

Abstract
The blood-brain barrier (BBB) is critical to central nervous system (CNS) health. Brain microvascular endothelial cells (BMECs) are often used as in vitro BBB models for studying BBB dysfunction and therapeutic screening applications. Human pluripotent stem cells (hPSCs) can be differentiated to cells having key BMEC barrier and transporter properties, offering a renewable, scalable source of human BMECs. hPSC-derived BMECs have been previously shown to respond to all-trans retinoic acid (RA), and the goal of this study was to identify the stages at which differentiating human induced pluripotent stem cells (iPSCs) respond to activation of RA receptors (RARs) to impart BBB phenotypes. Here we identified that RA application to iPSC-derived BMECs at days 6-8 of differentiation led to a substantial elevation in transendothelial electrical resistance and induction of VE-cadherin expression. Specific RAR agonists identified RARα, RARγ, and RXRα as receptors capable of inducing barrier phenotypes. Moreover, RAR/RXRα costimulation elevated VE-cadherin expression and improved barrier fidelity to levels that recapitulated the effects of RA. This study elucidates the roles of RA signaling in iPSC-derived BMEC differentiation, and identifies directed agonist approaches that can improve BMEC fidelity for drug screening studies while also distinguishing potential nuclear receptor targets to explore in BBB dysfunction and therapy.
Keywords: blood-brain barrier, brain microvascular endothelial cells, human pluripotent stem cells, nuclear hormone receptors, retinoic acid
Graphical abstract
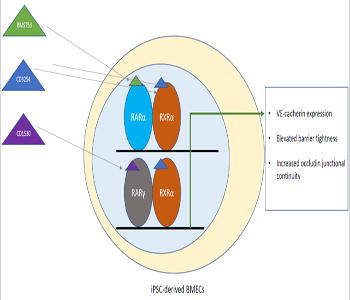
Brain microvascular endothelial cells, the cells the form the physical blood-brain barrier, can be used for drug screening studies and investigating blood-brain barrier contribution to central nervous system diseases. Human pluripotent stem cells can be differentiated into a scalable and renewable source of brain-like endothelial cells, and these cells exhibit increased barrier tightness when treated with all-trans retinoic acid. In this study, the authors examine the small molecule agonists against specific retinoic acid signaling receptors to identify receptors for future therapeutic applications. Through this work, they found that activation of the RARα, RARγ or RXRα receptors lead to earlier VE-cadherin expression–a mature endothelial cell protein, elevated barrier tightness, and increased junctional integrity of the tight junction protein Occludin. In addition, coactivation of the RAR/RXRα receptors using small molecule agonists exhibited appreciable improvements above single agonist treatment conditions.
1 Introduction
The blood-brain barrier (BBB) is a critical component for central nervous system (CNS) health. The selectively impermeable blood vessels that comprise the BBB regulate the exchange of material between the bloodstream and brain [1]. Brain microvascular endothelial cells (BMECs) form the principle barrier, and BMECs express a host of tight junction proteins, nutrient transporters, and efflux transporters. Recent studies have implicated BBB dysfunction early in CNS disease progression [2-6]. In addition, the BBB restricts many therapeutics from brain entry, creating a bottleneck in CNS disease treatment [7, 8]. Human in vitro BBB models offer the capability for high throughput screening of potential therapeutics and for the study of cellular mechanisms that drive human BBB health and disease. Recently, human pluripotent stem cell (hPSC)-sourced BMECs have been described that express BBB tight junction proteins, efflux transporters, and nutrient transporters while exhibiting functionally tight barriers [9, 10].
Differentiation of hPSCs to BMECs occurs in a four step process in which hPSCs are first seeded as single cells on Matrigel and expanded as hPSCs in mTeSR1 for three days [9, 11, 12] (D-3 to D0) (Figure 1A). Cells are subsequently cultured in unconditioned medium (UM) for six days (D0-D6), conditions that result in the codifferentiation of neural cells (NCs) and endothelial cells (ECs) that gain properties of BMECs. The BMEC population is expanded in EC medium for two days (D6-D8). Finally, BMECs are subcultured onto collagen/fibronectin-coated plates or filters as virtually pure monolayers (D8-D10) for assessing barrier properties and other BMEC phenotypes. BMECs express efflux transporter and tight junction proteins by D8 of the differentiation, but do not express VE-cadherin, a more mature EC marker, until after subculture [9].
Figure 1.
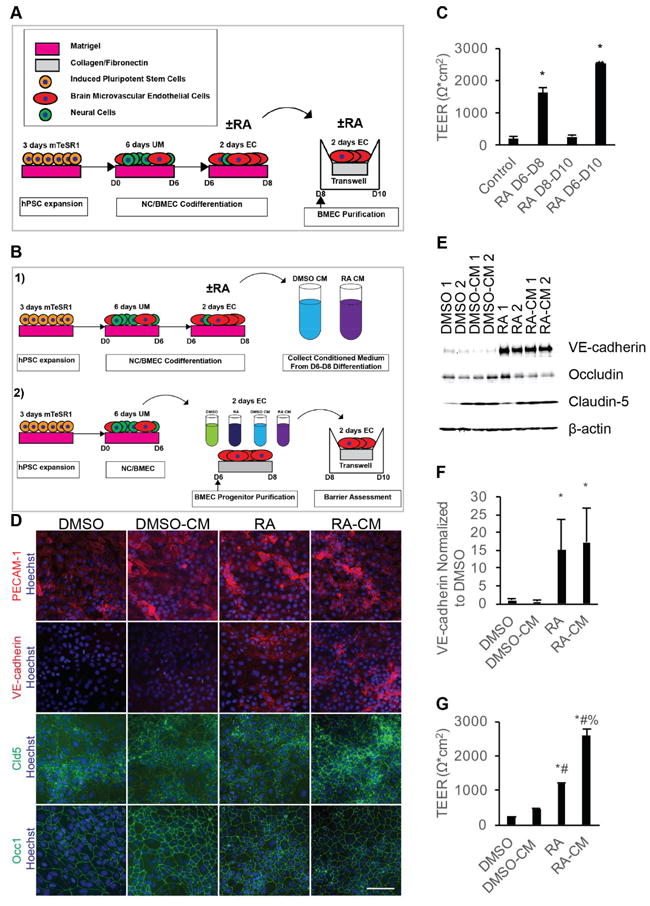
Temporal effects of RA signaling on TEER of iPSC-derived BMECs. A) Experimental timeline for 10 μM RA dosage during BMEC differentiation. B) Experimental timeline for BMEC progenitor purification and treatment. C) D10 TEER following 10 μM RA stimulation over the indicated two day increments. Error bars represent standard error of the mean; n = 3. ANOVA followed by Tukey HSD test; * p < 0.05 vs. control; # p < 0.05 vs. RA D6-D8. D) Representative images of D8 purified BMEC progenitors exposed to the indicated media from three independent differentiations. Scale bar represents 100 μm. E) Representative Western blot of D8 BMEC progenitors exposed to the indicated treatments from D6-D8 from three independent differentiations; n = 3. Replicates bands are technical replicates within a single differentiation. F) Quantification of VE-cadherin band intensities from three independent differentiations via densitometry of Western blots; n = 3. Error bars represent standard error of the mean. ANOVA followed by Dunnett post-hoc analysis. * p < 0.05 vs. DMSO. G) Representative D10 TEER from purified BMEC progenitors exposed to the indicated media from D6-D8 and passaging to filters at D10. TEER assessed in three independent differentiations, and values are technical replicates (N = 2) of a single representative differentiation. Error bars represent standard deviation. All groups were compared using ANOVA followed by Tukey HSD. * p < 0.05 vs. DMSO; # p < 0.05 vs. DMSO-CM; % p < 0.05 vs. RA.
