

Abstract
Currently, many strains of influenza A virus have developed resistance against anti-influenza drugs, and it is essential to find new chemicals to combat this virus. The influenza polymerase with three proteins, PA, PB1 and PB2, is a crucial component of the viral ribonucleoprotein (RNP) complex. Here, we report the identification of a hit compound 221 by surface plasmon resonance (SPR) direct binding screening on the C-terminal of PA (PAC). Compound 221 can subdue influenza RNP activities and attenuate influenza virus replication. Its analogs were subsequently investigated and twelve of them could attenuate RNP activities. One of the analogs, compound 312, impeded influenza A virus replication in Madin-Darby canine kidney cells with IC50 of 27.0 ± 16.8 μM. In vitro interaction assays showed that compound 312 bound directly to PAC with Kd of about 40 μM. Overall, the identification of novel PAC-targeting compounds provides new ground for drug design against influenza virus in the future.
Introduction
Influenza virus is a major threat to the health of the community. The annual deaths caused by seasonal influenza epidemics range from 250000 to 5000001. The ever-changing nature of influenza virus by antigenic variations has been challenging for the development of an effective influenza drug. Oseltamivir-resistant strains were identified from the outbreaks of 2009 H1N1 pandemic2 and 2013 H7N93. Recently, many human cases of avian influenza A H7N9 virus were confirmed in China4. Also, a highly pathogenic avian influenza A H5N8 virus has also been rapidly spreading since June 20165. All these call for attention to the urgency of developing new antivirals.
The influenza RNA-dependent RNA polymerase (RdRP), consisting PA, PB1 and PB2 subunits, is a component of the viral ribonucleoprotein (RNP) complex that is crucial for viral transcription and replication6. Nucleic acid polymerase activity resides in the PB1 subunit, which forms the core of the complex7,8. Both PA and PB2 are involved in various accessory functions that are essential for the transcription and replication of the viral genome. The highly conserved nature of RdRP makes it an attractive target for drug discovery9.
PA, one of the RdRP subunits, consists of two domains-the N-terminal endonuclease domain (residues 1–195) and the C-terminal domain (PAC) (residues 257–716). They are connected by an extensive linker that wraps around PB1 complex7,8. PAC contains a hydrophobic groove that interacts with the N-terminus of PB1 subunit. This PB1-binding groove is highly conserved among different viral strains10,11. PA-PB1 interaction is necessary for the assembly of the RdRP complex, and it is also involved in the nuclear import of both proteins12. Several studies have demonstrated that the disruption of PA-PB1 interaction could impede influenza virus replication13–20. Furthermore, PAC has been reported to have protease activity21. Mutational study has also revealed that several residues in PAC are important for the transcription and replication activity of influenza ribonucleoprotein (RNP)22. All these suggest that PAC could be a feasible target for drug screening.
In the present study, we have conducted a surface plasmon resonance (SPR) screening of an in-house library to identify hit compounds targeting PAC. SPR is a biophysical method for characterizing label-free macromolecular interaction. It is highly sensitive and could provide quantitative analysis of the interactions between protein and small molecules. Direct binding screening using SPR has been employed on various protein targets, using libraries of several hundred compounds to several thousand23–27. From our screening study, two hit compounds (compound 221 and 283) were discovered to attenuate RNP activities and inhibit influenza virus. Analogs of compound 221 were further evaluated and one of them (compound 312) was characterized as a promising inhibitor of influenza virus.
Results
SPR screening for compounds binding to PAC in vitro
One hundred and sixty-five compounds from our in-house library were screened against PAC by SPR using Biacore 3000. The RU responses of test compounds after DMSO calibration were recorded and ranked (Fig. 1). The top 10% (17 compounds) were selected for further investigation. Their structures are summarized in Supplementary Table 1.
Figure 1.

SPR screening for PAC. 165 compounds were screened against PAC by SPR using Biacore3000. 17 compounds with the highest RU responses were chosen for further examination.
Cytotoxicity of compounds
Cytotoxicity of selected compounds were evaluated in both 293T and MDCK cells. Most of them have CC50 over 100 μM. The cytotoxicity of the hit compounds is summarized in Supplementary Table 2.
Identification of hits that inhibit ribonucleoprotein (RNP) function and impede influenza virus replication
Next we employed an RNP reconstitution reporter assay to investigate if the hit compounds can reduce RNP transcriptional activities. The highest non-cytotoxic concentration of each compound was used in the assay. Among the 17 hit compounds, compound 221 and 283 caused a significant attenuation of RNP transcriptional activities (Fig. 2a). Both compounds exhibited dose dependent inhibition of RNP activity (Fig. 2b,c), with compound 221 having IC50 of 68.8 ± 1.9 μM and compound 283 having IC50 of 8.80 ± 2.4 μM. Furthermore, compound 221 and 283 could impede influenza virus (A/WSN/33) replication in viral yield reduction assay, with IC50 30.58 ± 21.37 and 1.67 ± 1.27 μM respectively (Fig. 2d,e).
Figure 2.

Evaluation of hit compounds. (a) 17 hit compounds obtained from SPR screening was evaluated by RNP assays. The highest non-cytotoxic concentration (in μM) of each compound is shown in bracket. Compounds 221 and 283 showed significant attenuation of RNP. Nucleozin was included as a positive control. (b) Compound 221 and (c) compound 283 elicited dose dependent inhibition of influenza RNP activity. (d) Compound 221 and (e) compound 283 showed dose dependent inhibition of influenza A/WSN/33 at MOI 0.01. Bar chart represents viral yield while XY curve represents cell viability. Results were obtained from three independent experiments.
Biological activity of compound 221 analogs
According to the chemical structure of compound 221, 10 analogs were designed and synthesized. Another 13 commercially available analogs were also purchased. Their structures are summarized in Table 1.
Table 1.
Chemical structures of 221 analogs.
