

Abstract
One way to preserve a rare book is to lock it away from all potential sources of damage. Of course, an inaccessible book is also of little use, and the paper and ink will continue to degrade with age in any case. Like a book, the information stored in our DNA needs to be read, but it is also subject to continuous assault. In this review, we examine how the replication stress response that is controlled by the kinase ataxia telangiectasia and Rad3-related (ATR) senses and resolves threats to DNA integrity so the DNA remains available to read in all of our cells. We discuss the multiple data that have revealed an elegant yet increasingly complex mechanism of ATR activation. These involve a core set of components that recruit ATR to stressed replication forks, stimulate kinase activity and amplify ATR signaling. We focus on the activities of ATR in control of cell cycle checkpoints, origin firing and replication fork stability, and how proper regulation of these processes is crucial to ensure faithful duplication of a challenging genome.
Introduction
During every cell cycle, human cells must accurately and efficiently replicate over six billion base pairs of DNA. Replicating a genome of this size that is packed into a very small nucleus constitutes an enormous logistical, spatial and energetic challenge. Furthermore, there are numerous impediments encountered by DNA replication forks that block their progression, including DNA damage, the transcription machinery, RNA–DNA hybrids and secondary DNA structures1. Not surprisingly, cells have robust replication stress [G] response mechanisms to ensure the entire genome is accurately replicated once every cell cycle.
The kinase ataxia telangiectasia and Rad3-related (ATR) orchestrates multiple branches of the replication stress response. It is essential for cell viability, and, emphasizing its importance, ATR-deficient embryos have shattered chromosomes2,3. Moreover, hypomorphic alleles [G] of ATR cause the developmental disorder Seckel syndrome, which is characterized by primordial dwarfism, microcephaly, craniofacial abnormalities and mental retardation4.
ATR is a phosphoinositide 3-kinase-related protein kinase5,6. It shares sequence and functional homology with two other DNA damage response kinases, ataxia telangiectasia mutated (ATM) and DNA protein kinase (DNA-PK). Both ATM and DNA-PK respond primarily to DNA double-stranded breaks (DSBs), and in contrast to ATR, neither is essential for cell survival7. This difference highlights the essential role of the replication stress response to ensure faithful duplication of a challenging genome. Although ATR has other functions, including during DSB repair8, inter-strand crosslink repair8 and meiosis9, as well as at telomeres10 and in response to mechanical and osmotic stresses11, in this Review we focus on the function of ATR in the replication stress response. We will begin with an overview of how cells detect replication stress and activate the checkpoint and later discuss the mechanisms by which the ATR signaling pathway helps replication forks overcome replication stress.
MECHANISMS OF ATR ACTIVATION
ATR responds to many types of genotoxic stress [G], including stress induced by ultraviolet radiation, DNA polymerase inhibitors, dNTP depletion, topoisomerase [G] poisons, base alkylating agents, and DNA crosslinkers12. A common theme linking these stressors is that they stall or slow DNA polymerases. Numerous factors work together to form the basic components to recruit and activate ATR at stressed replication forks.
Components of ATR pathway activation
Various types of damage and stress activate ATR, and many of these have been traced to a common DNA structure formed at the replication fork that ATR can recognize. This structure consists at least partly of single-stranded DNA (ssDNA), which is also the trigger for the SOS DNA damage response [G] in bacteria13. Like in bacteria, where a ssDNA binding protein (RecA) triggers the SOS response, the canonical ATR pathway is triggered by binding of the ssDNA binding protein complex replication protein A (RPA) to ssDNA. RPA–ssDNA interactions serve as a platform for the recruitment of many proteins including the ATR-interacting protein (ATRIP)14, which facilitates recruitment of ATR to stressed replication forks15 (Figure 1a).
Figure 1. Components of ATR activation pathways.

(a, Left) DNA polymerase stalling on the lagging strand generates a single-stranded DNA (ssDNA) gap that is bound by replication protein A (RPA), providing a platform for ataxia telangiectasia and Rad3-related (ATR) activation. The 5′-ended ssDNA–dsDNA junction formed at the Okazaki fragment adjacent to this ssDNA serves as the loading point for the RAD9–RAD1–HUS1 (9-1-1) clamp complex, which is loaded onto the DNA by RAD17–replication factor C subunits 2-5 (RFC2-5) clamp loader. (a, Right) The 9-1-1 complex with assistance from RHINO (RAD9–HUS1–RAD1 interacting nuclear orphan 1) and the MRE11–RAD50–NBS1 (MRN) complex recruits the ATR activator topoisomerase II binding protein (TOPBP1), thereby allowing stimulation of ATR and phosphorylation of specific downstream effectors, including checkpoint kinase 1 (CHK1). Ewings tumor associated antigen (ETAA1) bound to RPA activates ATR in a parallel pathway. (b) In budding yeast Dpb11, DNA damage checkpoint protein 1 (Ddc1) and Dna2 have a disordered ATR activating domain (AAD), which contains critical hydrophobic amino acids that are needed for Mec1 (ATR homologue) activation. In humans, the ATR activator TOPBP1 is a homolog of Dpb11, whereas ETAA1 is not related to any yeast protein, and unlike the other activators, contains two motifs (RPA70N and RPA32C) that interact with two domains of RPA23–25. (c) The ATR kinase domain is followed by two motifs that are needed for ATR activation: FATC, and PIKK-regulatory domain (PRD), which may directly contact the AAD17. ATR-interacting protein (ATRIP) contains an amino-terminal RPA-interaction domain (RPA70N), a coiled-coil dimerization domain (CC), a motif that is needed for AAD interaction and ATR activation, and a carboxy-terminal region that interacts with ATR17,163–166. (d) Model of ATR activation. ATR–ATRIP forms a dimer of dimers167. TOPBP1 or ETAA1 AAD binding likely induces a conformational change in ATR that reduces the Km of ATR for its substrates and thereby activates ATR168.
BRCT, BRCA1 C Terminus.
Although RPA–ssDNA is sufficient to recruit the ATR–ATRIP complex, it is not sufficient for ATR activation16. Kinase activation depends on some as yet undefined ATR conformational change that is mediated by the binding of an activator protein17. In budding yeast, there are three Mec1 (the homolog of ATR) activating proteins: the protein kinase activating protein Dpb11, the DNA damage checkpoint protein 1 (Ddc1) and the bifunctional ATP-dependent DNA helicase and ssDNA endodeoxyribonuclease Dna2 18–21 (Figure 1b). In vertebrates, two ATR activators have been identified. Topoisomerase II binding protein 1 (TOPBP1), which is a Dpb11 orthologue, contacts both ATR and ATRIP through its ATR-activation domain (AAD)22 (Figure 1b). The second ATR activator is Ewing tumor-associated antigen 1 (ETAA1), which is recruited to stressed replication forks through direct interactions with RPA23–25 (Figure 1a). ETAA1 is a large protein that also contains an AAD23–25. However, it does not share any sequence homology to the other yeast or human ATR activators outside of a small motif in the AAD (Figure 1b). TOPBP1 may be the more important activator since mutations in its AAD are lethal in mice26, whereas mutations in the ETAA1 AAD do not significantly alter the growth of U2OS or HEK293T cells23. However, TOPBP1 also has important functions during the initiation of DNA replication and whether mutations in its AAD disrupt TOPBP1 function at replication origins has not been ruled out27.
The ATR activators are thought to be recruited to the stalled replication fork [G] independently of ATR12. TOPBP1 recruitment is dependent on the presence of a 5′-ended ssDNA–dsDNA junction. This junction serves as the loading point for the RAD9–RAD1–HUS1 (9-1-1) checkpoint clamp complex, which is required for TOPBP1 recruitment and subsequent stimulation of ATR kinase activity28,29 (Figure 1a). The 9-1-1 clamp is structurally similar to the proliferating cell nuclear antigen (PCNA) sliding clamp and is loaded onto the DNA by the RAD17–replication factor C subunits 2-5 (RFC2-5) clamp loader30–32. An alternative clamp loader, CTF18–RFC2-5, interacts with DNA polymerase epsilon (Pol ε)33, which is the enzyme that synthesizes the leading strand at the replication fork. In budding yeast, this interaction is a key step in the activation of the downstream replication checkpoint response34,35. How this alternative clamp loader activates the checkpoint and whether this is important for checkpoint activation in human cells is unknown. Recruitment of TOPBP1 to the 9-1-1 clamp is also partially dependent on the MRE11–RAD50–NBS1 (MRN) complex36,37 and on RAD9, RAD1, HUS1 interacting nuclear orphan (RHINO)38,39, although the mechanisms by which these components help recruit TOPBP1 are unknown. Once it is bound to the 9-1-1 clamp, the TOPBP1 AAD interacts with ATR–ATRIP, and this stimulates ATR activation (Figure 1a,d).
Little is known about why there is more than one ATR activating protein, but current evidence suggests that ETAA1 and TOPBP1 function in parallel and distinct pathways of ATR activation23,24. ETAA1 and TOPBP1 may direct ATR towards distinct substrates, respond to different replication stress inputs, or serve to amplify or increase the robustness of the stress response.
Pathway-activating DNA structure(s)
Because ATR does not directly recognize the DNA lesion, one focus in the field has been to define the DNA structure that ATR recognizes and how it is formed. ATR-activating ssDNA can be generated through uncoupling of the helicase and DNA polymerase enzymatic activities of the replisome [G], such that the helicase continues to unwind the DNA ahead of the stalled polymerase40. This is not necessarily a physical uncoupling of the helicase and polymerase as the replisome likely stays intact, but instead a functional uncoupling of enzymatic activities that allows generation of ssDNA (Figure 2a). If the lagging-strand polymerase stalls at a DNA lesion, then the ssDNA that is generated may be no larger than the length of an Okazaki fragment since repriming events should allow the fork to simply bypass the lesion. However, the nascent strand gap that is left may be expanded by exonucleases to generate a larger ssDNA platform for ATR recruitment and activation. If the stalling event is on the leading strand template, then more extensive ssDNA could be generated directly by uncoupling. In the Xenopus laevis egg extract in vitro replication system extensive unwinding can occur, but this may not be the case in human cells. Electron microscopy analysis of ssDNA at individual stressed forks revealed that various genotoxic agents typically caused ssDNA stretches ranging from 70 to 500 bases41–43. It may be that the persistence of such relatively small amount of ssDNA is what is required to activate ATR.
Figure 2. Generation of the ATR-activating structure at stressed replication forks.