We recently found that differentiating hPSC-derived BMECs respond to all-trans retinoic acid (RA), a hormone implicated in CNS development and hindbrain patterning [13] and BBB development. Previous studies have identified RA production by astrocyte progenitor cells during embryonic BBB development and coordination with WNT signaling to promote BBB fidelity [14-16]. During hPSC differentiation to BMECs, administration of RA during the EC expansion phase (D6-D8) and the first 24 hours of the subculture phase (D8-D9) induced VE-cadherin expression at D8, and the resulting BMECs exhibited dramatically elevated transendothelial electrical resistance (TEER) at D10 of the differentiation, an indicator of BMEC barrier function [10]. Whether RA is essential during the codifferentiation phase when NCs are present, or if RA can act directly on BMECs following subculture remains unknown. Moreover, RA signaling occurs via all-trans RA binding to nuclear RA receptors (RARs), which bind to DNA response elements and regulate RA-targeted gene transcription [17, 18]. All-trans RA can directly activate three RAR family isoforms (α, β, and γ), and RAR members dimerize with retinoid X receptors (RXRs) to bind DNA [19, 20]. RXRs also exist as three separate isoforms (α, β, and γ) and exhibit different heterodimeric combinations with RAR receptors based on tissue-specific expression patterns of RAR and RXR nuclear hormone receptors [21]. Understanding the functional and temporal roles of RA signaling and the particular receptors that can impart BMEC phenotypes in differentiating hPSCs could identify potential signaling targets important in human BBB development and maintenance.
We therefore set out to elucidate the impact of RAR and RXR agonism on BMEC differentiation and BBB phenotype acquisition. RA treatment of differentiating human induced pluripotent stem cell (iPSC)-derived BMEC cultures imparted optimal barrier phenotypes when applied during the EC expansion phase (D6-D8). Agonism of RARα and RARγ upregulated VE-cadherin expression and enhanced BMEC barrier formation. Furthermore, we determined that RARα or RARγ could synergize with RXRα to impart BMEC phenotypes. This work establishes the role of RA in BMEC differentiation from iPSCs and identifies selective RA-associated nuclear hormone receptor agonists that are sufficient to improve iPSC-derived BMEC phenotypes while offering insight into RA signaling pathway roles in BBB development and maintenance.
2 Methods
2.1 Differentiation of iPSCs to a mixture of BMECs and NCs
iPSC culture and differentiation methods were described in detail in Stebbins et al. [12]. Briefly, IMR90-C4 iPSCs (WiCell) were maintained in adherent culture on Matrigel (BD Biosciences) coated plates. iPSCs were cultured in mTeSR1 with daily medium changes. When iPSC colony edges began to contact adjacent colonies, cells were passaged in colony format using Versene (Gibco).
Initial starting density for BMEC differentiation has a substantial impact on differentiation efficiency, with an ideal starting density of 3.0 × 104 cells/cm2 [11]. To achieve this target density, iPSCs were singularized 3 days prior to initiating a differentiation (D-3) using Accutase (Innovative Cell Technologies) and replated onto fresh Matrigel-coated plates in mTesR1 containing 10 μM Y27632 (Tocris; from 10 mM Y27632 solution diluted in sterile water). Cells were typically seeded between 7.5 × 103 cells/cm2 and 1.25 × 104 cells/cm2 at D-3 to achieve a cell density of 3.0 × 104 cells/cm2 at day 0 (D0). Cell medium was replaced 24 hours later (D-2) and 2 days after seeding (D-1) with mTeSR1 lacking Y27632. To initiate BMEC differentiation at D0, cell medium was replaced with UM (392.5 mL DMEM/F12, Gibco; 100 mL KnockOut Serum Replacement, Gibco; 5 mL MEM NEAA, Gibco; 2.5 mL GlutaMAX, Gibco; 3.5 μL β-mercaptoethanol, Millipore Sigma). Cell medium was replaced daily with fresh UM until D6 to differentiate iPSCs to a mixture of BMECs and NCs.
2.2 Probing the temporal effects of RA signaling during iPSC-derived BMEC differentiation
Cell medium at D6 was replaced with EC medium, containing Human Endothelial Serum Free Medium (HESFM; Gibco) supplemented with 20 ng/mL bFGF (100 μg/mL stock solution from Waisman Biomanufacturing) and 1% platelet-poor plasma derived serum (Alfa Aesar). Cells were treated with a final concentration of 10 μM RA (Tocris resuspended in dimethyl sulfoxide, DMSO, Millipore Sigma; 10 mM stock concentration dissolved in DMSO) or an equivalent volume DMSO. EC medium was not replaced at D7. At day 8 (D8), BMEC/NC cocultures were singularized using Accutase and replated in EC medium supplemented with DMSO or 10 μM RA (Figure 1A). Cells were replated onto 4:1:5 collagen IV (Millipore Sigma): fibronectin (Millipore Sigma): sterile Milli Q water coated Transwell filters (Corning) at a seeding density of 1.0 × 106 cells/cm2. Cell medium was replaced at D9 with EC medium supplemented with DMSO or 10 μM RA. 10 μM Y23732 was added at D9 to improve cell viability and increase experimental reproducibility because DMSO vehicle control cells exhibited cell detachment at 48 hours after replating. Note that bFGF was included in EC medium at D9, unlike previously published protocols, to preserve the same medium conditions that cells received from D6-D8 of the differentiation [9-12].
2.3 BMEC progenitor purification at D6
Prior to conducting a BMEC progenitor purification at D6, conditioned medium (CM) was harvested from BMEC/NC codifferentiation cocultures at D7 and D8 of the differentiation. To harvest CM, D6 BMEC/NC codifferentiation mixtures were treated with EC medium containing 10 μM RA or volume equivalent DMSO (Figure 1B panel 1). At D7, CM was collected from cells into a 15 mL conical tube, centrifuged for 5 minutes at 1000 RPM, and the supernatant transferred to a fresh 15 mL conical tube. The supernatant was snap frozen at -20°C. Media in BMEC/NC codifferentiations were replaced at D7 with fresh EC medium supplemented with 10 μM RA or volume equivalent DMSO. At D8, CM was harvested as described above.
To isolate BMEC progenitors at D6, a subsequent iPSC-derived BMEC/NC codifferentiation was dissociated at D6 with Accutase (Figure 1B panel 2). Cells were quenched 4:1 with DMEM/F12 and pelleted by centrifuging cells 5 minutes at 1000 RPM. Cells were replated at a split ratio of 4 6-wells to 6 12-wells onto 5x dilute collagen/fibronectin-coated 12-well plates. Cells were replated in the 1 of the 4 following conditions: “DMSO”, “RA”, “DMSO-CM”, or “RA-CM.” For “RA” and “DMSO” treatment conditions, cells were replated in EC medium containing 10 μM RA or volume equivalent DMSO. For “RA-CM” and “DMSO-CM” samples, cells were replated in a 25% fresh EC: 75% CM from D7 BMEC/NC codifferentiations. Fresh medium was mixed into CM to supplement CM with fresh nutrients. Fresh medium was supplemented with 10 μM RA or volume equivalent DMSO prior to mixing fresh medium with CM to adjust RA and DMSO concentrations to comparable levels as “RA” and “DMSO” conditions. At D7, media from “RA” and “DMSO” samples were replaced with fresh EC medium containing 10 μM RA or volume equivalent DMSO. “RA-CM” and “DMSO-CM” samples were treated with 75% D8 CM and 25% fresh EC medium as described above. All samples were supplemented with 10 μM Y27632 to increase cell survival. Cell medium was replaced at D7 to remove dead cell debris and increase cell survival following replating at D6.
Cells were replated at 6.6 × 105 cells/cm2 per Transwell filter at D8 onto collagen/fibronectin coated filters as described above. All sample conditions were replated in EC medium without RA. At D9, cell medium was switched to EC medium without bFGF supplemented with 10 μM Y27632 to increase cell viability and prevent cell detachment from filters.
2.4 Retinoid agonist treatment
Cell medium at D6 was replaced with EC medium containing activating concentrations of retinoid agonists (10 μM CD3254, BMS753, BMS453, or docohexaonic acid from stock 10 mM solutions dissolved in DMSO) or subactivating concentrations of these agonists (1 μM CD3254 or BMS753; diluted from stock 10 mM solutions diluted in DMSO). CD1530 is a potent RARγ agonist, but can activate RARα and RARβ at high concentrations. We, therefore, added CD1530 at a concentration below the IC50 for CD1530 activation of RARα and RARβ (1 μM CD1530 activating concentration from 10 mM stock dissolved in DMSO; 0.1 μM CD1530 subactivating concentration from 1 mM stock solution dissolved in DMSO). All retinoid agonists were purchased from Tocris. Cell medium was not changed at D7.
