

Abstract
Functionalized polyfluorinated aromatics have become an important group of molecules for pharmaceutical and industrial applications. However, facile access to such valuable molecules remains an unmet challenge. In this review, we present and discuss photocatalytic C–F functionalization, which is emerging as a straightforward and operationally simple path to access partially fluorinated aromatics.
Keywords: hydrodefluorination, C–F functionalization, photocatalysis, partially fluorinated aromatics
1. Introduction
Fluorinated organic compounds have become an extremely important and valued class of molecules with amplified use as pharmaceuticals1 and agrochemicals,2 and with a number of industrial applications such as organic photovoltaics (i.e., OLEDs)3 and liquid crystal molecules.4 Following the invention in the 1950s of the first fluorine-containing pharmaceutical, fludrocortisone,1a the field has grown rapidly. In the last few decades, the frequency of fluorine incorporation within drugs has risen sharply. Starting from 2% of drugs that contained fluorine in 1970, this proportion has grown to about 25% of the total number of drugs available today.1b Furthermore, among the small-molecule drugs that have been approved by the U.S. Food and Drug Administration (FDA) in 2013, 33% contained a C–F bond and several of them (i.e., Adempas®, Gilotrif®, Tafinlar®, Tivicay®) contain a fluoroarene moiety in their structure. Within the crop sciences, the percentage of molecules containing organofluorine is at least 30%.5
Upon close inspection of the structures of medicinally and industrially important fluoroaromatics, some common features can be identified (Figure 1). Almost all of them are (i) considerably functionalized in order to perform a desired role, (ii) partially fluorinated not perfluorinated, and (iii) the fluoroaromatic moiety in each serves as a terminating unit except in very few cases. This latter observation is perhaps due to the unavailability of complex polyfluorinated arenes that can serve to elaborate the molecules further. Recently, more efforts have been devoted to the development of selective fluorination strategies that can be employed to access polyfluorinated arenes. These strategies include C–H fluorination6 (Scheme 1, Part (a)) and cross-coupling reactions using both nucleophilic7 and electrophilic8 fluorine sources (Scheme 1, Part (b)). While these methods have significantly expanded the number of accessible fluorinated arenes, the need for arenes that contain either strategically prefunctionalized or specific directing groups in the starting arene limits the use of such methods. This limitation is most clear when polyfluorinated arenes are desired, which would require highly elaborated starting materials, or simply may not be accessible with directed fluorination.6b,9 An alternate approach would be to start with a simple perfluoroarene in which C–F bonds already exist in all of the desired locations, and develop selective C–F functionalization reactions that utilize the undesired C–F bonds to construct the molecule. If successful, the perfluoroarene would serve as a synthetic lynchpin of polyfluorinated arenes. For instance, current syntheses of fluorinated azole fungicides share a common synthetic intermediate, 2a,1a which is accessible in six steps from 1,3-dichlorobenzene (2j), a nonfluorinated commercially available starting material (Scheme 2).10 Conceivably, direct C–F functionalization of the corresponding perfluoroarene, 2k, followed by C–F reduction could significantly shorten the synthetic sequence. In this review, we present and discuss recent advances in the field of photocatalytic C–F reduction and functionalization of polyfluoroarenes, whereby new reactions are developed that can provide facile and rapid synthesis of functionalized polyfluorinated aromatics.
Figure 1.
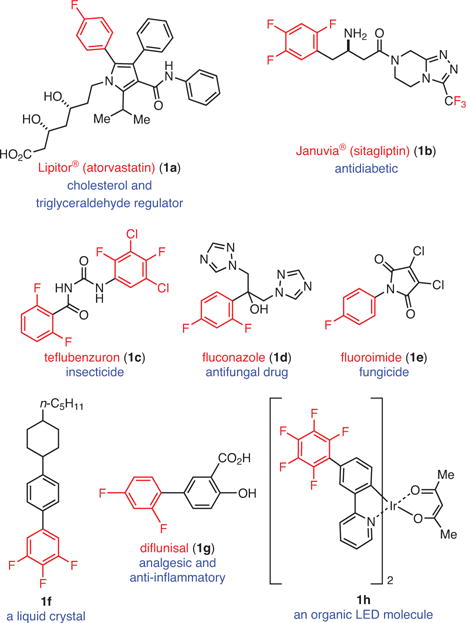
Pharmaceutically and Industrially Important Fluorinated Aromatics.
Scheme 1.
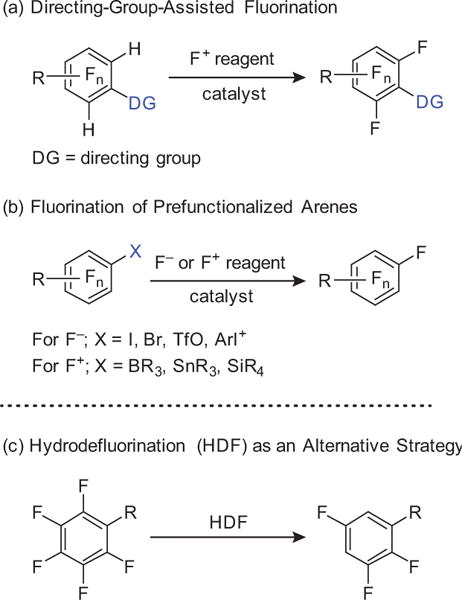
Selective Fluorination Strategies and Hydrodefluorination (HDF).
Scheme 2.
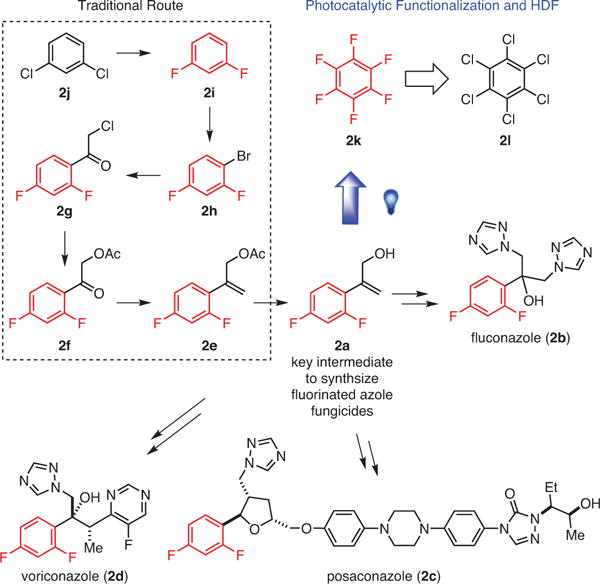
Actual and Potential Expedient Syntheses of Key Intermediate for Fluorinated Azole Fungicides. (Ref. 1a,10)
2. Hydrodefluorination
As an alternative to selective fluorination, hydrodefluorination (HDF) (Scheme 1, Part (c))5,11 has emerged as one way to access polyfluorinated arenes. While the potential advantages of the HDF approach as compared to existing selective fluorinations make it attractive, a number of challenges make its future less clear. Although the reduction of aryl iodides, bromides, and even chlorides is feasible,11 cleavage of short and strong Caryl–F bonds (Figure 2) has been a challenge. The extreme properties associated with C–F bonds make the design of efficient HDF catalysts quite challenging.