| Compound | R1 | R2 | R3 | |||
|---|---|---|---|---|---|---|
Synthetic compounds group 1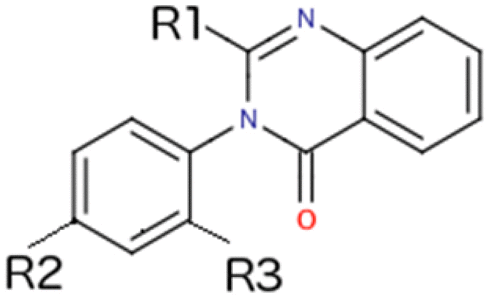
|
S1a |
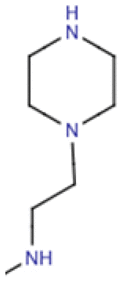
|
-H | -H | ||
| S1b |
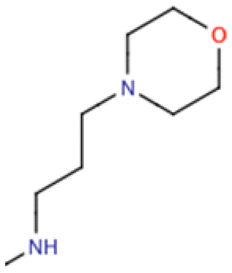
|
-H | -CH3 | |||
| S1c |
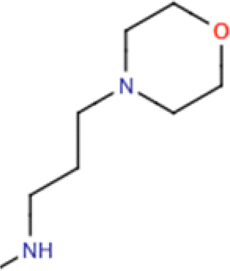
|
-CH3 | -H | |||
| S1d |
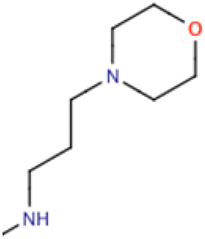
|
-F | -H | |||
Synthetic compounds group 2
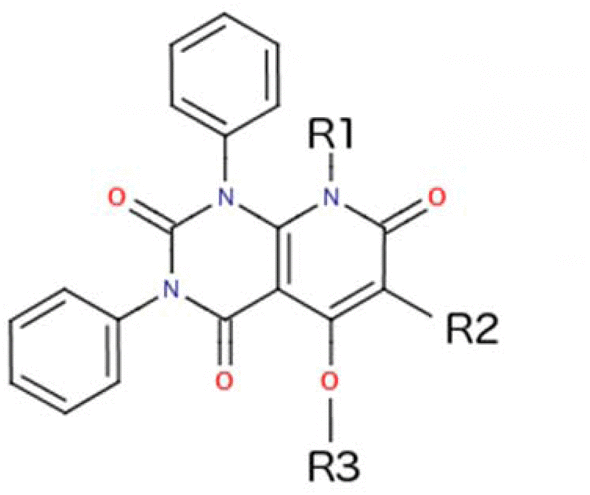
|
Compound | R1 | R2 | R3 | ||
| 221 | -CH3 |
|
-H | |||
| S2a | -CH2CH2OH |
|
-H | |||
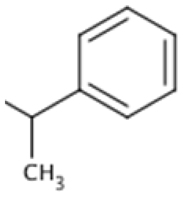
| S2b | -H |
|
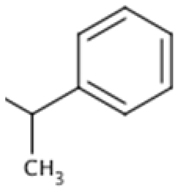
-H | |||
| S2c | -CH3 |
|
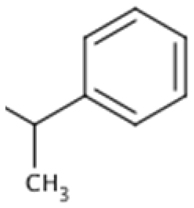
-H | |||
| S2d | -CH3 |
|
-CH3 | |||
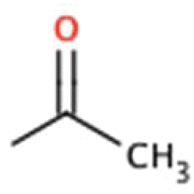
| S2e |
|
|
-H | |||
| S2f | -CH3 |
|
-H | |||
Commercially available analogs group 1
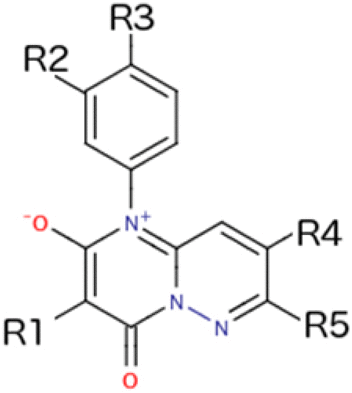
|
Compound | R1 | R2 | R3 | R4 | R5 |
| 390 |
|
-H | -H | -H | -CH3 | |
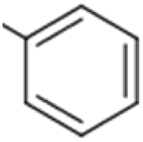
| 391 |
|
-H | -OCH3 | -OH |
|
|
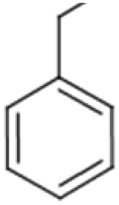
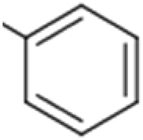
| 394 | -CH2CH3 | -H | -H | -H |
|
|
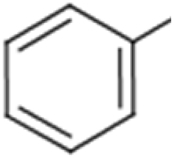
| 395 |
|
-CH3 | -H | -H |
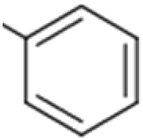
|
|
| 396 | -CH2CH3 | -H | -F | -H |
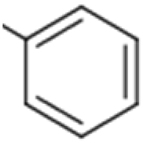
|
|
| 397 |
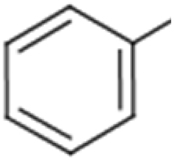
|
-H | -CH3 | -H | -CH3 | |
Commercially available analogs group 2
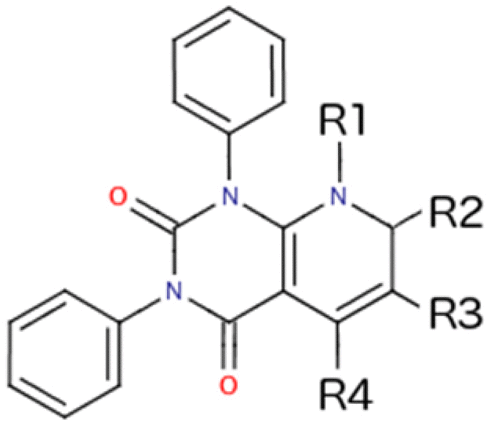
|
Compound | R1 | R2 | R3 | R4 | |
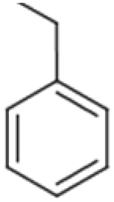
| 221 | -CH3 | =O |
|
-OH | ||
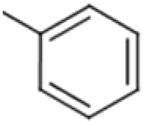
| 312 | -H | =O |
|
-OH | ||
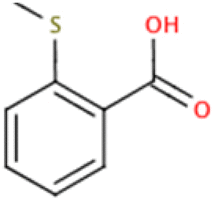
| 385 | -CH3 | =O |
|
-OH | ||
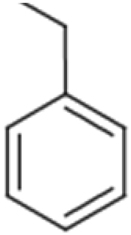
| 387 | -H | =O |
|
-OH | ||
| Uncategorized commercially available analogs | Compound | Chemical illucture | ||||
| 310 |
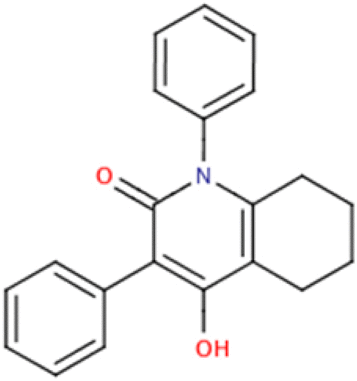
|
|||||
| 384 |
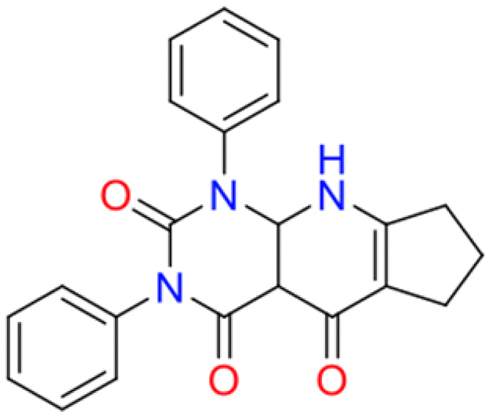
|
|||||
| 389 |
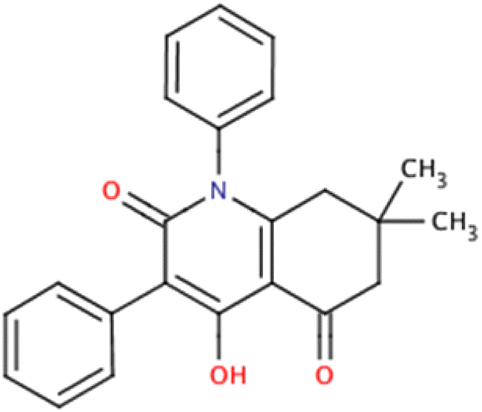
|
|||||
| 392 |
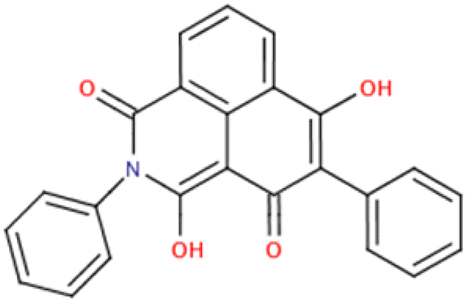
|
|||||
RNP screening showed that 12 out of the 23 analogs were effective in attenuating influenza RNP activities (Fig. 3a,b). All effective compounds exhibited dose dependent inhibition of RNP activity, most of them having IC50 below 100 μM (Table 2).