At day 8 (D8), BMEC/NC cocultures were singularized using Accutase and replated in EC medium. For screening experiments (Figure 2A), retinoid agonists were not added at D9. Retinoid agonists were added at D9 for all other experiments to assess maximum TEER response to small molecule agonists. Cells were replated onto 4:1:5 collagen IV: fibronectin: sterile Milli Q water-coated Transwell filters at a seeding density of 1.0 × 106 cells/cm2 per filter or 1.32 × 105 cells/cm2 per plate. 96 well plates typically exhibited lower plating efficiency than other cell culture plate sizes, and therefore required a plating density of 2.0 × 105 cells/well. At D9, cell medium was switched to EC without bFGF supplemented with 10 μM Y27632.
Figure 2.
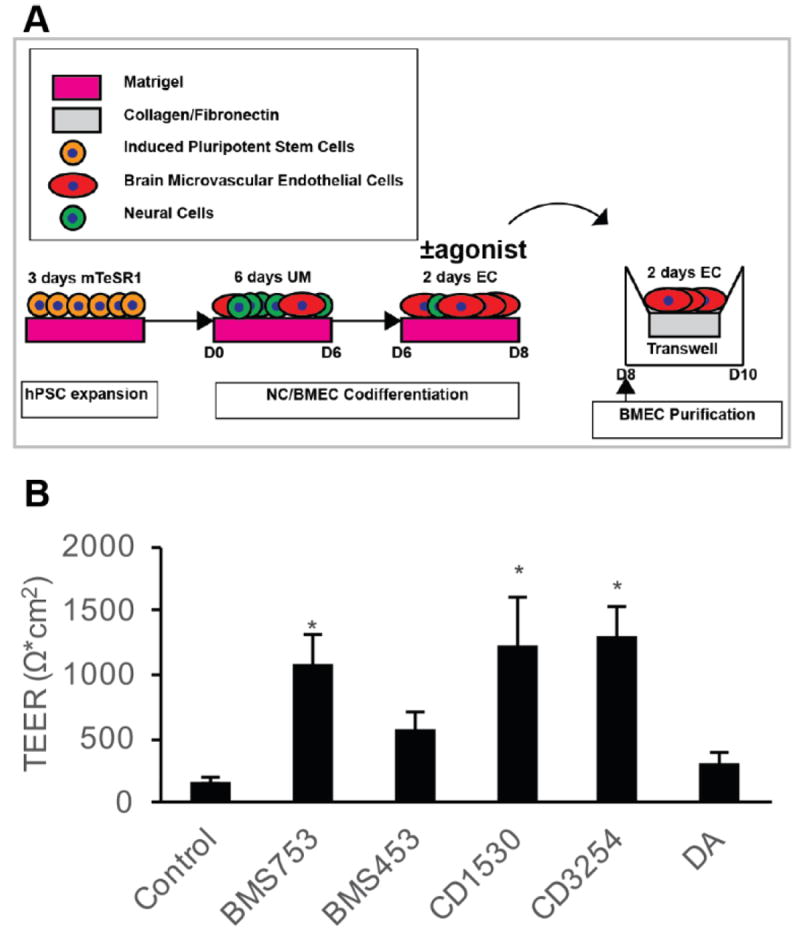
Effect of RA-associated nuclear hormone receptor agonists on BMEC barrier fidelity. A) Experimental timeline for application of RA-signaling associated nuclear hormone receptor agonists during iPSC differentiation to BMECs. B) D10 TEER following D6-D8 treatment with RA signaling associated nuclear hormone receptor agonists: 10 μM BMS753 (RARα agonist; n = 7), 10 μM BMS453 (RARβ agonist and RARα/γ antagonist; n = 7), 1 μM CD1530 (potent RARγ agonist; n = 7), 10 μM CD3254 (RXRα agonist; n = 7), 10 μM DA (pan-RXR agonist; n = 3). Error bars represent standard error of the mean. ANOVA followed by Dunnett’s test; * p < 0.05 vs. control.
2.5 Assessment of functional barrier tightness
BMEC TEER was measured using an Epithelial Volt/Ohm Meter (EVOM; World Precision Instruments) with chopstick electrodes on D9 and D10. Briefly, TEER was taken as a soon as Transwell plates were removed from the incubator, as TEER increases as temperature decreases. The short electrode and long electrode arms were inserted vertically into the apical and basolateral chambers respectively, taking care not to touch the Transwell filter or plate sides. To assess TEER, electrical resistance was measured in Ω, multiplied by the surface area of the Transwell filter, and subtracted from the TEER of a blank filter.
2.6 Efflux transporter substrate accumulation assay
Cell medium was aspirated and cells were incubated for 1 hour at 37°C in EC medium without bFGF ± 10 μM efflux transporter inhibitors (P-glycoprotein inhibitor cyclosporin A “CsA”, Millipore Sigma, 10 mM stock solution dissolved in DMSO; MRP inhibitor MK5371, Thermo Fisher, 10 mM stock solution dissolved in DMSO). Cells were subsequently incubated 2 hours at 37°C on a rotating platform at 35 RPM with efflux transporter assay substrate diluted in EC medium without bFGF ± efflux transporter inhibitor, washed 1 time with cold PBS, and lysed using RIPA buffer. Substrate accumulation was quantified using a Tecan plate reader at 485 nm excitation/530 nm emission. To assay P-glycoprotein activity, cells were incubated with or without 10 μM CsA then incubated with rhodamine 123 P-glycoprotein substrate (Millipore Sigma, from 10 mM stock solution dissolved in sterile water) ± 10 μM CsA. Carboxymethyl-2′,7′-dichlorofluorescein diacetate (DCFDA; Thermo Fisher, 10 mM stock solution dissolved in DMSO) was used as an MRP substrate along with 10 μM MK571 as an MRP inhibitor to determine MRP activity in cells treated with retinoid agonists. Fluorescent substrate accumulation levels were normalized to protein content determined by protein content as measured by BCA (Pierce BCA Protein Assay Kit) for each treatment condition.
2.7 Immunocytochemistry
Cells were fixed for 15 minutes with 100% ice-cold methanol (Millipore Sigma) and washed three times with Dulbecco’s phosphate buffered saline (DPBS; Millipore Sigma) without Ca2+ or Mg2+. Cells were subsequently blocked in DPBS with 10% goat serum (PBSG; Millipore Sigma) for one hour at room temperature and incubated overnight with primary antibodies in PBSG. The following day, cells were washed three times with DPBS and incubated 1 hour at room temperature with secondary antibodies. Cells were subsequently incubated for 15 minutes with 1:5000 Hoechst (Thermo Fisher) diluted in DPBS at room temperature, washed 3 times with DPBS, and stored in DPBS until ready to image. A detailed list of the primary and secondary antibodies as well as their dilutions can be found in Stebbins et al. [22].
2.8 Image quantification of tight junction continuity
Tight junctions exhibit a continuous network, which display maximum peak intensities above a low intensity background. The area fraction covered by tight junctions can be quantified by setting a threshold to remove background signal and determining the number of pixels above this threshold covered by tight junction proteins.
To assess tight junction continuity, images were analyzed using the FIJI software (Figure S1). Fluorescent images were first corrected to remove uneven illumination using the Background Correction Plugin. A Gaussian blur filter was applied to each image to reduce noise. Next, a line was drawn by the user, and the grey level intensity profile was plotted over line distance to determine a threshold value to eliminate background. Images were converted to a binary image and processed using the outline filter to count the tight junction perimeter in pixels encompassing each cell within a given field. The total number of tight junction perimeter pixels was normalized to the square root of the number of cells in each field, to account for shared perimeters between adjacent cells. Cell number for each field was determined using the FIJI Analyze Particle feature to count the number of cell nuclei per each tight junction image.