Figure 2.
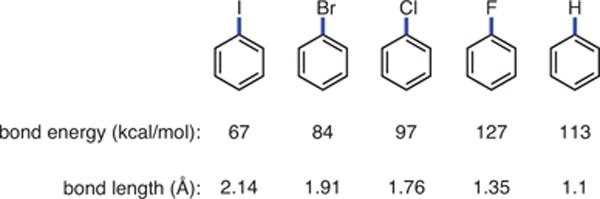
Comparison of Aryl–X Bond Lengths and Strengths.
Existing catalysts for HDF reactions often suffer from low turnover numbers (TONs) due to the formation of strong metal–fluorine bonds during the catalytic cycle. In their attempts to develop more robust HDF systems, chemists have utilized a number of strategies to deal with this dilemma. One solution is to use “fluorophilic” silyl12 or aluminum13 hydrides, in which the hydride-bearing atom itself can form a strong fluorine bond, liberating the catalyst from the thermodynamic well and increasing the overall exergonicity of the reaction. Nonetheless, most HDF systems still suffer from low TONs.14
2.1. Photocatalytic Hydrodefluorination
In recent years, photoredox catalysis15 has become popular as a way to introduce a single electron into a molecule. The beauty of photocatalyzed single-electron chemistry is the ability to make reactive species catalytically and in a controlled manner in situ. Given this and indirect evidence from Stephenson’s photocatalytic Caryl–I reduction,16 investigations have been undertaken to selectively reduce C–F bonds using visible light photocatalysis. The prediction was that fac-tris[2-phenylpyridinato-C2,N]iridium(III) [fac–Ir(ppy)3], which is a coordinatively saturated 18-electron complex and a potent reductant [Ir(III)/(II), −2.19 V vs SCE]15a,17a would be capable of transferring an electron to the perfluoroarene. This was expected to lead to fragmentation of the C–F bond to give a fluoride, perfluoroaryl radical, and ultimately the desired C–F reduced products after a hydrogen-atom transfer.17b,c Furthermore, the outer sphere nature of the electron transfer might avoid the problematic catalyst–fluoride intermediates and lead to higher TONs.
Pentafluoropyridine (3b) was chosen as the model substrate, because electron transfer was expected to be slightly exothermic between the reduced photocatalyst [Ir(III)/(II)] and 3b (3b, V = −2.12 vs SCE) (Figure 3).18 When the photocatalyst, 4a, absorbs a photon in the visible region (Scheme 3), it is promoted to an excited state (4b).15a From this excited state, the photocatalyst can act as either a reductant or an oxidant.15a It was proposed that a single-electron transfer from the tertiary aliphatic amine (4c) to the excited state of the catalyst (i.e., reductive quenching) results in an amine radical cation (4d) and the reduced photocatalyst (4e). Intermediate 4e engages in an outer-sphere electron transfer to the fluoroarene substrate, 3b, to generate a perfluoroaryl radical anion, 4f. Subsequent fluoride extrusion forms a perfluoroaryl radical (4g) which then abstracts a hydrogen atom from either the amine, 4c, or amine radical cation, 4d, leading to the desired reduced product, 4k. The other results discussed in this review arise from the interception of the versatile key radical intermediate 4g with π bonds of alkenes, arenes, and alkynes. The interception with alkynes has been employed to understand the underlying controlling factors of energy vs electron transfer, by which E- or Z-alkene products can be obtained by the judicious choice of photocatalyst. The use of the inexpensive and easy-to-handle N,N-diisopropylethylamine (i-Pr2NEt) as the reductant alleviates the need for fluorophilic metal hydrides, and makes this methodology operationally simple.
Figure 3.
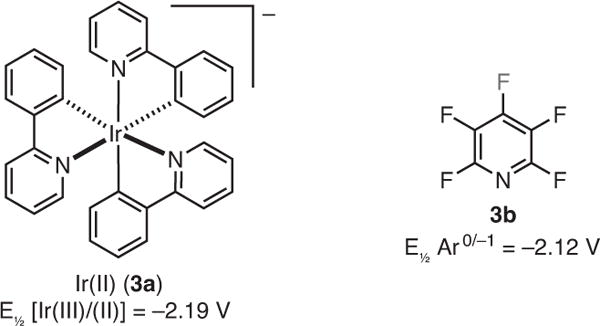
Comparison of Catalyst and Substrate Reduction Potentials. (Ref. 18)
Scheme 3.
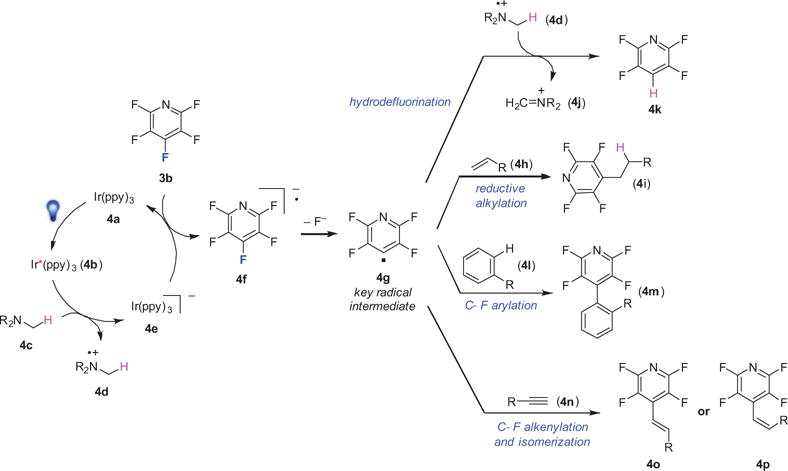
Plausible Mechanistic Pathways to HDF and Reductive Alkylation, Arylation, and Alkenylation.