(a) When the leading strand polymerase stalls, a 5′-ended ssDNA–dsDNA junction is not initially present. New primer synthesis ahead of the stalled leading-strand polymerase would create the ssDNA–dsDNA junction. DNA polymerase alpha (Pol α) and/or primase and DNA directed polymerase (PrimPol) (not shown) may catalyze primer synthesis ahead of the stalled polymerase. (b) Fork remodeling may be necessary to generate the ATR-activating structure when a DNA lesion, such as an inter-strand crosslink (ICL), stalls the fork entirely. In this situation, DNA translocases may reverse the fork. Once reversed, specialized helicases, such as Werner syndrome RecQ like helicase (WRN) or Fanconi anemia complementation group J (FANCJ) (not shown), can unwind the dsDNA of the reversed strands. Exonucleases, including DNA replication helicase/nuclease 2 (DNA2), can resect the DNA in the 5′-3′ direction as it is unwound by the helicase, generating both the ssDNA and the 5′-ended ssDNA–dsDNA junction required for ATR activation.
MCM2-7, minichromosome maintenance 2-7 complex; RPA, Replication protein A; ATRIP, ATR-interacting protein; TOBP1, topoisomerase II binding protein 1; 9-1-1, RAD9–RAD1–HUS1
At least for TOPBP1-dependent activation, ssDNA is not sufficient to activate ATR16. The loading of the 9-1-1 complex requires a free 5′-ended ssDNA–dsDNA junction, which is needed for ATR activation in X. laevis egg nuclear protein extracts when there isn’t a free 3′ DNA end for DNA extension16. A 5′-end junction with an RNA–DNA primer is naturally formed on the lagging strand when the polymerase stalls due to the discontinuous manner of DNA synthesis on this strand (Figure 1a). Likewise, primer synthesis on the leading strand could also generate the appropriate junction44 (Figure 2a). Thus, the uncoupling of helicase and polymerase activities accompanied by DNA primer synthesis may be sufficient for TOPBP1-dependent ATR activation.
This model for ATR activation at a stressed fork explains the response to several types of agents that stall the DNA polymerase, but what if the polymerase stalls without significant uncoupling of helicase and polymerase activities? For example, an inter-strand crosslink could block replication without generating significant amounts of ssDNA, because it stalls the helicase, which is positioned at the forefront of the replisome. In these cases, active fork remodeling and DNA processing such as fork reversal followed by nascent strand resection could contribute to generating the checkpoint-activating structure (Figure 2b).
Fork reversal is thought to be a common mechanism of fork protection and repair45, during which the nascent DNA [G] strands anneal to one another. An equilibrium between fork reversal and fork progression may be established in response to most types of replication stress41,46. If the nascent lagging strand is shorter than the leading strand then an appropriate 5′-ended ssDNA–dsDNA junction would be formed to activate ATR. However, if the nascent lagging strand is longer, enzymatic processing by a nuclease is needed to activate ATR. Indeed, DNA replication helicase/nuclease 2 (DNA2) may resect nascent DNA at a reversed fork in the 5′ to 3′ direction to generate the 5′-ended ssDNA–dsDNA junction (Figure 2b). Consistent with this activity creating a signal that activates ATR, DNA2 depletion impairs ATR signaling47. How much cells rely on fork reversal and resection to generate ssDNA as opposed to helicase-polymerase uncoupling likely depends on the type of damage or stress that impedes fork progression.
If fork reversal is a common mechanism in generating an ATR-activating signal, then deficiencies in fork reversal enzymes would be expected to cause ATR-activation defects. As yet there is little evidence for this, possibly because the enzymes needed to catalyze fork reversal are still poorly defined. Many enzymes including DNA translocases like SMARCAL1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a like 1), ZRANB3 (zinc finger RANBP2-type containing 3) and HLTF (helicase like transcription factor)48,49 as well as helicases including BLM (Bloom syndrome RecQ like helicase), FANCM (Fanconi anemia complementation group M), FANCJ and WRN (Werner syndrome RecQ like helicase) can catalyze fork regression in vitro45. Some combination of these enzymes is likely to cooperate with the RAD51 recombinase to generate reversed forks in cells45. Of these enzymes, genetic depletion of only the helicases FANCM, FANCJ and WRN has been linked to reduced ATR signaling47,50–57.
Other nucleases such as endonuclease/exonuclease/phosphatase family domain containing 1 (EEPD1) have also been implicated in generating single-stranded DNA at replication forks for ATR activation58, and ATR activating structures can be generated without DNA replication. For example, resection of a double-strand break59,60 or deprotection of a telomere can activate ATR10. Also, some repair-related DNA processing such as that taking place during excision repair may be sufficient to generate a transient ATR-activating DNA structure, especially if repair is not completed promptly61.
Although much has been discovered on the formation of ATR pathway activating structures, recent work has uncovered mechanisms that can shield some activating structures from ATR. For example, proteomic analysis and electron microscopy imaging of centromeric alpha-satellite DNA revealed the presence of DNA loops surrounded by a matrix of proteins that prevent RPA loading and ATR signaling at centromeres62. This suppression is needed to facilitate replication of these sequences. As half of the genome is made up of repetitive DNA sequences, it will be important to understand how and when exclusion of ATR is beneficial to replication and repair of alpha-satellite DNA and other repetitive DNA sequences.
Thresholds of activation
Conditional deletion of ATR2 or kinase inactivation with inhibitors63 causes cell lethality, suggesting ATR is active during each S phase. It is unknown how much of this is due to the continuous presence of mild replication stress or simply whether the process of replication inherently activates ATR. After all, the ATR-activating structure forms on the lagging strand during normal DNA synthesis, albeit transiently. Indeed, Mec1 phosphorylates and regulates numerous proteins during normal DNA replication, although its major effector Rad53 (the budding yeast functional homolog of checkpoint kinase 1 [CHK1]) and other stress-induced Mec1 substrates were notably not found to be phosphorylated in normal growth conditions64. It was suggested that this Mec1 activation occurs at the lagging strand of a moving replication fork, because it required the 9-1-1 clamp and the lagging strand factor Dna2. This is distinct from Mec1 activation induced by replication stress, which is primarily dependent on Dpb11 and not Dna264. More robust activation of ATR may require more persistence of the activating structure than would be possible during the rapid synthesis and processing happening on the lagging strand during normal replication elongation.
Similarly, in human cells, CHK1 is not strongly phosphorylated by ATR during normal DNA replication. Nevertheless, inhibition of ATR or CHK1 has effects on unperturbed cells, suggesting that ATR and CHK1 are active in normal conditions. Even in the absence of TOPBP1 or ETAA1, purified ATR and ATR–ATRIP complexes retain some kinase activity14,65. This basal level of activity may be sufficient to cause some substrate phosphorylation, particularly if ATR is directed to the substrate.
It is also possible that different biological responses require different levels of ATR activity. For example, common fragile sites (CFSs), which are specific chromosomal regions that are highly sensitive to replication stress, require ATR to maintain their stability under conditions of mild replication stress and even during unperturbed replication66, yet the major checkpoint effectors of ATR are not detectably phosphorylated under these conditions67. Importantly, chromosomal fragility at CFSs is detected and measured as breaks or gaps in metaphase chromosomes. Thus, conditions that hinder replication at CFSs, though they require ATR activity to prevent fragility, may not cause a detectable cell cycle arrest. This suggests that transient activation of ATR exists at individual forks that are intrinsically challenged, such as those at CFSs, and that ATR activity is crucial for the function of these forks.
Amplification of ATR signaling
Although it seems likely that different biological responses require different levels of ATR activity, cells also possess mechanisms to amplify ATR activation at individual replication forks. One way cells amplify ATR activity is by increasing the number of ssDNA–dsDNA junctions at a fork through continued primer synthesis68. For example, when a lesion stalls the leading strand polymerase and generates ssDNA ahead of the polymerase, cells are able to reinitiate DNA synthesis ahead of the stalled polymerase to create multiple short primers interspersed with ssDNA gaps (Figure 3a). ATR activation is greater, as measured by CHK1 phosphorylation, when there are more primers ahead of the stalled polymerase68, perhaps because re-priming creates multiple 5′-ended ssDNA–dsDNA junctions that can recruit multiple 9-1-1 checkpoint clamps and additional TOPBP1 to activate ATR (Figure 3a). Importantly, this mode of pathway amplification is not restricted to leading strand lesions, as continued primer synthesis also occurs on the lagging strand.
Figure 3. Amplification of ATR signaling.

There are multiple ways to amplify ATR signaling at individual replication forks. (a) Continued primer synthesis by DNA polymerase alpha (Pol α) ahead of the stalled leading-strand polymerase can generate multiple ssDNA–dsDNA junctions at a single fork. This would create multiple loading points for the RAD9–RAD1–HUS1 (9-1-1) complex and the ATR activator topoisomerase II binding protein 1 (TOPBP1), and, accordingly, would increase the number of ATR proteins that are activated at a single fork. (b) The E3 ubiquitin ligase PRP19 creates a feed-forward loop to amplify ATR activity. PRP19 ubiquitylates replication protein A (RPA), which increases ATR activity, which in turn boosts PRP19-dependent ubiquitylation of RPA. (c) Multiple post-translational modifications amplify ATR activation. These include protein kinase A (PKA) phosphorylation of ATR, ATR autophosphorylation, cyclin-dependent kinase 2 (CDK2) phosphorylation of ATR-interacting protein (ATRIP), ATR and/or ATM phosphorylation of TOPBP1, and potentially ATR phosphorylation of Ewing tumor-associated antigen 1 (ETAA1). ATRIP deacetylation by Sirtuin 2 (SIRT2) helps recruit ATR–ATRIP to the stalled replication fork. (d) The second ATR activator, ETAA1, can stimulate ATR activity independently of the presence of ssDNA–dsDNA junctions. Because ETAA1, ATR and ATRIP are recruited to RPA–ssDNA, longer stretches of ssDNA could recruit multiple ETAA1, ATR and ATRIP proteins to a single fork and produce an amplified ATR-mediated response.
CHK1, checkpoint kinase 1
We can envision at least two ways to create these primers. First, the recently described primase and DNA directed polymerase (PrimPol) can prime new synthesis ahead of ultraviolet-induced lesions and hydroxyurea-stalled forks69–72, although it has yet to be demonstrated that this amplifies ATR activation. Second, these primers may result from the priming activity of Pol α, which is a component of the replisome that provides primase (priming) activity on the lagging strand and initiates replication on the leading strand68,73. Some evidence suggests that Pol α stimulates loading of the 9-1-1 complex73, and this may occur through primer synthesis along the ssDNA to generate multiple ssDNA–dsDNA junctions (Figure 3a).
