

Abstract
Background
The bacterial pathogen Mycoplasma synoviae can cause subclinical respiratory disease, synovitis, airsacculitis and reproductive tract disease in poultry and is a major cause of economic loss worldwide. The M. synoviae strain MS-H was developed by chemical mutagenesis of an Australian isolate and has been used as a live attenuated vaccine in many countries over the past two decades. As a result it may now be the most prevalent strain of M. synoviae globally. Differentiation of the MS-H vaccine from local field strains is important for epidemiological investigations and is often required for registration of the vaccine.
Results
The complete genomic sequence of the MS-H strain was determined using a combination of Illumina and Nanopore methods and compared to WVU-1853, the M. synoviae type strain isolated in the USA 30 years before the parent strain of MS-H, and MS53, a more recent isolate from Brazil. The vaccine strain genome had a slightly larger number of pseudogenes than the two other strains and contained a unique 55 kb chromosomal inversion partially affecting a putative genomic island. Variations in gene content were also noted, including a deoxyribose-phosphate aldolase (deoC) fragment and an ATP-dependent DNA helicase gene found only in MS-H. Some of these sequences may have been acquired horizontally from other avian mycoplasma species.
Conclusions
MS-H was somewhat more similar to WVU-1853 than to MS53. The genome sequence of MS-H will enable identification of vaccine-specific genetic markers for use as diagnostic and epidemiological tools to better control M. synoviae.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4501-8) contains supplementary material, which is available to authorized users.
Keywords: Mycoplasma synoviae, MS-H vaccine strain, Complete genome sequencing, Comparative genetic analysis, Large chromosomal inversion, Vaccine-specific genetic markers
Background
The avian pathogen Mycoplasma synoviae is a member of the class Mollicutes, a group of bacteria that are characterised by their very small size, lack of cell wall, complex nutritional requirements and ability to persist in their hosts and establish chronic infections [1]. M. synoviae strains appear to have varying tissue tropisms and virulence, although these characteristics may depend on the route of infection [2]. It causes subclinical upper respiratory tract infections in chickens, turkeys and other birds [3]. It can also disseminate further into the host, leading to synovitis or egg defects [4]. It is transmitted horizontally via direct contact, or vertically via fertile eggs [1]. Although M. synoviae is rarely associated with bird mortality, its impact on avian health is significant. As a result of the implementation of adequate control programs for M. gallisepticum, the other major mycoplasmal pathogen of poultry, M. synoviae may now be the most important bacterial cause of economic loss in the poultry industry [5]. Vaccination is commonly used to control M. synoviae infection in commercial flocks in many countries with significant commercial poultry industries. The temperature-sensitive strain MS-H was produced by N-nitro-N’methyl-N-nitrosoguanidine (NTG) chemical mutagenesis of an Australian field isolate, 86,079/7NS [6, 7]. MS-H does not grow at the core body temperature of birds, colonises only their upper respiratory tract and establishes solid protection against wild type M. synoviae. MS-H was first registered as a live attenuated vaccine in Australia in 1996 (Vaxsafe MS; Bioproperties Pty. Ltd., Ringwood, Victoria, Australia) and is now formally registered and used for vaccination of commercial poultry in 26 different countries across 6 continents. In addition, MS-H is used in several other countries where formal registration is not required (personal communications with Dr. Ross Henderson and Dr. Chris Morrow, Bioproperties Australia Pty. Ltd.). The international use of the MS-H vaccine suggests that it may now be the most common strain of M. synoviae globally and highlights the importance of assays developed to differentiate local endemic strains from the vaccine [8]. However, little information is available about the genetic relatedness of MS-H and other M. synoviae field strains found in poultry. Moderately virulent M. synoviae strains have been isolated from flocks previously vaccinated with MS-H [9], raising the question of the origin of these organisms and prompting interest in the sequence of the MS-H genome. Such knowledge would help to define molecular markers for tracking MS-H in vaccinated flocks and assessing any variation in the level of cross-protection against local strains of M. synoviae. The sequences of the M. synoviae field strain MS53, isolated around 2003 from a broiler flock in Brazil [10], and the type strain WVU-1853, isolated in 1957 from a hock joint of chicken in the USA [11], are the only two complete genomes determined thus far for this species. This is possibly due to difficulties experienced in assembling next generation sequencing (NGS) data for M. synoviae, which contains large, low complexity, repeat-rich regions. Here, MS-H was completely sequenced by combining short, accurate Illumina sequence data with long reads generated using the Oxford Nanopore technology. This genome sequence was compared to the complete genome sequences of strains WVU-1853 or MS53 with the aim of identifying features unique to MS-H, WVU-1853 or MS53, and assessing the degree of overall similarity between the three genomes and identifying MS-H specific features that could be targeted to develop genotyping assays and differentiate the vaccine from field strains, enabling improved assessment of disease control strategies.