2.2. Mono(hydrodefluorination)
Under the optimized conditions, both electron-deficient perfluoroheteroarenes and electron-rich fluorinated heterocycles underwent smooth mono-HDF to form 5a, 5f, and 5h, respectively (eq 1).19 The reaction demonstrated remarkable functional group tolerance, and had a broad substrate scope. It is worth noting that the chlorine atom in 3-chlorotetrafluoropyridine was preferentially fragmented leading to 5f, while the distant bromines in the precursor to 5i survived the HDF with only a trace amount of bromine loss. Tertiary aliphatic amines (5h) and even phosphines (5e) also survived the hydrodefluorination reaction. During the optimization trials, it was found that the two other, significantly less reducing photocatalysts—Ru(bpy)3Cl2 (V = −1.33 vs SCE) and Ru(bpm)3Cl2 (V = −0.91 vs SCE)20 (bpy = 2,2′-bipyridine, bpm = 2,2′-bipyrimidine)—also facilitated HDF (leading to 5a), albeit at a much slower rate. This phenomenon suggests that there are other unexplored factors governing these electron transfers apart from the reduction potentials alone.
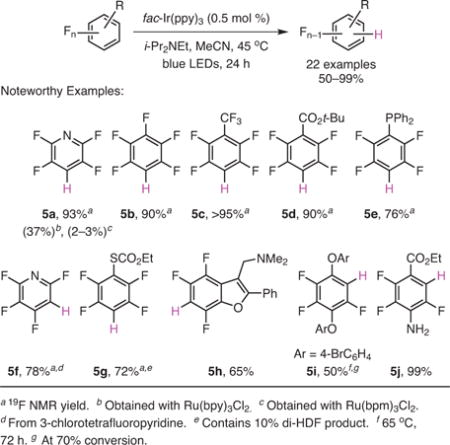 |
eq 1 |
(Ref. 19)
2.3. Di- and Tri(hydrodefluorination)
Often, the rates between the first and second reductions were substantially different such that one could obtain either mono-, or di-HDF products by simply varying the amount of the amine reductant and reaction time (eq 2).19 The regiochemistry of HDF is primarily dictated by the electronics of the aromatic system in the starting material, though it is worth noting that there can be a steric contribution, as seen by comparing the rate difference between the di-HDF of the methyl (6d) and tert-butyl (6e) esters of perfluorobenzoic acid.
Our group also investigated the robustness of the catalytic system. Repeated additions of pentafluoropyridine and diisopropylethylamine to the photocatalytic system achieved an unprecedented TON of 22,550— the highest among the TONs for all of the HDF systems reported to date. In addition, by utilizing pentafluoropyridine and octafluoronaphthalene, we have demonstrated that the kinetics of the reaction could be further enhanced by utilizing a flow system.19 Collectively, the ability to perform the photocatalytic reaction in flow and at very low loading of a commercially available catalyst might allow the process to be scaled.
3. Alkylation of Fluoroarenes
Having demonstrated that photoredox catalysis can be employed to break the robust C–F bond and access the relatively unexplored perfluoroaryl radical, we attempted to exploit this understanding to develop more elaborate C–F functionalization reactions. To this end, the first photocatalytic C–F alkylation was published in 2015.21 Carbon-centered radicals possess a remarkable bond-forming capability22 with unactivated π bonds that are sterically congested and are generally considered inert under most reaction conditions. Key to utilizing radicals in this manner is their controlled generation. In general, the idea was to intercept with alkenes the perfluoroaryl radical as it is formed. This would result in a more stable, longer-lived alkyl radical, which would then undergo the subsequent H-atom abstraction to afford a net hydroperfluoroarylation of the alkene. Given the vast number of alkenes available, this approach was expected to lead to a large variety of alkylated polyfluoroarenes.
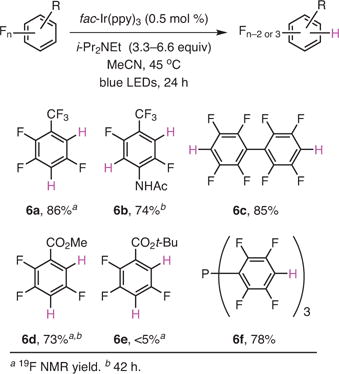 |
eq 2 |
(Ref. 19)
In the last few years, several groups have successfully alkylated highly fluorinated arenes. Love and co-workers have demonstrated the directing-group-assisted ortho alkylation of polyfluoroarenes with Pt23 and later with Ni24 based catalysts in the presence of a benzyl imine directing group (Scheme 4, Parts (a) and (b)). In 2014, Li’s group showed that the direct addition of alkyl Grignard reagents to perfluoroarenes was also possible (Scheme 4, Part (c)).25 More recently, Wu’s team developed a regioselective alkyl transfer from phosphonium ylides to perfluoroarenes (Scheme 4, Part (d)).26 While these approaches are making inroads toward selective C–F alkylation, there is still an urgent need to develop new synthetic methods that provide access to complex alkylated fluoroarenes.
Scheme 4.
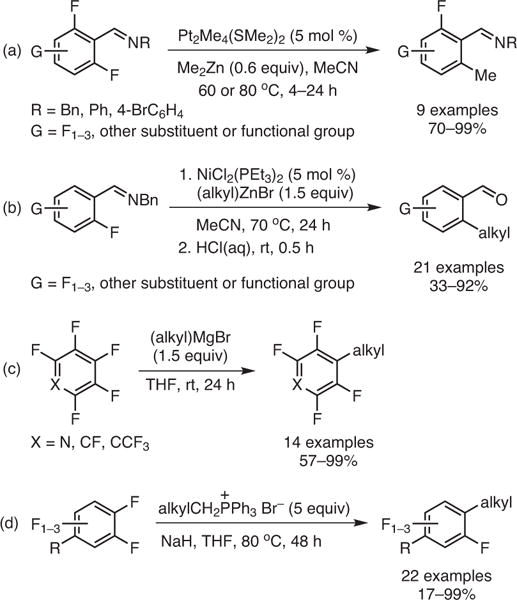
Previous Alkylation Strategies of Fluoroaromatics. (Ref. 23–26)
3.1. Photocatalytic C–F Alkylation of Fluoroarenes
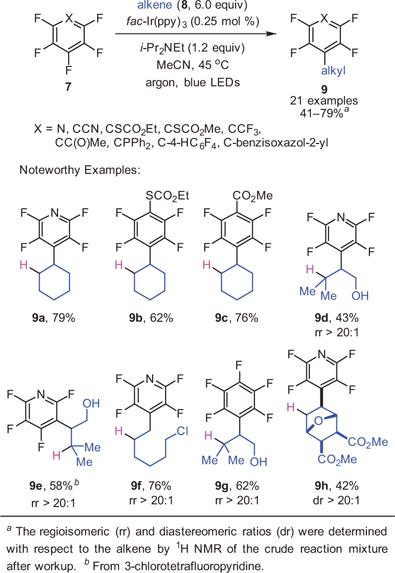
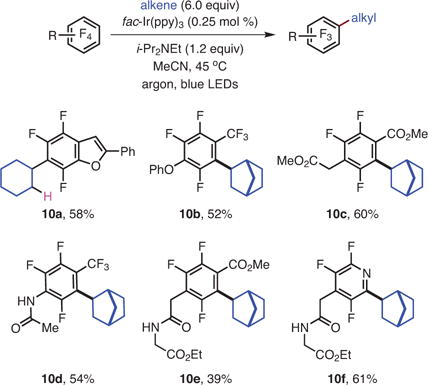
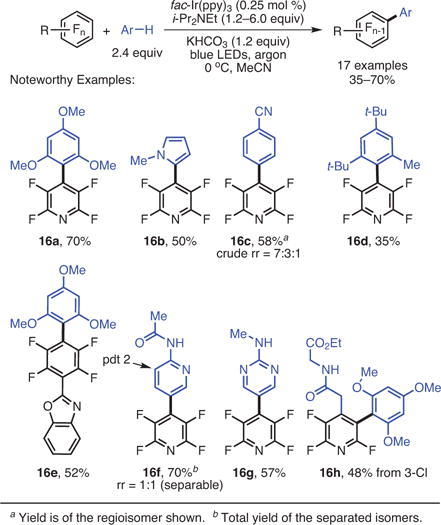
With the goal of developing selective C–F alkylations, our group found that a variety of perfluoroarenes engaged unactivated alkenes to give alkylated products.21 In general, since the perfluoroaryl radical was anticipated to be extremely unstable and consequently highly reactive,27 one might have expected that the reaction would be poorly regioselective with respect to the alkene. The addition, however, takes place with excellent selectivity when there are differences in the substitution patterns of the alkene, with addition occurring at the less substituted carbon (9d–f) of the alkene (eq 3). The complementary reactivity of SNAr chemistry and photocatalysis was demonstrated by subjecting 3-chlorotetrafluoropyridine to the photocatalytic reductive alkylation reaction, which results in the functionalization of the 3-chloro position while keeping the 4-fluoro intact (9e). In contrast, SNAr substitution on this same precursor would be expected to occur at the C-4 position.28 Survival of the remote alkyl chloride of 9f is also noteworthy, and speaks to the functional group compatibility of the photocatalytic reaction.