2.9 Flow cytometry
To assess the percentage of VE-cadherin+ cells per sample condition, adherent live cells were incubated for 30 minute at room temperature with PBSG, and subsequently incubated for 1 hour with VE-cadherin (Millipore Clone BV9, 8 μg/mL) or equivalent concentration of mouse IgG2a,κ isotype control (BD Biosciences) diluted in PBSG. Cells were washed three times with DPBS and incubated for 30 minutes at room temperature in secondary antibodies (Thermo Fisher Alexa 488 Goat Anti-Mouse IgG, 1:200) diluted in PBSG. Cells were subsequently washed 3 times with DPBS and dissociated at 37°C using Accutase until 90% of cells began to dissociate from the plate. Cells were sprayed 3 to 5 times to singularize cell clumps, passed through a 40 μm nylon filter to remove cell aggregates, and quenched with DMEM/F12. Cells were spun down 5 minutes at 200g, resuspended in 4% paraformaldehyde (16% paraformaldehyde from Electron Microscopy Sciences diluted in DPBS), and incubated 15 minutes at room temperature. Cells were spun down 5 minutes at 200g, resuspended in 1% BSA diluted in DPBS and run immediately on a BD FACSCalibur flow cytometer.
Cells stained for occludin and claudin-5 were first prepared by rinsing cells with Versene and subsequently incubating cells in Versene at 37°C until 90% of cells dissociated from the plate. Cells were passed through a 40 μm nylon mesh strainer and incubated in 4% paraformaldehyde as previously described. Cells were blocked 30 minutes in 10% donkey serum (Millipore Sigma) diluted in DPBS (PBSDo) with 0.1% Triton-X 100 (Millipore Sigma). Cells were resuspended in PBSDo with primary antibody and incubated overnight at 4°C (occludin, Invitrogen, OC-3F10, IgG1, 10 μg/1e6 cells; claudin-5, Invitrogen, 4C3C2, IgG1, 10 μg/1e6 cells). The following day, cells were washed 2 times in 1% BSA in PBS (flow buffer) and incubated 30 minutes at room temperature with secondary antibody diluted in PBSDo. Cells were washed 2 times with flow buffer and analyzed on a BD FACSCalibur flow cytometer.
Cells stained for PECAM-1 were dissociated using Accutase until 90% of cells were dissociated from the plate. Cells were passed through a 40 μm nylon mesh strainer and incubated 15 minutes with 1% paraformaldehyde (from 16% paraformaldehyde solution diluted in PBS). Cells were subsequently permeabilized in 100% ice-cold methanol for 15 minutes at room temperature. Cells were washed 3 times with 0.5% BSA (Millipore Sigma) + 0.3% Triton-X 100 diluted in DPBS (wash buffer). Cells were incubated overnight with primary antibody (PECAM, LabVison, RB-1033, rabbit polyclonal, 18 μg/1e6 cells) or IgG control (Cell Signaling Technologies) at 4°C. The following day, cells were washed 3 times with wash buffer and incubated for 30 minutes at room temperature with secondary antibodies (Thermo Fisher goat anti-rabbit Alexa 488, 1:200). Cells were subsequently washed and quantified on a BD FACSCalibur flow cytometer.
2.10 Western blotting
Cells were lysed using RIPA buffer (Rockland) including protease/phosphatase inhibitor cocktail (Pierce) and protein concentration quantified via BCA. Lysates were denatured at 95°C and loaded with equal protein content into 4-12% Tris-glycine 15-well WedgeWell SDS-PAGE gels (Thermo Fisher) under reducing conditions. Lysates were resolved at 125 V for 90 minutes. Protein was transferred to nitrocellulose membranes, and membranes were blocked 1 hour with 5% milk in TBS + 0.1% Tween-20 (Thermo Fisher) (TBST). Membranes were incubated overnight at 4°C with primary antibodies diluted in 5% milk in TBST. Membranes were probed for VE-cadherin (Santa Cruz BV9, 1:200), claudin-5 (Thermofisher 4C3C2, 1:250), occludin (Thermofisher OC-3F10), and β-actin (Cell Signaling Technology D59D7, 1:1000). The following day, membranes were washed 5 times with TBST and incubated 1 hour with secondary antibodies (Licor IRDye 800CW donkey anti-mouse IgG; Licor IRDye 680RD donkey anti-rabbit IgG) diluted 1:5000 in 5% milk + TBST. Membranes were washed 5 times with TBST and imaged on a Licor Odyssey Classic Scanner.
2.11 Statistical analysis
Student t-test was used for comparing one experimental condition to control, while ANOVA analysis was used for comparing more than 2 experimental conditions followed by Dunnett’s post-hoc analysis for comparing all experimental conditions only to the control or Tukey’s Honest Significant Difference (HSD) for comparing all experimental conditions against each other. All error is reported as standard error of the mean unless otherwise indicated. A lower case “n” denotes that data represent the average of the means of “n” independent differentiations. An uppercase “N” denotes “N” number of technical replicates averaged together across one representative differentiation, unless otherwise indicated.
3 Results
3.1 Temporal effects of RA signaling on iPSC-derived BMECs
To determine if RA treatment during the EC expansion phase of the NC/BMEC codifferentiation (D6-D8) or the BMEC subculture phase (D8-D10) imparts BMEC-specific phenotypes, all-trans RA was administered during these two timeframes and TEER was measured at D10, the time point previously shown to exhibit maximum barrier tightness (Figure 1A) [10, 11]. Whereas D6-D8 RA treatment led to a striking elevation in TEER (1634 ± 160 Ω*cm2; p<0.05) compared to DMSO vehicle control (200 ± 57 Ω*cm2), RA treatment of purified BMECs (D8-D10 treatment) did not lead to a statistically significant change in TEER (274 ± 41 Ω*cm2, Figure 1C) compared to control. RA administration during both the EC expansion phase and BMEC subculture (D6-D10) improved TEER (2528 ± 34 Ω*cm2; p<0.05) above cells treated only during the EC expansion phase (D6-D8, Figure 1C). These data demonstrate that RA treatment during EC expansion phase is essential for RA-mediated TEER elevation, yet subcultured BMECs can also respond to RA treatment if previously primed with RA during the EC expansion phase.
The D6-D8 RA activation window may be critical because the ECs are more responsive at this differentiation stage, because the codifferentiating NCs may be sensitive to RA induction at this stage thereby inducing BMEC properties, or both. To explore these possibilities, differentiating PECAM-1+ BMEC progenitors were subcultured onto collagen/fibronectin plates at D6 to permit investigation of RA effects on purified BMECs (Figure 1B and D). First, the purified D6 BMEC progenitors were expanded for an additional two days in the presence or absence of RA to investigate if RA can directly affect differentiating BMECs during the D6-D8 window. To additionally test if RA effects are reliant on the codifferentiating NCs, conditioned media from control and RA-treated Day 6-8 codifferentiating cultures (containing NC/BMEC mixtures) were added to D6 purified BMEC progenitors (Figure 1B). At D8, purified BMEC progenitors treated with either RA or RA CM exhibited increased VE-cadherin expression (Figure 1 D-F, 15 ± 9-fold improvement and 17 ± 10-fold improvement over controls, respectively, p < 0.05). To determine if RA or CM from RA-treated cells affected induction of barrier properties, we passaged purified BMEC progenitors and D6-8 treated BMECs onto collagen/fibronectin-coated filters and measured TEER at D10. Notably, in contrast to D8-10 treatment of purified BMECs (Figure 1C), D6-8 RA treatment of D6 BMEC progenitors elevated TEER (1179 ± 52 Ω*cm2) (Figure 1G). While control CM promoted minimal TEER improvement (464 ± 14 Ω*cm2), we found that RA CM improved TEER beyond that observed with direct RA treatment of purified BMEC progenitors (Figure 1G, 2590 ± 179 Ω*cm2, p < 0.05). All treatment groups expressed junctionally localized claudin-5 and occludin, and neither RA nor RA CM treatment impacted the total protein levels of claudin-5 or occludin at D8 of the differentiation (Figure 1D and E, Figure S2A and B). Collectively, these data demonstrate that BMECs can respond directly to RA during the D6-D8 EC expansion differentiation stage as evidenced by VE-cadherin induction and barrier formation, and suggest that additional components present in the RA-CM from the codifferentiating NC/BMEC cell mixture can further improve barrier properties.