Another way to amplify ATR activation is through feed-forward signaling loops, which is a common mechanism of amplification of signal transduction. For example, the E3 ubiquitin ligase PRP19 is recruited to stalled replication forks, where it binds RPA-coated ssDNA and ubiquitylates RPA74. RPA ubiquitylation helps recruit ATRIP–ATR and promotes ATR activation. Importantly, ATR activation facilitates further PRP19 recruitment to RPA-coated ssDNA, forming a feed-forward loop for robust activation of the ATR pathway74 (Figure 3b). Sumoylation of ATRIP may also prime the ATR pathway for activation and boost protein interactions among the upstream ATR activators75. A second feed-forward regulatory mechanism may involve phosphorylation of ATR-activating proteins. Phosphorylation of TOPBP1 by ATM increases its ability to activate ATR76. Similarly, in budding yeast, Dpb11 is phosphorylated by Mec1, and this increases its ability to activate Mec1 in vitro19. ETAA1 is also phosphorylated in response to replication stress, although whether this affects ATR activation is not known23.
Post-translational modifications of ATR and ATRIP also contribute to ATR regulation (Figure 3c). ATRIP is both phosphorylated and acetylated77–79; ATRIP phosphorylation by cyclin-dependent kinase 2 (CDK2) increases ATR signaling whereas ATRIP acetylation decreases it. Deacetylation of ATRIP by sirtuin 2 (SIRT2) following replication stress increases the affinity of ATRIP for RPA79. ATR is also phosphorylated on multiple residues, including on Thr198980,81, which may be autophosphorylated, although it is not part of the preferred consensus sequence recognized by ATR. Thr1989 phosphorylation may increase the ability of ATR to bind and be activated by TOPBP1 and to phosphorylate other substrates. Thus, ATR autophosphorylation could potentiate ATR activation, similar to a model proposed for ATM autophosphorylation82. However, the importance of both ATM and ATR autophosphorylation in signaling is controversial since a mutation in the ATM autophosphorylation site did not impair ATM signaling in mouse models83,84, nor was ATR signaling impaired in human cells expressing only the non-phosphorylatable ATRT1989A mutant81. Other ATR phosphorylation sites such as Ser428 and the nearby protein kinase A (PKA)-mediated phosphorylation site (Ser435) may be important regulators in specific stress conditions85.
Emerging themes in ATR pathway activation
Although we have a clear grasp of how the basic components function to activate ATR following replication stress, new and interesting themes have begun to emerge. Of interest is the role of the NIMA-related kinase (NEK) family in ATR activation. There are at least eleven members in humans, and many have reported roles in facilitating activation of the ATR pathway or downstream checkpoints86, raising the intriguing possibility that the NEK family evolved in part to modulate the ATR pathway.
The discovery of multiple ATR activators provides an additional way to regulate or amplify ATR signaling quantitatively. The persistent ssDNA generated by replication stress provides a platform for recruitment of more ATR–ATRIP and ETAA1 to the stalled fork. Since current evidence suggests that ETAA1 does not require the 9-1-1 complex to activate ATR, it is possible that it amplifies ATR signaling even without the generation of additional 5′-ended junctions (Figure 3d). What makes ATR more active when it is bound by an AAD is not known, but all the yeast and vertebrate ATR activating proteins identified to date share a similar motif within their AAD that contains two large hydrophobic amino acids within a relatively unstructured region, which can interact with the ATR–ATRIP complex and promote its activation87. A mutation in ATR that prevents TOPBP1 binding and activation also impairs ETAA1 binding and activation17,23. Furthermore, in vitro studies have shown that TOPBP1 and ETAA1 confer overlapping substrate specificity to activated ATR. These findings suggest that the mechanism of kinase activation of all ATR-activating proteins is similar, and may involve an allosteric change in ATR conformation that translates into a difference in the ability of ATR to bind substrates17. Thus, the abundance of the ATR activating protein in proximity to ATR could tune the quantity of ATR signaling.
It is also possible that there are qualitative differences in the signaling of the ETAA1–ATR complex vs. the TOPBP1–ATR complex. Unlike TOPBP1, ETAA1 may not need a 5′ ssDNA–dsDNA junction to activate ATR since it can bind directly to RPA (Figure 3d). Thus, it is possible that active ETAA1–ATR and TOPBP1–ATR complexes form on different DNA substrates and that their proximity to different substrates leads to differences in signaling. ETAA1 and TOPBP1 could also provide qualitative differences in signaling by directly binding to different ATR substrates. Much more work on these alternative ATR activators is needed to understand how having more than one is advantageous.
Finally, recent work has revealed a new mode of ATR activation at the nuclear envelope in response to mechanical stress11. Although the molecular mechanism underlying ATR activation in this context is unclear, it does not require RPA, TOPBP1 or RAD17, strongly suggesting this activation is not triggered by ssDNA—dsDNA junctions11. However, given the mechanical forces associated with DNA replication and chromosome condensation, especially where chromatin is attached to the nuclear envelope, it is likely that ATR activation in response to mechanical stress contributes to the integrity of the genome. Continued work on this emerging aspect of ATR activation is needed before a clear model can be developed, but the activation of ATR following mechanical stress seems to indicate there may be other mechanisms of ATR activation.
ATR FUNCTIONS DURING DNA REPLICATION
Once activated at stressed replication forks, ATR orchestrates a multifaceted response that protects the integrity of the genome. Instrumental to this response is the downstream effector, CHK1, which is a kinase that is activated by phosphorylation by ATR12. Together, ATR and CHK1 function to arrest the cell cycle, suppress origin firing [G], stabilize replication forks and promote fork repair and restart. By coupling cell cycle arrest to fork stabilization and restart, ATR and CHK1 likely ensure that cells do not enter mitosis when replication is perturbed.
Cell cycle arrest
One crucial function of the ATR pathway is to arrest the cell cycle following DNA damage in S phase. This arrest is initiated by the phosphorylation of CHK1 by ATR, a reaction mediated by Claspin, which helps to bring CHK1 to the replication fork and into proximity with ATR88. Once activated, CHK1 phosphorylates and inactivates the cell division cycle 25 (CDC25) phosphatases, including CDC25A, CDC25B and CDC25C89. These phosphatases remove inhibitory phosphorylations from CDK2 and CDK1, and are required to activate the cyclin–CDK complexes necessary for cell cycle progression90. The mechanisms of checkpoint-mediated CDC25 inactivation vary. CHK1-dependent phosphorylation of CDC25A triggers its rapid degradation in S phase91,92. By contrast, CDC25C phosphorylation leads to its association with 14-3-3 signaling modifier proteins and sequestration into the cytoplasm93,94. Although all three phosphatases are negatively regulated by the ATR–CHK1 pathway following DNA damage, evidence from a genome-wide CRISPR screen in haploid stem cells suggests that CDC25A is the most important target and a major determinant of sensitivity to ATR inhibition63.
Regulation of origin firing
Origin firing is tightly-regulated and occurs in an orderly fashion, both in terms of the timing and spacing of initiation events95. Depending on the cell type, replication origins within specific chromosome domains fire exclusively in early, mid, or late S phase. ATR and CHK1 are negative regulators of origin firing and prevent excessive origin firing even during an unperturbed S phase. Early studies in X. laevis egg extracts indicate that loss of ATR leads to excessive origin firing, particularly in early S phase96,97. Furthermore, loss of CHK1 activity in mammalian cells results in late origin firing in early S phase, in part due to premature activation of the cyclin A–CDK1 complex, which is thought to promote late origin firing98. This function of the ATR–CHK1 pathway may limit the density of active replication forks throughout the genome so that replication forks have a sufficient supply of DNA precursors and replication factors for optimal fork progression.
ATR also has a crucial role in limiting DNA replication by blocking initiation in response to replication stress99–101. ATR prevents origin firing by blocking recruitment of CDC45 to the minichromosome maintenance 2-7 complex (MCM2-7), which is a heterohexameric helicase complex that unwinds DNA at the replication fork (Box 1). Helicase activation requires CDC45 binding, which occurs following CDK-dependent phosphorylation of Treslin (Sld3 in budding yeast)102 and DBF4-dependent kinase (DDK)- mediated phosphorylation of the MCM 2–7 complex103 (Figure 4a). Accordingly, one way the ATR–CHK1 pathway may prevent CDC45 loading and helicase activation is by down-regulating the kinase activities of CDK and DDK. Indeed, in yeast, the CHK1 homolog Rad53 phosphorylates Dbf4 to suppress DDK activity in response to replication stress104,105, and human CHK1 phosphorylates CDC25A, thereby triggering its rapid degradation and CDK inhibition91,106.
Box 1. Origin firing.
Initiation of DNA replication at origins is a two-step process, which involves origin licensing and origin firing95. Both steps are strictly regulated and occur during separate phases of the cell cycle. First, origins are licensed in late mitosis and early G1 by loading on chromatin the pre-replicative complex (pre-RC), which includes, among other factors, the origin recognition complex (ORC) and the core replicative helicase minichromosome maintenance 2-7 complex (MCM2-7; see the figure). MCM2-7 helicase activation follows when origins fire in S phase. Helicase activation requires the loading of the pre-initiation complex (pre-IC), which includes Treslin, cell division cycle 45 (CDC45), the GINS complex (SLD5–PSF1–PSF2–PSF3), topoisomerase II binding protein 1 (TOPBP1), a DNA polymerase, and other replication factors (see the figure). Recruitment of the pre-IC is dependent on the activities of the DBF4-dependent kinase (DDK) and the S phase cyclin-dependent kinase (CDK). The two kinases phosphorylate several factors needed to recruit the pre-IC and to activate the MCM helicase to unwind the DNA and initiate replication.
Throughout the genome, clusters of replication origins fire simultaneously, and the space between these origins is typically 50–150 kilobases. Some clusters fire earlier in S phase than others. Whereas an average of 4–6 origins fire in a typical cluster, the number of licensed origins within a cluster is thought to be approximately 20-fold higher95. Some of these unused origins are referred to as dormant origins, and are only used if the replication forks emanating from fired origins are stalled or slowed162. Accordingly, dormant origins are often thought of as back-up origins that facilitate the completion of DNA synthesis within a cluster when the progression of neighboring replication forks is impeded.
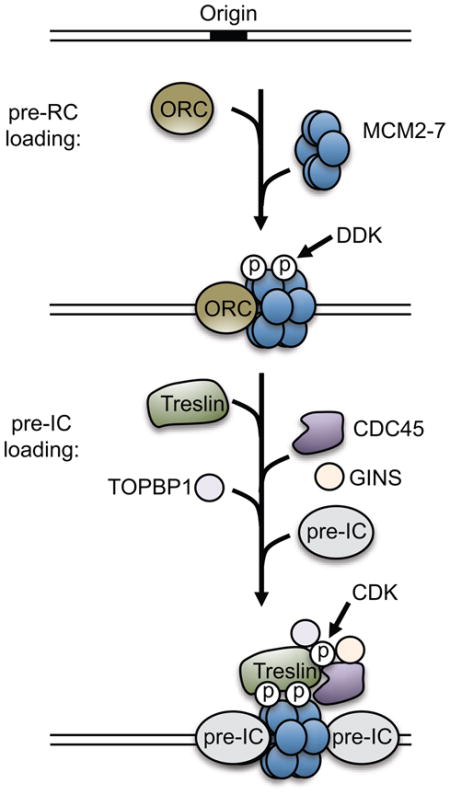
Figure 4. Pathways regulated by ATR to suppress origin firing.