A common disadvantage of previous C–F alkylation reactions was the incremental increase in complexity of the final products, which arises as a result of alkylating reagents that are of low complexity. In this regard, the use of unmodified alkenes presents enormous opportunity to access coupled products that are stereochemically dense in a single step, since sophisticated alkenes are ubiquitous. This feature makes the photocatalytic reductive alkylation reaction extraordinarily versatile. For instance, a [4 + 2] adduct derived from furan has been satisfactorily coupled with pentafluoropyridine, yielding the product, 9h, which contains five stereocenters—two of them base-labile—three cycles, and a bridging oxygen.
The SNAr reaction takes advantage of the highly fluorinated nature of perfluoroarenes to simultaneously elaborate the molecules and reduce their fluorine content to access sophisticated fluoroarenes with just 2–3 fluorines. It was initially suspected that the fluorines on the arene ring activate the substrate towards reduction and that the removal of fluorine would deactivate the substrate towards reduction, and thus it was not clear that subsequent photochemical C–F functionalizations would take place with sufficient rates to be useful on substrates with fewer fluorine atoms. A series of substrates containing rings with three fluorine atoms were synthesized and subjected to the reaction. After substitution of the most electronically activated 4 position, photocatalytic functionalization moved to the C–F bond adjacent to the electron-withdrawing group or atom (eq 4).21 The ability to perform photocatalytic C–F functionalizations on previously elaborated substrates would thus allow rapid access to structurally complex, polyfluorinated arenes with diverse functionality.
 |
eq 3 |
(Ref. 21)
The photochemical functionalization and SNAr reactions of perfluoroarenes exhibit complementary selectivities (Scheme 5).21,28,29 This phenomenon was demonstrated by subjecting substituted fluoroarenes 11a and 11b, which are themselves products of SNAr chemistry,28 to the photochemical alkylation conditions (Scheme 6).21
Scheme 5.
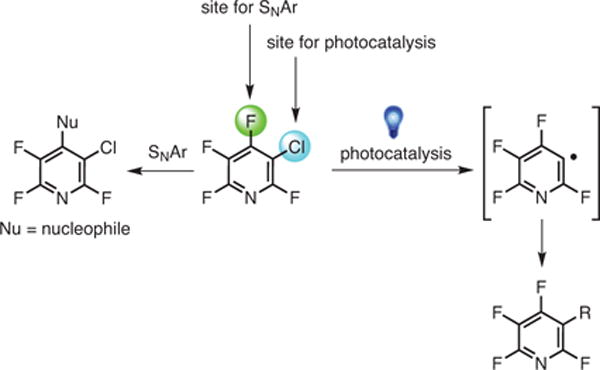
Complementary Nature of SNAr and Photocatalysis. (Ref. 21,28,29)
Scheme 6.
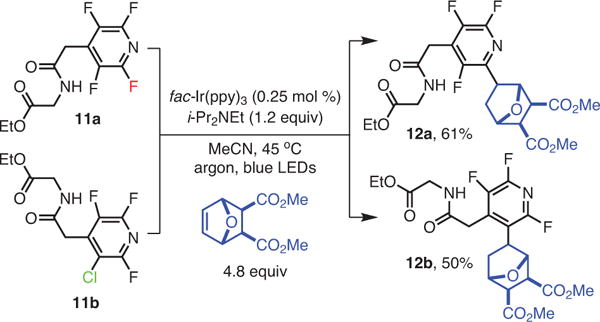
Accessing Complementary Regioisomers by Using Differential Reactivities of C–X Bonds. (Ref. 21)
Densely functionalized arenes with a reduced number of fluorines are a challenging target in drug discovery.1c Subsequent photocatalytic HDF of the products could provide access to additional valuable partially fluorinated arenes. Thus, commercially available perfluoroarenes (13a–d) were subjected to the SNAr reaction, giving rise to elaborated perfluoroarenes (Scheme 7).21,28 Next, the elaborated substrates were photocatalytically functionalized to obtain alkylated arenes. Finally, they were subjected to photocatalytic HDF to further reduce the fluorine content, supplying structurally complex difluorinated arenes (13e–j). The sequence took place with acceptable yields and with excellent regio- and diastereoselectivities, and demonstrated how structurally complex difluoro aromatics can be obtained in a straightforward manner from commercially available perfluoroarenes.
 |
eq 4 |
(Ref. 21)
Scheme 7.
