N-terminal HA tag or a C-terminal myc tag.23–25 Bacterial display efficiency on E. coli is typically measured using a commercially antibody that binds to a C-terminal myc tag. To permit direct comparative analysis, bacteria (E. coli) were engineered to display a small (~15 kDa) well behaved protein (monomeric streptavidin, mSA2), with flanking N-terminal and C-terminal BC2T and myc tags, respectively (Figure 3a). Yeast were engineered to display a HA-mSA2-BC2T-myc fusion (Figure 3b).
Figure 3.
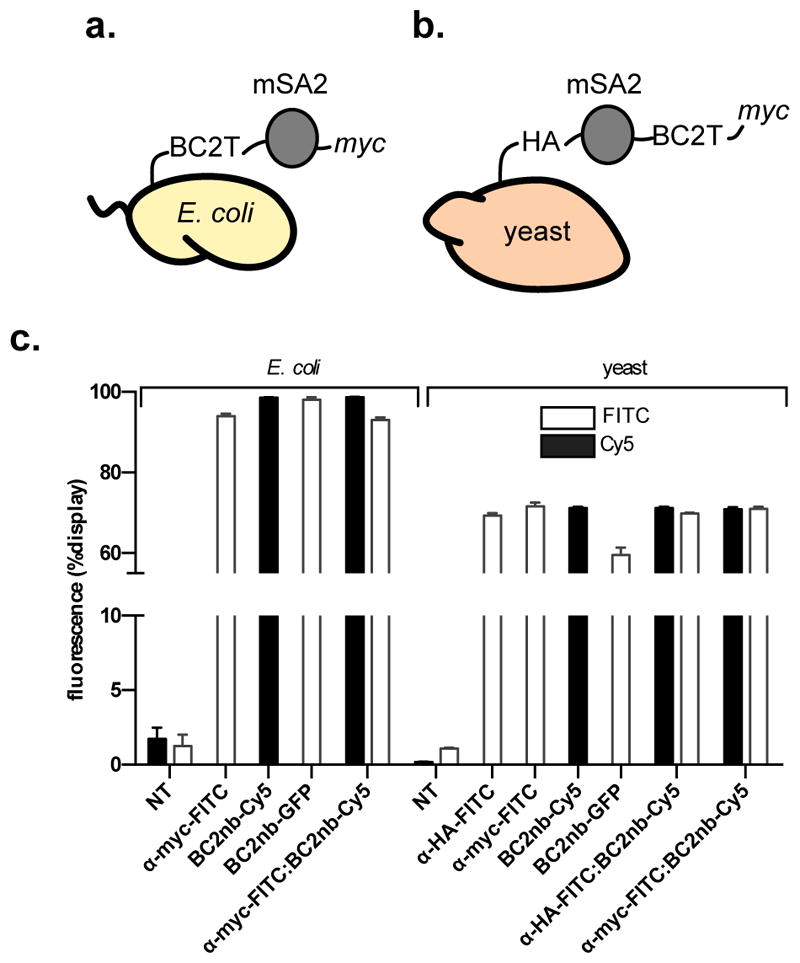
(a) Representation of E. coli engineered for flow cytometry experiments. (b) Representation of yeast engineered for flow cytometry experiments. (c) Flow cytometry detection of displayed monomeric streptavidin (mSA2) on the surface of E. coli or yeast, as determined by commercially available antibody α-myc-FITC, or nanobody reagents BC2nb-Cy5, or BC2nb-GFP (for E. coli), or commercially available antibodies α-myc-FITC, or α-HA-FITC, or nanobody reagents BC2nb-Cy5, or BC2nb-GFP (for yeast). All experiments were performed in triplicate. Error bars represent standard deviation of three experiments. α = anti; NT = no treatment.