Methods
Preparation of genomic DNA and sequencing
M. synoviae strain MS-H was inoculated into mycoplasma culture medium containing 10% swine serum and 0.01% (w/v) nicotinamide adenine dinucleotide [12] and grown until late logarithmic phase (pH of approximately 6.8) at 37 °C in a 50 mL final volume. Cells were collected by centrifugation at 10000×g for 30 min. Genomic DNA was prepared by proteinase K digestion of the pellet, phenol-chloroform extraction and ethanol precipitation [13, 14]. An Illumina paired-end (300 bp insert size) genomic DNA library was prepared and sequenced by the Micromon DNA Sequencing Facility (Monash University, Australia) using a Genome Analyzer IIx system. An Oxford Nanopore long read genomic DNA library was prepared with the sequencing kit SQK-NSK007 (Oxford Nanopore Technologies, Oxford, OX4 4GA, UK) according to the manufacturer’s instructions using a mixture of 1.2 μg of genomic DNA sheared using a Covaris-g TUBE and 0.8 μg of unsheared genomic DNA. Sequencing data were generated on a MinION MK-I device fitted with a FLO-MIN104 flowcell (R9 chemistry) and processed using the cloud-based Metrichor workflow 2D base caller RNN SQK-NSK_007 rev 1.107. Oligonucleotides (Geneworks, Australia) were designed using Primer3 version 2.3.4 and PCR amplicons were sequenced using the Sanger method at the Micromon DNA Sequencing Facility (Monash University, Melbourne, Australia).
De novo sequence assembly, gene prediction and annotation of MS-H
Paired-end Illumina reads were filtered to select those with quality values above 20 and a minimal read length of 91 after trimming of adapter sequences. The quality of the filtered reads was confirmed with the FastQC [15]. The Illumina reads were de novo assembled using Velvet [16] (version 1.2.10) using k-mer values of 81 and 91 and coverage cut-off values of 5, 10, 20, 50, 100 and 200. Nanopore 2D reads with lengths > 2500 bp were extracted from the set of fast5 files returned in the “pass” folder by the Metrichor basecaller and converted into .fasta format using Poretools [17]. The nanopore reads were de novo assembled with Canu 1.2 [18] using the parameters genomesize = 0.9 m and errorRate = 0.1. The Illumina read datasets were mapped against the Nanopore assembly and the consensus sequence extracted using the Geneious version 7.0.6 sequence manipulation suite. Automatic annotation of the MS-H sequence was performed on the RAST webserver with default parameters for mycoplasmas [19]. Provisional locus tag numbers were assigned and the corresponding nucleotide positions are listed in Additional file 1: Table S1.
