

Summary
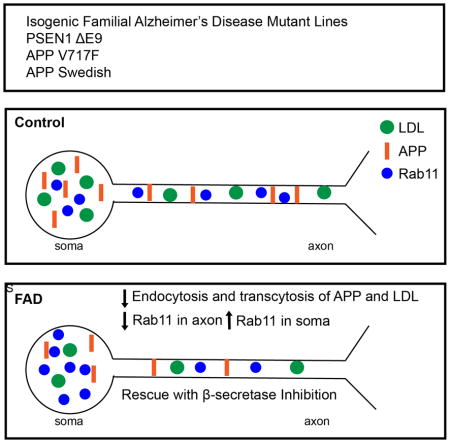
We investigated early phenotypes caused by familial Alzheimer’s Disease (fAD) mutations in isogenic human iPSC-derived neurons. Analysis of neurons carrying fAD PS1 or APP mutations introduced using genome editing technology at the endogenous loci revealed that fAD mutant neurons had previously unreported defects in the recycling state of endocytosis and soma-to-axon transcytosis of APP and lipoproteins. The endocytosis reduction could be rescued through treatment with a β-secretase inhibitor. Our data suggest that accumulation of β-CTF fragments of APP, but not Aβ, slow vesicle formation from an endocytic recycling compartment marked by the transcytotic GTPase Rab11. We confirm previous results that endocytosis is affected in AD, and extend these to uncover a neuron-specific defect. Decreased lipoprotein endocytosis and transcytosis to the axon suggests that a neuron-specific impairment in endocytic axonal delivery of lipoproteins and other key materials might compromise synaptic maintenance in fAD.
eTOC Blurb
Woodruff et al find that FAD mutant neurons display abnormal endocytosis and transcytosis of APP and lipoproteins that is mediated by Rab11. Defective endocytosis and transcytosis of lipoproteins is rescued by β-secretase inhibition.
Introduction
Alzheimer’s Disease (AD) is a progressive and devastating neurodegenerative disorder that affects more than 30 million people worldwide including 11% of those over 65 years of age and 32% of those over 85 (Fargo and Bleiler, 2014). AD is characterized by progressive cerebral dysfunction, memory loss, synapse loss, and neuronal impairment leading to cell death. To date, there are no disease-modifying treatments that can cure or reduce the progression of AD. Genetically, AD is segmented into two populations: sporadic AD (sAD) where the underlying primary cause is not known and rare, autosomal-dominant mutations causing familial AD (fAD) (Gatz et al., 2006). The common pathological features of sAD and fAD patients are the accumulation of senile plaques composed of aggregated amyloid-β (Aβ) and neurofibrillary tangles (NFT), composed of hyper-phosphorylated tau (Spires-Jones and Hyman, 2014). Importantly, many experimental findings regarding AD have come from overexpression studies or studies of non-neuronal cells that lack the unique polarization and compartmentalization of neurons.
The amyloid cascade hypothesis of AD posits that extracellular Aβ fragments of proteolytically processed amyloid precursor protein (APP) and intraneuronal tau accumulate abnormally in sAD and fAD. These accumulations are proposed to drive cellular stress, neurotoxicity, and ultimately synapse loss and neurodegeneration (Toyn and Ahlijanian, 2014). Previous work also reported apparent defects in early endocytosis in post-mortem brains of AD patients (Cataldo et al., 2001; Ginsberg et al., 2010). Complementary investigations in overexpression and non-neuronal cellular models also pointed to defects in early endocytosis and endocytic dysfunction driven not by Aβ, but instead by β C-terminal fragments (β-CTF) of APP (Von Bartheld, 2004; Cataldo et al., 2000; Ginsberg et al., 2010; Karch and Goate, 2014; Lee et al., 2010; Maxfield, 2014). In addition, unbiased screens in non-neuronal cells consistently identified regulators of endocytic trafficking as key to normal levels of APP processing (Cataldo et al., 2000; Ginsberg et al., 2010; Treusch et al., 2011). However, many of the key investigations did not utilize normal expression levels of AD-related proteins or did not fully examine sorting pathways in highly polarized neurons. Thus, while several important insights on the role of endocytosis in regulating APP processing and sorting have been obtained from various cell models, there is little known about endocytic-dependent trafficking in polarized human neurons with mutations expressed at normal levels.
We tested whether isogenic iPSC-derived human neurons with fAD PS1 and APP mutations induced by genome editing mutagenesis (TALEN and CRISPR/Cas9) at the endogenous loci display amyloid-independent cellular trafficking and transport phenotypes, beyond the known effects on APP processing, that could account for the development of fAD (Von Bartheld, 2004; Cataldo et al., 2000; Ginsberg et al., 2010; Karch and Goate, 2014; Lee et al., 2010; Maxfield, 2014). Human neurons expressing fAD mutant PS1 and APP at normal expression levels from the endogenous loci, alter subcellular distribution and trafficking of APP and internalized lipoprotein, leading to elevated levels of APP in the soma and reduced levels in the axons. The redistribution of APP is accompanied by the concurrent accumulation of Rab11 endosomal vesicles in the neuronal soma and reduced Rab11 axonal staining, suggesting that the reduction in axonal APP and lipoproteins can be explained by reduced Rab11-dependent soma-to-axon transcytosis and defects in the recycling endosome (Ascaño et al., 2009; Von Bartheld, 2004; Buggia-Prévot et al., 2014). Knockdown of Rab11 also leads to reductions in APP axonal density and lipoprotein endocytosis and transcytosis. Our study reveals that a common early defect among fAD PS1 and APP mutations is APP β-CTF accumulation-induced impairment of a key neuron-specific traffic pathway, soma to axon transcytosis caused by defects in the recycling endosome.
Results
The PS1ΔE9 Mutation Increases APP in the Soma of Human Neurons and Decreases APP in Axons
PS1 has been reported to have a role in APP trafficking in primary neurons and in non-neuronal cell types (Burns et al., 2003; Cai et al., 2003; Gandy et al., 2007; Zhang et al., 2006). PS1 knockout has been reported to increase cell surface APP (Leem et al., 2002), while PS1 fAD mutations have been shown to delay APP arrival at the cell surface (Cai et al., 2003; Gandy et al., 2007). To determine whether the PS1ΔE9 mutation affects APP localization in human neurons, we generated purified neurons from isogenic iPSC lines from wild-type (PS1wt/wt) and lines heterozygous (PS1wt/ΔE9) and homozygous (PS1ΔE9/ΔE9) for the PS1ΔE9 mutation (Woodruff et al., 2013). We stained with an APP antibody, which has minimal staining in cells from APP knockout mice (Guo et al., 2012; Weissmiller et al., 2015). We found that PS1wt/ΔE9 and PS1ΔE9/ΔE9 neurons exhibited a modest but significant increase in intracellular APP staining in the cell body (Figure 1A). To determine whether APP was also increased on the surface of PS1ΔE9 neurons, we stained unpermeabilized purified neurons with an N-terminal APP antibody that recognizes the extracellular portion of APP (22C11) (Figure 1B). We observed increased APP on the soma surface of PS1ΔE9 neurons (Figure 1B). As a control, we also stained unpermeabilized neurons with a C-terminal antibody and observed minimal staining, as would be expected since the C-terminus of APP would not be present on the cell surface (Figure 1C).
Figure 1. PS1ΔE9 iPSC-derived neurons exhibit altered sub-cellular distribution of APP.

A) PS1ΔE9 neurons have a gene-dose dependent increase in soma APP intensity. Data are represented as mean ± SEM of all cell values with 10 PS1wt/wt, 6 PS1wt/ΔE9 and 10 PS1ΔE9/ΔE9 biological replicates. (PS1wt/ΔE9 p<0.05 and PS1ΔE9/ΔE9 p<0.0001) Scale bar = 20μm B) Surface intensity of APP is increased in PS1wt/ΔE9 and PS1ΔE9/ΔE9 (p<0.01) neurons compared to PS1wt/wt. Scale bar = 5μm C) PS1wt/wt neurons exhibited minimal APP CTF staining in unpermeabilized cells and dramatically increased NTF staining when permeabilized. Data represent the mean ± SEM from 2 immunofluorescence experiments with 2 biological replicates per control (p = 0.0262) Scale bar = 5μm D) Average density of APP (PS1wt/ΔE9 p<0.05 and PS1ΔE9/ΔE9 p<0.0001) and individual puncta intensities (PS1wt/ΔE9 p<0.0001 and PS1ΔE9/ΔE9 p<0.0001) were decreased in a gene-dose dependent manner in PS1ΔE9 axons. Data represent the pooled median values with Tukey whiskers from 3 immunofluorescence experiments representing 7 PS1wt/wt, 4 PS1wt/ΔE9 and 6 PS1ΔE9/ΔE9 biological replicates. Scale bar = 5μm E) A schematic of the microfluidic devices. F) sAPPα/β ratios in soma were not statistically different (p=0.9614), but the ratio of axonally secreted sAPPα/β was increased in PS1ΔE9/ΔE9 axons (p=0.0199). Relative amounts of secreted sAPPα and sAPPβ in PS1ΔE9/ΔE9 axons compared to PS1wt/wt. sAPPβ levels are reduced (p=0.0056), but not sAPPα levels (p=0.0540) in PS1ΔE9/ΔE9 axons. Data represent 4 PS1wt/wt and 4 PS1ΔE9/ΔE9 biological replicates. See also Figure S1.