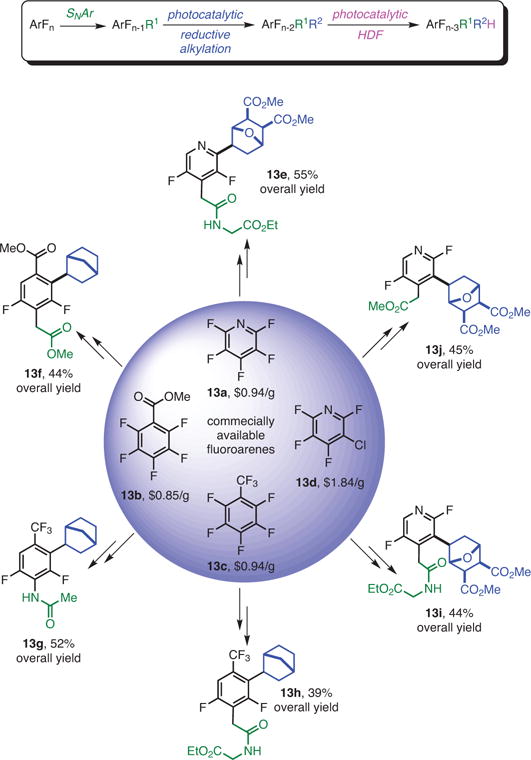
Sequential SNAr and Photocatalysis as a Succinct and Versatile Way to Access Complex, Partially Fluorinated Arenes. (Ref. 21,28)
4. Arylation of Fluoroarenes
Having demonstrated the ability to perform the reductive alkylation of a C–F bond, our group investigated next the possibility of performing an oxidation of the incipient alkyl radical rather than the hydrogen-atom transfer that takes place in the alkylation reaction. It was expected that accomplishing an oxidation might be difficult, since the conditions used to accomplish the C–F fragmentation must produce a strong reductant, at least transiently. As a starting point, hydrogen arenes were chosen, and were anticipated to re-aromatize after temporary loss of aromaticity, which was expected to serve as a driving force for the oxidation. Careful consideration of electronic factors suggested that oxidation of intermediate 14 could happen either before (Path A) or after (Path B) the deprotonation step (Scheme 8).29 The actual path taken would depend on the nature of the two arene partners. Starting with the HDF conditions, the reaction was optimized to produce appreciable amounts of the C–F arylation product, demonstrating the first catalytic dual C–F, C–H functionalization to obtain polyfluorinated biaryls,29 though other less direct methods exist (eq 5).30
Scheme 8.
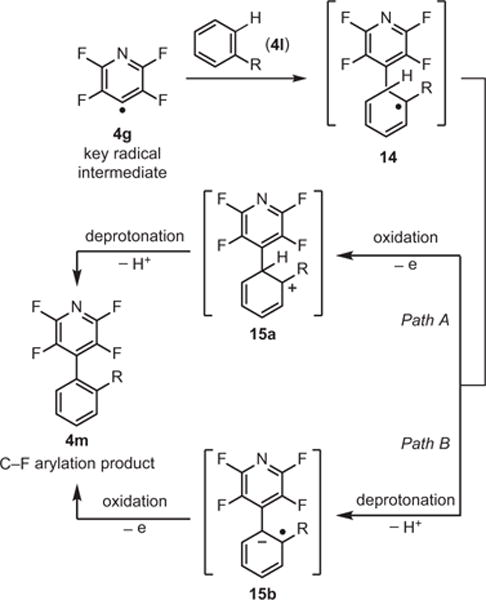
Proposed Mechanism for C–F Arylation. (Ref. 29)
4.1. Photocatalytic Arylation
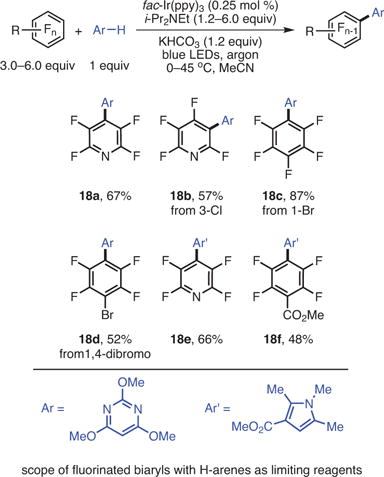
Unlike reductive alkylation, the desired arylation of fluoroarenes is expected to end with oxidative re-aromatization. Since there is no hydrogen-atom abstraction during the generation of the desired product, it was speculated that the amine might simply serve as a transient electron donor and a base. Consequently, substoichiometric amounts of amine base would be sufficient for the reduction of the catalyst, fac-Ir(ppy)3, if it were liberated from the HF salt. Given that the free amine, i-Pr2NEt, can act as a hydrogen atom source as well, the amount of the HDF product could be controlled by keeping the amine content low and lowering the temperature. An inorganic base, KHCO3, was found to be optimal for scavenging the HF byproduct. Good-to-modest yields were obtained for a variety of fluoroarenes and electron-rich and electron-poor H-arenes that were coupled together (eq 6).29 The ability of the perfluoroaryl radical to form highly sterically congested C–C bonds (16d) is particularly noteworthy and is rivaled by few methods. Consistent with that observed in the photocatalytic HDF (5f) and photochemical C–F alkylation chemistry (9e), C–Cl fragmentation (16h) takes place selectively over C–F fragmentation. Interestingly, some basic heterocycles showed anti-Minisci selectivity (16f,g) demonstrating a divergence from typical radical addition to basic heterocycles (Figure 4).29 This difference may be due to the differences in pH between the photocatalytic C–F, C–H arylation and typical Minisci conditions, which are strongly acidic. The acidic matrix protonates the basic heterocycles and causes a polarity reversal.
Figure 4.
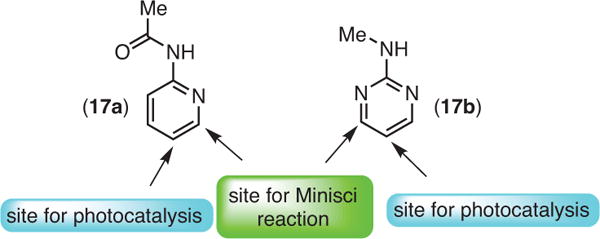
Observed anti-Minisci Selectivity. (Ref. 29)
Given the mildness of the reaction and the ability to construct sterically congested biaryls, it was envisioned that the reaction would be ideal for late-stage functionalization. This could ease the difficult fluoroarene installation and offer an alternative route to some of the lengthy procedures to access fluorinated biaryls. In the case of heavily functionalized H-arenes, which would likely be the more valuable component of the reaction mixture, it is logical to use the perfluoroarene as the excess reagent (eq 7).29
Recall that the further photocatalytic alkylations and reductions were possible on the substrates with a reduced number of fluorines (see Scheme 7). Similarly, we wanted to demonstrate the ability to access di-fluorinated biaryls. The commercially available fluoroarenes were subjected to sequential substitution, arylation, and HDF. Electronically determined regioselectivity was observed during the HDF reaction, while some other competing phenomena gave rise to minor regioisomers (Scheme 9).29
Scheme 9.