(a) (Left) DBF4-dependent kinase (DDK) and cyclin-dependent kinase (CDK) activities promote origin firing. DDK phosphorylates the minichromosome maintenance 2-7 complex helicase (MCM), and CDK phosphorylates Treslin. These phosphorylations promote recruitment of cell division cycle 45 (CDC45) and other pre-initiation complex (pre-IC) factors to activate the helicase.(Right) ATR may suppress origin firing through at least two distinct pathways. The first is through phosphorylation and stabilization of myeloid/lymphoid or mixed-lineage leukemia (MLL). This promotes MLL association with chromatin where it methylates histone H3 Lys4 (H3K4me). This chromatin modification blocks CDC45 loading onto nearby origins. The second pathway is through ATR-dependent activation of checkpoint kinase 1 (CHK1), which negatively regulates CDK-dependent phosphorylations at origins, thereby blocking the loading of CDC45 and other pre-IC factors. CHK1 also directly phosphorylates Treslin, which limits CDC45 binding to origins. (b and c) ATR allows local dormant origins to fire in response to replication stress. (b) When a replication fork is stalled, nearby dormant origins fire to help complete DNA synthesis in its vicinity. At the same time, cells also block origin firing in later replicating regions. This prevents the accumulation of additional replication stress and the potential depletion of replication factors or nucleotides. (c) It is unclear how ATR allows dormant origins to fire locally. One proposed mechanism involves inhibition of CHK1 activity in the vicinity of the stalled polymerase. ATR activated at the stalled fork can phosphorylates MCM2 locally, primarily at nearby unfired origins. MCM2 phosphorylation creates a docking site for polo-like kinase 1 (PLK1), which suppresses activation of CHK1 and allows recruitment of CDC45 to nearby origins.
ORC, origin recognition complex
Another mechanism by which the intra-S phase checkpoint blocks CDC45 recruitment is through ATR-dependent phosphorylation of the histone methyltransferase myeloid/lymphoid or mixed-lineage leukemia (MLL)107. This leads to MLL stabilization and its accumulation on chromatin, where it methylates histone H3 Lys 4. This modification prevents loading of CDC45 at nearby replication origins, and thus suppresses origin firing (Figure 4a). In budding yeast, downstream of Mec1, Rad53 suppresses origin firing in response to DNA damage by phosphorylating Sld3105,108. This phosphorylation also blocks loading of Cdc45 at origins and thereby enforces the intra-S checkpoint. A similar pathway is found in humans, where CHK1 phosphorylates Treslin, the functional homolog of Sld3, to block CDC45 loading and suppress origin firing109 (Figure 4a).
A key aspect of the ATR-dependent checkpoint is that, under conditions of replication stress, it suppresses origin firing globally, yet allows dormant origin firing locally (Figure 4b)110. The firing of dormant origins within an actively replicating region supports the completion of DNA replication within these regions. At the same time, the global suppression of new origin firing minimizes widespread DNA polymerase stalling and prevents problematic replication at yet to be replicated regions of the genome.
Within a chromatin domain, clusters of origins fire concurrently. Genetic disruption or chemical inhibition of the ATR–CHK1 pathway significantly decreases both the rate of fork progression and the inter-origin distance (IOD) at local origin clusters111–116, suggesting that ATR and CHK1 modulate replication initiation events locally, perhaps through direct regulation of the CDC25–CDK pathway. However, this effect within local domains may be indirect, as dormant origins may fire passively if loss of ATR or CHK1 causes slower fork progression. Partial inhibition of CDK activity restored the IOD and the rate of fork progression in CHK1-inhibited cells114. CDK inhibition would prevent further origin firing and reduce the number of active forks. This in turn would be predicted to increase the pool of available dNTPs and correspondingly increase the rate of fork progression. Indeed, addition of dNTP precursors increased both the fork progression rate and the IOD in CHK1-inhibited cells116. Therefore, the decreased IOD in CHK1-inhibited cells may be a consequence of slower fork progression. Importantly, nucleoside addition did not increase the distances between initiation events in ATR-inhibited cells, suggesting that ATR modulates the IOD in a manner distinct from CHK1.
Although advantageous to cells, it remains enigmatic how the checkpoint can simultaneously suppress global origin firing, yet allow local dormant origins to fire102. One possibility to explain how local dormant origins escape checkpoint inhibition is that CDC45 is already loaded at dormant origins within actively replicating regions. If so, these origins would be beyond the activation step controlled by the checkpoint. Indeed, in X. laevis egg extracts, modulating CDK activity does not appear to induce dormant origin firing within actively replicating regions117. ATR may also block the checkpoint inhibition of nearby dormant origins by phosphorylating MCM2118. In X. laevis egg extracts, MCM2 phosphorylation recruits polo-like kinase 1 (Plk1), which inhibits Chk1 activity. This could allow origins near a stalled fork, where ATR is active, to fire119 (Figure 4c). Finally, recent data suggest that ATR regulation of global and local origin firing is even more complex, in that it depends on the level of replication stress. In mild to moderate levels of replication stress, FANCI binds unfired origins and directs DDK-dependent phosphorylation of the MCM helicase and subsequent dormant origin firing120. However, when replication stress is high, ATR phosphorylates FANCI and blocks FANCI-mediated dormant origin firing. Clearly, the regulation of origin initiation is complex and further studies are needed to assemble all the regulatory mechanisms into a unified model.
Maintaining replication-fork stability
It is well established that ATR is essential for stabilizing stressed replication forks67,121–123 (Figure 5). Replication fork stabilization is loosely defined as maintaining the ability of stalled polymerases to restart DNA synthesis following removal or bypass of a block to replication elongation. An unstable fork cannot resume DNA replication and is said to have collapsed, a process that at the molecular level often involves formation of a DSB at replication forks (Figure 5a). How this precisely occurs has been a matter of debate. Initially, it was proposed that fork collapse involved dissociation of the replisome, a conclusion based primarily on the study of a few replication forks in the budding yeast genome124; however, recent large scale genomic data in budding yeast and proteomic experiments in human cells suggest that the replisome itself is stable in ATR-deficient cells125,126. Instead, fork collapse may be an active process driven by structure-specific nucleolytic enzymes that catalyze cleavage of structured DNA formed at stalled replication forks or remodeled forks. DSBs may also be an intermediate generated in a process of recombination-based replication restart.
Figure 5. Proposed mechanisms by which ATR maintains replication-fork stability.

(a) ATR prevents fork collapse, which is illustrated here as double-strand DNA break (DSB) formation at the fork. In this example, replication of the leading strand was blocked by a DNA lesion. (b) ATR phosphorylates SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a like 1 (SMARCAL1) at stressed replication forks, thereby suppressing its activity and limiting fork reversal. As structure-specific endonuclease subunit (SLX4)-dependent nucleases can cleave reversed forks, ATR-dependent inhibition of SMARCAL1 is a mechanism by which ATR stabilizes the fork. (c) ATR phosphorylates several proteins at the replication fork to modify replisome function. ATR mediates the recruitment of Fanconi anemia complementation group D2 (FANCD2) to the fork, which may occur in response to ATR-dependent phosphorylation of the MCM2-7 helicase and FANCD2. FANCD2 minimizes the accumulation of ssDNA caused by meiotic recombination 11 homolog A (MRE11)-dependent resection of the nascent DNA. FANCD2 also slows DNA polymerases at stressed forks, and fork slowing may prevent collapse. (d) ATR prevents exhaustion of replication protein A (RPA) availability and subsequently replication catastrophe. This indirect function of ATR is mediated partly through its role in suppressing origin firing in response to hydroxyurea (HU)- or aphidicolin (APH)-induced replication stress.
Inhibition of nuclease-dependent fork collapse
ATR function is crucial to prevent the nuclease-mediated cleavage of replication forks. As yet, however, there is little evidence that ATR directly regulates structure-specific nucleases; instead, fork cleavage may result from the failure of ATR to regulate fork processing events such as fork reversal. Indeed, in budding yeast, Rad53 restrains fork reversal and prevents nucleolytic processing of stressed replication forks43,127. Moreover, in human cells, the putative fork reversal enzyme SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A-like protein 1 (SMARCAL1) is phosphorylated by ATR, thereby decreasing its ability to reverse forks in vitro, and suppressing fork collapse in cells115 (Figure 5b). Loss of this regulation by ATR may lead to excessive fork reversal in cells, and subsequently to DNA cleavage. In fact, loss of structure-specific endonuclease subunit (SLX4), which is a scaffold protein that binds to several structure-specific nucleases, reduces the formation of DSBs in ATR-inhibited cells, suggesting that an SLX4-dependent nuclease may process the four-way junction of the reversed replication fork115,128 (Figure 5b).
The effects of ATR loss on fork stability may also be indirectly tied to its effects on CDK activity and cell cycle progression. Some of the structure-specific endonuclease complexes are CDK-regulated and become activated in late G2 or mitosis to resolve Holliday junctions [G] and under-replicated loci129. This is an important process in mitosis as it is necessary for the separation of sister chromatids during anaphase. However, if these nucleases become prematurely activated in S phase due to increased CDK activity, they could process stalled replication forks into DSBs. This is observed when the G2/M checkpoint kinase WEE1 is inhibited in S phase130,131 and may also be the case in ATR-deficient cells. Of course, some structure-specific nucleases, such as the EME2—MUS81 endonuclease complex, are active in S phase and promote restart of stalled replication forks132. Thus, nuclease-dependent DSB formation in ATR-inhibited cells may be an attempt to restart inactivated replication forks.
Downstream of ATR, CHK1 also stabilizes stressed replication forks. CHK1 inhibition leads to endonuclease-dependent fork collapse, which is catalyzed by MUS81116,133,134. In vitro, MUS81 can cleave stalled replication forks, and in the absence of MUS81 many fork-associated breaks do not form135. However, MUS81 is not essential for fork collapse in ATR-inhibited cells115,128. Furthermore, CHK1 inhibition alone causes massive fork collapse in S phase cells and cell death, whereas ATR inhibition alone does not. This could reflect redundancy with the other DNA damage response kinases, including DNA-PK and ATM, which may phosphorylate and partially activate CHK1 in ATR-deficient cells136. However, CHK1 could also have essential activity in the absence of these upstream kinases, and basal CHK1 could be very important to stabilize replication forks.