We previously published that PS1ΔE9 human neurons do not exhibit changes in total levels of full-length APP though there are increases in the APP CTFs (Woodruff et al., 2013). The increase in soma APP suggests that APP CTFs are accumulating in the soma of purified neurons and/or that APP is missorted possibly at the expense of axonal APP. We therefore assessed APP staining in the axons of PS1ΔE9 neurons. To ensure that we were quantifying staining in axons and not dendritic processes, we made use of microfluidic compartments, which separate axons from the bulk somatodendritic population (Figure 1E) (Niederst et al., 2015; Selfridge et al., 2015; Taylor et al., 2006). Neurons grown in microfluidic devices extend long processes into the axonal space that do not stain for the somatodendritic marker Map2 and that do stain for the axonal marker neurofilament-H (SMI31) (Selfride et al., 2015) (Figure 1E and Figure S1A). We observed that PS1ΔE9 axons grown in microfluidic devices have decreased axonal APP puncta and APP puncta intensity (Figure 1D); this decrease is sensitive to PS1ΔE9 gene dose.
To further characterize APP fragments that are present in axons, we also measured soluble APP (sAPP) fragments, sAPPα and sAPPβ. To quantify secreted sAPP, we plated neurons in compartments and allowed them to extend their axons into the axonal space. We then performed a full media change, kept the axons in fluidic isolation, and harvested media from the soma and axon sides. When we measured the sAPPα/sAPPβ ratio from the soma side of the cultures we observed no significant differences between PS1wt/wt and PS1ΔE9/ΔE9 genotypes (Figure 1F). Additionally, there were no significant differences in the total levels of sAPPα or sAPPβ from the soma side (Figure S1B). However, when we quantified the sAPPα/sAPPβ ratio from axons only, we observed a significant increase from the PS1ΔE9/ΔE9 axons compared to PS1wt/wt (Figure 1F). Upon quantification of the total levels of sAPPα and sAPPβ, we determined that the ratio was increased in PS1ΔE9/ΔE9 axons primarily due to a reduction in sAPPβ (Figure 1F). To test whether the phenotypic differences that we observed in PS1ΔE9 neurons might be due to differences in neuronal subtypes in our cultures we stained neurons of each genotype with the neuronal subtype markers GABA, GAD65/67 and vGlut (Figure S1C). We found no significant differences in the proportion of cells that stained positive for GABA, GAD65/67 or vGlut between different genotypes (Figure S1C).
Rab11 Distribution is Altered in PS1ΔE9 Neurons
There are at least two pathways by which APP can be delivered to the axon. The first is by direct delivery from the trans Golgi network (TGN) and the second is by an indirect pathway where APP first arrives at the cell surface of the somatodendritic compartment, then undergoes endocytosis and sorting to the axon. The indirect pathway is a process known as transcytosis and multiple cargo including TrkA (Ascaño et al., 2009) and L1/NgCAM (Anderson et al., 2005; Yap et al., 2008) have been demonstrated to be sorted along this pathway in neurons.
An endocytic regulator that functions at the intersection of the TGN and transcytotic pathways is the Rab GTPase Rab11 (Welz et al., 2014). Rab11 has a well-established role in mediating recycling of many receptors including transferrin receptor (Ullrich et al., 1996), and LDL receptors (Sakane et al., 2010; Takahashi et al., 2007). In addition to its role in recycling, Rab11 has also been shown to mediate transcytosis in epithelial cells and neurons. Specifically, in neurons, TrkA receptors undergo Rab11-dependent transcytosis to the axon (Ascaño et al., 2009; Lazo et al., 2013). Rab11 may also be involved in trafficking of BACE1 to the axon (Buggia-Prévot et al., 2014), colocalizes with some APP in axons (Niederst et al., 2015), and was recently identified in an unbiased Rab GTPase screen in non-neuronal cells as a regulator of Aβ and sAPPβ production (Udayar et al., 2013). Intriguingly, presenilins have also been reported to interact directly with Rab11 through their hydrophilic loop (Dumanchin et al., 1999).
To determine if Rab11 could be playing a role in the reduction of axonal APP in PS1ΔE9 cells, we stained neurons with a Rab11 antibody and measured Rab11 in the somatodendritic and axonal compartments. The Rab11 staining was reminiscent of the altered APP distribution such that PS1ΔE9/ΔE9 neurons exhibited increased soma Rab11 intensity, Rab11 puncta, and Rab11 puncta area (Figure 2A). In axons, both the PS1wt/ΔE9 and PS1ΔE9/ΔE9 genotypes had decreased Rab11 puncta density and puncta intensity (Figure 2B). In support of a role of Rab11-dependent trafficking of APP to the axon, shRNA-mediated knockdown of Rab11 on the soma-side of neurons grown in microfluidic devices resulted in a ~40% reduction in APP axonal density (Figure 2C). In keeping with a transcytotic route of APP to the axon, somatodendritic inhibition of endocytosis with Dynasore led to reduced APP (~25% less) and Rab11 (~40% less) density in axons (Figure 2D).
Figure 2. PS1ΔE9/ΔE9 neurons exhibit altered Rab11 distribution.

A) PS1wt/wt neurons exhibited distinct punctate pattern of Rab11 staining in soma, PS1ΔE9/ΔE9 neurons had a marked accumulation of Rab11 in soma (arrowheads). Quantification of staining showed that soma Rab11 intensity (PS1wt/ΔE9 p=0.3215 and PS1ΔE9/ΔE9 p<0.0001), puncta count (PS1wt/ΔE9 p=0.5876 and PS1ΔE9/ΔE9 p<0.0001), and puncta area (PS1wt/ΔE9 p=0.6791 and PS1ΔE9/ΔE9 p<0.0001) were all significantly increased in PS1ΔE9/ΔE9 neurons. Data represent the mean ± SEM or median with Tukey whiskers from 4 PS1wt/wt, 4 PS1wt/ΔE9 and 4 PS1ΔE9/ΔE9 biological replicates. Scale bar = 20μm B) Quantification of Rab11 in axons. Rab11 density (PS1wt/ΔE9 p=0.0003 and PS1ΔE9/ΔE9 p<0.0001) and puncta intensity (PS1wt/ΔE9 p<0.0001 and PS1ΔE9/ΔE9 p<0.0001) were reduced in a dose-dependent manner in PS1ΔE9 neurons. Data represent the mean ± SEM or median with Tukey whiskers of 6 PS1wt/wt, 4 PS1wt/ΔE9 and 5 PS1ΔE9/ΔE9 biological replicates. Scale bar = 5μm C) Knockdown of Rab11a diminished APP to ~60% of PS1wt/wt levels. Data represent the average of 4 untreated PS1wt/wt and 3 Rab11 shRNA-treated PS1wt/wt biological replicates. (Rab11 density p <0.0001, Rab11 intensity p <0.0001, APP density p <0.0001, APP intensity p <0.0001) D) Inhibition of soma endocytosis resulted in ~50% decrease in Rab11 density (first inset) and approximately a 20% decrease in APP density (second inset). Data represent the mean ± SEM or median with Tukey whiskers from axons of 4 untreated PS1wt/wt and 3 Rab11 shRNA-treated PS1wt/wt biological replicates. (Rab11 density p <0.0001, Rab11 intensity p <0.0001, APP density p =0.0018, APP intensity p <0.0001). See also Figure S2.