3.2 Agonism of RA-associated nuclear receptors can improve BMEC barrier fidelity
Next, to identify individual RA-signaling associated nuclear hormone receptors that are capable of driving the elevated TEER observed in the differentiating BMEC cultures from D6-D8, a series of small molecule RAR and RXR agonists were tested (Figure 2A). Several individual RA receptor agonists elevated TEER above vehicle treated controls (172 ± 24 Ω*cm2), including BMS753 (RARα agonist; 1071 ± 226 Ω*cm2; p < 0.05), CD1530 (RARγ agonist; 1249 ± 355 Ω*cm2; p < 0.05), and CD3254 (RXRα agonist; 1278 ± 236 Ω*cm2; p < 0.05) (Figure 2B).While BMS453 (RARβ agonist) and docosahexaenoic acid (DA; RXR family pan agonist) modestly improved TEER above DMSO controls (585 ± 125 Ω*cm2 and 305 ± 94 Ω*cm2, respectively), these treatments did not reach statistical significance (p>0.05). Conversely, compared to cells treated with RA (1283 ± 266 Ω*cm2), pan-antagonism of the RAR receptor family with BMS493 reduced the effect of RA-mediated TEER elevation (406 ± 166 Ω*cm2; p=0.057) (Figure S3). Neither individual RARα antagonist BMS195615 nor individual RARγ antagonist MM11253 significantly affected the RA-mediated TEER elevation (Figure S3). These data collectively indicate that while RARα, RARγ and RXRα activation are sufficient for increased barrier function in differentiating BMECs, RARα and RARγ are individually nonessential for mediating RA-associated TEER elevation, suggesting compensatory effects.
3.3 Nuclear receptor activation in differentiating BMECs can mimic the effects of RA treatment on VE-cadherin expression
We subsequently examined whether RARα, RARγ, or RXRα activation via small molecule agonists could improve other BMEC properties associated with RA enhancement, including induction of VE-cadherin expression and improved occludin expression and localization [10]. Compared to vehicle controls, samples treated from D6-D8 of the differentiation with CD3254 (RXRα), BMS753 (RARα), and CD1530 (RARγ) contained pockets of VE-cadherin+ cells by D8 (Figure 3A). Quantitation across multiple differentiations indicated that BMS753, CD1530 and CD3254 led to the largest changes (17-45-fold) in VE-cadherin protein expression and percentage of cells expressing VE-cadherin at Day 8 of differentiation (Figure 3B and C). Neither RA nor individual small molecule agonist treatment affected the percentage of PECAM-1+ ECs in the differentiating cultures, and therefore, the changes in VE-cadherin expression observed reflect differences in EC maturation rather than a change in the differentiation fate (Figure S4).
Figure 3.
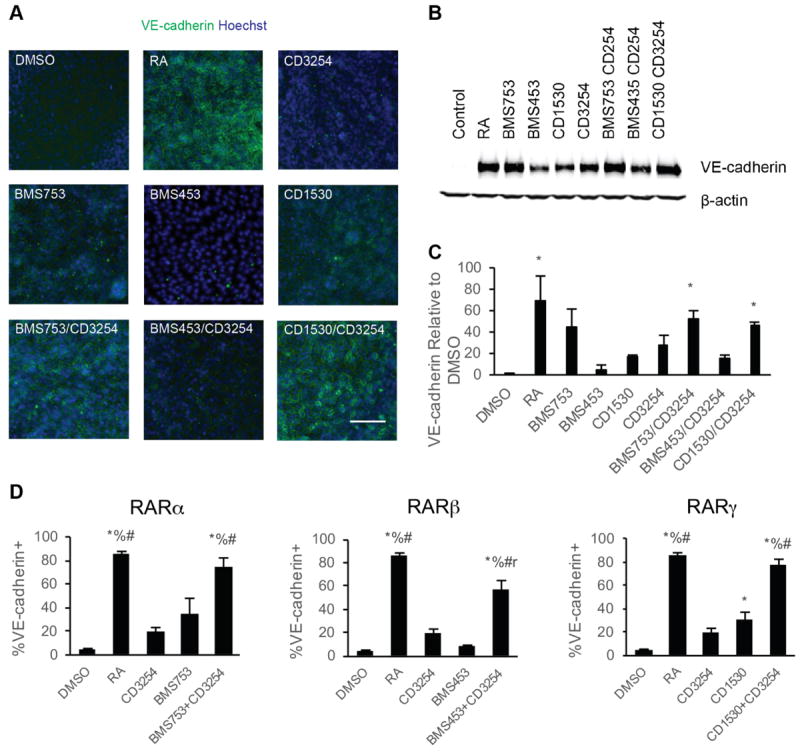
Impact of RA signaling associated nuclear hormone activation on iPSC differentiation to BMECs. A) Representative VE-cadherin immunocytochemistry images of BMEC/NC codifferentiations at D8 following D6-D8 treatment with the indicated RA-signaling associated nuclear hormone receptor agonists; n = 6. VE-cadherin is indicated in green and Hoechst counterstain is indicated in blue. Scale bar represents 100 μm. B) VE-cadherin and β-actin signal from representative Western blots of D8 cells following D6-D8 application of the indicated RA-signaling associated nuclear hormone receptor agonists from three independent differentiations; n = 3. The β-actin signal is additionally shown as a control in Figure 5A. C) Quantification of VE-cadherin protein levels over three independent differentiations via densitometry of Western blots. Error bars represent standard error of the mean. ANOVA followed by Dunnett’s test; * p < 0.05 vs. control. D) Percentage of VE-cadherin+ cells determined by flow cytometry analysis of D8 BMEC/NC codifferentiating population following D6-D8 treatment with the indicated RA-signaling associated nuclear hormone receptor agonists; n = 2. Error bars represent standard error of the mean. Right panel: comparison for RARα agonism to coRARα/RXRα and controls. Middle: comparison for RARβ agonism to coRARβ/RXRα and controls. Left: comparison for RARγ agonism to coRARγ/RXRα and controls. ANOVA followed by Tukey HSD test. * p < 0.05 vs. DMSO vehicle control; % p < 0.05 vs. CD3254; # p < 0.05 vs. RAR agonist; r p < 0.05 vs. RA.
We next investigated whether dual RXRα - RAR agonism may contribute to additive phenotypic enhancement above single agonist controls. Coactivation of RXRα/RARα or RXRα/RARγ via CD3254/BMS753 or CD3254/CD1530, respectively induced a more substantial increase in VE-cadherin expression (Figure 3B, C, 46-53 fold, p<0.05) as well as the percentage of cells expressing VE-cadherin (Figure 3D, 73-78%, p<0.05) than either agonist alone. In fact, these two coactivation scenarios led to indistinguishable responses in VE-cadherin induction compared with RA treatment (Figure 3D, p>0.05). By contrast, costimulation of RXRα/RARβ receptors via CD3254/BMS453 treatment promoted only a 15 ± 3-fold increase in VE-cadherin expression above vehicle control that was not statistically significant (Figure 3B, C, p>0.05), and the percentage of VE-cadherin expressing cells was below that induced by RA treatment (Figure 3D).
3.4 Nuclear receptor activation in differentiating BMECs can mimic the effects of RA treatment on barrier formation
Next BMEC barrier function was assessed after RARα/RXRα and RARγ/RXRα costimulation. We dosed RARα and RXRα agonists from D6-D9 of differentiation, the timeline for maximal RA-responsiveness to TEER (Figure 1A and C, and refs [10, 11]). When added at the activating concentration at which individual agonists increased TEER, RARα/RXRα costimulation demonstrated increased TEER over either single agonist (Figure 4A, 3096 ± 374 Ω*cm2; p<0.05). Similarly, costimulation of RARγ and RXRα at activating concentrations increased TEER over either single agonist, although this change was not statistically significant from single agonist controls (Figure 4A, 2741 ± 548 Ω*cm2, p>0.05). To further explore whether RAR/RXR costimulation could result in synergistic effects, the agonists were instead dosed at 10-fold lower concentrations which yielded minimal activation by either individual agonist (Figure 4B). While these lowered doses of RARα and RXRα agonists induced modest barrier tightening, costimulation resulted in synergistic increases, elevating TEER to 2254 ± 157 Ω*cm2 (Figure 4B, p<0.05) that was not statistically significantly different from RA treatment (2740 ± 582 Ω*cm2, p > 0.05). Similarly, costimulation of RARγ and RXRα led to a synergistic increase in TEER to 1750 ± 151 Ω*cm2 (Figure 4B, p<0.05), but did not reach the levels of RA treatment.