Control of replisome function
Although the replisome may remain stable when ATR is inhibited, other aspects of its function may be regulated by ATR. Indeed, ATR phosphorylates many replisome components following replication stress, including several DNA polymerases, the clamp-loader [G] RFC, the MCM helicase, RPA, and the Claspin-Timeless-Tipin-And1 complex12,137. Consistent with this idea, in budding yeast Mec1 phosphorylates several components of the replicative helicase, including DNA replication protein Psf1, to alter replisome progression125. Moreover, in human cells ATR recruits FANCD2 to the replication fork. FANCD2 recruitment may be mediated by binding the MCM helicase, and correlates with MCM phosphorylation by ATR138 (Figure 5c). FANCD2 slows the progression of the DNA polymerase and presumably the helicase, and importantly, minimizes ssDNA formation and MRE11-dependent resection of DNA at stressed forks138. It is unknown if slowing fork progression is ATR-dependent. DNA lesions themselves slow polymerase progression, but in budding yeast this effect was found to be independent of Mec1 and Rad53122. The CHK1 homolog in fission yeast, Cds1, was suggested to slow fork movement in response to DNA damage139. Similarly, in human cells CHK1 was shown to slow fork elongation rates following camptothecin-induced DNA damage140,141. Although the precise mechanisms have yet to be identified, these results suggest that ATR modifies the function of the replisome to prevent unwanted DNA resection or unwinding at the fork, and perhaps to slow replication elongation (Figure 5c).
Exhaustion of replication factors
ATR may also stabilize replication forks indirectly, by suppressing late-origin firing and preventing the depletion or “exhaustion” of RPA. RPA exhaustion is observed when ATR inhibition is combined with replication inhibition by hydroxyurea or aphidicolin142. Under these replication stress conditions, the accumulation of ssDNA at replication forks as a result of excessive origin firing, helicase–polymerase uncoupling and DNA-end resection, apparently exceeds the availability of RPA142. RPA is needed to regulate the recruitment and activities of DNA repair and fork reversal enzymes; therefore, its exhaustion leads to widespread fork collapse (Figure 5d).
The suppression of origin firing in ATR-inhibited cells delays RPA exhaustion, suggesting that the pool of available RPA is initially sufficient to coat and protect the ssDNA formed at active replication forks. Accordingly, RPA exhaustion may be particularly detrimental to replication forks emanating from the aberrantly-fired origins in ATR inhibited cells. Indeed, non-homologous end-joining repair proteins accumulate at aberrantly-fired origins in ATR-inhibited cells, instead of the normal homologous recombination factors that promote fork restart [G] 126. Intriguingly, suppressing new origin firing also results in the retention of homologous recombination factors, including RAD51 and BRCA1, at stressed forks. This may suggest that, like RPA, homologous recombination factors could also become exhausted when ATR inhibition is combined with replication stress. However, as ATR phosphorylates RPA143,144 and the homologous recombination factors partner and localizer of BRCA2 (PALB2)145,146 and X-ray repair cross complementing 3 (XRCC3)147, it is also possible that homologous recombination factors in ATR inhibited-cells are not properly activated. As fork restart can involve a RAD51 recombination-dependent process148, a functional loss of homologous recombination may also contribute to the fork instability seen in ATR-deficient cells.
Regulation of replication fork restart
Another aspect of ATR-mediated fork stabilization involves the regulation of pathways that actively promote replication fork restart in addition to those that prevent fork collapse (Figure 6). Multiple processes have been suggested to promote restart1. For example, fork restart may involve repriming ahead of the stalled polymerase by PrimPol. Other processes that promote fork restart involve the DNA damage tolerance [G] pathways, which facilitate continued DNA synthesis without repairing the lesion. This can occur through direct bypass of damage, a process that involves translesion polymerases. Alternatively, template switching [G] may occur, which allows the use of the undamaged sister chromatid as a template for replication. Replication restart may also occur though fork reversal processes45. Fork reversal can place a polymerase-blocking lesion back into the context of dsDNA so that repair can occur. Replication can then continue once the reversed fork is reset. Lastly, endonucleases may also cleave a reversed or stalled fork to facilitate homologous recombination-mediated mechanisms of fork restart149 (Figure 6).
Figure 6. Proposed roles of ATR in promoting replication-fork restart.

In addition to stabilizing the replication fork, ATR is thought to promote restart of stalled forks. There are several pathways to restart a stalled fork, including repriming ahead of the lesion by primase and DNA directed polymerase (PrimPol), lesion bypass through translesion synthesis (TLS), lesion bypass through template switching, and fork reversal and lesion repair. If a stalled fork collapses into a double-strand DNA break (DSB), homologous recombination-dependent pathways can restart the fork. It is unknown whether ATR regulates PrimPol activity to restart replication forks, but ATR does phosphorylate two TLS polymerases, REV1 and DNA polymerase eta (Pol η), and may promote lesion bypass. ATR also phosphorylates several proteins that promote radiation sensitive 51 (RAD51)-dependent replication restart pathways, including template switching, fork reversal and repair, and homologous recombination. These include X-ray repair cross complementing 3 (XRCC3), partner and localizer of BRCA2 (PALB2), replication protein A (RPA), Werner syndrome RecQ like helicase (WRN) and Bloom syndrome RecQ like helicase (BLM).
How precisely ATR functions in these processes is not clear, but it likely contributes to several of them. ATR phosphorylates two of the translesion polymerases, REV1 and Pol η150,151, suggesting that ATR may control lesion bypass. Other fork restart pathways, including template switching, fork reversal and homologous recombination, require RAD5145,149. Importantly, ATR likely regulates RAD51-dependent fork restart pathways, as several factors that act in these pathways are ATR substrates and are required for ATR-mediated fork restart. For example, ATR phosphorylates RPA, PALB2 and XRCC3 and these modifications are thought to promote RAD51 recruitment to stalled forks or DSBs at collapsed forks143–147. ATR also phosphorylates the helicases BLM and WRN, which may promote replication restart by processing repair intermediates152,153 (Figure 6). However, until we have good methods to distinguish between the effect of ATR on fork stabilization and on fork restart, it will be difficult to clearly delineate the molecular mechanisms and processes that occur when fork stall and restart.
Ensuring dNTP availability
In addition to maintaining the stability of stalled forks, ATR also actively functions to prevent fork stalling from happening in the first place by ensuring that dNTP levels in replicating cells are sufficient. Early genetic studies in budding yeast demonstrated that the lethality caused by Mec1 loss could be rescued by increasing the activity of ribonucleotide reductase (RnR), which is the rate limiting enzyme in dNTP production154,155. By overexpressing RnR or deleting an RnR inhibitor, nucleotide levels are increased in the Mec1-deficient yeast, thereby yielding fewer stalling forks and reducing the need for replication stress signaling. Furthermore, Mec1 regulates RnR activity in yeast by multiple mechanisms156.
Similarly, in human cells ATR activity is needed for efficient transcription factor E2F1-dependent expression of ribonucleoside-diphosphate reductase subunit M2 (RRM2)136. ATR-mediated RRM2 expression may be especially important in the early stages of S phase, where loss of ATR leads to the formation of greater levels of ssDNA. ATR also boosts RnR activity when DNA damage levels (and consequently the need for dNTPs) are high by preventing CDK and cyclin F-dependent RRM2 degradation157.
The importance of ATR in maintaining RRM2 expression and consequently dNTP levels was recently explored in a mouse model of Seckel syndrome158. Seckel mice have an Atr mutation that leads to reduced levels of ATR, similar to what is observed in humans with ATR mutations. These mice recapitulate many of the ATR mutation phenotypes seen in humans, including craniofacial abnormalities and dwarfism159. Strikingly, crossing the Seckel mouse with mice genetically engineered to express supra-physiological levels of Rrm2 resulted in a considerable increase in the overall size and lifespan of the mice. This elegant genetic study strongly supports the importance of ATR in regulation of dNTP biosynthesis158.
Conclusions and future directions
The past few years have revealed complexities in ATR regulation and function that emphasize its essential functions in maintaining genome integrity. The use of new reagents such as highly potent ATR inhibitors has lead to new discoveries, and such reagents are being developed for cancer therapies160. Cancer cells have elevated levels of replication stress and may be more dependent than normal cells on ATR function. ATR inhibition also sensitizes cells to many current agents that target DNA repair and replication. Thus, combining ATR inhibition with therapies that induce replication stress or targeting cancers with high levels of replication stress may be useful clinical strategies. Finding the right individuals that will benefit from these drugs as well as biomarkers that predict drug responses remain essential areas of investigation.
The identification of the second ATR activator ETAA123–25 illustrates that much is left to learn about the basic mechanisms of ATR regulation. Unanswered questions include: Why do cells need multiple ATR activators? How is ATR signaling tuned to yield different cellular responses? What is the complete constellation of ATR targets that are needed to execute an effective replication stress response?
New approaches such as high-resolution cryo-EM will hopefully reveal how ATR is activated at atomic resolution. A high-resolution structure may explain how post-translational modifications and other processes generate different levels of ATR signaling. The recent biochemical reconstitution of origin-dependent DNA replication161 also provides an opportunity to build fully-defined in vitro systems to study replication stress responses.
The genome cannot be fully protected from exogenous and endogenous sources of damage, which threaten its stability. Fortunately, ATR solves many of these genome maintenance challenges to ensure to the integrity of the information stored in the DNA.
Online Summary.
ATR is an essential kinase that is active in S phase, senses stressed replication forks and orchestrates a multifaceted response to DNA replication stress. This response helps ensure completion of DNA replication and maintains the integrity of the genome.
ATR and its binding partner, ATRIP, are recruited to stalled forks through direct interactions with the RPA–ssDNA complex that forms at stressed replication forks. Once bound to ssDNA, the kinase activity of ATR is stimulated by the ATR-activating domains of TOPBP1 or ETAA1, which are independently recruited to ssDNA–dsDNA junctions or to RPA–ssDNA, respectively.
ATR activity can be amplified by generating more ssDNA–dsDNA junctions at individual replication forks, through feed-forward signaling loops, and by post-translational modifications of the signaling complexes.
Once activated, ATR directs the replication stress response to arrest the cell cycle, block origin of replication firing, and stabilize and repair stalled replication forks.
ATR and its effector CHK1 are active during both an unperturbed S phase, to prevent excessive origin firing and in response to replication stress, to slow DNA replication. However, this negative regulation of replication initiation does not prevent the firing of dormant origins within a replication domain, which can rescue replication completion without requiring the damaged fork to restart.