For E. coli, cells were induced to express the displayed protein/tag fusion (as a fusion to OmpX – an E. coli cell surface protein typically used for bacterial display), then treated with either a commercially available anti-myc-FITC antibody-fluorescent dye conjugate, an ‘in house’ prepared BC2 nanobody-Cy5 conjugate (BC2nb-Cy5), or a BC2 nanobody-GFP fusion protein (BC2nb-GFP) (Supporting Information, Figure S-5). Following washing steps to remove unbound material, cells were analyzed by flow cytometry, using a laser/detection channel specific to either Cy5 or FITC (GFP). Both the BC2nb-Cy5 conjugate and BC2nb-GFP fusion compared favorably to the anti-myc-FITC antibody-fluorescent dye conjugate (~98% display efficiency for each, Figure 3c). Co-treatment with equal parts anti-myc-FITC and BC2nb-Cy5 show essentially identical fluorescence (recognition of their respective displayed peptide tag (Figure 3c)). Representative flow cytometry histograms are provided in the Supporting Information (Figure S-3, Table S-1).
For yeast, cells were induced to express the displayed protein/tag fusion at the C-terminus of Aga2 (a yeast cell surface protein typically used for yeast display), then treated with either a commercially available anti-myc-FITC, anti-HA-FITC antibody-fluorescent dye conjugate, BC2nb-Cy5 conjugate, or BC2nb-GFP fusion. Again, the nanobody reagents compared favorably to commercially available antibody reagents. Individual treatment, or co-treatment with equal parts anti-myc-FITC and BC2nb-Cy5, or anti-HA-FITC and BC2nb-Cy5 show essentially identical fluorescence (recognition of their respective displayed peptide tag, Figure 3c). Representative flow cytometry histograms are provided in the Supporting Information (Figure S-3, Table S-2).
As a final evaluative measure, the utility of the BC2 nano-body/BC2T platform was assessed in a Western blot – a commonly used technique to measure the presence of a specific protein (such as a tagged protein) in cell lysate. Execution of a Western blot typically requires: (1) denaturation of proteins from cell lysate; (2) separation of proteins based on their size via SDS-PolyAcrylamide Gel Electrophoresis (SDS-PAGE); (3) electrophoretic transfer of separated proteins to a membrane; (4) treatment of the protein-bound membrane with a primary antibody that either recognizes a specific protein, or a specific peptide tag, and; (5) treatment with a secondary antibody-dye conjugate, which serves to illuminate the primary antibody-bound protein. To function in this context, the BC2 nanobody must recognize the BC2T tag following a chemical denaturation step (and subsequent denaturation of the protein to which it is attached). For this reason, many antibodies (and nanobodies) are not suitable for Western blot analysis.
For comparison to IR dye 790-labelled commercially available secondary antibody, an IR dye 800-labelled BC2 nanobody conjugate was prepared by reaction between a C-terminal cysteine and commercially available dye-maleimide. First, 5μM of GFP lacking the BC2T peptide, or GFP-BC2T was run on a polyacrylamide gel, transferred to PVDF membrane, and treated with BC2nb-IR800 reagent. Only GFP-BC2T was detected, but not GFP lacking BC2T peptide, indicating that recognition relies entirely on the nanobody-tag recognition, in this context (Supporting Information, Figure S-4). Next, purified GFP-HA, GFP-myc, or GFP-BC2T were ran in duplicate on a polyacrylamide gel at 20 μM, 10 μM, 5 μM, and 1 μM concentrations. Following PAGE, one gel was stained by coomassie to determine protein purity. Proteins embedded in the other gel were transferred onto a PVDF membrane. Membranes containing GFP-HA or GFP-myc were first treated with commercially available anti-HA or anti-myc primary antibodies suggested for Western blot experiments. Next, these membranes were treated with a secondary antibody-Alexa Fluor 790 dye. Following washing steps, membranes were imaged on a Li-Cor Odyssey instrument. All three proteins (GFP-HA, GFP-myc, or GFP-BC2T) were found to be pure, as determined by Coomassie staining (Figure 4a–c, left gels). As expected, both anti-HA and anti-myc antibodies recognize HA or myc tagged proteins in the Western blot (Figure 4a–b, right gels). Satisfyingly, the BC2 nanobody IR800 dye conjugate recognized GFP-BC2T with excellent potency and selectivity (Figure 4c, right gels). In fact, the BC2nb/BC2T pair generated a more robust and cleaner signal, in comparison to the HA and myc platforms.
Figure 4.