Previous publications have implicated alterations in early endosomes and lysosomes in PS1 knockout cells (Coen et al., 2012; Lee et al., 2010; Neely et al., 2011). We, therefore, looked at the early endosome effector, EEA1, and the lysosomal marker, Lamp2, even though they are not thought to traffic substantially to axons (Wilson et al., 2000). Despite evidence in the literature that early endosomes and lysosomes are dramatically affected in fAD and sAD, EEA1 and Lamp2 staining in PS1ΔE9 neurons was not obviously different suggesting that they are not playing an active role in sorting APP to the axon (Figure S2 A and B).
The PS1ΔE9 Mutation Decreases Endocytosis and Transcytosis of APP and LDL
Since we observed alterations in the subcellular distribution of both APP and Rab11 in PS1ΔE9 neurons, we investigated whether endocytosis and/or transcytosis could account for the APP localization changes. To assess endocytosis of APP we treated purified neurons with an N-terminal APP antibody (22C11) and allowed cells to internalize antibody for 30 minutes, 2 hours or 4 hours and fixed cells at each of those time points. We then stained with a secondary antibody and quantified the amount of APP endocytosis at each time point (Figure 3A). We observed a decrease in APP puncta in the PS1ΔE9/ΔE9 genotype starting at 30 minutes compared to PS1wt/wt and this decrease was more prominent at both 2 and 4 hours (Figure 3A). The PS1wt/ΔE9 genotype also exhibited decreased APP puncta compared to PS1wt/wt at the 2 and 4 hour time points. As a control to confirm whether the APP antibody was being endocytosed uniquely via antigen binding, we performed an endocytosis assay with a non-specific antibody and found minimal uptake (Figure S3A). To assess whether this endocytosis defect was specific to APP or common to other cargo, we also measured uptake of fluorescently labeled LDL at 30 minutes, 1 hour, 2 hours and 4 hours (Figure 3B). Similar to what was observed with APP, LDL puncta intensity and LDL puncta density were reduced in PS1wt/ΔE9 and PS1ΔE9/ΔE9 neurons at 2 hours and 4 hours (Figure 3B). To test if the differences observed were due to a non-specific defect in endocytosis, we also quantified uptake of fluorescently labeled dextran as a marker of bulk endocytosis. We did not observe any significant differences in dextran endocytosis at any time point (Figure 3C). Additionally, we quantified endocytosis of fluorescently labeled transferrin and similar to dextran we did not observe any significant differences (Figure S3B and C)
Figure 3. PS1 ΔE9 Neurons Exhibit Reduced Endocytosis and Transcytosis of APP and LDL.

A) Example images of internalization of APP 22C11 antibody at 30 minutes in PS1wt/wt and PS1ΔE9/ΔE9 neurons. Bar graphs depict average APP puncta count and average intensity normalized to cell area at time 30min (PS1wt/ΔE9 p=0.1341 and PS1ΔE9/ΔE9 p<0.0001), 120min (PS1wt/ΔE9 p<0.0001 and PS1ΔE9/ΔE9 p<0.0001), and 240min (PS1wt/ΔE9 p=0.0024 and PS1ΔE9/ΔE9 p<0.0001). Data represent the mean ± SEM of a minimum of 50 neurons per time point from 4 PS1wt/wt, 4 PS1wt/ΔE9 and 4 PS1ΔE9/ΔE9 biological replicates. Scale bar = 10μm B) Example images of LDL-BODIPY labeling in PS1wt/wt and PS1ΔE9/ΔE9 neurons. Graphs depict quantification of LDL puncta count and intensity normalized to cell area. As shown, PS1wt/ΔE9 and PS1ΔE9/ΔE9 neurons have reduced APP and LDL soma endocytosis, with the LDL defect appearing most prominently at 240min. Puncta data represent the pooled mean ± SEM or median with Tukey whiskers of over 140 cells per time point and 4–6 biological replicates per genotype per time point. 30 min (PS1wt/ΔE9 p=0.0671 and PS1ΔE9/ΔE9 p=0.1637), 60 min (PS1wt/ΔE9 p=0.0013 and PS1ΔE9/ΔE9 p<0.0001), 120 min (PS1wt/ΔE9 p<0.0001 and PS1ΔE9/ΔE9 p<0.0001), and 240 min (PS1wt/ΔE9 p<0.0001 and PS1ΔE9/ΔE9 p<0.0001). Scale bar = 20μm C) Example images and a graph of intensity quantification are depicted for endocytosis of the fluid phase fixable marker, Dextran-TetramethylRhodamine. Data represent the mean ± SEM of soma intensities across 23–28 images per time point representing 8 PS1wt/wt, 8 PS1wt/ΔE9 and 8 PS1ΔE9/ΔE9 biological replicates. 30 min (PS1wt/ΔE9 p=0.1683 and PS1ΔE9/ΔE9 p=0.6532), 60 min (PS1wt/ΔE9 p=0.6265 and PS1ΔE9/ΔE9 p=0.8126), 120 min (PS1wt/ΔE9 p=0.9117 and PS1ΔE9/ΔE9 p=0.5285), and 240 min (PS1wt/ΔE9 p=0.1595 and PS1ΔE9/ΔE9 p=0.6465). Scale bar = 20μm D and E) Neurons were grown in microfluidic devices and then allowed to internalized either APP antibody (densities PS1wt/ΔE9 p <0.0001 and PS1ΔE9/ΔE9 p = 0.0011) or LDL-BODIPY (densities PS1wt/ΔE9 p <0.0001 and PS1ΔE9/ΔE9 p = 0.0024) on the soma side with the axons in fluidic isolation. All intensity comparisons were p<0.0001. Axons that passed through the channels were imaged and puncta densities and intensities were evaluated. Data represent the mean ± SEM of 4 PS1wt/wt, 3 PS1wt/ΔE9 and 4 PS1ΔE9/ΔE9 biological replicates per stain. Scale bar = 5μm. See also Figure S3.
We measured transcytosis of APP and LDL by growing neurons in microfluidic compartments. The axon side of the compartment was kept in fluidic isolation from the soma side and APP antibody or labeled LDL was added only to the soma side. Transcytotic delivery of cargo from the soma to the axon is a relatively slow process since internalized cargo has to travel long distances (on the order of millimeters in cultured neurons). After 4 hours of endocytosis, cells were fixed and we quantified the amount of anterogradely transcytosed APP by using an anti-mouse secondary antibody; fluorescent LDL was measured directly (Figure 3D, E). We observed that both the PS1wt/ΔE9 and PS1ΔE9/ΔE9 genotypes exhibited decreased APP axonal density and APP intensity after 4 hours of transcytosis (Figure 3D). Similarly, LDL axonal density and intensity were also decreased in PS1wt/ΔE9 and PS1ΔE9/ΔE9 axons after 4 hours (Figure 3E). These results suggest that the reduction in basal axonal APP in PS1ΔE9 neurons is due to a decrease in a constitutive transcytosis pathway.
Rab11 Mediates Transcytosis of APP and LDL
To further investigate the role of Rab11 in endocytosis of LDL and transcytosis of APP and LDL, we used shRNA-mediated knockdown of Rab11 in wild type neurons (Figure 4B). Knockdown of Rab11 in PS1wt/wt neurons almost completely abolished LDL endocytosis in the somatodendritic region (Figure 4A) and transcytosed LDL was undetectable under Rab11 knockdown conditions (Figure 4C and D). Further support for a role of Rab11 mediated transcytosis of APP comes from co-staining experiments where axonal transcytosed APP (22C11) were stained with Rab11. As seen in Figure S4, 35% of Rab11 overlapped with transcytosed APP in PS1wt/wt axons and about 25% of transcytosed APP overlapped with Rab11 in PS1wt/wt axons. The percent of Rab11 with transcytosed 22C11 was reduced in PS1ΔE9/ΔE9 axons (as expected given the reduced density of transcytosed 22C11), but the percent of 22C11 with Rab11 remained steady (Figure S4A).
Figure 4. Rab11 Mediates Endocytosis and Transcytosis of LDL.

A) PS1wt/wt neurons were transduced with lentivirus containing control (CTRL) or Rab11 shRNA (Rab11 KD) and then treated with LDL for 4 hours. Rab11 KD data represent mean ± SEM of soma obtained from 3 biological replicates of PS1wt/wt (p <0.0001). Scale bar = 10μm B) Western blot of cells transduced with control or one of 3 different Rab11 shRNAs. C) PS1wt/wt axons and PS1wt/wt axons with Rab11 knockdown and co-stains. Scale bar = 5μm D) Vesicle densities of Rab11 and LDL, under control and Rab11 knockdown conditions. For LDL density graph, Rab11 knockdown (KD) resulted in non-detectable (N.D.) levels of axonal LDL. Puncta seen in Rab11 KD were the result of background auto fluorescence. LDL transyctosis data represent the mean ± SEM of 3 biological replicates. See also Figure S4.