ATR phosphorylates numerous replisome proteins and repair factors that prevent fork collapse and the formation of DNA breaks. These post-translational modifications regulate the remodeling of replication forks and subsequent nuclease-dependent cleavage and/or resection of forks. They also regulate pathways needed to repair stalled forks and restart DNA synthesis.
Acknowledgments
This work was supported by a Postdoctoral Fellowship from the American Cancer Society (PF-15-165-01 – DMC) and a Postdoctoral Enrichment Program Award from the Burroughs Wellcome Fund to J.C.S., an NIH grant (CA102729) to D.C., and grants from the NIH (GM100489 and ES016486) to K.A.C.
Glossary
- Replication stress
the slowing or stalling of replication fork progression and/or DNA synthesis
- Hypomorphic alleles
mutated genes that encode proteins with reduced function
- Genotoxic stress
broadly refers to any agent that damages cellular DNA
- Topoisomerase
an enzyme that relieves torsional stress caused by DNA supercoiling during replication or transcription
- SOS DNA damage response
a bacterial DNA damage response that arrests the cell cycle and induces error prone DNA repair
- Stalled replication fork
A replication fork that has prematurely halted DNA synthesis
- Replisome
a multiprotein complex that unwinds double-stranded DNA and catalyzes both leading and lagging strand DNA synthesis
- Nascent DNA
the newly synthesized strands of DNA during DNA replication
- Origin firing
refers to the initiation of DNA replication at an origin
- Holliday junctions
branched structures that contain a four-way junction of double-stranded DNA
- Clamp-loader
a protein complex that catalyzes the loading of clamp complex onto DNA
- Fork restart
the process of restarting DNA replication at a fork that had been stalled
- DNA damage tolerance
a set of pathways that allow cells to replicate damaged DNA, including translesion synthesis and template switching
- Template switching
Switching of the DNA polymerase to the newly synthesized DNA as a template for DNA replication, when the parental template is damaged
Biographies
Joshua C. Saldivar is a postdoctoral fellow in the Department of Chemical and Systems Biology at the Stanford University School of Medicine. He was trained in molecular genetics at the Ohio State University, where he investigated the mechanisms that initiate genomic instability in precancerous cells. His research currently focuses on the ATR pathway in transcriptional regulation and control of cell cycle transitions. He is also exploring novel approaches to enhance selective killing of cancer cells with ATR inhibitors.
David Cortez received his Ph.D. from Duke University and performed postdoctoral work at the Baylor College of Medicine in Houston, TX, USA. He is currently a Professor of Biochemistry and Ingram Professor of Cancer Research at Vanderbilt University School of Medicine, Nashville, TN, USA. His research interests focus on the mechanisms of replication stress responses with active projects on checkpoint signaling, fork remodeling, fork protection, and replication-associated DNA repair.
Karlene A. Cimprich is a professor in the Department of Chemical and Systems Biology at the Stanford University School of Medicine. Her lab focuses on understanding the mechanisms by which a cell maintains genome stability, particularly in the context of DNA replication. She identified the ATR checkpoint kinase as a postdoctoral fellow and has continued to study its activation and cellular functions. Her laboratory also studies DNA damage tolerance pathways, and the mechanism by which transcription causes DNA damage during S phase.
Footnotes
Competing interests statement
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nature Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. Demonstrated that ATR is essential for viability in vivo. [PMC free article] [PubMed] [Google Scholar]
- 3.de Klein A, et al. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–482. doi: 10.1016/s0960-9822(00)00447-4. Demonstrated that ATR is essential for viability in vivo. [DOI] [PubMed] [Google Scholar]
- 4.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nature Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 5.Cimprich KA, Shin TB, Keith CT, Schreiber SL. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci USA. 1996;93:2850–2855. doi: 10.1073/pnas.93.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bentley NJ, et al. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J. 1996;15:6641–6651. [PMC free article] [PubMed] [Google Scholar]
- 7.Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol. 2001;13:225–231. doi: 10.1016/s0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 8.Sirbu BM, Cortez D. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol. 2013;5:a012724. doi: 10.1101/cshperspect.a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Royo H, et al. ATR acts stage specifically to regulate multiple aspects of mammalian meiotic silencing. Genes Dev. 2013;27:1484–1494. doi: 10.1101/gad.219477.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maciejowski J, de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nature Rev Mol Cell Biol. 2017;18:175–186. doi: 10.1038/nrm.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar A, et al. ATR Mediates a Checkpoint at the Nuclear Envelope in Response to Mechanical Stress. Cell. 2014;158:633–646. doi: 10.1016/j.cell.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nature Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutton MD, Smith BT, Godoy VG, Walker GC. The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu Rev Genet. 2000;34:479–497. doi: 10.1146/annurev.genet.34.1.479. [DOI] [PubMed] [Google Scholar]
- 14.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. Identified critical components of ATR recruitment to ssDNA. [DOI] [PubMed] [Google Scholar]
- 15.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. Identified critical components of ATR recruitment to ssDNA. [DOI] [PubMed] [Google Scholar]
- 16.MacDougall CA, Byun TS, Van C, Yee M-C, Cimprich KA. The structural determinants of checkpoint activation. Genes Dev. 2007;21:898–903. doi: 10.1101/gad.1522607. Used defined DNA substrates in X. laevis egg extracts to show that ssDNA and a ssDNA–dsDNA junction are suffient to activate ATR signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mordes DA, Glick GG, Zhao R, Cortez D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008;22:1478–1489. doi: 10.1101/gad.1666208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar S, Burgers PM. Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev. 2013;27:313–321. doi: 10.1101/gad.204750.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mordes DA, Nam EA, Cortez D. Dpb11 activates the Mec1-Ddc2 complex. Proc Natl Acad Sci USA. 2008;105:18730–18734. doi: 10.1073/pnas.0806621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navadgi-Patil VM, Burgers PM. Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. J Biol Chem. 2008;283:35853–35859. doi: 10.1074/jbc.M807435200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Navadgi-Patil VM, Burgers PM. The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol Cell. 2009;36:743–753. doi: 10.1016/j.molcel.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. Identified TopBP1 as a direct ATR activator. [DOI] [PubMed] [Google Scholar]
- 23.Bass TE, et al. ETAA1 acts at stalled replication forks to maintain genome integrity. Nature Cell Biol. 2016;18:1185–1195. doi: 10.1038/ncb3415. Identified ETAA1 as a direct ATR activator. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haahr P, et al. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nature Cell Biol. 2016;18:1196–1207. doi: 10.1038/ncb3422. Identified ETAA1 as a direct ATR activator. [DOI] [PubMed] [Google Scholar]
- 25.Lee Y-C, Zhou Q, Chen J, Yuan J. RPA-Binding Protein ETAA1 Is an ATR Activator Involved in DNA Replication Stress Response. Curr Biol. 2016;26:3257–3268. doi: 10.1016/j.cub.2016.10.030. Identified ETAA1 as a direct ATR activator. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou ZW, et al. An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. PLoS Genet. 2013;9:e1003702. doi: 10.1371/journal.pgen.1003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia V, Furuya K, Carr AM. Identification and functional analysis of TopBP1 and its homologs. DNA Repair. 2005;4:1227–1239. doi: 10.1016/j.dnarep.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Delacroix S, Wagner JM, Kobayashi M, Yamamoto KI, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J, Kumagai A, Dunphy WG. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem. 2007;282:28036–28044. doi: 10.1074/jbc.M704635200. [DOI] [PubMed] [Google Scholar]
- 30.Bermudez VP, et al. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc Natl Acad Sci USA. 2003;100:1633–1638. doi: 10.1073/pnas.0437927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci USA. 2003;100:13827–13832. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ellison V, Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol. 2003;1:E33. doi: 10.1371/journal.pbio.0000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murakami T, et al. Stable interaction between the human proliferating cell nuclear antigen loader complex Ctf18-replication factor C (RFC) and DNA polymerase ε is mediated by the cohesion-specific subunits, Ctf18, Dcc1, and Ctf8. J Biol Chem. 2010;285:34608–34615. doi: 10.1074/jbc.M110.166710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crabbé L, et al. Analysis of replication profiles reveals key role of RFC-Ctf18 in yeast replication stress response. Nature Struct Mol Biol. 2010;17:1391–1397. doi: 10.1038/nsmb.1932. [DOI] [PubMed] [Google Scholar]
- 35.García-Rodríguez LJ, et al. A conserved Polε binding module in Ctf18-RFC is required for S-phase checkpoint activation downstream of Mec1. Nucleic Acids Res. 2015;43:8830–8838. doi: 10.1093/nar/gkv799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duursma AM, Driscoll R, Elias JE, Cimprich KA. A Role for the MRN Complex in ATR Activation via TOPBP1 Recruitment. Mol Cell. 2013;50:116–122. doi: 10.1016/j.molcel.2013.03.006. Identified additional components for ATR activation, though still poorly understood mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee J, Dunphy WG. The Mre11-Rad50-Nbs1 (MRN) complex has a specific role in the activation of Chk1 in response to stalled replication forks. Mol Biol Cell. 2013;24:1343–1353. doi: 10.1091/mbc.E13-01-0025. Identified additional components for ATR activation, though still poorly understood mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cotta-Ramusino C, et al. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science. 2011;332:1313–1317. doi: 10.1126/science.1203430. Identified additional components for ATR activation, though still poorly understood mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindsey-Boltz LA, Kemp MG, Capp C, Sancar A. RHINO forms a stoichiometric complex with the 9-1-1 checkpoint clamp and mediates ATR-Chk1 signaling. Cell Cycle. 2015;14:99–108. doi: 10.4161/15384101.2014.967076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Byun TS, Pacek M, Yee M-C, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. Identified activities that generate ssDNA to activate ATR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zellweger R, et al. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol. 2015;208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nature Struct Mol Biol. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 44.Michael WM, Ott R, Fanning E, Newport J. Activation of the DNA replication checkpoint through RNA synthesis by primase. Science. 2000;289:2133–2137. doi: 10.1126/science.289.5487.2133. [DOI] [PubMed] [Google Scholar]