Genome comparison and analysis
To ensure consistency in the analysis, the genome sequences of M. synoviae strains MS53 and WVU-1853 (GenBank accession numbers AE017245 and CP011096) were re-annotated using the RAST website with default parameters as above. Genome characteristics of each M. synoviae strain were analysed using Artemis [20]. Whole genome sequence alignments were performed using the Mauve Aligner and LASTZ tools in Geneious version 7.0.6. Pairwise alignments of nucleotide and amino acid sequences of individual genes were performed using the generic Geneious alignment tool with the default parameters. The search for pathogenicity genomic islands was conducted using the IslandViewer3 server (http://www.pathogenomics.sfu.ca/islandviewer) and the SIGI-HMM and IslandPath-DIMOB methods [21]. The PHAge Search Tool (PHAST) (http://phast.wishartlab.com) was used to detect complete and/or incomplete prophage sequences [22]. Clustered regularly interspersed short palindromic repeats (CRISPR) sequences were detected using CRISPRfinder (http://crispr.i2bc.paris-saclay.fr) [23]. Insertion sequences (IS) were analysed using the ISFinder tool (http://www-is.biotoul.fr) [24].
Results and Discussion
Assembling MS-H genome sequence and resolving the highly repeated vlhA region
Extensive assembly attempts using Velvet with a set of 30,345,486 MS-H Illumina paired-end reads with various K-mer and coverage cutoff values produced 659 contigs with sizes ranging from 181 bp to 134,673 bp. These contigs were aligned to the strain MS53 genome to obtain assembly parameters. De novo assembly of the MS-H genome was then attempted. However, many genomic regions could not be assembled solely from the Illumina datasets. Sanger sequencing of 28 PCR products and scaffolding of the Velvet data produced a 766,314 bp contig, representing an almost complete genome, but failed to assemble the vlhA pseudogene region, a 50 kb locus containing a cluster of highly repeated sequences. To resolve the vlhA region and complete the MS-H genome, a set of 29,015 Nanopore reads containing a total of 209,108,300 nt were independently assembled and circularised into a single 810,924 bp contig. Because Nanopore sequences are error-prone, the Illumina reads were then mapped against the contig, providing an average coverage depth of 3661 ± 829, and the consensus sequence was then extracted from the aligned reads. This approach generated a fully assembled genome encompassing the entire vlhA cluster and all the other regions that were difficult to assemble solely from the Illumina reads. The final MS-H genome was then verified by aligning the previously obtained Velvet contigs and Sanger sequenced PCR products, where available, with the new assembly. All nucleotide discrepancies between the two datasets were resolved manually by inspecting the alignments of Illumina reads to verify the quality of the consensus. The results from the Illumina/Nanopore hybrid approach was found to be accurate in all cases, except for a dinucleotide repeat sequence, (CT)15, which was correctly re-interpreted as (CT)13. These results demonstrate that a strategy combining error-prone but long single-molecule reads (Nanopore) with accurate short next generation sequencing data (Illumina) can generate a high quality, complete mycoplasma genome sequence. This approach is particularly well adapted to Mollicutes, which often contain repeated regions that are difficult to assemble. As expected, the MS-H-specific mutation in the obg gene [25], previously reported as a marker of the vaccine strain, was identified. The whole-genome sequence of MS-H has been deposited in the National Centre for Biotechnology Information (NCBI) under accession number CP021129.