Figure 3.

Evaluation of analogs of compound 221. (a) RNP screening of analogs of 221 obtained by chemical synthesis. Ten analogs of compound 221 were designed and synthesized. Six of them exhibited significant reduction of RNP activities. The highest non-cytotoxic concentration (in μM) of each compound is shown below its name. (b) Thirteen commercially available analogs of compound 221 were evaluated by RNP assays. Six of them exhibited significant reduction of RNP activities. The highest non-cytotoxic concentration of each compound is shown belowits name. Dose dependent viral yield reduction assays of (c) S1b, (d) S2a, (e) S2b, (f) S2d, (g) S2e, (h) S2f, (i) 310, (j) 312, (k) 384, (l) 387, (m) 389, (n) 392. Bar chart represents viral yield while XY curve represents cell viability. Nucleozin (Nu) at 25 μM was used as positive control. Results were obtained from three independent experiments.
Table 2.
Properties of analogs of compound 221.
| Compound | RNP assay | Viral Yield Reduction Assay | MST | SPR | |||
|---|---|---|---|---|---|---|---|
| IC50a (μM) | CC50b (μM) | IC50c (μM) | CC50d (μM) | Kde (μM) | Kdf (μM) | ||
| 221 | 68.8 ± 1.9 | >100 | 30.58 ± 21.37 | >100 | N.D. | N.D. | |
| Analogs obtained through chemical modification | S1b | 87.8 ± 21.8 | >100 | >100 | >100 | N.D. | N.D. |
| S2a | 9.69 ± 3.00 | 84.6 ± 17.30 | 7.74 ± 6.02 | >100 | 1.77 ± 0.78 | N.D. | |
| S2b | 14.2 ± 3.6 | >100 | 4.96 ± 3.84 | 42.8 ± 9.0 | 16.2 ± 8.3 | N.D. | |
| S2d | >12.5 | 66.6 ± 6.09 | 3.18 ± 2.44 | 35.6 ± 9.24 | >25 | N.D. | |
| S2e | 6.69 ± 7.49 | >100 | 5.11 ± 4.22 | 53.1 ± 9.25 | 50.9 ± 25.5 | N.D. | |
| S2f | 62.4 ± 22.4 | >100 | >100 | >100 | N.D. | N.D. | |
| Commercially available analogs | 310 | 98.45 ± 3.6 | >100 | >100 | >100 | N.D. | N.D. |
| 312 | 35.37 ± 4.3 | >100 | 27.0 ± 16.8 | >100 | 38.2 ± 5.5 | 37.7 ± 4.6 | |
| 384 | >100 | >100 | >100 | >100 | N.D. | N.D. | |
| 389 | 96.54 ± 28.7 | >100 | >100 | >100 | N.D. | N.D. | |
| 387 | 15.45 ± 2.8 | >100 | 6.45 ± 5.63 | >100 | 46.3 ± 27.4 | N.D. | |
| 392 | >6.25 | 62.66 ± 18.7 | 4.78 ± 5.63 | 87.03 ± 7.22 | 6.03 ± 1.05 | N.D. | |
aIC50 is the concentration of test compound which produces 50% inhibition of RNP activity compared with DMSO control; reported values represent means ± standard deviation of data from three independent experiments.
b293T cells were incubated with test compounds for 24 hrs; CC50 is the concentration of test compounds which produces 50% cytotoxicity as determined by MTT assays. Reported values represent means ± standard deviation of data from three independent experiments.
cIC50 is the concentration of test compound which produces 50% inhibition of viral yield compared with DMSO control; reported values represent means ± standard deviation of data from three independent experiments.
dMDCK cells were incubated with test compounds for 24 hrs; CC50 is the concentration of test compounds which produces 50% cytotoxicity as determined by MTT assays. Reported values represent means ± standard deviation of data from three independent experiments.
eKd were measured by MST and calculated by NanoTemper MO.Control using ‘Kd model’. Reported values represent Kd ± confidence level calculated by the software.
fKd were measured by SPR and calculated by Biacore BiaEvaluation v 4.1 using ‘1:1 Langmuir binding model’. Reported values represent mean ± standard deviation of data from three independent experiments.
Viral yield reduction study with A/WSN/33 (0.01 MOI) shows that compound S2a, S2b, S2d, S2e, 312, 387 and 392 exhibited dose-dependent inhibition of influenza virus (Fig. 3c–n). Most of their IC50 fall within low micromolar range (Table 2). However, the dose dependent effect of compounds S2b, S2d and S2e might not be reliable due to their correlation with cytotoxicity (Fig. 3e–g). Compound S2a and 387 exhibited slight cytotoxicity at their highest tested concentration (Fig. 3d,l).
For compounds with anti-influenza effect, their interactions with PAC were further confirmed and measured by microscale thermophoresis (MST) assay. Except for compound S2d, the Kd of other compounds all lie below 100 μM (Table 2 and Supplementary Figure S1–S6). As compound S2d showed significant intrinsic fluorescence which could interfere with MST measurement, we were unable to measure its MST signal at a concentration higher than 25 μM.
As compound 312 exhibited good dose-dependent inhibition of A/WSN/33 (IC50: 27.0 ± 16.8 μM) without showing any cytotoxicity (i.e. non-cytotoxic at 100 μM) (Fig. 3j), it was selected for further investigation.
Biological activity of compound 312
RNP from various influenza strains (A/WSN/33 (H1N1), A/Japan/305/1957(H2N2), A/HK/1/68(H3N2) and A/HK/156/97(H5N1)) were used to evaluate compound 312. Dose dependent inhibition of RNP activities is observed in all strains. The potencies of compound 312 on H1N1, H2N2 and H3N2 were similar, while it was significantly weaker on H5N1 RNP (Fig. 4a). Next, besides A/WSN/33, we also did viral yield assay with A/PR/8/34 virus. Compound 312 exhibited significant inhibition of A/PR/8/34 virus, reducing viral yield for about 57 fold at 100 μM (Fig. 4b).
Figure 4.