Figure 4.
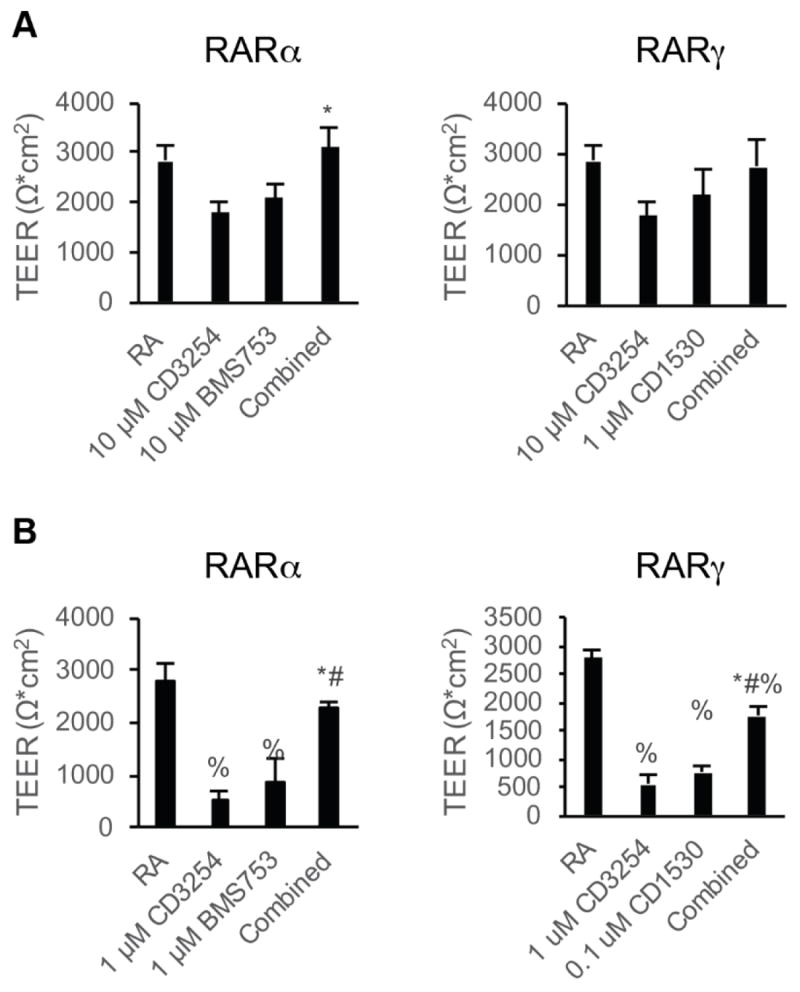
Effect of RAR/RXR coactivation on BMEC barrier function. Error bars represent standard error of the mean. ANOVA followed by Tukey HSD test. A) D10 TEER in iPSC-derived BMECs following treatment with the indicated dosages with RARα/γ agonists in combination with RXRα agonism during D6-D9 of the differentiation; n = 6. Left: comparison of 10 μM CD3254 and 10 μM BMS753 treatment to combined 10 μM CD3254/10 μM BMS753. * p < 0.05 vs. CD3254. Right: comparison of 10 μM CD3254 and 1 μM CD1530 treatment to combined 10 μM CD3254/1 μM CD1530. ANOVA failed. B) D10 TEER in iPSC-derived BMECs following subactivating dosages with the indicated RARα/γ agonists in combination with RXRα agonism during D6-D9 of the differentiation; at least three independent differentiations (n ≥ 3) included per condition * p < 0.05 vs. CD3254; # p < 0.05 vs. RAR agonist; % p < 0.05 vs. RA. Left: comparison of 1 μM CD3254 and 1 μM BMS753 treatment to combined 1 μM CD3254/1 μM BMS753. * p < 0.05 vs. CD3254. Right: comparison of 1 μM CD3254 and 0.1 μM CD1530 treatment to combined 1 μM CD3254/0.1 μM CD1530.
We next explored whether changes in tight junction protein expression or continuity were observed upon BMEC barrier tightening induced by RAR and RXRα agonism. Occludin expression was not increased by individual agonist treatments or by costimulation as measured by Western blot and flow cytometry, nor was the percentage of BMECs expressing occludin changed by agonist treatment (Figure 5A-C). However, noticeable discontinuities in junctional occludin were present in the untreated controls, and the discontinuities lessened upon agonist treatment (Figure 5E, F). Costimulation of either RARα/RXRα or RARγ/RXRα was sufficient to yield junctional continuity that mimicked that provided by RA stimulation (Figure 5E, F). This effect was limited to occludin localization as no differences in claudin-5 junctional continuity were observed nor were there differences in claudin-5 positive cells percentages or expression levels (Figure S5). Finally, the effects of nuclear receptor costimulation on P-glycoprotein and MRP activity were measured and no differences in efflux activity were observed for either the RARα/RXRα or RARγ/RXRα costimulation groups compared to untreated control cells (Figure S6).
Figure 5.
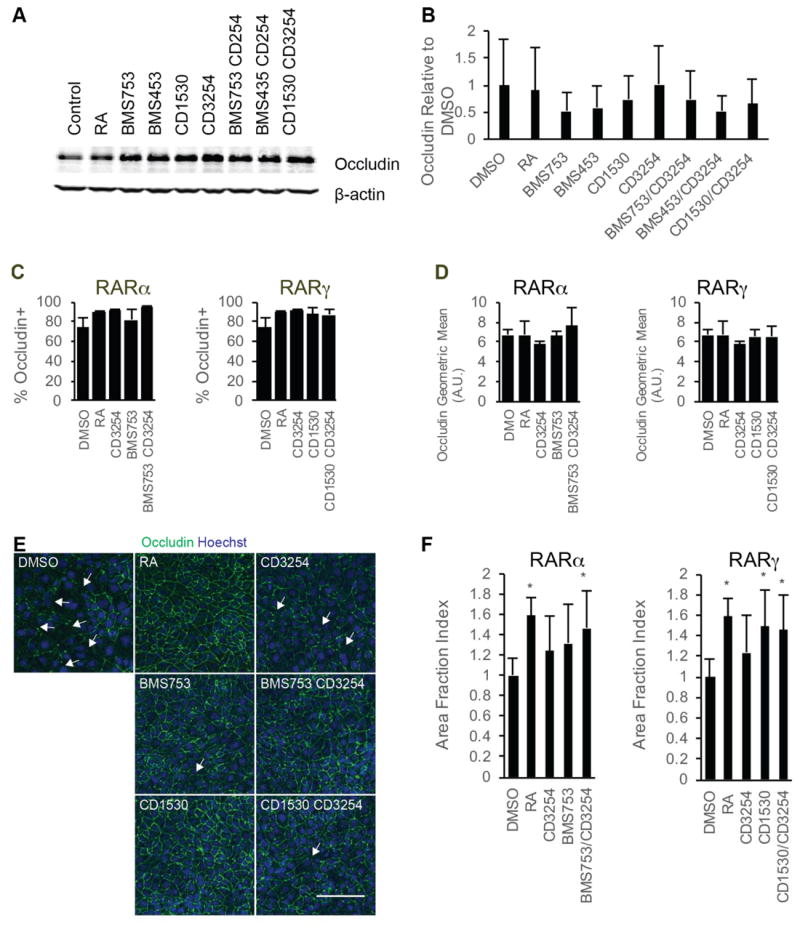
Changes in tight junction proteins following RAR/RXRα coactivation. A) Occludin- and β-actin signals from representative Western blots of D8 cell lysates following D6-D8 treatment with the indicated compounds from three independent differentiations; n = 3. The β-actin signal is additionally shown as a control in Figure 3B. B) Quantification of occludin protein levels over three independent differentiations via densitometry of Western blots. Error bar represents standard error of the mean. ANOVA test failed. C) Percentage of occludin+ cells as quantified by flow cytometry in D10 iPSC-derived BMECs following D6-D9 treatment with the indicated RA-signaling associated nuclear hormone receptor agonists; n = 3. Error bars represent standard error of the mean. Right panel: comparison for RARα agonism to coRARα/RXRα and controls. Left panel: comparison for RARγ agonism to coRARγ/RXRα and controls. ANOVA tests failed. D) Geometric mean of occludin fluorescence levels in occludin+ cells as measured by flow cytometry in D10 iPSC-derived BMECs following D6-D9 treatment with the indicated RA-signaling associated nuclear hormone receptor agonists; n = 3. Error bars represent standard error of the mean. Right panel comparison for RARα agonism to coRARα/RXRα and controls. Left panel comparison for RARγ agonism to coRARγ/RXRα and controls. ANOVA failed. E) Representative occludin immunocytochemistry images for iPSC-derived BMECs at D10 following D6-D9 treatment with the indicated RA-signaling associated nuclear hormone receptor agonists; n = 3. occludin is shown in green and Hoechst counterstain is indicated in blue. Scale bar represents 100 μm. F) Area fraction index quantification of occludin immunostaining images. Error bars represent standard deviation. N = 11 independent fields over three differentiations with at least three independent fields per differentiation. Right panel comparison for RARα agonism to coRARα/RXRα and controls. Left panel comparison for RARγ agonism to coRARγ/RXRα and controls. ANOVA followed by Dunnett’s test. * p < 0.05 vs. DMSO vehicle control.