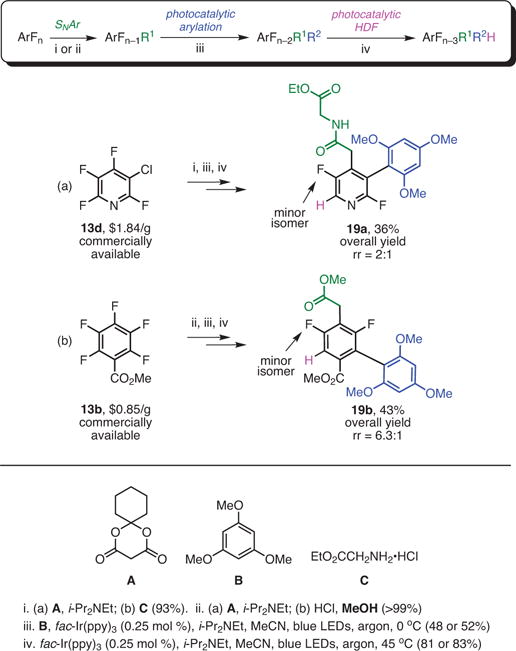
Elaboration via Synergistic SNAr and Photocatalysis. (Ref. 29)
5. Photocatalytic Alkenylation and Energy Transfer
The work described so far in this review is a consequence of photocatalyst-induced electron transfer that results in either C–F reductions or C–F functionalizations, leading to new C–C bonds. However, it has been shown that the excited state of the Ir-based photocatalysts can also be quenched via an energy-transfer process rather than electron transfer in cases where a styrenyl-like system is present.31 However, the reactions employing both electron transfer and energy transfer are rather rare, presumably due to a diminutive understanding of the factors that govern these two fundamentally different processes. Among the very few examples of sequential electron and energy transfers,32 chemists have utilized strategies such as a solvent change to favor electron transfer or a switch to photocatalysts of insufficient energy to prevent energy transfer.32,33 In order to gain a better insight into how to switch between these two mechanistically distinct photoquenching processes, a novel hydrofluoroarylation reaction of alkynes was conceived. Toward that end, the work on selective energy transfer was integrated with the ability to perform C–F functionalization via photocatalytic electron transfer.32,34 The proposed reaction was ideal, because the available photocatalysts that were sufficiently reducing to induce C–F fragmentation were also sufficiently energetic to facilitate the isomerization event, effectively removing the known strategies for separating these two mechanistic processes.
 |
eq 5 |
(Ref. 30)
Additionally, we were interested in investigating an underexplored facet of photocatalytic energy transfer, specifically, the rate of energy transfer as a function of intermolecular distance between the substrate and photocatalyst.35 Given that both Förster’s36 and Dexter’s37 energy-transfer processes are known to be highly dependent on internuclear distance, we anticipated that the steric volume of the photocatalyst could potentially serve as a handle that would allow us to turn energy transfer on or off. The key radical intermediate, 4g, generated via electron transfer would add to the alkyne to give a vinyl radical, 20a, followed by H-atom abstraction to give the alkenylated product, 20b (Scheme 10).35 An energy-transfer process could then lead to a selective double-bond isomerization to obtain the Z isomer, 4p, preferentially.
 |
eq 6 |
(Ref. 29)
 |
eq 7 |
(Ref. 29)
Scheme 10.
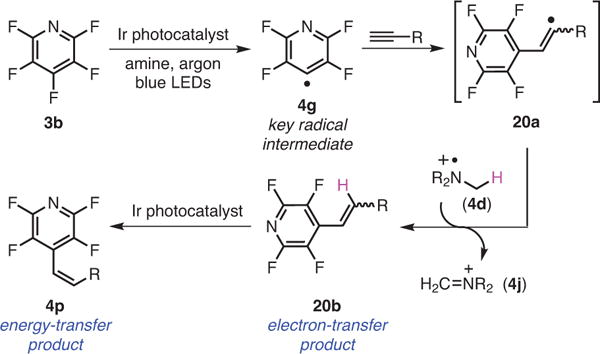
Photocatalytic Alkenylation and Isomerization via Subsequent Electron- and Energy-Transfer Processes. (Ref. 35)
First, we carried out optimizations that focused on achieving high yields of the alkenylated product, regardless of E or Z geometry. Incremental addition of amine and reduced temperature suppressed the formation of the undesired (HDF) product.
Next, to probe the mechanistic details, a bulky alkyne, tert-butylacetylene (21, Scheme 11) was used because of its strong kinetic preference for the E isomer (22a) and large preference for the Z isomer (22b) at the photostationary state.
Scheme 11.
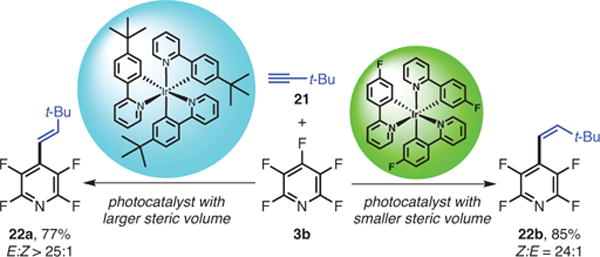
Observed Alkenylation E or Z Switch by Switching Photocatalysts with Different Steric Volumes. (Ref. 35)
The observation of sensitized isomerization of stilbenes with planar sensitizers, such as benzophenone and xanthone, had revealed a strong correlation between the emissive energy of the sensitizer and the Z:E ratio.38 In contrast to such planar sensitizers, Ir-based photocatalysts with three bidentate ligands are approximately spherical, and a plot of log(Z:E) vs the photocatalyst’s emissive energy revealed no correlation.38 The plot of log(Z:E) as a function of the radius of the catalyst approximated as a sphere shows a linear correlation, suggesting that the propensity to undergo energy transfer diminishes as the volume of the catalyst increases. This valuable observation should lead chemists to consider the size of photocatalysts as a sensitive parameter that can be employed to switch between photocatalyzed processes regardless of emissive energy.
6. Conclusions and Outlook
In summary, photocatalysis is becoming a powerful tool to help solve the central problem of C–F functionalization as it pertains to accessing polyfluorinated arenes. It has been shown that photocatalysis provides access to the reactive perfluoroaryl radical, and we have demonstrated several strategies for intercepting the radical with a variety of coupling partners. The use of a commercially available photocatalyst and a tertiary aliphatic amine furnished the highest reported TON for an HDF reaction without any need for metal hydrides. The photocatalytic reactions discussed in this review take place under mild conditions and display excellent functional group compatibility and broad substrate scope. The reactions open a new avenue to synthesize complex polyfluorinated arenes. While some yields of the C–F functionalization products are modest, they are offset by the fact that the reagents need no prefunctionalization and come directly from commercial sources, allowing an unprecedented level of complexity to be achieved in just a few synthetic steps, and creating many opportunities for chemists who are involved in lead discovery programs.