Evaluation of compound 312. (a) Compound 312 elicited dose dependent inhibition of RNP activities of various influenza subtypes. (b) Compound 312 showed inhibition of influenza A/PR/8/34. (c) Viral growth curve with compound 312. Compound 312 could impede influenza A/WSN/33 propagation. (d) MDCK cells were infected with 0.01 MOI A/WSN/33 and incubated with 100 μM compound 312 at various time points. Compound 312 was able to attenuate virus when added post infection. (e) SPR study of compound 312 with PAC. Using the ‘1:1 Langmuir binding’ model by BIAevaluation v. 4.1 software, Kd value was estimated to be 37.7 ± 4.6 μM. Data shown represent the mean ± SD of three independent experiments. *Represents p < 0.05, **Represents p < 0.01. (f) MST study of compound 312 with PAC. Kd value was estimated to be 38.2 ± 5.5 μM by MO.Control software using Kd model.
To further confirm the effects of compound 312 on viral replication, we compared the viral growth curve in the presence or absence of compound 312. MDCK cells were infected with 0.001 MOI H1N1 (A/WSN/33) virus and incubated with test compounds for 24, 48 and 72 hrs. The growth curves of influenza virus at 50 and 100 μM of compound 312 showed that it can significantly suppress viral growth (Fig. 4c). Suppression of influenza virus replication was also observed by the addition of compound 312 after viral infection (Fig. 4d).
The binding kinetics of compound 312 was further studied with SPR. Compound 312 elicited an increase of RU in a concentration dependent manner (Fig. 4e). Using the ‘1:1 Langmuir binding’ model by BIAevaluation v. 4.1 software, Kd value was estimated to be 37.7 ± 4.6 μM. The Kd measured by SPR is consistent with the Kd obtained from MST assay (38.2 ± 5.5 μM) (Fig. 4f).
Discussion
In this study, we performed an SPR screening for PAC. Among the 165 compounds, we have identified two hits that can attenuate viral RNP activities. Both compounds showed dose dependent inhibition of RNP and viral yield with IC50 at micromolar level.
Compound 221 was selected for further investigation due to its structural simplicity and better solubility. Based on the structure of compound 221, ten analogs were designed and synthesized (Table 1). Compound S2a, S2b, S2c, S2d, S2e and S2f all have three aromatic groups attached to the core structure, while S1a, S1b, S1c and S1d have two of the aromatic groups removed.
All S1 compounds showed no cytotoxicity at 100 μM. However, all had lost their abilities to inhibit RNP activities except for S1b. Although all four of them retain a central two-ring core attached with an aromatic side group, their structural changes are more drastic than other synthetic compounds. Two aromatic rings were removed and a side group was added to position R1 (Table 1), which was not present in the structure of compound 221. These significant changes might explain the loss of activity.
On the other hand, in the class of S2 compounds, S2a, S2b, S2c, S2d and S2e highly resemble the original compound 221 (Table 1). Except for S2c, all had marked improvement in their potencies against RNP. However, they also exhibited increased cytotoxicity. Compounds S2b, S2d and S2e had CC50 at low micromolar level in MDCK cells. In contrast, compound S2a showed only slight increase in cytotoxicity at 100 μM. By inspecting the structures of these analogs, it can be shown S2a resembles compound 221 at position R2, having a phenyl side group without a methyl group attaching to it. In contrast, compounds S2b, S2d and S2e all have an extra methyl group. This might cause the discrepancy in cytotoxicity between these analogs. As for compound S2f, the side chain at position R2 has a carbon atom replaced by a nitrogen atom. This structural change might also explain its inability to attenuate influenza virus.
Next, 13 commercially available analogs of compound 221 were found in the Pubchem database using Tanimoto similarity index. Based on their core structures, these analogs can mostly be categorized into two groups (Table 1). Group 1 includes compounds 390, 391, 394, 395, 396 and 397. Group 2 includes compounds 312, 385 and 387. The remaining compounds 310, 384, 389 and 392 cannot be fitted into both groups.
All the analogs in group 1 do not inhibit RNP activity. Its core structure has charges in it while the core structure of compound 221 is non-polar. This could explain why even some of these analogs highly resemble compound 221 (i.e. compounds 391 and 395), yet they did not inhibit RNP.
For the uncategorized compounds (310, 384, 389 and 392), all four were able to inhibit RNP activities. Compound 384 has one aromatic ring removed, and the original two-ring core structure has a five-member ring fused to it. Although having one side group removed, it still retained the capability to inhibit RNP, albeit to a less extent. The case is similar with compound 310 and compound 389. Both have a two-ring core structure with only two aromatic side groups. Compound 310 and 389 inhibit RNP activities with IC50 of 98.45 ± 3.6 and 96.54 ± 28.7 μM respectively, both being less potent than compound 221. In light of this, it could be conjectured that the aromatic group which the three (310, 384 and 389) lack is non-essential but beneficial for their inhibitory effects. Furthermore, all three compounds were unable to inhibit influenza viral yield at 100 μM. Their inabilities to inhibit viral replication are consistent with their mild inhibition on RNP activities. The last uncategorized compound, 392, has a tri-ring core attached with two aromatic side groups. It is more potent than compound 221, inhibiting influenza virus with IC50 of 4.78 ± 5.63 μM, but exhibiting higher cytotoxicity in both 293T and MDCK cells.
In group 2, the core structure is identical to that of compound 221. Among the three analogs, two of them (Compounds 312 and 387) attenuate RNP activities. Compounds 312 and 387 both highly resemble compound 221. Compound 387 differs from 221 by the removal of a methyl group from its core structure, while compound 312 replaces a benzyl group with a phenyl group. Both retain three aromatic side groups. Compound 312 and compound 387 inhibited RNP activities with IC50 of 35.37 ± 4.3 and 15.45 ± 2.8 μM respectively. Compound 387 is slightly cytotoxic in MDCK cells at above 50 μM (Fig. 3l). The failure of compound 385 to inhibit RNP activities could be due to its drastically different group at position R3.
Although it is evident that compound 221 and 312 can act on RNP and bind to PAC, their detail mechanism of action remains obscure. PAC has been shown to interact with: (1) PB1N-terminal10,11, (2) PB1 and PB2 at other positions7,8, (3) 5′ promoter region of viral RNA7,8, (4) small viral RNA (svRNA) and promote genome replication in a segment-specific manner28, (5) C-terminal domain of RNA polymerase II, which allows the RNP to recruit 5′-capped primers from nascent Pol II transcripts for viral genome transcription29.
The discrepancy between the inhibition of H5N1 RNP activities and that of other influenza strains might shed some lights on the mechanism of action by compound 312. Sequence alignment of A/WSN/33 (H1N1), A/Japan/305/1957 (H2N2), A/HK/1/68 (H3N2) and A/HK/156/97 (H5N1) PAC reveals that there are 17 residues where H5N1 PAC are distinct from the other three (Fig. 5 & Supplementary Fig. S7). The three-dimensional structure of bat influenza A RdRP complex (PDB: 4WSB) was inspected to understand the distribution and location of these residues7. It should be noted that residues 349–353 of H1N1, H2N2, H3N2 and H5N1 PA are not present in the H17N10 bat influenza polymerase, and the amino acid numbering of the later is different from the formers. Among the 17 residues, R497, T552 and R615 are in close proximity to the Pol II interacting residues (Residues 289, 454, 636 and 638; the interacting site is circled in pink); M367, E382, H394 and A651 are located at the viral promoter and svRNA binding region (circled in yellow); I602 and A651 reside in the PB1-binding cavity (circled in orange). Other residues scatter mainly around the head domain of PAC. It is possible that compound 312 binds to PAC near these residues, resulting in the discrepancy between the inhibition of H5N1 RNP activities and the inhibition of other strains. Further investigation is needed to reveal the exact mechanism of action by compound 312.