4 Discussion
Here, we sought to elucidate the differentiation stage-specific effects of RA on iPSC-derived BMEC phenotypes and determine how individual RAR and RXR retinoid nuclear hormone receptors can affect these phenotypes. We found that the D6-D8 differentiation window was critical for RA-mediated TEER elevation. During this differentiation phase both BMEC progenitors and NCs are present as the BMEC population is expanding [9, 11]. D6 BMEC progenitors autonomously respond to RA when compared to D6 BMEC progenitors treated with vehicle control. These responses included induction of VE-cadherin expression and higher TEER compared to vehicle control-treated cells. Notably, BMECs purified at D8 of differentiation did not exhibit elevated TEER following D8-D10 RA treatment. While D6 BMEC progenitors can directly respond to RA treatment, RA alone did not completely account for enhanced barrier properties, as CM from RA-treated cocultures induced higher TEER elevation in purified BMEC progenitors compared to purified BMEC progenitors treated with RA alone. These findings reveal that BMEC-autonomous RA effects are differentiation-stage dependent, with greatest induction when BMECs are in a more immature state. Thus, the observed improvements may be due to synergistic effects of direct RA activation of the BMEC progenitors and paracrine signaling factors provided by RA-stimulated NCs. Taken together, these data map onto in vivo evidence of RA-EC autonomous and non-autonomous effects on BBB fidelity [15].
Neural progenitor cells secrete VEGF to promote cerebral vascular angiogenesis, and WNT 7A and 7B ligands to induce BBB properties during embryonic BBB development [23, 24]. We previously observed WNT7A and WNT7B transcript expression in NCs during iPSC codifferentiation to NCs and BMECs, and RA signaling was recently demonstrated to improve WNT signaling in neural progenitor cells and reduce WNT signaling activity in BMECs [9, 15]. WNT signaling during embryonic development is important for BBB formation, and RA signaling enhances barrier function in BMECs by initially inducing then reducing WNT signaling activity [25]. Data presented here motivate future studies to examine how specific NC paracrine factors, such as VEGF or WNTs, may interact with RA signaling to induce BBB properties in iPSC-derived BMECs.
We next sought to determine specific RA-associated nuclear hormone receptors that are capable of driving acquisition of BMEC phenotypes since RAR and RXR receptor isoforms can exhibit tissue specific effects [22]. RXRα (CD3254), RARα (BMS753), and RARγ (CD1530) agonism alone induced significantly tighter barriers than vehicle control when dosed during the crucial D6-D8 differentiation stage, demonstrating that activation of single retinoid receptor isoforms can elicit barrier tightening. BMS453 modestly improved TEER, yet when compared to BMS753, CD1530, or CD3254, this TEER improvement was not statistically significant. As reported by Mizee et al., hCMEC/D3 immortalized human BMECs stimulated with BMS453 exhibited a small but significant increase in TEER (1.3-fold increase from baseline), consistent with modest improvements in TEER observed with BMS453 in differentiating iPSC-derived BMECs [14]. In vivo comparisons between BMECs and peripheral ECs identified BMEC specificity for RA signaling proteins, including retinol binding protein and its membrane transporter STRA6 [24]. Transcriptomics analysis of mouse brain endothelium identified enrichment of RA-associated signaling pathways compared to peripheral endothelium [25]. Global reduction of RA production in mice leads to embryonic cerebral hemorrhaging, and global genetic knockout of RARα and RARγ, two RA receptors, recapitulates these effects [26]. In the current study, pan-RAR antagonism diminished RA-mediated effects and demonstrated the necessity of RAR in RA-mediated responses. However, while we identified both RARα and RARγ receptors as sufficient to induce TEER response in BMECs, antagonist experiments demonstrated that neither isoform was necessary for mediating RA-dependent TEER induction in iPSC-derived BMECs. RAR isoform compensatory mechanisms are well established and may account for these observations [21]. Thus, future studies may require multiple RAR isoform deletion in brain ECs to investigate essential RAR isoforms in BBB development and maintenance.
To further understand how individual retinoid receptor stimulation can affect iPSC-derived BMEC properties, we investigated RA induction of VE-cadherin [10, 14]. Single RAR isoform agonists and CD3254 induced VE-cadherin expression without affecting the total percentage of PECAM-1+ ECs in the differentiation. This suggests that RA matures ECs rather than promotes endothelial expansion during the D6-D8 codifferentiation phase. Dual RXRα/RAR agonism synergized to elevate VE-cadherin levels and the percentage of VE-cadherin+ cells to levels that mimicked those produced by RA treatment. We also identified synergistic effects of RXRα/RAR agonism in barrier formation as determined by TEER elevation. These findings are consistent with earlier reports demonstrating the synergistic nature of RAR and RXR partners to activate RA-targeted gene transcription [29]. Furthermore, increased TEER correlated with more continuous junctional occludin as previously described for RA treated iPSC-derived BMECs [10]. Neither RA nor retinoid receptor agonists elevated occludin levels, contrasting with previous observations of RA-mediated increases in occludin expression in hPSC-derived BMECs and hCMECs treated with RA [10, 14]. Recent differentiation protocol adaptions, including standardized cell seeding densities [11], may have improved basal occludin protein levels in iPSC-derived BMECs thereby mitigating occludin expression level effects of RA treatment. In addition, claudin-5 and occludin promoter regions lack a RA response element, and therefore, RA effects described in previous reports may have been a result of indirect regulation [9, 14, 30, 31]. Finally, neither RA nor RA-associated nuclear hormone receptor agonists affected P-glycoprotein or MRP activity, although previous observations in vitro and in vivo have suggested that RA can increase efflux transporter expression and activity [10, 14, 32]. In conclusion, this work demonstrates that RA signaling impacts iPSC-derived BMEC differentiation and demonstrated that RAR/RXR small molecule activation could mimic RA-mediated barrier improvements. Since all-trans RA is a pleiotropic signaling molecule, this could provide an alternative, directed approach to supplying RA-mediated cues to the BMEC differentiation process. In addition, the identification of specific RAR and RXR isoforms that are sufficient to induce BBB properties motivates further investigation in vivo, and suggests possible targeted approaches to restore BBB dysfunction in pathological states.
Supplementary Material
Acknowledgments
The authors wish to acknowledge Dr. Xiaoping Bao and Dr. Scott Canfield for their helpful discussions of RA signaling. The authors also would like to acknowledge Dr. Brandon Kim for his helpful discussions regarding statistical analyses. The authors wish to acknowledge the following funding sources: M.J.S. is a recipient of the Biotechnology Training Program Fellowship. E.S.L was a recipient of the Chemical Biology Interface Training Program Fellowship. M.G.F. is a recipient of the Hilldale Undergraduate Fellowship. This work was supported by National Institutes of Health Grant NS083688
Abbreviations
- BBB
blood-brain barrier
- BMECs
brain microvascular endothelial cells
- CM
conditioned medium
- CNS
central nervous system
- CsA
cyclosporin A
- DCFDA
Carboxymethyl-2′,7′-dichlorofluorescein diacetate
- DMSO
dimethyl sulfoxide
- EC
endothelial cell
- hPSCs
human pluripotent stem cells
- HSD
honest significant difference
- iPSCs
induced pluripotent stem cells
- NCs
neural cells
- RA
retinoic acid
- RAR
retinoic acid receptor
- RXR
retinoid X receptor
- TEER
transendothelial electrical resistance
- UM
unconditioned medium
Footnotes
Conflict of Interest
The authors declare no financial or commercial conflict of interest.