PS1ΔE9 Impairs Recycling of LRP1
The absence of a lipoprotein endocytic defect at early time-points in PS1ΔE9 neurons suggested that transcription, levels, degradation, or recycling of the LDL receptor may be driving the reduction in transcytosis. While there are many potential LDL receptors, LRP1 was an attractive candidate because of its high expression in brain and neuronal samples (Zhang et al., 2014). To determine if transcription or degradation of LRP1 was affected in PS1ΔE9 neurons, we treated purified human neurons with unlabeled LDL and harvested neurons for mRNA and protein. We did not observe differences in LRP1 mRNA or protein levels (Figure 5A) at baseline or after LDL treatment, suggesting that transcription, total levels, or degradation of LRP1 after LDL treatment are not playing a role in the reduced endocytosis of LDL. Though PS1 has been hypothesized to drive reduced degradation of proteins because of alterations in lysosomal pH (Nixon and Mcbrayer, 2013; Nixon and Yang, 2011), we did not observe changes in lysosomal pH in the PS1ΔE9 neurons as assessed by the two ratiometric pH probes LysoSensor Yellow/Blue Dextran and fluorescein-tetramethylrhodamine Dextran (Figure S5A). In addition, degradation of full-length APP is not different in PS1ΔE9/ΔE9 neurons treated with cycloheximide (Figure S5B) or when lysosome degradation is blocked with chloroquine.
Figure 5. PS1ΔE9 Impairs Recycling of LRP1.

A) LRP1 protein and mRNA levels were measured before and after 4 hours of LDL treatment. PS1ΔE9 LRP1 protein were not different without (p = 0.1064) and with 4h LDL treatment (p = 0.4395). Nor were PS1ΔE9 LRP1 mRNA levels in untreated (p = 1.0) and 4h LDL-treated neurons (p = 0.4627). Data represent mean ± SEM of 4 biological replicates. B) Surface LDL levels were not different in PS1ΔE9 neurons after 4h of LDL treatment (p = 0.5793). Data represent mean ± SEM of soma values from 12 PS1wt/wt, 12 PS1wt/ΔE9 and 12 PS1ΔE9/ΔE9 biological replicates. Scale bar = 20μm C) PS1ΔE9/ΔE9 LRP1 surface levels, identified in the pull-down (PD) lane, were not different at baseline, but trended to lower compared to PS1wt/wt after 4 hours LDL treatment, though results did not reach significance (p = 0.129) Whiskers depict mean ± SEM of 3 biological replicates per condition. See also Figure S5.
To determine if the amount of lipoprotein receptors at the somatodendritic surface are driving endocytosis defects at later timepoints, we measured LDL receptor amount at baseline and after 4 hours of LDL treatment using two different methods. Purified neurons were incubated at 4°C with labeled LDL for 30 minutes and then fixed (0h time point, this gives a measure of total LDL receptors at the cell surface (Figure 5B). Neurons were also incubated for 4 hours with unlabeled LDL, then brought to 4°C, unlabeled LDL was washed off and then cells were incubated with labeled LDL followed by fixation. This gives a measure of LDL receptors at the surface after 4 hours of endocytosis (Figure 5B). Under these conditions, there were no differences in surface LDL puncta count in PS1ΔE9/ΔE9 neurons compared to PS1wt/wt neurons (Figure 5B). Since LDL could be binding non-specifically to surface proteins, we evaluated specific receptor populations by biotinylating the surface of neurons with a cleavable biotin and used streptavidin beads to pull down surface proteins before and after LDL treatment. Probing for LRP1 demonstrated that the surface levels of LRP1 at baseline were not significantly different between PS1wt/wt and PS1ΔE9/ΔE9. However, after 4 hours of LDL treatment, LRP1 receptor levels trend to decreased in PS1ΔE9/ΔE9 (p= 0.1293) (Figure 5C). Thus, the decrease in LDL endocytosis we observed in PS1 ΔE9/ΔE9 neurons (Figure 3B) can in part be accounted for by the modest (~50%) LRP1 reduction at the neuronal surface after 4 hours of LDL treamtment.
LDL Endocytosis Defects in PS1ΔE9 Neurons are Rescued by β-secretase Inhibition
We previously demonstrated that the PS1ΔE9 mutation impairs γ-secretase activity and APP CTFs accumulate in human PS1ΔE9 neurons (Woodruff et al., 2013). A previous study also demonstrated that γ-secretase inhibition in MEFs reduces LDL endocytosis (Tamboli et al., 2008). To determine if γ-secretase activity or if the β-CTF fragment might be responsible for the impaired LDL endocytosis in human neurons, we treated PS1wt/wt neurons with β- and γ-secretase inhibitors and measured LDL endocytosis (Figure 6A and B). Inhibition of γ-secretase severely decreased LDL endocytosis at 4 hours while β-secretase inhibition had no significant effect (Figure 6A and B). Treatment with both β- and γ-secretase inhibitors caused a marked accumulation of α-CTFs while ablating β-CTFs (Figure 6C), but had no effect on LDL endocytosis at 4 hours (Figure 6B). These results suggest that the β-CTF, not α-CTF, of APP is responsible for impairing LDL endocytosis in ΔE9 neurons. To further test this possibility, we treated PS1ΔE9 neurons with a β-secretase inhibitor and measured LDL endocytosis (Figure 6A and D). We observed that upon treatment with a β-secretase inhibitor, PS1ΔE9/ΔE9 neurons were rescued in the ability to endocytose LDL (Figure 6D). Treatment of PS1ΔE9/ΔE9 neurons with both β- and γ-secretase inhibitors, which abolishes β-CTF and increases α-CTF, also rescued the LDL endocytosis defect (Figure 6D), which suggests that accumulation of only the β-CTF is responsible for impaired LDL endocytosis. To test whether β-secretase inhibition could rescue the transcytosis defect that we observed, we treated PS1ΔE9/ΔE9 neurons grown in microfluidic compartments with a β-secretase inhibitor and then measured LDL transcytosis (Figure 6E and F). Similar to the endocytosis result, we also observed that β-secretase inhibition significantly increased the amount of transcytosed LDL in PS1ΔE9/ΔE9 neurons (Figure 6E).
Figure 6. LDL Endocytosis Defects in PS1ΔE9 Neurons are Rescued by β-Secretase Inhibition.

A) Example images from drug-treated PS1wt/wt, PS1wt/ΔE9 and PS1ΔE9/ΔE9 neurons. Scale bar = 20μm B) Quantification of LDL puncta count per soma in 4 hour LDL-treated PS1wt/wt neurons. Treatment with a γ-secretase inhibitor (GSI) resulted in a decrease in LDL endocytosis in PS1wt/wt neurons, while treatment with a β-secretase inhibitor (BSI) had no effect. Data represent the mean ± SEM 12 PS1wt/wt, 6 GSI 100nM (p <0.0001), 6 GSI 1uM (p <0.0001), 12 BSI 4uM (n.s.), and 4 GSI 1uM + BSI 4uM (n.s) biological replicates. C) Example Western blot of neurons treated with β- and γ-secretase inhibitors. α-CTFs and β-CTFs are indicated by arrows. D) Quantification of LDL endocytosis in PS1 ΔE9/ΔE9 neurons treated with inhibitors. N= 10 PS1ΔE9/ΔE9 for VEH, 12 12 PS1 ΔE9/ΔE9 for BSI, and 4 PS1ΔE9/ΔE9 for 4uM BSI + 1uM GSI biological replicates (*** p<0.0001 compared to vehicle). E) Quantification of LDL transcytosis in PS1 ΔE9/ΔE9 under control and β-secretase inhibition. Treatment with 4uM of BSI increased levels of transcytosed, axonal LDL compared to untreated PS1ΔE9/ΔE9 from an average density of ~0.33 count/um to ~0.43 count/um (p = 0.0044). F) Example images of PS1 ΔE9/ΔE9 axons with and without β-secretase inhibition after 4 hours of LDL transcytosis. Axons were co-stained with the axonal marker SMI31 for neurofilament-H.