Figure 5.

Location of the distinct residues of H5N1 PAC. Cartoon representation of bat influenza A polymerase complex structure (PDB: 4WSB). PA, PB1 and PB2 are colored in green, cyan and pink respectively. Seventeen residues where H5N1 PAC is distinct from H1N1, H2N2 and H3N2 PAC are shown as red spheres. The Pol II binding site on PAC is circled in pink, the viral promoter binding site is circled in yellow and the PB1 interacting site is circled in orange.
In summary, we have identified a hit compound (compound 221) targeting influenza PAC domain by SPR screening. It can inhibit influenza RNP activities and impede viral replication. Twenty-three analogs of compound 221 were obtained by chemical synthesis or from purchase. Twelves of them could inhibit influenza RNP activity, among which compound 312 was identified as the most promising analog. Compound 312 could inhibit RNP, attenuate viral yield and bind directly to PAC at micromolar range.
Methods
Cell, virus and commercially available compound
293T and MDCK cells were routinely cultured in Minimum Essential Medium (MEM, Gibco) supplemented with 10% (v/V) fetal bovine serum (FBS, Gibco) at 37 °C in a humidified 5% CO2 incubator. Influenza virus A/WSN/33 and A/PR/8/34 were used for viral yield reduction assays. All commercially available compounds were purchased mainly from InterBioScreen (Moscow, Russia) and SPEC (Zoetermeer, Netherlands).
Bioactivity assay
Protein expression and purification
Residues 256–716 of A/WSN/33 PAC was cloned into vector pET28a and expressed in Escherichia coli BL21(DE3) pLysS for 16 hrs at 16 °C. Cell pellet was resuspended with 20 mM Tris, 200 mM NaCl, 1% Glycerol, 1 mM TCEP, pH 8.0. The suspension was then lysed by sonication, and the lysate was centrifuged at 16,000 g for 1 hr at 4 °C. The supernatant was passed through a HisTrap column (GE Healthcare) for purification. The bound protein was eluted with 20 mM sodium phosphate, 200 mM NaCl and 500 mM imidazole. The eluate was then loaded to Superdex 200 (GE Healthcare) in 20 mM Tris, 200 mM NaCl, 1% Glycerol, pH 8.0 for further purification. The protein was purified to >95% purity, as assessed by polyacrylamide gel electrophoresis.
Surface plasmon resonance (SPR) for screening and kinetic study
Purified PAC was diluted to 50 μg/ml by immobilization buffer (Phosphate buffer saline, pH 7.4) and was immobilized onto CM5 sensor chips with their primary amide groups using Amine Coupling Kit (Pharmacia). The CM5 sensor chip was activated by injecting 100 μl of 1:1 of EDC (0.4 μM 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide in water) and NHS (0.1 μM N-hydroxysuccinimide in water) at 5 μl/min. 50 μg/ml PAC was then injected into the flow cell at 5 μl/min. 50 μl of 1 M ethanolamine (pH 8.5) was then injected at 5 μl/min to inactivate the excess reactive group on the sensor surface. A control uncoupled sensor surface was also treated similarly without the addition of protein. The chip was then equilibrated with running buffer (Phosphate buffer saline, 5% DMSO, pH 7.4) before measurements. In both screening study and kinetic study, the flow rate was fixed at 40 μl/min and temperature was fixed at 25 °C.
For screening study, 4781 RU of purified PAC is immobilized to the CM5 chip. Compounds from DMSO stock were prepared and diluted in running buffer. Depending on the solubility of chemical, final concentration of chemical spanned from 25 to 100 μM. Using the “binding analysis” program, samples were injected into both coupled and uncoupled flow cells for 1 min. Binding surface was regenerated by the injection of 2 pulses of regeneration buffer (25 mM NaOH, 50 mM NaCl) onto chip surfaces. In addition, solvent control was injected for double referencing. DMSO calibration was implemented to correct for solvent effect.
For kinetic measurements, 3383 RU of purified PAC is immobilized to the CM5 chip. Compound 312 was serially diluted in running buffer and injected into both coupled and uncoupled flow cells for 2 mins. Binding surface was regenerated by regeneration buffer (25 mM NaOH, 50 mM NaCl). The association constant, dissociation constant and the affinity of the interaction are calculated by BIAevaluation v. 4.1 using the ‘1:1 Langmuir binding’ model.
Cytotoxicity of the selected compounds
Cytotoxicity of compounds were assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were seeded on a 96-well microtiter plate in MEM supplemented with 10% FBS. After overnight culture, cells were treated with the compounds. On the day of harvest, MTT (USB Corporation) in phosphate buffered saline was freshly prepared. MTT solution was added to each well and the plates were incubated at 37 °C for 2 hrs. DMSO was then added to dissolve the formazan. Absorbance at 540 nm was measured by VICTOR 3 Multilabel plate reader (Perkin Elmer).
RNP reconstitution assay
Plasmids pcDNA-PB1, pcDNA-PB2, pcDNA-PA, pcDNA-NP, pEGFP and pPolI-Luc-RT have been described previously30,31. 1 × 105 293T cells were seeded on a 96-well microtiter plate overnight. 125ng of pcDNA-PB1, pcDNA-PB2, pcDNA-PA, pcDNA-NP, pPolI-Luc-RT and pEGFP were co-transfected to 293T cells to reconstitute the RNP complexes with Lipofectamine 2000 (Invitrogen). After 6 hrs of transfection, growth medium with candidate compounds were added. 24 hrs later, the luciferase activity was assayed by Steady-Glo luciferase substrate (Promega). The fluorescence from GFP expression and luminescence were read with VICTOR 3 Multilabel plate reader (Perkin Elmer).
Viral yield assay
MDCK cells were seeded into 24-well plate at 1 × 105 cells/well in MEM medium, supplemented with 10% FBS, and incubated at 37 °C for 24 hrs. Cells were washed with PBS and infected with 100 µl influenza A/WSN/33 or A/PR/8/34 virus at MOI of 0.01. After incubating at 37 °C for 1 hr, the virus particles were removed and the cell monolayer was washed with PBS. Medium with various concentration of chemical compounds was then added to cells and incubated at 37 °C for 24 hrs. Viral titer was determined by standard plaque assay with MDCK cells. In vitro cell infection experiments with A/WSN/33 or A/PR/8/34 viruses were performed under biosafety level 2 (BSL-2) laboratory conditions.
Microscale thermophoresis
Microscope Thermophoresis experiments were performed using 1:1 mixture of PAC and compounds in standard grade capillaries (Nanotemper Technologies). Samples were incubated at 25 °C within the capillaries for 5 mins prior to measurement. All measurements were conducted using Monolith NT.LabelFree instrument (NanoTemper Technologies) at 25 °C. Assays were conducted at 20% IR-laser power and Medium MST powers. The buffer used for the experiment was 20 mM Tris, 200 mM NaCl and 15% Glycerol, pH 8. Stock solutions of test compounds were first diluted in buffer to their respective initial concentrations. A two-fold dilution series of the compounds were then prepared for MST measurement.