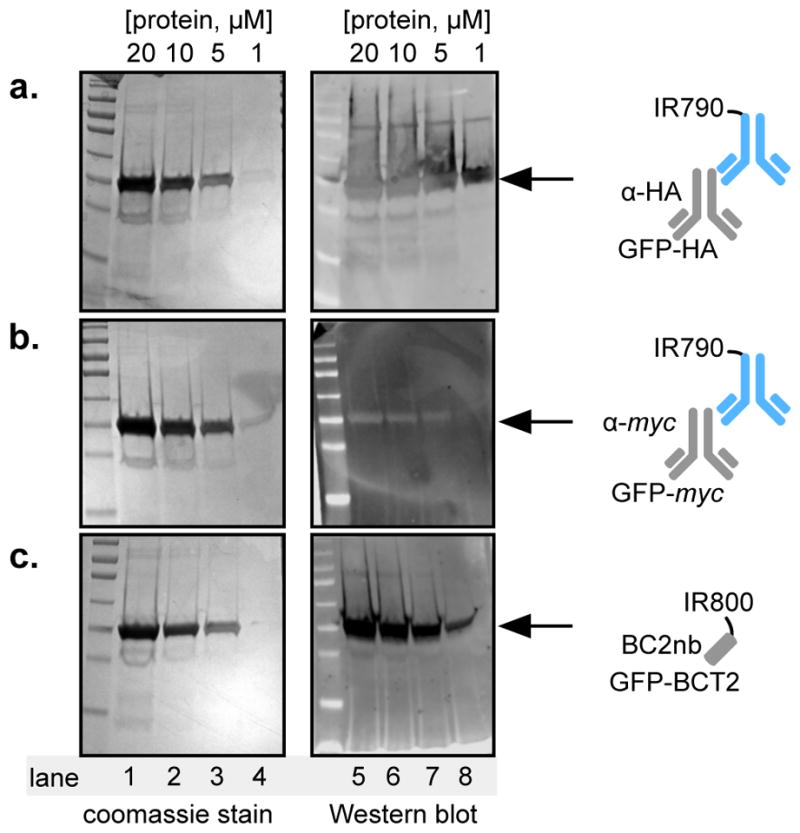
(a–c, left gel) Coomassie stained polyacrylamide gels following loading with 20, 10, 5, or 1 μM GFP-HA, GFP-myc, or GFP-BCT2, and electrophoresis. (a–c, right gel) Western blot data for the GFP-HA/anti-HA; GFP-myc/anti-myc, or; GFP-BC2T/BC2nb pairs, respectively. α = anti.
In conclusion, antibodies and their conjugates play a central role in a multitude of bioanalytical methods and sensor platforms. However, their cost and complexity add to challenges with their use. Researchers have developed minimalist protein architectures, which mimic structural features found in the antigen binding region of antibodies. One minimalist scaffold is the nanobody – a camelid-derived protein that can be evolved to bind virtually any target, including relatively short peptides. In contrast to antibodies, nanobodies express well in E. coli and can be easily manipulated – such as conjugation to a small-molecule dye or genetic fusion to a reporter protein. Collectively, these features make nanobody-based reagents an attractive alternative to antibodies and their conjugates. Using a recently reported BC2 nanobody/BC2T peptide tag pair, and ‘in house’ prepared nanobody conjugates and fusion proteins, comparative analysis to commercially available antibodies and antibody-conjugates has been conducted. In every platform tested (ELISA, flow cytometry, and Western blot), nanobody-based reagents compare favorably to, or outperform, antibody-based reagents. We hope these findings encourage the use of nanobody-based reagents in bioanalytical methods, and lead to the evolution of new nanobody/peptide tag binding pairs.
Supplementary Material
Acknowledgments
We acknowledge funding from the NIH/NIGMS (R01GM107520).
Footnotes
Supporting Information and detailed experimental procedures is available free of charge on the ACS Publications website.