LDL Endocytosis Defects are Common to fAD APP Mutations
Accumulation of APP β-CTFs is a phenotype shared by many APP and PS1 fAD mutations (Chang and Suh, 2005; De Jonghe et al., 2001; McPhie et al., 1997; Sinha and Lieberburg, 1999; Wiley et al., 2005). To assess whether other fAD mutations might share a common phenotype of impaired LDL endocytosis, we generated additional isogenic cell lines harboring either the APP V717F (APPV717F) or APP Swedish (APPswe) mutations (Sherrington et al., 1995). In neurons homozygous for the APPV717F mutation, there was significantly reduced LDL endocytosis after 4 hours and this defect was rescued by β-secretase inhibition (Figure 7A and B). Similar to both the PS1ΔE9 and APPV717F mutations, neurons homozygous for the APPswe mutation also exhibited decreased LDL endocytosis (Figure 7A and B). In contrast to the other fAD mutations, β-secretase inhibition in the APPswe neurons did not rescue the LDL endocytosis defect (Figure 7A and B). However, mutant APP Swedish protein has previously been reported to decrease the potency of β-secretase inhibitors (Yamakawa et al., 2009), which could explain why in our study β-secretase inhibition did not rescue the APPswe neurons. These findings demonstrate that the reduction in LDL endocytosis is not specific to the PS1ΔE9 mutation, but is a phenotype common to fAD APP mutations. Similar to PS1ΔE9 axons, we also observed reduced basal levels of axonal APP and Rab11 densities and reduced endocytosis of 22C11 at 4h in APPV717F neurons (Figure S6). Thus, endocytosis defects, and perhaps transcytosis defects, are common among at least one fAD PS1 mutation and two fAD APP mutations.
Figure 7. LDL Endocytosis Defects are Common to Other fAD Mutations.

A) Representative images of LDL uptake in APPv717f and APPswe neurons and co-stained with the somatodendritic marker Map2. Scale bar = 20μm B) Quantification of LDL puncta count after 4 hours of LDL treatment in APPwt/wt, APPV717F/V717F, and APPswe/swe neurons with vehicle and 4uM BSI treatment demonstrates that APP mutations also result in decreased LDL uptake (APPV717F/V717F p = 0.0009, APPswe/swe p<.0001), which is rescued with BSI treatment in APPV717F (APPV717F/V717F p = 0.5545) but not APPswe. Data represent the mean ± SEM of 6 APPwt/wt, 3 APPV717F/V717F and 3 APPswe/swe biological replicates. C) Model of APP and lipoprotein transcytosis in neurons. C1) FL-APP is internalized in Rab5+ sorting endosomes that contain LDL associated with LRP1 as well as β-secretase (BACE1) and possibly PS1. C2) As vesicles become more acidic along the endocytic pathway, LDL dissociates from LRP1 and FL-APP gets cleaved by β-secretase. A proportion of the cleaved APP population resides in Rab11+ endosomes containing LRP1, β-CTF and LDL. C3) When β-CTF gets cleaved by γ-secretase, LRP1 can be recycled back to cell surface and transcytosis of LDL and FL-APP to the axon can occur. fAD mutations increase β-CTFs either by enhancing β-Secretase processing or impairing γ-secretase-mediated cleavage of APP. β-CTFs impair recycling of LRP1 and decrease transcytosis of APP and lipoproteins, possibly by directly sequestering LRP1 in a Rab11+ endosome. See also Figure S6.
Discussion
Here we demonstrate that in human neurons with endogenous expression of fAD mutations, induced with genome editing technology, increased β-CTF of APP alters the subcellular localization of APP, the distribution of Rab11, and decreases endocytosis and soma-to-axon transcytosis of LDL. LDL endocytosis and transcytosis defects are rescued by β-Secretase inhibition in at least some of the fAD mutations. Our results show that impaired LDL endocytosis and transcytosis is present in multiple types of fAD mutations (Figure 7C) and they together define an apparent defect in the Rab11 recycling endosome. Epidemiologic evidence implicating cholesterol as a major player in AD also dovetails with these molecular and cellular findings (reviewed in Fonseca et al., 2010; Wolozin, 2004).
We demonstrate that APP β-CTFs may cause impaired LDL uptake by reducing recycling of LRP1 receptors back to the cell surface. One possible mechanism is that β-CTFs bind to LRP1 (Kounnas et al., 1995; Pietrzik et al., 2002; Tamboli et al., 2008), or other LDL receptors and retain LDL receptors in a Rab11 containing compartment until the β-CTF is cleaved by γ-secretase. This possibility would explain why γ-secretase inhibition impairs LDL uptake and why that defect can be rescued by β-Secretase inhibition. The observation that basal Rab11 is reduced in axons of fAD mutant neurons suggests that a common constitutive recycling/transcytotic pathway is impaired and raises the possibility that modulating Rab11 activity could also rescue fAD phenotypes. While we cannot rule out proteolysis of the 22C11 or labeled LDL probe following endocytosis as a possible explanation for the reduced transcytosis, our data are most consistent with the possibility reported in previous work that early endocytosis is affected (Cataldo et al., 2000).
The finding that fAD neurons have defects in lipoprotein transcytosis is intriguing in light of previous work showing that neurons are dependent on uptake of extracellular cholesterol from lipoprotein particles to perform functions such as axon elongation and synapse formation and maintenance (Barres and Smith, 2001; Lane-Donovan et al., 2014; Mauch et al., 2001; Nägler et al., 2001; Pfrieger, 2003; Pierrot et al., 2013). In fact, glia-derived cholesterol was reported to enhance synaptogenesis of adult rat CNS (Mauch et al., 2001; Nägler et al., 2001) suggesting that a defect in endocytosis and transcytosis of extracellularly-derived cholesterol could have long-term functional consequences leading to impaired neurotransmitter release and synaptic function. For instance, many studies of APOE function have focused on its potential role in mediating Aβ clearance, but APOE has also been identified as the major lipoprotein carrier in the brain and the e4 allele is less efficient in transporting brain cholesterol (Liu et al., 2012). Interestingly, post-mortem studies comparing sAD patients to age-matched controls found that brain cholesterol levels are reduced in the areas of learning and memory, the hippocampus and cortex (Svennerholm and Gottfries, 1994).
Previous work implicated early endosome dysfunction in sAD and some forms of fAD (Cataldo et al., 2001; Ginsberg et al., 2010). In contrast to previous studies, we report abnormalities in a Rab11 marked recycling endosome in fAD mutations, which is consistent with previous work on transcytosis that identified Rab11 as an endocytic regulator with a unique role in polarized cells including neurons (Ascaño et al., 2009; Buggia-Prévot et al., 2014; Welz et al., 2014). Thus, we describe a defect in a neuron-specific pathway that could contribute to the reported early endosomal defects, and could produce AD pathology. In contrast, other recent findings have suggested that lysosomal pathology is the major driver of pathology in PS1 fAD mutations (Lee et al., 2010; Lee et al., 2015). Our data suggests that dysfunction in endosomal sorting could drive changes in lysosomal function, rather than an original defect in the lysosome that drives such changes (Peric and Annaert, 2015).
We thus propose that transcytotic trafficking defects could be at the root of many types of fAD and potentially sAD. In fact, endocytic trafficking changes have been reported for APOE4 relative to APOE3 and 2 (Chen et al., 2010). These types of defects could lead to previously reported axonal amyloid-dependent and independent transport defects (Kim et al., 2015; Stokin et al., 2008; Vossel et al., 2015). Similarly, many fAD mutations thought to act solely by changing Aβ production may in fact also act by changing sorting and trafficking signals in β-CTF of APP leading to changes in constituents of axonal vesicles derived by transcytotic trafficking of lipoproteins and other key synaptic constituents. Although it may seem counter-intuitive that cleavage of β-CTF is needed to generate axonal vesicles containing β-CTF or full-length APP, we suggest that β-CTF cleavage happens in the portion of the recycling endosome that remains in the soma and that buds off axonal vesicles with APP, lipoproteins, and other components (Figure 7C). Finally, we note that the defects we report occur in the absence of overexpression of any of the proteins involved and thus may accurately reflect the earliest changes from normal behavior generated by fAD.
Experimental Procedures
Statistical Methods
Statistics were performed using GraphPad Prism. Normality for each data set was assessed using D’Agostino-Pearson test. When data were normally distributed, a two-way ANOVA with a post hoc Tukey test was used to compare genotypes. For precise p-value comparisons, a multiple t-test was done after ANOVA calculations. Most immunofluorescence data were non-normally distributed and a nonparametric Kruskal-Wallis test with Dunn’s multiple comparison was used to compare genotype medians. Data are depicted with bar graphs of the mean ± SEM of all values in an experiment or box plots where the median is depicted with a line and whiskers indicate the Tukey distribution as determined by GraphPad Prism.