Statistical analysis
Errors bars are given as the standard deviation. Significance of differences were analyzed using two-tailed t-test. All analyzes were performed using Microsoft excel. Statistics with p value < 0.05 were considered as significant.
Data availability
Procedures for chemical synthesis of compound 221 analogs and data generated or analysed in this study are included in this published article (and its Supplementary material).
Chemicals
2-[(2-(Piperazino)ethyl)amino]-3-phenylquinazolin-4(3H)-one (S1a)
A mixture of phenylisocyanate (0.01 mol) solution in toluene and 1.67 g (0.01 mol) 2- parathesin was refluxed for 5 hrs. After cooling to room temperature, the mixture was filtered and the residue was washed by toluene to afford product as a solid. A mixture of Ethyl 2-(3-phenylureido)benzoate (0.01 mol), ethanol (40 ml), and triethylamine (0.03 mol) was refluxed for 2 hrs. After cooling to room temperature, the mixture was filtered and the residue was washed by ethanol to afford product as a solid. A mixture of 3-Phenylquinazoline-2,4(1H,3H)-dione (0.001 mol) and phosphorus oxychloride (0.02 mol) was refluxed for 8 hrs. After cooling to ambient temperature, the solution was evaporated in vacuo. The residue was dissolved in dichloromethane (DCM) and purified by column chromatography on silica gel to get the product. A mixture of 2-chloro-3-phenylquinazolin-4(3H)-one (0.001 mol), 3- (morpholino)propanamine or 2-(piperazino)ethanamine (0.001 mol), triethylamine (0.003 mol) and n-butyl alcohol (3 ml) was refluxed for 6 hrs. After cooling to room temperature, the mixture was filtered and the residue was dried to afford 2-[(2-(piperazino)ethyl)amino]-3-phenylquinazolin-4(3H)-one (S1a).
2-[(3-Morpholinopropyl)amino]-3-(2-methylphenyl)quinazolin-4(3H)-one (S1b)
A mixture of 2-methylphenyl isocyanate (0.01 mol) solution in toluene and 1.67 g (0.01 mol) 2- parathesin was refluxed for 5 hrs. After cooling to room temperature, the mixture was filtered and the residue was washed by toluene to afford the product as a solid. A mixture of Ethyl 2-[3-(2-methylphenyl)ureido]benzoate (0.01 mol), ethanol (40 ml), and triethylamine (0.03 mol) was refluxed for 2 hrs. After cooling to room temperature, the mixture was filtered and the residue was washed by ethanol to afford the product as a solid. A mixture of 3-(2-methylphenyl)quinazoline-2,4(1H,3H)-dione (0.001 mol) and phosphorus oxychloride (0.02 mol) was refluxed for 8 hrs. After cooling to ambient temperature, the solution was evaporated in vacuo. The residue was dissolved in DCM and purified by column chromatography on silica gel to get the product. A mixture of 2- Chloro-3-(2-methylphenyl)quinazolin-4(3H)-one (0.001 mol), 3- (morpholino)propanamine or 2-(piperazino)ethanamine (0.001 mol), triethylamine (0.003 mol) and n-butyl alcohol (3 ml) was refluxed for 6 hrs. After cooling to room temperature, the mixture was filtered and the residue was dried to 2-[(3-morpholinopropyl)amino]-3-(2-methylphenyl)quinazolin-4(3H)-one (S1b).
2-[(3-Morpholinopropyl)amino]-3-(4-methylphenyl)quinazolin-4(3H)-one (S1c)
A mixture of 4-methylphenyl isocyanate (0.01 mol) solution in toluene and 1.67 g (0.01 mol) 2-parathesin was refluxed for 5 hrs. After cooling to room temperature, the mixture was filtered and the residue was washed by toluene to afford the product as a solid. A mixture of Ethyl 2-[3-(4-methylphenyl)ureido]benzoate (0.01 mol), ethanol (40 ml), and triethylamine (0.03 mol) was refluxed for 2 hrs. After cooling to room temperature, the mixture was filtered and the residue was washed by ethanol to afford the product as a solid. A mixture of 3-(4-methylphenyl)quinazoline-2,4(1H,3H)-dione (0.001 mol) and phosphorus oxychloride (0.02 mol) was refluxed for 8 hrs. After cooling to ambient temperature, the solution was evaporated in vacuo. The residue was dissolved in DCM and purified by column chromatography on silica gel to get the product. A mixture of 2-Chloro-3-(4-methylphenyl)quinazolin-4(3H)-one (0.001 mol), 3- (morpholino)propanamine or 2-(piperazino)ethanamine (0.001 mol), triethylamine (0.003 mol) and n-butyl alcohol (3 ml) was refluxed for 6 hrs. After cooling to room temperature, the mixture was filtered and the residue was dried to afford 2-[(3-morpholinopropyl)amino]-3-(4-methylphenyl)quinazolin-4(3H)-one (S1c).
3-(4-Fluorophenyl)-2-((3-morpholinopropyl)amino)quinazolin-4(3H)-one (S1d)
A mixture of 4-fluorophenyl isocyanate (0.01 mol) solution in toluene and 1.67 g (0.01 mol) 2-parathesin was refluxed for 5 hrs. After cooling to room temperature, the mixture was filtered and the residue was washed by toluene to afford the product as a solid. A mixture of Ethyl 2-[3-(4-fluorophenyl)ureido]benzoate (0.01 mol), ethanol (40 ml), and triethylamine (0.03 mol) was refluxed for 2 hrs. After cooling to room temperature, the mixture was filtered and the residue was washed by ethanol to afford the product as a solid. A mixture of 3-(4-fluorophenyl)quinazoline- 2,4(1H,3H)-dione (0.001 mol) and phosphorus oxychloride (0.02 mol) was refluxed for 8 hrs. After cooling to ambient temperature, the solution was evaporated in vacuo. The residue was dissolved in DCM and purified by column chromatography on silica gel to get the product. A mixture 2-Chloro-3-(4-fluorophenyl)quinazolin-4(3H)-one (0.001 mol), 3- (morpholino)propanamine or 2-(piperazino)ethanamine (0.001 mol), triethylamine (0.003 mol) and n-butyl alcohol (3 ml) was refluxed for 6 hrs. After cooling to room temperature, the mixture was filtered and the residue was dried to afford 3-(4-Fluorophenyl)-2-((3-morpholinopropyl)amino)quinazolin-4(3H)-one (S1d).
Diethyl 2-(1-phenylethyl)malonate (1)
To a solution of diethyl malonate (1 eq) in dimethylformamide (DMF) in with ice bath, NaH(1.2 eq) was added and stirring was continued for 30 mins at 0 °C. (1-bromoethyl)benzene (1 eq) was then added. The reaction mixture was stirred for 5 hrs at room temperature. The reaction was then quenched with NH4Cl aq. extraction with ethyl acetate (EA), crude product was obtained, and was purified by silica gel column chromatography to afford compound (1).