- 45.Neelsen KJ, Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nature Rev Mol Cell Biol. 2015;16:207–220. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- 46.Couch FB, Cortez D. Fork reversal, too much of a good thing. Cell Cycle. 2014;13:1049–1050. doi: 10.4161/cc.28212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thangavel S, et al. DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol. 2015;208:545–562. doi: 10.1083/jcb.201406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bétous R, et al. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blastyák A, Hajdú I, Unk I, Haracska L. Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol. 2010;30:684–693. doi: 10.1128/MCB.00863-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwab RA, Blackford AN, Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. 2010;29:806–818. doi: 10.1038/emboj.2009.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Collis SJ, et al. FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell. 2008;32:313–324. doi: 10.1016/j.molcel.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 52.Singh TR, et al. ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res. 2013;73:4300–4310. doi: 10.1158/0008-5472.CAN-12-3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gong Z, Kim JE, Leung CCY, Glover JNM, Chen J. BACH1/FANCJ Acts with TopBP1 and Participates Early in DNA Replication Checkpoint Control. Mol Cell. 2010;37:438–446. doi: 10.1016/j.molcel.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie J, et al. FANCJ/BACH1 acetylation at lysine 1249 regulates the DNA damage response. PLoS Genet. 2012;8:e1002786. doi: 10.1371/journal.pgen.1002786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blackford AN, et al. The DNA translocase activity of FANCM protects stalled replication forks. Hum Mol Genet. 2012;21:2005–2016. doi: 10.1093/hmg/dds013. [DOI] [PubMed] [Google Scholar]
- 56.Patro BS, Frøhlich R, Bohr VA, Stevnsner T. WRN helicase regulates the ATR-CHK1-induced S-phase checkpoint pathway in response to topoisomerase-I-DNA covalent complexes. J Cell Sci. 2011;124:3967–3979. doi: 10.1242/jcs.081372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Basile G, Leuzzi G, Pichierri P, Franchitto A. Checkpoint-dependent and independent roles of the Werner syndrome protein in preserving genome integrity in response to mild replication stress. Nucleic Acids Res. 2014;42:12628–12639. doi: 10.1093/nar/gku1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu Y, et al. EEPD1 Rescues Stressed Replication Forks and Maintains Genome Stability by Promoting End Resection and Homologous Recombination Repair. PLoS Genet. 2015;11:e1005675. doi: 10.1371/journal.pgen.1005675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jazayeri A, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nature Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 60.Gong Y, Handa N, Kowalczykowski SC, de Lange T. PHF11 promotes DSB resection, ATR signaling, and HR. Genes Dev. 2017;31:46–58. doi: 10.1101/gad.291807.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lindsey-Boltz LA. Bringing It All Together: Coupling Excision Repair to the DNA Damage Checkpoint. Photochem Photobiol. 2017;93:238–244. doi: 10.1111/php.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aze A, Sannino V, Soffientini P, Bachi A, Costanzo V. Centromeric DNA replication reconstitution reveals DNA loops and ATR checkpoint suppression. Nature Cell Biol. 2016;18:684–691. doi: 10.1038/ncb3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruiz S, et al. A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol Cell. 2016;62:307–313. doi: 10.1016/j.molcel.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Oliveira FMB, et al. Phosphoproteomics Reveals Distinct Modes of Mec1/ATR Signaling during DNA Replication. Mol Cell. 2015;57:1124–1132. doi: 10.1016/j.molcel.2015.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cliby WA, et al. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- 67.Koundrioukoff S, et al. Stepwise Activation of the ATR Signaling Pathway upon Increasing Replication Stress Impacts Fragile Site Integrity. PLoS Genet. 2013;9:e1003643. doi: 10.1371/journal.pgen.1003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van C, Yan S, Michael WM, Waga S, Cimprich KA. Continued primer synthesis at stalled replication forks contributes to checkpoint activation. J Cell Biol. 2010;189:233–246. doi: 10.1083/jcb.200909105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mourón S, et al. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nature Struct Mol Biol. 2013;20:1383–1389. doi: 10.1038/nsmb.2719. [DOI] [PubMed] [Google Scholar]
- 70.Bianchi J, et al. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell. 2013;52:566–573. doi: 10.1016/j.molcel.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.García-Gómez S, et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell. 2013;52:541–553. doi: 10.1016/j.molcel.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wan L, et al. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013;14:1104–1112. doi: 10.1038/embor.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yan S, Michael WM. TopBP1 and DNA polymerase-α directly recruit the 9-1-1 complex to stalled DNA replication forks. J Cell Biol. 2009;184:793–804. doi: 10.1083/jcb.200810185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maréchal A, et al. PRP19 Transforms into a Sensor of RPA-ssDNA after DNA Damage and Drives ATR Activation via a Ubiquitin-Mediated Circuitry. Mol Cell. 2014;53:235–246. doi: 10.1016/j.molcel.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu CS, et al. SUMOylation of ATRIP potentiates DNA damage signaling by boosting multiple protein interactions in the ATR pathway. Genes Dev. 2014;28:1472–1484. doi: 10.1101/gad.238535.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoo HY, Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. Ataxia-telangiectasia mutated (ATM)-dependent activation of ATR occurs through phosphorylation of TopBP1 by ATM. J Biol Chem. 2007;282:17501–17506. doi: 10.1074/jbc.M701770200. [DOI] [PubMed] [Google Scholar]
- 77.Venere M, Snyder A, Zgheib O, Halazonetis TD. Phosphorylation of ATR-interacting protein on Ser239 mediates an interaction with breast-ovarian cancer susceptibility 1 and checkpoint function. Cancer Res. 2007;67:6100–6105. doi: 10.1158/0008-5472.CAN-07-0369. [DOI] [PubMed] [Google Scholar]
- 78.Myers JS, Zhao R, Xu X, Ham AJL, Cortez D. Cyclin-dependent kinase 2 dependent phosphorylation of ATRIP regulates the G2-M checkpoint response to DNA damage. Cancer Res. 2007;67:6685–6690. doi: 10.1158/0008-5472.CAN-07-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang H, et al. ATRIP Deacetylation by SIRT2 Drives ATR Checkpoint Activation by Promoting Binding to RPA-ssDNA. Cell Rep. 2016;14:1435–1447. doi: 10.1016/j.celrep.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu S, et al. ATR Autophosphorylation as a Molecular Switch for Checkpoint Activation. Mol Cell. 2011;43:192–202. doi: 10.1016/j.molcel.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nam EA, et al. Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. J Biol Chem. 2011;286:28707–28714. doi: 10.1074/jbc.M111.248914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 83.Daniel JA, et al. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J Cell Biol. 2008;183:777–783. doi: 10.1083/jcb.200805154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pellegrini M, et al. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 2006;443:222–225. doi: 10.1038/nature05112. [DOI] [PubMed] [Google Scholar]
- 85.Jarrett SG, et al. PKA-mediated phosphorylation of ATR promotes recruitment of XPA to UV-induced DNA damage. Mol Cell. 2014;54:999–1011. doi: 10.1016/j.molcel.2014.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 86.Meirelles GV, et al. ‘Stop Ne(c)king around’: How interactomics contributes to functionally characterize Nek family kinases. World J Biol Chem. 2014;5:141–160. doi: 10.4331/wjbc.v5.i2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Navadgi-Patil VM, Burgers PM. Cell-cycle-specific activators of the Mec1/ATR checkpoint kinase. Biochem Soc Trans. 2011;39:600–605. doi: 10.1042/BST0390600. [DOI] [PubMed] [Google Scholar]
- 88.Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell. 2000;6:839–849. doi: 10.1016/s1097-2765(05)00092-4. [DOI] [PubMed] [Google Scholar]
- 89.Karlsson-Rosenthal C, Millar JBA. Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006;16:285–292. doi: 10.1016/j.tcb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 90.Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol. 2006;18:185–191. doi: 10.1016/j.ceb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 91.Mailand N, et al. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 92.Sørensen CS, et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell. 2003;3:247–258. doi: 10.1016/s1535-6108(03)00048-5. [DOI] [PubMed] [Google Scholar]
- 93.Peng CY, et al. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 94.Sanchez Y, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 95.Fragkos M, Ganier O, Coulombe P, Méchali M. DNA replication origin activation in space and time. Nature Rev Mol Cell Biol. 2015;16:360–374. doi: 10.1038/nrm4002. [DOI] [PubMed] [Google Scholar]
- 96.Shechter D, Costanzo V, Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nature Cell Biol. 2004;6:648–655. doi: 10.1038/ncb1145. [DOI] [PubMed] [Google Scholar]
- 97.Marheineke K, Hyrien O. Control of replication origin density and firing time in Xenopus egg extracts: role of a caffeine-sensitive, ATR-dependent checkpoint. J Biol Chem. 2004;279:28071–28081. doi: 10.1074/jbc.M401574200. [DOI] [PubMed] [Google Scholar]
- 98.Katsuno Y, et al. Cyclin A–Cdk1 regulates the origin firing program in mammalian cells. Proc Natl Acad Sci USA. 2009;106:3184–3189. doi: 10.1073/pnas.0809350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Santocanale C, Diffley JF. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature. 1998;395:615–618. doi: 10.1038/27001. The first study to show the replication checkpoint regulates origin firing. [DOI] [PubMed] [Google Scholar]
- 100.Costanzo V, et al. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell. 2003;11:203–213. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- 101.Karnani N, Dutta A. The effect of the intra-S-phase checkpoint on origins of replication in human cells. Genes Dev. 2011;25:621–633. doi: 10.1101/gad.2029711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yekezare M, Gómez-González B, Diffley JFX. Controlling DNA replication origins in response to DNA damage - inhibit globally, activate locally. J Cell Sci. 2013;126:1297–1306. doi: 10.1242/jcs.096701. [DOI] [PubMed] [Google Scholar]
- 103.Deegan TD, Yeeles JT, Diffley JF. Phosphopeptide binding by Sld3 links Dbf4-dependent kinase to MCM replicative helicase activation. EMBO J. 2016;35:961–973. doi: 10.15252/embj.201593552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Heffernan TP, et al. Cdc7-Dbf4 and the human S checkpoint response to UVC. J Biol Chem. 2007;282:9458–9468. doi: 10.1074/jbc.M611292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zegerman P, Diffley JFX. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature. 2010;467:474–478. doi: 10.1038/nature09373. Identified key replication checkpoint targets and phosphorylation events for inhibiting origin firing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhao H, Watkins JL, Piwnica-Worms H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci USA. 2002;99:14795–14800. doi: 10.1073/pnas.182557299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu H, et al. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature. 2010;467:343–346. doi: 10.1038/nature09350. Identified key replication checkpoint targets and phosphorylation events for inhibiting origin firing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lopez-Mosqueda J, et al. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature. 2010;467:479–483. doi: 10.1038/nature09377. Identified key replication checkpoint targets and phosphorylation events for inhibiting origin firing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guo C, et al. Interaction of Chk1 with Treslin Negatively Regulates the Initiation of Chromosomal DNA Replication. Mol Cell. 2015;57:1–14. doi: 10.1016/j.molcel.2014.12.003. Identified key replication checkpoint targets and phosphorylation events for inhibiting origin firing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ge XQ, Blow JJ. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol. 2010;191:1285–1297. doi: 10.1083/jcb.201007074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Petermann E, et al. Chk1 Requirement for High Global Rates of Replication Fork Progression during Normal Vertebrate S Phase. Mol Cell Biol. 2006;26:3319–3326. doi: 10.1128/MCB.26.8.3319-3326.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Petermann E, Caldecott KW. Evidence that the ATR/Chk1 pathway maintains normal replication fork progression during unperturbed S phase. Cell Cycle. 2006;5:2203–2209. doi: 10.4161/cc.5.19.3256. [DOI] [PubMed] [Google Scholar]