Biographies
Sameera Senaweera received his B.Sc. degree in chemistry in 2010 from the University of Kelaniya, Sri Lanka. After spending the following year teaching at the same university, Sameera moved to the U.S.A., and commenced his graduate studies at Oklahoma State University under the supervision of Professor Jimmie Weaver. Currently, his research in the Weaver laboratory is focused on the development of novel and facile methodologies, such as photocatalytic reduction and functionalization of perfluoroarenes, to access pharmaceutically and industrially important, partially fluorinated arene building blocks.

Jimmie D. Weaver completed his B.S. degree in chemistry with math and physics minors in 2004 at Southern Nazarene University. He spent the following year working in an immunology lab under the guidance of Professor William Hildebrand at the University of Oklahoma Health Sciences Center. Jimmie then enrolled in the graduate program at The University of Kansas under the guidance of Professor Jon Tunge, where he developed palladium-mediated decarboxylative coupling reactions. After earning his Ph.D. degree in 2010, he accepted a postdoctoral position in Professor Jonathan A. Ellman’s laboratory at Yale University. In the fall of 2012, Jimmie joined the chemistry department faculty at Oklahoma State University as an assistant professor. His research program focuses on the catalytic generation and exploitation of reactive intermediates for a variety of applications such as C–H functionalization, C–F functionalization, cross-coupling of perfluoroarenes, and uphill catalysis in which light is used to drive endergonic transformations.

Footnotes
Trademarks. Adempas® (Bayer Aktiengesellschaft); Gilotrif® (Boehringer Ingelheim International GmbH); Januvia® (Merck Sharp & Dohme Corp.); Lipitor® (Pfizer Ireland Pharmaceuticals); Tafinlar® (Novartis Pharma AG); Tivicay® (ViiV Healthcare Company).
References
- 1.(a) Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]; (b) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (c) Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 2.(a) Selby TP, Bereznak JF, Bisaha JJ, Ding AX, Gopalsamuthiram V, Hanagan MA, Long JK, Taggi AE. Fungicidal Substituted Azoles. WO 2009137651. World Pat Appl. 2010 Mar 18;A8; (b) Gregory V, Taggi AE. Fungicidal Mixtures. WO 2011056463. World Pat Appl. 2011 May 12;A2
- 3.(a) Tsuzuki T, Shirasawa N, Suzuki T, Tokito S. Adv Mater. 2003;15:1455. [Google Scholar]; (b) Sakamoto Y, Suzuki T, Miura A, Fujikawa H, Tokito S, Taga Y. J Am Chem Soc. 2000;122:1832. [Google Scholar]
- 4.Hird M. Chem Soc Rev. 2007;36:2070. doi: 10.1039/b610738a. [DOI] [PubMed] [Google Scholar]
- 5.Amii H, Uneyama K. Chem Rev. 2009;109:2119. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]
- 6.(a) Fier PS, Hartwig JF. Science. 2013;342:956. doi: 10.1126/science.1243759. [DOI] [PubMed] [Google Scholar]; (b) Chan KSL, Wasa M, Wang X, Yu JQ. Angew Chem, Int Ed. 2011;50:9081. doi: 10.1002/anie.201102985. [DOI] [PubMed] [Google Scholar]
- 7.(a) Watson DA, Su M, Teverovskiy G, Zhang Y, García-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661. doi: 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ichiishi N, Canty AJ, Yates BF, Sanford MS. Org Lett. 2013;15:5134. doi: 10.1021/ol4025716. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ye Y, Schimler SD, Hanley PS, Sanford MS. J Am Chem Soc. 2013;135:16292. doi: 10.1021/ja408607r. [DOI] [PubMed] [Google Scholar]
- 8.(a) Fier PS, Luo J, Hartwig JF. J Am Chem Soc. 2013;135:2552. doi: 10.1021/ja310909q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tang P, Furuya T, Ritter T. J Am Chem Soc. 2010;132:12150. doi: 10.1021/ja105834t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weaver J, Senaweera S. Tetrahedron. 2014;70:7413. [Google Scholar]
- 10.(a) Blundell P, Ganguly AK, Girijavallabhan VM. Synlett. 1994;263 [Google Scholar]; (b) Saksena AK, Girijavallabhan VM, Lovey RG, Pike RE, Desai JA, Ganguly AK, Hare RS, Loebenberg D, Cacciapuoti A, Parmegiani RM. Bioorg Med Chem Lett. 1994;4:2023. [Google Scholar]
- 11.Blanksby SJ, Ellison GB. Acc Chem Res. 2003;36:255. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- 12.Aizenberg M, Milstein D. Science. 1994;265:359. doi: 10.1126/science.265.5170.359. [DOI] [PubMed] [Google Scholar]
- 13.Arndt P, Spannenberg A, Baumann W, Burlakov VV, Rosenthal U, Becke S, Weiss T. Organometallics. 2004;23:4792. [Google Scholar]
- 14.Kuehnel MF, Lentz D, Braun T. Angew Chem, Int Ed. 2013;52:3328. doi: 10.1002/anie.201205260. [DOI] [PubMed] [Google Scholar]
- 15.(a) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yoon TP, Ischay MA, Du J. Nat Chem. 2010;2:527. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]; (c) Ischay MA, Yoon TP. Eur J Org Chem. 2012;3359 [Google Scholar]; (d) Tucker JW, Stephenson CRJ. J Org Chem. 2012;77:1617. doi: 10.1021/jo202538x. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen JD, D’Amato EM, Narayanam JMR, Stephenson CRJ. Nat Chem. 2012;4:854. doi: 10.1038/nchem.1452. [DOI] [PubMed] [Google Scholar]
- 17.(a) Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top Curr Chem. 2007;281:143. [Google Scholar]; (b) Konovalov VV, Laev SS, Beregovaya IV, Shchegoleva LN, Shteingarts VD, Tsvetkov YD, Bilkis I. J Phys Chem A. 2000;104:352. [Google Scholar]; (c) Shteingarts VD. J Fluorine Chem. 2007;128:797. [Google Scholar]