1-bromo-2-(methoxymethoxy)ethane (2)
To a solution of 2-bromoethanol (1 eq) in DCM with ice bath, N,N-diisopropylethylamine (2 eq) was added. Methoxymethyl chloride (1.5 eq) was added slowly and stirred for 4 hrs at room temperature. The reaction was quenched with a saturated aqueous NaHCO3 solution and extracted with DCM. Crude product was purified by silica gel column chromatography to afford compound (2).
1,3-diphenylpyrimidine-2,4,6(1H,3H,5H)-trione (3)
To a solution of 1,3-diphenylurea(1 eq) in EtOH, NaOEt (1.3 eq) was added. Diethyl malonate (1.2 eq) was then added and stirred for 1 hr at room temperature and then reflux overnight. The reaction was quenched with 1M HCl at 0 °C, extracted with EA. Crude product was purified by silica gel column chromatography to afford compound (3).
6-chloro-1,3-diphenylpyrimidine-2,4(1H,3H)-dione (4)
Compound (3) was added to POCl3 (10 eq) and water (1.1 eq) at room temperature and stirred for 50 min, and then stirred for 5 hrs at 110 °C. Reaction mixture was cooled to room temperature, ice water and saturated aqueous NaHCO3 solution was added, and extracted with EA. Crude product was purified by silica gel column chromatography to afford compound (4).
6-azido-1,3-diphenylpyrimidine-2,4(1H,3H)-dione (5)
To a solution of compound (4) (1 eq) in DMF, 1.25 eq NaN3 was added and stirred overnight at room temperature. The reaction was then quenched with water, and extracted with EA. Crude product was purified by silica gel column chromatography to afford compound (5).
6-amino-1,3-diphenylpyrimidine-2,4(1H,3H)-dione (6)
To a solution of compound (5) in MeOH, Palladium on carbon (Pd/C) (10%) was added, it was then hydrogenated for 5 hrs. Then Pd/C was filtered from the solution, the filtrate was concentrated to obtain the product (6).
6-(methylamino)-1,3-diphenylpyrimidine-2,4(1H,3H)-dione (7)
To a solution of compound (4) (1 eq) in EtOH, MeNH2/MeOH (1.5 eq) was added, the reaction mixture was stirred at 80 °C overnight. The reaction was concentrated to obtain the product (7).
6-benzyl-5-hydroxy-1,3-diphenylpyrido[2,3-d]pyrimidine- 2,4,7(1H,3H,8H)-trione (8)
To a solution of compound (6) (1 eq) in PhOPh, diethyl 2-benzylmalonate (1 eq) was added. The mixture was heated to 220–250 °C, stirred overnight, and cooled to room temperature. The reaction was then quenched with water, and extracted with EA. Crude product was purified by silica gel column chromatography to afford compound (8).
6-benzyl-5-hydroxy-8-(2-(methoxymethoxy)ethyl)-1,3-diphenylpyrido[2,3- d]pyrimidine-2,4,7(1H,3H,8H)-trione (9)
To a solution of compound (8) (1 eq) in DMF, K2CO3 (1.5 eq) was added, stirred for 30 min. 1-bromo-2-(methoxymethoxy)ethane (1.3 eq) was added, and the reaction was stirred for 4 hrs. The reaction was then quenched with water, and extracted with EA, Crude product was purified by silica gel column chromatography to afford compound (9).
5-hydroxy-8-methyl-1,3-diphenylpyrido[2,3-d]pyrimidine- 2,4,7(1H,3H,8H)-trione (10)
To a solution of compound (7) (1 eq) in PhOPh, diethyl malonate (1 eq) was added, then the mixture was heated to 220–250 °C. It was stirred overnight, and cooled to room temperature. The reaction was then quenched with water, and extracted with EA. Crude product was purified by silica gel column chromatography to afford compound (10).
6-benzyl-5-hydroxy-8-(2-hydroxyethyl)-1,3-diphenylpyrido[2,3- d]pyrimidine-2,4,7(1H,3H,8H)-trione (S2a)
To a solution of compound (9) in MeOH, HCl/MeOH was added and stirred for 4 hrs. The reaction was concentrated to obtain the product (S2a).
5-hydroxy-1,3-diphenyl-6-(1-phenylethyl)pyrido[2,3-d]pyrimidine- 2,4,7(1H,3H,8H)-trione (S2b)
To a solution of compound (6) (1 eq) in PhOPh, compound (1) (1 eq) was added, then the mixture was heated to 220–250 °C, stirred overnight, and cooled to room temperature. The reaction was then quenched with water, and extracted with EA. Crude product was purified by silica gel column chromatography to afford compound (S2b).
5-hydroxy-8-methyl-1,3-diphenyl-6-(1-phenylethyl)pyrido[2,3- d]pyrimidine-2,4,7(1H,3H,8H)-trione (S2c)
To a solution of compound (7) (1 eq) in PhOPh, compound (1) (1 eq) was added. The mixture was heated to 220–250 °C, stirred overnight, and cooled to room temperature. The reaction was then quenched with water, and extracted with EA. The crude product was purified by silica gel column chromatography to afford compound (S2c).
5-methoxy-8-methyl-1,3-diphenyl-6-(1-phenylethyl)pyrido[2,3- d]pyrimidine-2,4,7(1H,3H,8H)-trione (S2d)
To a solution of compound (S2c) in DMF, K2CO3 (1.5 eq) was added and stirred for 30 min. MeI(1.3 eq) was then added, and the reaction was stirred for 4 hrs. The reaction was then quenched with water, and extracted with EA. Crude product was purified by silica gel column chromatography to afford compound (S2d).
8-acetyl-5-hydroxy-1,3-diphenyl-6-(1-phenylethyl)pyrido[2,3- d]pyrimidine-2,4,7(1H,3H,8H)-trione (S2e)
To a solution of compound (S2b) (1 eq) in EtOH, acetic anhydride (1.05 eq) was added at room temperature. The solution was stirred for 15 min then concentrated in vacuo to give compound (S2e).
5-hydroxy-8-methyl-1,3-diphenyl-6-(phenylamino)pyrido[2,3- d]pyrimidine-2,4,7(1H,3H,8H)-trione (S2f)
To a solution of compound (10) in H2O under N2, PhI(OAc)2(1 eq) and Na2CO3(2 eq) was added. The reaction was stirred for 4 hrs at room temperature. The reaction was then quenched with water, and extracted with EA. Crude product was obtained.
The crude product was dissolved in toluene, aniline (1 eq) was added, the reaction mixture was refluxed overnight after evaporation of the solvent and was purified by silica gel column chromatography to afford compound (S2f).
Electronic supplementary material
Acknowledgements
This work was supported by a Health and Medical Research Fund (Project No. 13120052), an Area of Excellence Scheme (Project No. AoE/M-12/06) and a Theme-based grant (Project No. T11-705/14N) of the Research Grants Council of Hong Kong HKSAR.
Author Contributions
C.Y.L. and P.C.S. designed the project. C.Y.L. and O.T.W. conducted the experiments. W.P.T., C.H. and G.X.W. synthesized chemical compounds. C.Y.L., C.H., G.X.W. and P.C.S. wrote the manuscript. C.H., G.X.W., J.C.K.N., D.C.C.W., L.L.M.P. and P.C.S. supervised and discussed the data. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-20772-9.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO. Influenza (Seasonal) Fact sheet. http://www.who.int/mediacentre/factsheets/fs211/en/ (2016).