Microfluidic Compartments
Microfluidic compartments were made in house as previously described (Niederst et al., 2015). Briefly, Sylgard 182 (Ellsworth Adhesives, Germantown, WI) was used to mold devices. Once cured, devices were cut and then washed with isopropanol, water, and 70% ethanol. Devices were plasma treated and mounted onto glass coverslips. The mounted device was this coated with PO/L as described above.
sAPP Measurements
sAPP was measured from conditioned media from microfluidic compartments from the soma side (contains soma, axons, dendrites) and the axon side (axons only) 48 hours after a full media change in which the axons were kept in fluidic isolation. sAPP was measured using the MSD sAPPα/sAPPβ human kit (Meso Scale Discovery). The kit has sensitivity down to 120 pg/mL for sAPPα and 52 pg/mL for sAPPβ, and all measurements were in the linear range.
Immunofluorescence
Purified neurons were grown in 384-well imaging plates at a density of 25,000 cells per well for 7–9 days post-sort. Neurons were fixed in 4% paraformaldehyde and PBS for 30 min at 37°C, permeabilized with 0.1% Triton X-100, and blocked (10% donkey serum, 3% BSA, 0.1% Triton X-100 in PBS). For surface labeling experiments, neurons were not permeabilized. For compartment experiments, PDMS microfluidic devices were plasma bonded directly onto glass coverslips (Niederst et al., 2015). Neurons were seeded at a density of 1–3 million cells per compartment and grown for 7–10 days (until axons passed through the channels). Compartments were then fixed as above and imaged. Antibodies used for immunofluorescence experiments were anti-Rab11 (1:1000, Life Technologies 71-5300), anti-APP Y188 (1:200, Abcam ab32136), anti-APP 22C11 (1:100, EMD Millipore, MAB348), anti-EEA1 (1:100, BD 610457), anti-NF-H (1:1000, Biolegend SMI-31r), and anti-Map2 (1:500, ab5392). Secondary antibodies were Alexa Fluor donkey anti-mouse and anti-rabbit IgG (Invitrogen) and Dylight 405 donkey anti-chicken IgY from (Jackson ImmunoResearch, 703-475-155) were used at 1:200. Images were acquired on a Zeiss confocal microscope.
Endocytosis and Transcytosis
For constitutive uptake endocytosis assays, neurons were incubated with LDL-BODIPY (20 μg/ml, Invitrogen L3483) or Dextran-tetramethylrhodamine (TMR) MW 10,000 (250 μg/ml, Invitrogen D1817), at 37°C for indicated times, fixed, and imaged. For all fixed endocytosis assays, a custom ImageJ program was used to identify Map2-positive soma and automatically generate region of interests (ROI) corresponding to soma. Mean intensity and puncta count per soma were determined and averaged across images and experiments. All endocytosis assays were repeated at least three times. In experiments where secretase inhibitors were used, cells were treated with the inhibitor 24 hours prior to the experiment and kept in the presence of the inhibitor for the duration of the experiment. Compound E (GSI, EMD Chemicals) was used at 200nM in endocytosis assays and at 100nM and 1μM for Western blot. βIV Inhibitor (β Inh, Millipore) was used at 4μM.
For transcyctosis experiments, neurons were grown in compartments and treated on the soma side with LDL-Bodipy (20 μg/ml), LDL-DiI (12.5 μg/ml), or mouse anti-22C11 (1:100) for 4 hours with axons in fluidic isolation. Axonal puncta analysis was done as previously described (Szpankowski et al., 2012). Axons were imaged at 100x and a custom Guassian-fitting colocalization package in MATLAB (MathWorks) was used to calculate axonal density, puncta intensity, and percent colocalization per axon.
Surface Biotinylation Assay
Neuronal media was changed to warm fresh media or media supplanted with 12.5 μg/ml of unlabeled LDL for 4 hours. At the end of the incubation, neurons were washed twice with ice-cold PBS and then incubated at 4C with 2mM EZ-Link™ Sulfo-NHS-SS-Biotin (Life Technologies) in PBS for 30 minutes. Cells were then lysed in equal volumes of RIPA buffer. For pulldown experiments, 200 μg of harvested protein at 0.5 μg/μl was incubated with 50 μl of pre-washed Pierce Streptavidin Magnetic Beads (Life Technologies, 88817) overnight at 4°C. The next day, beads were washed to remove residual, unbound proteins and biotinylated proteins were released from the streptavidin beads by boiling samples in loading buffer at 100°C. Westerns were run with 5% of input, 5% of supernatant, and 50% of pull down. Quantification of recycling was determined based on input signal(Pull down/Input).
Supplementary Material
Highlights.
FAD mutations impair endocytosis and transcytosis of APP and lipoproteins
Reduced lipoprotein endocytosis and transcytosis is rescued by β-secretase inhibition
Acknowledgments
Thanks to Angels Almenar for helpful suggestions and scientific advice. G.W. was supported by an institutional training grant (2T32AG000216-21). S.M.R. was supported by NIH predoctoral training grant T32 GM008666, a CIRM predoctoral training grant, and a Tina Nova scholarship. R.v.d.K. is supported by an ERC Marie Curie International Outgoing Fellowship and an Alzheimer Netherlands Fellowship This work was supported by California Institute of Regenerative Medicine (RT2-01927) awarded to L.S.B.G. and NIA RF1 AG048083 also to L.S.B.G.
Footnotes
Author Contributions:
S.M.R., G.W. and L.S.B.G. wrote the manuscript and designed the experiments. G.W., J.A.C., J.E.Y., and E.A.R. designed and generated the isogenic iPSC lines. S.M.R., G.W., M.D., and R.K. performed the experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson E, Maday S, Sfakianos J, Hull M, Winckler B, Sheff D, Fölsch H, Mellman I. Transcytosis of NgCAM in epithelial cells reflects differential signal recognition on the endocytic and secretory pathways. J Cell Biol. 2005;170:595–605. doi: 10.1083/jcb.200506051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony SM, Granick S. Image analysis with rapid and accurate two-dimensional Gaussian fitting. Langmuir. 2009;25:8152–8160. doi: 10.1021/la900393v. [DOI] [PubMed] [Google Scholar]
- Antonell A, Lladó A, Altirriba J, Botta-Orfila T, Balasa M, Fernández M, Ferrer I, Sánchez-Valle R, Molinuevo JL. A preliminary study of the whole- genome expression profile of sporadic and monogenic early-onset Alzheimer’s disease. Neurobiol Aging. 2013;34:1772–1778. doi: 10.1016/j.neurobiolaging.2012.12.026. [DOI] [PubMed] [Google Scholar]
- Arold S, Sullivan P, Bilousova T, Teng E, Miller CA, Poon WW, Vinters HV, Cornwell LB, Saing T, Cole GM, et al. Apolipoprotein e level and cholesterol are associated with reduced synaptic amyloid beta in Alzheimer’s disease and apoE TR mouse cortex. Acta Neuropathol. 2012;123:39–52. doi: 10.1007/s00401-011-0892-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascaño M, Richmond A, Borden P, Kuruvilla R. Axonal targeting of Trk receptors via transcytosis regulates sensitivity to neurotrophin responses. J Neurosci. 2009;29:11674–11685. doi: 10.1523/JNEUROSCI.1542-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres Ba, Smith SJ. Cation Between Nerve Cells At Meet- Purified Neurons To Form Synaps- Science (80-) 2001;294:1296–1297. [Google Scholar]
- Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Brodbeck J, McGuire J, Liu Z, Meyer-Franke A, Balestra ME, Jeong DE, Pleiss M, McComas C, Hess F, Witter D, et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J Biol Chem. 2011;286:17217–17226. doi: 10.1074/jbc.M110.217380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggia-Prévot V, Fernandez CG, Riordan S, Vetrivel KS, Roseman J, Waters J, Bindokas VP, Vassar R, Thinakaran G. Axonal BACE1 dynamics and targeting in hippocampal neurons: a role for Rab11 GTPase. Mol Neurodegener. 2014;9:1. doi: 10.1186/1750-1326-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M, Gaynor K, Olm V, Mercken M, LaFrancois J, Wang L, Mathews PM, Noble W, Matsuoka Y, Duff K. Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J Neurosci. 2003;23:5645–5649. doi: 10.1523/JNEUROSCI.23-13-05645.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Leem JY, Greenfield JP, Wang P, Kim BS, Wang R, Lopes KO, Kim SH, Zheng H, Greengard P, et al. Presenilin-1 regulates intracellular trafficking and cell surface delivery of β-amyloid precursor protein. J Biol Chem. 2003;278:3446–3454. doi: 10.1074/jbc.M209065200. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic Pathway Abnormalities Precede Amyloid β Deposition in Sporadic Alzheimer’s Disease and Down Syndrome. Am J Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo A, Rebeck G, Ghetti B, Hulette C, Lippa C, Van Brockhoven C, Van Duijn C, Cras P, Bogdanovic N, Bird T, et al. Endocytic disturbances distinguish among subtypes of Alzheimer’s disease and related disorders. Ann Neurol. 2001;50:661–665. [PubMed] [Google Scholar]