References
- 1.Byrne B, Stack E, Gilmartin N, O’Kennedy R. Sensors (Basel) 2009;9:4407–4445. doi: 10.3390/s90604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holford TR, Davis F, Higson SP. Biosens Bioelectron. 2012;34:12–24. doi: 10.1016/j.bios.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 3.Liu ZQ, Mahmood T, Yang PC. N Am J Med Sci. 2014;6:160. doi: 10.4103/1947-2714.128482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bull World Health Organ. 1976;54:129–139. [PMC free article] [PubMed] [Google Scholar]
- 5.Laerum OD, Farsund T. Cytometry. 1981;2:1–13. doi: 10.1002/cyto.990020102. [DOI] [PubMed] [Google Scholar]
- 6.Carlsson HE, Hurvell B, Lindberg AA. Acta Pathol Microbiol Scand C. 1976;84:168–176. doi: 10.1111/j.1699-0463.1976.tb00016.x. [DOI] [PubMed] [Google Scholar]
- 7.Einhauer A, Jungbauer A. J Biochem Biophys Methods. 2001;49:455–465. doi: 10.1016/s0165-022x(01)00213-5. [DOI] [PubMed] [Google Scholar]
- 8.Evan GI, Lewis GK, Ramsay G, Bishop JM. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt T, Skerra A. Methods Mol Biol. 2015;1286:83–95. doi: 10.1007/978-1-4939-2447-9_8. [DOI] [PubMed] [Google Scholar]
- 10.Wilson IA, Niman HL, Houghten RA, Cherenson AR, Connolly ML, Lerner RA. Cell. 1984;37:767–778. doi: 10.1016/0092-8674(84)90412-4. [DOI] [PubMed] [Google Scholar]
- 11.Bruce VJ, Ta AN, McNaughton BR. Chembiochem. 2016;17:1892–1899. doi: 10.1002/cbic.201600303. [DOI] [PubMed] [Google Scholar]
- 12.Muyldermans S. Annu Rev Biochem. 2013;82:775–797. doi: 10.1146/annurev-biochem-063011-092449. [DOI] [PubMed] [Google Scholar]
- 13.Gray MA, Tao RN, DePorter SM, Spiegel DA, McNaughton BR. Chembiochem. 2016;17:155–158. doi: 10.1002/cbic.201500591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoonooghe S, Laoui D, Van Ginderachter JA, Devoogdt N, Lahoutte T, De Baetselier P, Raes G. Immunobiology. 2012;217:1266–1272. doi: 10.1016/j.imbio.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Buchfellner A, Yurlova L, Nuske S, Scholz AM, Bogner J, Ruf B, Zolghadr K, Drexler SE, Drexler GA, Girst S, Greubel C, Reindl J, Siebenwirth C, Romer T, Friedl AA, Rothbauer U. PLoS One. 2016;11:e0151041. doi: 10.1371/journal.pone.0151041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J, He QH, Xu Y, Fu JH, Li YP, Tu Z, Wang D, Shu M, Qiu YL, Yang HW, Liu YY. Talanta. 2016;147:523–530. doi: 10.1016/j.talanta.2015.10.027. [DOI] [PubMed] [Google Scholar]
- 17.Braun MB, Traenkle B, Koch PA, Emele F, Weiss F, Poetz O, Stehle T, Rothbauer U. Sci Rep. 2016;6:19211. doi: 10.1038/srep19211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubala MH, Kovtun O, Alexandrov K, Collins BM. Protein Sci. 2010;19:2389–2401. doi: 10.1002/pro.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. ACS Chem Biol. 2012;7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lofblom J. Biotechnol J. 2011;6:1115–1129. doi: 10.1002/biot.201100129. [DOI] [PubMed] [Google Scholar]
- 21.Daugherty PS. Curr Opin Struct Biol. 2007;17:474–480. doi: 10.1016/j.sbi.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Laplagne DA, Zylberman V, Ainciart N, Steward MW, Sciutto E, Fossati CA, Goldbaum FA. Proteins. 2004;57:820–828. doi: 10.1002/prot.20248. [DOI] [PubMed] [Google Scholar]
- 23.Boder ET, Wittrup KD. Methods Enzymol. 2000;328:430–444. doi: 10.1016/s0076-6879(00)28410-3. [DOI] [PubMed] [Google Scholar]
- 24.Kieke MC, Shusta EV, Boder ET, Teyton L, Wittrup KD, Kranz DM. Proc Natl Acad Sci U S A. 1999;96:5651–5656. doi: 10.1073/pnas.96.10.5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boder ET, Wittrup KD. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.