- 2.Chen H, et al. Oseltamivir-resistant influenza A pandemic (H1N1) 2009 Virus, Hong Kong, China. Emerg. Infect. Dis. 2009;15:1970–1972. doi: 10.3201/eid1512.091057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Y, et al. Association between adverse clinical outcome in human disease caused by novel influenza A H7N9 virus and sustained viral shedding and emergence of antiviral resistance. Lancet. 2013;381:2273–2279. doi: 10.1016/S0140-6736(13)61125-3. [DOI] [PubMed] [Google Scholar]
- 4.WHO. Human infection with avian influenza A(H7N9) virus-China. http://www.who.int/csr/don/17-january-2017-ah7n9-china/en/ (2017).
- 5.WHO. Assessment of risk associated with influenza A(H5N8) virus. http://www.who.int/influenza/human_animal_interface/avian_influenza/riskassessment_AH5N8_201611/en/ (2016).
- 6.Palese, P. & Shaw, M. Orthomyxoviridae: the viruses and their replication in Fields Virology 5th edition (ed. D. M. Knipe & P. M. Howley), 1647–1689 (Philadelphia: Lippincott Williams & Wilkins, 2007)
- 7.Pflug A, Guilligay D, Reich S, Cusack S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature. 2014;516:355–360. doi: 10.1038/nature14008. [DOI] [PubMed] [Google Scholar]
- 8.Reich S, et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature. 2014;516:361–366. doi: 10.1038/nature14009. [DOI] [PubMed] [Google Scholar]
- 9.Stevaert A, Naesens L. The influenza virus polymerase complex: an update on its structure, functions, and significance for antiviral drug design. Med. Res. Rev. 2016;36:1127–1173. doi: 10.1002/med.21401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obayashi E, et al. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature. 2008;454:1127–1131. doi: 10.1038/nature07225. [DOI] [PubMed] [Google Scholar]
- 11.He X, et al. Crystal structure of the polymerase PAC–PB1N complex from an avian influenza H5N1 virus. Nature. 2008;454:1123–1126. doi: 10.1038/nature07120. [DOI] [PubMed] [Google Scholar]
- 12.Deng T, Sharps J, Fodor E, Brownlee GG. In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza A virus polymerase subunits into a functional trimeric complex. J. Virol. 2005;79:8669–8674. doi: 10.1128/JVI.79.13.8669-8674.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wunderlich K, et al. Identification of a PA-binding peptide with inhibitory activity against influenza A and B virus replication. PLoS One. 2009;4:e7517. doi: 10.1371/journal.pone.0007517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muratore G, et al. Small molecule inhibitors of influenza A and B viruses that act by disrupting subunit interactions of the viral polymerase. Proc. Natl. Acad. Sci. 2012;109:6247–6252. doi: 10.1073/pnas.1119817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukuoka M, et al. Structure-based discovery of anti-influenza virus A compounds among medicines. Biochim. Biophys. Acta-Gen. Subj. 2012;1820:90–95. doi: 10.1016/j.bbagen.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Massari S, et al. Structural investigation of cycloheptathiophene-3-carboxamide derivatives targeting influenza virus polymerase assembly. J. Med. Chem. 2013;56:10118–10131. doi: 10.1021/jm401560v. [DOI] [PubMed] [Google Scholar]
- 17.Lepri S, et al. Optimization of small-molecule inhibitors of influenza virus polymerase: from thiophene-3-carboxamide to polyamido scaffolds. J. Med. Chem. 2014;57:4337–4350. doi: 10.1021/jm500300r. [DOI] [PubMed] [Google Scholar]
- 18.Massari S, et al. A broad anti-influenza hybrid small molecule that potently disrupts the interaction of polymerase acidic protein-basic protein 1 (PA-PB1) Subunits. J. Med. Chem. 2015;58:3830–42. doi: 10.1021/acs.jmedchem.5b00012. [DOI] [PubMed] [Google Scholar]
- 19.Trist IML, et al. 4,6-diphenylpyridines as promising novel anti-influenza agents targeting the PA-PB1 protein-protein interaction: structure-activity relationships exploration with the aid of molecular modeling. J. Med. Chem. 2016;59:2688–703. doi: 10.1021/acs.jmedchem.5b01935. [DOI] [PubMed] [Google Scholar]
- 20.Yuan S, et al. Identification of a small-molecule inhibitor of influenza virus via disrupting the subunits interaction of the viral polymerase. Antiviral Res. 2016;125:34–42. doi: 10.1016/j.antiviral.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Hara K, et al. Influenza virus RNA polymerase PA subunit is a novel serine protease with Ser624 at the active site. Genes Cells. 2001;6:87–97. doi: 10.1046/j.1365-2443.2001.00399.x. [DOI] [PubMed] [Google Scholar]
- 22.Hu J, Liu X. Crucial role of PA in virus life cycle and host adaptation of influenza A virus. Med. Microbiol. Immunol. 2015;204:137–149. doi: 10.1007/s00430-014-0349-y. [DOI] [PubMed] [Google Scholar]
- 23.Nordström H, et al. Identification of MMP-12 inhibitors by using biosensor-based screening of a fragment library. J. Med. Chem. 2008;51:3449–3459. doi: 10.1021/jm8000289. [DOI] [PubMed] [Google Scholar]
- 24.Perspicace S, et al. Fragment-based screening using surface plasmon resonance technology. J. Biomol. Screen. 2009;14:337–349. doi: 10.1177/1087057109332595. [DOI] [PubMed] [Google Scholar]
- 25.Elinder M, et al. Screening for NNRTIs with slow dissociation and high affinity for a panel of HIV-1 RT variants. J. Biomol. Screen. 2009;14:395–403. doi: 10.1177/1087057109333977. [DOI] [PubMed] [Google Scholar]
- 26.Rich RL, Myszka DG. Kinetic analysis and fragment screening with Fujifilm AP-3000. Anal. Biochem. 2010;402:170–178. doi: 10.1016/j.ab.2010.03.043. [DOI] [PubMed] [Google Scholar]
- 27.Lo CY, et al. Identification of lead inhibitors targeting influenza A virus nucleoprotein through surface plasmon resonance screening. Biodesign. 2015;3:131–137. [Google Scholar]
- 28.Perez JT, et al. A small-RNA enhancer of viral polymerase activity. J. Virol. 2012;86:13475–13485. doi: 10.1128/JVI.02295-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukarska M, et al. Structural basis of an essential interaction between influenza polymerase and Pol II CTD. Nature. 2017;541:117–121. doi: 10.1038/nature20594. [DOI] [PubMed] [Google Scholar]
- 30.Fodor E, et al. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 2002;76:8989–9001. doi: 10.1128/JVI.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li OTW, et al. Full factorial analysis of mammalian and avian influenza polymerase subunits suggests a role of an efficient polymerase for virus adaptation. PLoS One. 2009;4:e5658. doi: 10.1371/journal.pone.0005658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Procedures for chemical synthesis of compound 221 analogs and data generated or analysed in this study are included in this published article (and its Supplementary material).