- Chang KA, Suh YH. Pathophysiological roles of amyloidogenic carboxy-terminal fragments of the beta-amyloid precursor protein in Alzheimer’s disease. J Pharmacol Sci. 2005;97:461–471. doi: 10.1254/jphs.cr0050014. [DOI] [PubMed] [Google Scholar]
- Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A. 2010;107:12011–12016. doi: 10.1073/pnas.0914984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen K, Flannagan RS, Baron S, Carraro-Lacroix LR, Wang D, Vermeire W, Michiels C, Munck S, Baert V, Sugita S, et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. Journal of Cell Biology. 2012;198:23–35. doi: 10.1083/jcb.201201076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform–specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwu Z, Chen CS, Zhang C, Klaubert DH, Haugland RP. A novel acidotropic pH indicator and its potential application in labeling acidic organelles of live cells. Chem Biol. 1999;6:411–418. doi: 10.1016/s1074-5521(99)80059-3. [DOI] [PubMed] [Google Scholar]
- Dumanchin C, Czech C, Campion D, Cuif MH, Poyot T, Martin C, Charbonnier F, Goud B, Pradier L, Frebourg T. Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum Mol Genet. 1999;8:1263–1269. doi: 10.1093/hmg/8.7.1263. [DOI] [PubMed] [Google Scholar]
- Encalada SE, Szpankowski L, Xia CH, Goldstein LSB. Stable kinesin and dynein assemblies drive the axonal transport of mammalian prion protein vesicles. Cell. 2011;144:551–565. doi: 10.1016/j.cell.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fargo K, Bleiler L. Alzheimer’s Association Report: 2014 Alzheimers disease facts and figures. Alzheimer’s Dement. 2014;10:e47–e92. doi: 10.1016/j.jalz.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Fonseca ACRG, Resende R, Oliveira CR, Pereira CMF. Cholesterol and statins in Alzheimer’s disease: current controversies. Exp Neurol. 2010;223:282–293. doi: 10.1016/j.expneurol.2009.09.013. [DOI] [PubMed] [Google Scholar]
- Gandy S, Zhang YW, Ikin A, Schmidt SD, Levy E, Sheffield R, Nixon Ra, Liao FF, Mathews PM, Xu H, et al. Alzheimer’s presenilin 1 modulates sorting of APP and its carboxyl-terminal fragments in cerebral neurons in vivo. J Neurochem. 2007;102:619–626. doi: 10.1111/j.1471-4159.2007.04587.x. [DOI] [PubMed] [Google Scholar]
- Gatz M, Reynolds Ca, Fratiglioni L, Johansson B, Mortimer Ja, Berg S, Fiske A, Pedersen NL. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, Wuu J, Chao MV, Mufson EJ, Nixon RA, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry. 2010;68:885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulian M, Simon SM. Tracking single proteins within cells. Biophysical journal. 2000;79:2188–2198. doi: 10.1016/S0006-3495(00)76467-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Li H, Gaddam SSK, Justice NJ, Robertson CS, Zheng H. Amyloid precursor protein revisited: Neuron-specific expression and highly stable nature of soluble derivatives. J Biol Chem. 2012;287:2437–2445. doi: 10.1074/jbc.M111.315051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel Ma, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nature methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LW, Hua DH, Shie FS, Maezawa I, Sopher B, Martin GM. Novel tricyclic pyrone compounds prevent intracellular APP C99-induced cell death. J Mol Neurosci. 2002;19:57–61. doi: 10.1007/s12031-002-0011-9. [DOI] [PubMed] [Google Scholar]
- Karch CM, Goate AM. Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis. Biol Psychiatry. 2014;77:43–51. doi: 10.1016/j.biopsych.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini a, Rigoglioso a, Jiang Y, Nixon Ra. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol Psychiatry. 2015:1–10. doi: 10.1038/mp.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounnas MZ, Moir RD, Rebeck GW, Bush AI, Argraves WS, Tanzi RE, Hyman BT, Strickland DK. LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted β-amyloid precursor protein and mediates its degradation. Cell. 1995;82:331–340. doi: 10.1016/0092-8674(95)90320-8. [DOI] [PubMed] [Google Scholar]
- Lane-Donovan C, Philips GT, Herz J. More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron. 2014;83:771–787. doi: 10.1016/j.neuron.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo OM, Gonzalez a, Ascano M, Kuruvilla R, Couve a, Bronfman FC. BDNF Regulates Rab11-Mediated Recycling Endosome Dynamics to Induce Dendritic Branching. J Neurosci. 2013;33:6112–6122. doi: 10.1523/JNEUROSCI.4630-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, McBrayer M, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Pearl PY, Mohan P, Coffey EE, Uday K, et al. Presenilin 1 maintains lysosomal Ca2+ homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 2015;12:1430–1444. doi: 10.1016/j.celrep.2015.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem JY, Saura CA, Pietrzik C, Christianson J, Wanamaker C, King LT, Veselits ML, Tomita T, Gasparini L, Iwatsubo T, et al. A Role for Presenilin 1 in Regulating the Delivery of Amyloid Precursor Protein to the Cell Surface. Neurobiol Dis. 2002;11:64–82. doi: 10.1006/nbdi.2002.0546. [DOI] [PubMed] [Google Scholar]
- Li J, Kanekiyo T, Shinohara M, Zhang Y, LaDu MJ, Xu H, Bu G. Differential regulation of amyloid-β endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J Biol Chem. 2012;287:44593–44601. doi: 10.1074/jbc.M112.420224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms, and therapy. Changes. 2012;29:997–1003. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto a, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Maxfield FR. Role of endosomes and lysosomes in human disease. Cold Spring Harb Perspect Biol. 2014:6. doi: 10.1101/cshperspect.a016931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägler K, Mauch DH, Pfrieger FW. Glia-derived signals induce synapse formation in neurones of the rat central nervous system. J Physiol. 2001;533:665–679. doi: 10.1111/j.1469-7793.2001.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely KM, Green KN, LaFerla FM. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a γ-secretase-independent manner. J Neurosci. 2011;31:2781–2791. doi: 10.1523/JNEUROSCI.5156-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederst E, Reyna S, Goldstein LSB. Axonal Amyloid Precursor Protein and its Fragments Undergo Somatodendritic Endocytosis and Processing. Mol Biol Cell. 2015;26:205–17. doi: 10.1091/mbc.E14-06-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon Ra, Mcbrayer M. Lysosome and Calcium Dysregulation in Alzheimer’s disease - Partners in Crime. Biochem Soc Trans. 2013;41:1495–1502. doi: 10.1042/BST20130201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Yang DS. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol Dis. 2011;43:38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric A, Annaert W. Early etiology of Alzheimer’s disease: tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol. 2015;129(3):363–381. doi: 10.1007/s00401-014-1379-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW. Role of cholesterol in synapse formation and function. Biochim Biophys Acta - Biomembr. 2003;1610:271–280. doi: 10.1016/s0005-2736(03)00024-5. [DOI] [PubMed] [Google Scholar]