References
- 1.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 2.Winkler E, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, Sengillo JD, Hillman S, Kong P, Nelson AR, Sullivan JS, Zhao Z, Meiselman HJ, Wenby RB, Soto J, Abel ED, Makshanoff J, Zuniga E, De Vivo DC, Zlokovic BV. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci. 2015;18:521–530. doi: 10.1038/nn.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zlokovic BV. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6:393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erickson MA, Banks WA. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab. 2013;33:1500–13. doi: 10.1038/jcbfm.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L, Zhang ZG, Chopp M. The neurovascular unit and combination treatment strategies for stroke. TRENDS Pharmacol Sci. 2012;33:415–422. doi: 10.1016/j.tips.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lajoie JM, Shusta EV. Targeting receptor-mediated transport for delivery of biologics across the blood-brain barrier. Annu Rev Pharmacol Toxicol. 2015;55:613–31. doi: 10.1146/annurev-pharmtox-010814-124852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones AR, Shusta EV. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm Res. 2007;24:1759–1771. doi: 10.1007/s11095-007-9379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lippmann ES, Azarin SM, Kay JE, Nessler Ra, Wilson HK, Al-Ahmad A, Palecek SP, Shusta EV. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. 2012;30:783–91. doi: 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep. 2014;4:4160. doi: 10.1038/srep04160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson HK, Canfield SG, Hjortness MK, Palecek SP, Shusta EV. Exploring the effects of cell seeding density on the differentiation of human pluripotent stem cells to brain microvascular endothelial cells. Fluids Barriers CNS. 2015;12:13. doi: 10.1186/s12987-015-0007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stebbins MJ, Wilson HK, Canfield SG, Qian T, Palecek SP, Shusta EV. Differentiation and characterization of human pluripotent stem cell-derived brain microvascular endothelial cells. Methods. 2016;101:93–102. doi: 10.1016/j.ymeth.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maden M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci. 2007;8:755–65. doi: 10.1038/nrn2212. [DOI] [PubMed] [Google Scholar]
- 14.Mizee MR, Wooldrik D, Lakeman KAM, van Het Hof B, Drexhage JAR, Geerts D, Bugiani M, Aronica E, Mebius RE, Prat A, de Vries HE, Reijerkerk A. Retinoic Acid Induces Blood-Brain Barrier Development. J Neurosci. 2013;33:1660–1671. doi: 10.1523/JNEUROSCI.1338-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonney S, Harrison-Uy S, Mishra S, MacPherson AM, Choe Y, Li D, Jaminet S-C, Fruttiger M, Pleasure SJ, Siegenthaler JA. Diverse Functions of Retinoic Acid in Brain Vascular Development. J Neurosci. 2016;36:7786–7801. doi: 10.1523/JNEUROSCI.3952-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ziegler N, Awwad K, Fisslthaler B, Reis M, Devraj K, Corada M, Minardi SP, Dejana E, Plate KH, Fleming I, Liebner S. beta-Catenin Is Required for Endothelial Cyp1b1 Regulation Influencing Metabolic Barrier Function. J Neurosci. 2016;36:8921–8935. doi: 10.1523/JNEUROSCI.0148-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurokawa R, DiRenzo J, Boehm M, Sugarman J, Gloss B, Rosenfeld MG, Heyman RA, Glass CK. Regulation of retinoid signalling by receptor polarity and allosteric control of ligand binding. Nature. 1994;371:528–531. doi: 10.1038/371528a0. [DOI] [PubMed] [Google Scholar]
- 18.Al Tanoury Z, Piskunov A, Rochette-Egly C. Vitamin A and retinoid signaling: genomic and nongenomic effects. J Lipid Res. 2013;54:1761–75. doi: 10.1194/jlr.R030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leid M, Kastner P, Lyons R, Nakshatri H, Saunders M, Zacharewski T, Chen J-Y, Staub A, Garnier J-M, Mader S, Chambon P. Purification, cloning, and RXR identity of the HeLa cell factor with which RAR or TR heterodimerizes to bind target sequences efficiently. Cell. 1992;68:377–395. doi: 10.1016/0092-8674(92)90478-u. [DOI] [PubMed] [Google Scholar]
- 20.Yu VC, Delsert C, Andersen B, Holloway JM, Devary OV, Naar AM, Kim SY, Boutin JM, Glass CK, Rosenfeld MG. RXR beta: a coregulator that enhances binding of retinoic acid, thyroid hormone, and vitamin D receptors to their cognate response elements. Cell. 1991;67:1251–1266. doi: 10.1016/0092-8674(91)90301-e. [DOI] [PubMed] [Google Scholar]
- 21.Mark M, Ghyselinck NB, Chambon P. Function of retinoic acid receptors during embryonic development. Nucl Recept Signal. 2009;7:e002. doi: 10.1621/nrs.07002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helms HC, Abbott NJ, Burek M, Cecchelli R, Couraud P-O, Deli MA, Forster C, Galla HJ, Romero IA, Shusta EV, Stebbins MJ, Vandenhaute E, Weksler B, Brodin B. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J Cereb Blood Flow Metab. 2016;36:862–890. doi: 10.1177/0271678X16630991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogan KA, Ambler CA, Chapman DL, Bautch VL. The neural tube patterns vessels developmentally using the VEGF signaling pathway. Development. 2004;131:1503–1513. doi: 10.1242/dev.01039. [DOI] [PubMed] [Google Scholar]
- 24.Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–50. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 25.Ma S, Kwon HJ, Johng H, Zang K, Huang Z. Radial glial neural progenitors regulate nascent brain vascular network stabilization via inhibition of Wnt signaling. PLoS Biol. 2013;11:e1001469. doi: 10.1371/journal.pbio.1001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouillet P, Sapin V, Chazaud C, Messaddeq N, Décimo D, Dollé P, Chambon P. Developmental expression pattern of Stra6, a retinoic acid-responsive gene encoding a new type of membrane protein. Mech Dev. 1997;63:173–86. doi: 10.1016/s0925-4773(97)00039-7. [DOI] [PubMed] [Google Scholar]
- 27.Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The Mouse Blood-Brain Barrier Transcriptome: A New Resource for Understanding the Development and Function of Brain Endothelial Cells. PLoS One. 2010;5:e13741–e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lohnes D, Mark M, Mendelsohn C, Dollé P, Dierich A, Gorry P, Gransmuller A, Chambon P. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994;120:2749–71. doi: 10.1242/dev.120.10.2749. [DOI] [PubMed] [Google Scholar]
- 29.Roy B, Taneja R, Chambon P. Synergistic activation of retinoic acid (RA)-responsive genes and induction of embryonal carcinoma cell differentiation by an RA receptor alpha (RAR alpha)-, RAR beta-, or RAR gamma-selective ligand in combination with a retinoid X receptor-specific ligan. Mol Cell Biol. 1995;15:6481–6487. doi: 10.1128/mcb.15.12.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie X, Rigor P, Baldi P. MotifMap: a human genome-wide map of candidate regulatory motif sites. Bioinformatics. 2009;25:167–174. doi: 10.1093/bioinformatics/btn605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daily K, Patel VR, Rigor P, Xie X, Baldi P. MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinformatics. 2011;12:495. doi: 10.1186/1471-2105-12-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El Hafny B, Chappey O, Piciotti M, Debray M, Boval B, Roux F. Modulation of P-glycoprotein activity by glial factors and retinoic acid in an immortalized rat brain microvessel endothelial cell line. Neurosci Lett. 1997;236:107–111. doi: 10.1016/s0304-3940(97)00679-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.