- 113.Maya-Mendoza A, Petermann E, Gillespie DAF, Caldecott KW, Jackson DA. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J. 2007;26:2719–2731. doi: 10.1038/sj.emboj.7601714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Petermann E, Woodcock M, Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci USA. 2010;107:16090–16095. doi: 10.1073/pnas.1005031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Couch FB, et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610–1623. doi: 10.1101/gad.214080.113. Demonstrated that ATR regulation of SMARCAL1 is needed to prevent excessive fork remodeling that results in SLX4-dependent fork breakage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Técher H, et al. Signaling from Mus81-Eme2-Dependent DNA Damage Elicited by Chk1 Deficiency Modulates Replication Fork Speed and Origin Usage. Cell Rep. 2016;14:1114–1127. doi: 10.1016/j.celrep.2015.12.093. [DOI] [PubMed] [Google Scholar]
- 117.Thomson AM, Gillespie PJ, Blow JJ. Replication factory activation can be decoupled from the replication timing program by modulating Cdk levels. J Cell Biol. 2010;188:209–221. doi: 10.1083/jcb.200911037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cortez D, Glick G, Elledge SJ. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci USA. 2004;101:10078–10083. doi: 10.1073/pnas.0403410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Trenz K, Errico A, Costanzo V. Plx1 is required for chromosomal DNA replication under stressful conditions. EMBO J. 2008;27:876–885. doi: 10.1038/emboj.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen YH, et al. ATR-Mediated Phosphorylation of FANCI Regulates Dormant Origin Firing in Response to Replication Stress. Mol Cell. 2015;58:323–338. doi: 10.1016/j.molcel.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lopes M, et al. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–561. doi: 10.1038/35087613. Showed in S. cerevisiae that the ATR pathway regulates both fork stability and origin firing. [DOI] [PubMed] [Google Scholar]
- 122.Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–557. doi: 10.1038/35087607. Showed in S. cerevisiae that the ATR pathway regulates both fork stability and origin firing. [DOI] [PubMed] [Google Scholar]
- 123.Chanoux RA, et al. ATR and H2AX cooperate in maintaining genome stability under replication stress. J Biol Chem. 2009;284:5994–6003. doi: 10.1074/jbc.M806739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM. DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J. 2003;22:4325–4336. doi: 10.1093/emboj/cdg391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.De Piccoli G, et al. Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol Cell. 2012;45:696–704. doi: 10.1016/j.molcel.2012.01.007. Provided evidence that replisome stability is independent of the replication checkpoint. [DOI] [PubMed] [Google Scholar]
- 126.Dungrawala H, et al. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol Cell. 2015;59:998–1010. doi: 10.1016/j.molcel.2015.07.030. Provided evidence that replisome stability is independent of the replication checkpoint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cotta-Ramusino C, et al. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell. 2005;17:153–159. doi: 10.1016/j.molcel.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 128.Ragland RL, et al. RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev. 2013;27:2259–2273. doi: 10.1101/gad.223180.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wyatt HDM, Sarbajna S, Matos J, West SC. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol Cell. 2013;52:234–247. doi: 10.1016/j.molcel.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 130.Duda H, et al. A Mechanism for Controlled Breakage of Under-replicated Chromosomes during Mitosis. Dev Cell. 2016;39:740–755. doi: 10.1016/j.devcel.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 131.Domínguez-Kelly R, et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194:567–579. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pepe A, West SC. MUS81-EME2 promotes replication fork restart. Cell Rep. 2014;7:1048–1055. doi: 10.1016/j.celrep.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Forment JV, Blasius M, Guerini I, Jackson SP. Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PLoS ONE. 2011;6:e23517. doi: 10.1371/journal.pone.0023517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Murfuni I, et al. Survival of the replication checkpoint deficient cells requires MUS81-RAD52 function. PLoS Genet. 2013;9:e1003910. doi: 10.1371/journal.pgen.1003910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hanada K, et al. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nature Struct Mol Biol. 2007;14:1096–1104. doi: 10.1038/nsmb1313. [DOI] [PubMed] [Google Scholar]
- 136.Buisson R, Boisvert JL, Benes CH, Zou L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol Cell. 2015;59:1011–1024. doi: 10.1016/j.molcel.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Errico A, Costanzo V. Mechanisms of replication fork protection: a safeguard for genome stability. Crit Rev Biochem Mol Biol. 2012;47:222–235. doi: 10.3109/10409238.2012.655374. [DOI] [PubMed] [Google Scholar]
- 138.Lossaint G, et al. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell. 2013;51:678–690. doi: 10.1016/j.molcel.2013.07.023. Showed that ATR signaling regulates the S-phase checkpoint in part by promoting an interaction of FANCD2 with the MCM helicase. [DOI] [PubMed] [Google Scholar]
- 139.Kumar S, Huberman JA. Checkpoint-dependent regulation of origin firing and replication fork movement in response to DNA damage in fission yeast. Mol Cell Biol. 2009;29:602–611. doi: 10.1128/MCB.01319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Seiler JA, Conti C, Syed A, Aladjem MI, Pommier Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol Cell Biol. 2007;27:5806–5818. doi: 10.1128/MCB.02278-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Conti C, Seiler JA, Pommier Y. The mammalian DNA replication elongation checkpoint: implication of Chk1 and relationship with origin firing as determined by single DNA molecule and single cell analyses. Cell Cycle. 2007;6:2760–2767. doi: 10.4161/cc.6.22.4932. [DOI] [PubMed] [Google Scholar]
- 142.Toledo LI, et al. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell. 2013;155:1088–1103. doi: 10.1016/j.cell.2013.10.043. Showed that in conditions of high replicative stress, ATR is needed to prevent excessive accumulation of ssDNA, which can exhaust the availability of RPA. [DOI] [PubMed] [Google Scholar]
- 143.Murphy AK, et al. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol. 2014;206:493–507. doi: 10.1083/jcb.201404111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J Cell Sci. 2009;122:4070–4080. doi: 10.1242/jcs.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Buisson R, et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol Cell. 2017;65:336–346. doi: 10.1016/j.molcel.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ahlskog JK, Larsen BD, Achanta K, Sørensen CS. ATM/ATR-mediated phosphorylation of PALB2 promotes RAD51 function. EMBO Rep. 2016;17:671–681. doi: 10.15252/embr.201541455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Somyajit K, Basavaraju S, Scully R, Nagaraju G. ATM- and ATR-mediated phosphorylation of XRCC3 regulates DNA double-strand break-induced checkpoint activation and repair. Mol Cell Biol. 2013;33:1830–1844. doi: 10.1128/MCB.01521-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kolinjivadi AM, et al. Moonlighting at replication forks - a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett. 2017;591:1083–1100. doi: 10.1002/1873-3468.12556. [DOI] [PubMed] [Google Scholar]
- 150.Sale JE. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb Perspect Biol. 2013;5:a012708. doi: 10.1101/cshperspect.a012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Göhler T, Sabbioneda S, Green CM, Lehmann AR. ATR-mediated phosphorylation of DNA polymerase η is needed for efficient recovery from UV damage. J Cell Biol. 2011;192:219–227. doi: 10.1083/jcb.201008076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Davies SL, North PS, Dart A, Lakin ND, Hickson ID. Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol. 2004;24:1279–1291. doi: 10.1128/MCB.24.3.1279-1291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ammazzalorso F, Pirzio LM, Bignami M, Franchitto A, Pichierri P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010;29:3156–3169. doi: 10.1038/emboj.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zhao X, Muller EG, Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 155.Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998;94:595–605. doi: 10.1016/s0092-8674(00)81601-3. [DOI] [PubMed] [Google Scholar]
- 156.Sanvisens N, de Llanos R, Puig S. Function and regulation of yeast ribonucleotide reductase: cell cycle, genotoxic stress, and iron bioavailability. Biomed J. 2013;36:51– 58. doi: 10.4103/2319-4170.110398. [DOI] [PubMed] [Google Scholar]
- 157.D’Angiolella V, et al. Cyclin F-Mediated Degradation of Ribonucleotide Reductase M2 Controls Genome Integrity and DNA Repair. Cell. 2012;149:1023–1034. doi: 10.1016/j.cell.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lopez-Contreras AJ, et al. Increased Rrm2 gene dosage reduces fragile site breakage and prolongs survival of ATR mutant mice. Genes Dev. 2015;29:690–695. doi: 10.1101/gad.256958.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Murga M, et al. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nature Genet. 2009;41:891–898. doi: 10.1038/ng.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Karnitz LM, Zou L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin Cancer Res. 2015;21:4780–4785. doi: 10.1158/1078-0432.CCR-15-0479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yeeles JTP, Deegan TD, Janska A, Early A, Diffley JFX. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature. 2015;519:431–435. doi: 10.1038/nature14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tuduri S, Tourrière H, Pasero P. Defining replication origin efficiency using DNA fiber assays. Chromosome Res. 2010;18:91–102. doi: 10.1007/s10577-009-9098-y. [DOI] [PubMed] [Google Scholar]
- 163.Ball HL, Myers JS, Cortez D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol Biol Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ball HL, Cortez D. ATRIP oligomerization is required for ATR-dependent checkpoint signaling. J Biol Chem. 2005;280:31390–31396. doi: 10.1074/jbc.M504961200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ball HL, et al. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Mol Cell Biol. 2007;27:3367–3377. doi: 10.1128/MCB.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Itakura E, Sawada I, Matsuura A. Dimerization of the ATRIP protein through the coiled-coil motif and its implication to the maintenance of stalled replication forks. Mol Biol Cell. 2005;16:5551–5562. doi: 10.1091/mbc.E05-05-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sawicka M, et al. The Dimeric Architecture of Checkpoint Kinases Mec1ATR and Tel1ATM Reveal a Common Structural Organization. J Biol Chem. 2016;291:13436–13447. doi: 10.1074/jbc.M115.708263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mordes DA, Cortez D. Activation of ATR and related PIKKs. Cell Cycle. 2008;7:2809–2812. doi: 10.4161/cc.7.18.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]