- Pierrot N, Tyteca D, D’auria L, Dewachter I, Gailly P, Hendrickx A, Tasiaux B, El Haylani L, Muls N, N’Kuli F, et al. Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol Med. 2013;5:608–625. doi: 10.1002/emmm.201202215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzik CU, Busse T, Merriam DE, Weggen S, Koo EH. The cytoplasmic domain of the LDL receptor-related protein regulates multiple steps in APP processing. EMBO J. 2002;21:5691–5700. doi: 10.1093/emboj/cdf568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimplikar SW, Nixon Ra, Robakis NK, Shen J, Tsai LH. Amyloid- independent mechanisms in Alzheimer’s disease pathogenesis. J Neurosci. 2010;30:14946– 14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Polydoro M, Maury Ea, Nicholls SB, Reddy SM, Wegmann S, William C, Saqran L, Cagsal-Getkin O, Pitstick R, et al. Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol Commun. 2015;3:1–11. doi: 10.1186/s40478-015-0199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robakis NK. Mechanisms of AD neurodegeneration may be independent of Aβ and its derivatives. Neurobiol Aging. 2011;32:372–379. doi: 10.1016/j.neurobiolaging.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane H, Yamamoto H, Kikuchi A. LRP6 is internalized by Dkk1 to suppress its phosphorylation in the lipid raft and is recycled for reuse. J Cell Sci. 2010;123:360–368. doi: 10.1242/jcs.058008. [DOI] [PubMed] [Google Scholar]
- Selfridge A, Hyun N, Chiang CC, Reyna SM, Weissmiller AM, Shi LZ, Preece D, Mobley WC, Berns MW. Rat embryonic hippocampus and induced pluripotent stem cell derived cultured neurons recover from laser-induced subaxotomy. Neurophotonics. 2015;2:015006. doi: 10.1117/1.NPh.2.1.015006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva Ea, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Shioi J, Georgakopoulos A, Mehta P, Kouchi Z, Litterst CM, Baki L, Robakis NK. FAD mutants unable to increase neurotoxic Aβ 42 suggest that mutation effects on eurodegeneration may be independent of effects on Aβ. J Neurochem. 2007;101:674–681. doi: 10.1111/j.1471-4159.2006.04391.x. [DOI] [PubMed] [Google Scholar]
- Sinha S, Lieberburg I. Cellular mechanisms of beta-amyloid production and secretion. Proc Natl Acad Sci U S A. 1999;96:11049–11053. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soejitno A, Tjan A, Purwata T. Alzheimer’s Disease: Lessons Learned from Amyloidocentric Clinical Trials. CNS Drugs. 2015;29:487–502. doi: 10.1007/s40263-015-0257-8. [DOI] [PubMed] [Google Scholar]
- Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82:756–771. doi: 10.1016/j.neuron.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282– 1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Almenar-Queralt A, Gunawardena S, Rodrigues EM, Falzone T, Kim J, Lillo C, Mount SL, Roberts Ea, McGowan E, et al. Amyloid precursor protein-induced axonopathies are independent of amyloid-β peptides. Hum Mol Genet. 2008;17:3474–3486. doi: 10.1093/hmg/ddn240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino H, Watanabe A, Amada N, Yamamoto M, Ohgi Y, Kostic D, Sanchez R. Global Trends in Alzheimer Disease Clinical Development: Increasing the Probability of Success. Clin Ther. 2015 doi: 10.1016/j.clinthera.2015.07.006. Epub Ahead of Print. [DOI] [PubMed] [Google Scholar]
- Svennerholm L, Gottfries CG. Membrane lipids, selectively diminished in Alzheimer brains, suggest synapse loss as a primary event in early-onset form (type I) and demyelination in late-onset form (type II) J Neurochem. 1994;62:1039–1047. doi: 10.1046/j.1471-4159.1994.62031039.x. [DOI] [PubMed] [Google Scholar]
- Szpankowski L, Encalada SE, Goldstein LSB. Subpixel colocalization reveals amyloid precursor protein-dependent kinesin-1 and dynein association with axonal vesicles. Proc Natl Acad Sci. 2012;109:8582–8587. doi: 10.1073/pnas.1120510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Murate M, Fukuda M, Satoshi SB, Ohta A, Kobayashi T. Cholesterol Controls Lipid Endocytosis through Rab11. Mol Biol Cell. 2007;18:2667– 2677. doi: 10.1091/mbc.E06-10-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamboli IY, Prager K, Thal DR, Thelen KM, Dewachter I, Pietrzik CU, St George-Hyslop P, Sisodia SS, De Strooper B, Heneka MT, et al. Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J Neurosci. 2008;28:12097–12106. doi: 10.1523/JNEUROSCI.2635-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Blurton-jones M, Rhee SW, Cribbs DH, Carl W. A Microfluidic Culture Platform for CNS Axonal Injury, Regeneration, and Transport. Nat Methods. 2006;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom V, Princen K, De Witte K, Griffioen G, Geenslaan G, Leuven B. Derailed intraneuronal signalling drives pathogenesis in sporadic and familial. Alzheimer’s disease. 2014;2014:1–24. doi: 10.1155/2014/167024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyn JH, Ahlijanian MK. Interpreting Alzheimer’s disease clinical trials in light of the effects on amyloid-β. Alzheimers Res Ther. 2014;6:14. doi: 10.1186/alzrt244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treusch S, Hamamichi S, Goodman JL, Matlack KES, Chung CY, Baru V, Shulman JM, Parrado A, Bevis BJ, Valastyan JS, et al. Functional Links Between Aβ Toxicity. 2011;334:1241–1245. doi: 10.1126/science.1213210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udayar V, Buggia-Prévot V, Guerreiro RL, Siegel G, Rambabu N, Soohoo AL, Ponnusamy M, Siegenthaler B, Bali J, Simons M, et al. A Paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of β-amyloid production. Cell Rep. 2013;5:1536–1551. doi: 10.1016/j.celrep.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Reinsch S, Urbé S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110:E1807–E1816. doi: 10.1073/pnas.1220484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Bartheld CS. Axonal Transport and Neuronal Transcytosis of Trophic Factors, Tracers, and Pathogens. J Neurobiol. 2004;58:295–314. doi: 10.1002/neu.10315. [DOI] [PubMed] [Google Scholar]
- Vossel KA, Xu JC, Fomenko V, Miyamoto T, Suberbielle E, Knox JA, Ho K, Kim DH, Yu GQ, Mucke L. Tau reduction prevents Aβ-induced axonal transport deficits by blocking activation of GSK3β. J Cell Biol. 2015;209:419–433. doi: 10.1083/jcb.201407065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, van Echten-Deckert G. Cross-talk of membrane lipids and Alzheimer-related proteins. Mol Neurodegener. 2013;8:34. doi: 10.1186/1750-1326-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmiller AM, Natera-Naranjo O, Reyna SM, Pearn ML, Zhao X, Nguyen P, Cheng S, Goldstein LSB, Tanzi RE, Wagner SL, et al. A γ-Secretase Inhibitor, but Not a γ-Secretase Modulator, Induced Defects in BDNF Axonal Trafficking and Signaling: Evidence for a Role for APP. PLoS One. 2015;10:e0118379. doi: 10.1371/journal.pone.0118379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welz T, Wellbourne-Wood J, Kerkhoff E. Orchestration of cell surface proteins by Rab11. Trends Cell Biol. 2014;24:407–415. doi: 10.1016/j.tcb.2014.02.004. [DOI] [PubMed] [Google Scholar]
- Wilson JM, de Hoop M, Zorzi N, Toh BH, Dotti CG, Parton RG. EEA1, a tethering protein of the early sorting endosome, shows a polarized distribution in hippocampal neurons, epithelial cells, and fibroblasts. Mol Biol Cell. 2000;11:2657–2671. doi: 10.1091/mbc.11.8.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B. Cholesterol and the Biology of Alzheimer’s Disease. Neuron. 2004;41:7– 10. doi: 10.1016/s0896-6273(03)00840-7. [DOI] [PubMed] [Google Scholar]
- Woodruff G, Young JE, Martinez FJ, Buen F, Gore A, Kinaga J, Li Z, Yuan SH, Zhang K, Goldstein LSB. The Presenilin-1 δE9 Mutation Results in Reduced γ-Secretase Activity, but Not Total Loss of PS1 Function, in Isogenic Human Stem Cells. Cell Rep. 2013;5:974–985. doi: 10.1016/j.celrep.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap CC, Wisco D, Kujala P, Lasiecka ZM, Cannon JT, Chang MC, Hirling H, Klumperman J, Winckler B. The somatodendritic endosomal regulator NEEP21 facilitates axonal targeting of L1/NgCAM. J Cell Biol. 2008;180:827–842. doi: 10.1083/jcb.200707143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan SH, Martin J, Elia J, Flippin J, Paramban RI, Hefferan MP, Vidal JG, Mu Y, Killian RL, Israel Ma, et al. Cell-surface marker signatures for the Isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One. 2011:6. doi: 10.1371/journal.pone.0017540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Haapasalo A, Kim DY, Ingano LaM, Pettingell WH, Kovacs DM. Presenilin/gamma-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J. 2006;20:1176–1178. doi: 10.1096/fj.05-5531fje. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan Sa, Bennett ML, Scholze AR, Keeffe SO, Phatnani HP, Guarnieri XP, Caneda C, Ruderisch N, et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia. Neurons, and Vascular Cells of the Cerebral Cortex. 2014;34:1–19. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.