- 18.Beletskaya IP, Artamkina GA, Mil’chenko AY, Sazonov PK, Shtern MM. J Phys Org Chem. 1996;9:319. [Google Scholar]
- 19.Senaweera SM, Singh A, Weaver JD. J Am Chem Soc. 2014;136:3002. doi: 10.1021/ja500031m. [DOI] [PubMed] [Google Scholar]
- 20.Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, von Zelewsky A. Coord Chem Rev. 1988;84:85. [Google Scholar]
- 21.Singh A, Kubik JJ, Weaver JD. Chem Sci. 2015;6:7206. doi: 10.1039/c5sc03013g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lalevée J, Allonas X, Fouassier J-P. J Org Chem. 2005;70:814. doi: 10.1021/jo048381c. [DOI] [PubMed] [Google Scholar]
- 23.(a) Wang T, Alfonso BJ, Love JA. Org Lett. 2007;9:5629. doi: 10.1021/ol702591b. [DOI] [PubMed] [Google Scholar]; (b) Sun AD, Love JA. J Fluorine Chem. 2010;131:1237. [Google Scholar]
- 24.Sun AD, Leung K, Restivo AD, LaBerge NA, Takasaki H, Love JA. Chem—Eur J. 2014;20:3162. doi: 10.1002/chem.201303809. [DOI] [PubMed] [Google Scholar]
- 25.Sun Y, Sun H, Jia J, Du A, Li X. Organometallics. 2014;33:1079. [Google Scholar]
- 26.Lu W, Gao J, Yang JK, Liu L, Zhao Y, Wu HC. Chem Sci. 2014;5:1934. [Google Scholar]
- 27.Birchall JM, Hazard R, Haszeldine RN, Wakalski AW. J Chem Soc (c) 1967;47 [Google Scholar]
- 28.Senaweera SM, Weaver JD. J Org Chem. 2014;79:10466. doi: 10.1021/jo502075p. [DOI] [PubMed] [Google Scholar]
- 29.Senaweera S, Weaver JD. J Am Chem Soc. 2016;138:2520. doi: 10.1021/jacs.5b13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) DePasquale RJ, Tamborski C. J Org Chem. 1969;34:1736. [Google Scholar]; (b) Shang R, Fu Y, Wang Y, Xu Q, Yu HZ, Liu L. Angew Chem, Int Ed. 2009;48:9350. doi: 10.1002/anie.200904916. [DOI] [PubMed] [Google Scholar]; (c) Shang R, Xu Q, Jiang YY, Wang Y, Liu L. Org Lett. 2010;12:1000. doi: 10.1021/ol100008q. [DOI] [PubMed] [Google Scholar]; (d) Frohn HJ, Adonin NY, Bardin VV, Starichenko VF. J Fluorine Chem. 2002;117:115. [Google Scholar]; (e) Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Korenaga T, Kosaki T, Fukumura R, Ema T, Sakai T. Org Lett. 2005;7:4915. doi: 10.1021/ol051866i. [DOI] [PubMed] [Google Scholar]; (g) Wei Y, Kan J, Wang M, Su W, Hong M. Org Lett. 2009;11:3346. doi: 10.1021/ol901200g. [DOI] [PubMed] [Google Scholar]; (h) Lindner S, Bräse S, Masters K-S. J Fluorine Chem. 2015;179:102. [Google Scholar]; (i) Li H, Liu J, Sun CL, Li BJ, Shi ZJ. Org Lett. 2011;13:276. doi: 10.1021/ol102688e. [DOI] [PubMed] [Google Scholar]; (j) Wei Y, Su W. J Am Chem Soc. 2010;132:16377. doi: 10.1021/ja109383e. [DOI] [PubMed] [Google Scholar]; (k) Meyer AU, Slanina T, Yao C-J, König B. ACS Catal. 2016;6:369. [Google Scholar]; (l) Yoshikai N, Mashima H, Nakamura E. J Am Chem Soc. 2005;127:17978. doi: 10.1021/ja056327n. [DOI] [PubMed] [Google Scholar]; (m) Nakamura Y, Yoshikai N, Ilies L, Nakamura E. Org Lett. 2012;14:3316. doi: 10.1021/ol301195x. [DOI] [PubMed] [Google Scholar]; (n) Saijo H, Sakaguchi H, Ohashi M, Ogoshi S. Organometallics. 2014;33:3669. [Google Scholar]; (o) Sun AD, Love JA. Org Lett. 2011;13:2750. doi: 10.1021/ol200860t. [DOI] [PubMed] [Google Scholar]
- 31.(a) Metternich JB, Gilmour R. J Am Chem Soc. 2016;138:1040. doi: 10.1021/jacs.5b12081. [DOI] [PubMed] [Google Scholar]; (b) Kumarasamy E, Raghunathan R, Jockusch S, Ugrinov A, Sivaguru J. J Am Chem Soc. 2014;136:8729. doi: 10.1021/ja5034638. [DOI] [PubMed] [Google Scholar]; (c) Arceo E, Montroni E, Melchiorre P. Angew Chem, Int Ed. 2014;53:12064. doi: 10.1002/anie.201406450. [DOI] [PubMed] [Google Scholar]; (d) Farney EP, Yoon TP. Angew Chem, Int Ed. 2014;53:793. doi: 10.1002/anie.201308820. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zou YQ, Duan SW, Meng XG, Hu XQ, Gao S, Chen JR, Xiao WJ. Tetrahedron. 2012;68:6914. [Google Scholar]; (f) Lu Z, Yoon TP. Angew Chem, Int Ed. 2012;51:10329. doi: 10.1002/anie.201204835. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Chen Y, Kamlet AS, Steinman JB, Liu DR. Nat Chem. 2011;3:146. doi: 10.1038/nchem.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin QY, Xu XH, Qing FL. J Org Chem. 2014;79:10434. doi: 10.1021/jo502040t. [DOI] [PubMed] [Google Scholar]
- 33.Noble A, MacMillan DWC. J Am Chem Soc. 2014;136:11602. doi: 10.1021/ja506094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Singh K, Staig SJ, Weaver JD. J Am Chem Soc. 2014;136:5275. doi: 10.1021/ja5019749. [DOI] [PubMed] [Google Scholar]; (b) Fabry DC, Ronge MA, Rueping M. Chem—Eur J. 2015;21:5350. doi: 10.1002/chem.201406653. [DOI] [PubMed] [Google Scholar]; (c) Rackl D, Kreitmeier P, Reiser O. Green Chem. 2016;18:214. [Google Scholar]
- 35.Singh A, Fennell CJ, Weaver JD. Chem Sci. 2016 doi: 10.1039/C6SC02422J. Advance Article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Förster T. Ann Phys (Berlin, Ger) 1948;437:55. [Google Scholar]
- 37.Dexter DL. J Chem Phys. 1953;21:836. [Google Scholar]
- 38.(a) Hammond GS, Saltiel J, Lamola AA, Turro NJ, Bradshaw JS, Cowan DO, Counsell RC, Vogt V, Dalton C. J Am Chem Soc. 1964;86:3197. [Google Scholar]; (b) Klán P, Wirz J. Photochemistry of Organic Compounds: From Concepts to Practice. Wiley; Chichester, U.K.: 2009. Introduction; p. 1. [Google Scholar]