General comparisons of M. synoviae genomes and identification of a large chromosomal inversion in MS-H
The general features of the MS-H, MS53 and WVU-1853 genomes are listed in Table 1. The M. synoviae MS-H genome was 818,848 bp long with an overall GC content of 28.2%. The DNA-DNA sequence identities between MS-H and WVU-1853 and between MS-H and MS53, (excluding the vlhA locus region) were 92.1% and 91.3% respectively. All 3 strains had similar average gene lengths, ranging from 992 to 1002 bp, average gene densities of approximately 90%, and numbers of putatively encoded proteins, ranging from 723 to 764, of which 541 to 573 had predicted functions. In each strain, 34 tRNAs and 7 rRNAs, consisting of three copies of 5S, two copies of 23S, and two copies of the 16S rRNA subunits, were identified. The strains had most of their genes or gene products in common, sharing 92.5% - 100% nucleotide sequence identities and 88.0% - 100% amino acid sequence identities. Comparisons of open reading frames (ORFs) revealed that 5 of the MS-H, 7 of the WVU-1853 and 3 of the MS53 ORFs have insertions or deletions in multiples of 3 bp, resulting in slightly longer or shorter proteins without frameshifts (Table 2). Analysis of the genomes of the 3 strains using MAUVE revealed that their chromosomes were collinear, with the remarkable exception of a unique 55 kb inversion in MS-H (Fig. 1) (Additional file 1: Table S1). Large chromosomal inversions have not been reported before in M. synoviae, but have been seen occasionally in other mollicutes, including M. hyopneumoniae [10], and M. bovis [26]. The molecular mechanism underlying this inversion, which is delimited by a tRNAGly and an IS30 (Fig. 2), is unclear. It has been proposed that the partial lack of DNA replication and repair functions in Mycoplasma species could prevent the formation of large genomic inversions [27]. The NTG-mutagenesis used to create the MS-H vaccine generates point mutations and is therefore unlikely to have caused this rearrangement. Whether this genomic inversion occurred naturally in the lineage of MS-H and is a common feature amongst Australian field strains, or was induced by mutagenesis will require further genomic sequencing from the parental strain of the vaccine, namely 86,079/7NS. Repeated sequences can contribute to chromosomal rearrangements, including inversions [28, 29]. The genomic structures of Mycoplasma bovis and Mycoplasma agalactiae have high syntenies except for a 142 kb inversion, which may be related to an ISMbov1 element adjacent to this region in M. bovis [26]. A similar mechanism may have generated the chromosomal rearrangement in MS-H. Apart from the inversion, the genomic organisation of MS-H was generally similar to those of MS53 and WVU-1853.
Table 1.
General characteristic of the genomes of 3 M. synoviae strains MS-H, MS53 and WVU-1853
| Characteristic | MS-H | MS53 | WVU-1853 |
|---|---|---|---|
| Total length (base pairs) | 818,848 | 799,476 | 846,495 |
| G + C content (%) | 28.2 | 28.5 | 28.3 |
| Gene density (%) | 90.0 | 90.4 | 90.2 |
| Average gene length (base pairs) | 1002 | 994 | 992 |
| No. features (genes) | 775 | 764 | 805 |
| No. coding sequences | 734 | 723 | 764 |
| No. CDS with predicted function | 541 | 543 | 573 |
| No. rRNAs | |||
| 16S | 2 | 2 | 2 |
| 23S | 2 | 2 | 2 |
| 5S | 3 | 3 | 3 |
| No. of tRNAs | 34 | 34 | 34 |
Table 2.
Unique amino acid deletions/insertions in M. synoviae strains MS-H, MS53 and WVU-1853
| Features (Locus tag) | MS-H | MS53 | WVU-1853 |
|---|---|---|---|
| Cell division protein FtsZ (MS53_0340) | − 7 | ||
| Conserved hypothetical protein (MS53_0547) | − 4 | ||
| Conserved hypothetical protein (MS53_0590) | − 10 | ||
| Hypothetical protein (VY93_01870) | − 4 | ||
| Hypothetical protein (VY93_02525) | + 3 | ||
| Hypothetical protein (VY93_02550) | − 29 | ||
| Peptidase C1 (VY93_03235) | − 13 | ||
| Signal peptidase I (VY93_00225) | − 6 | ||
| PTS sugar transporter subunit IIABC (VY93_00785) | + 8 | ||
| ABC transporter ATP-binding protein (VY93_03350) | − 2 | ||
| Choline kinase family (MSH_00560) | − 5 | ||
| Hypothetical protein (MSH_02310) | − 3 | ||
| Hypothetical protein (MSH_04020) | − 5 | ||
| Siderophore-mediated iron transport protein (MSH_05040) | − 20 | ||
| Hypothetical protein (MSH_05940) | + 4 |
“+” indicates amino acid insertion into the protein and “-” indicates amino acid deletion from the protein, relative to the other two strains. Nucleotide positions corresponding to the locus tags of MS-H are listed in Additional file 1: Table S1
Fig. 1.
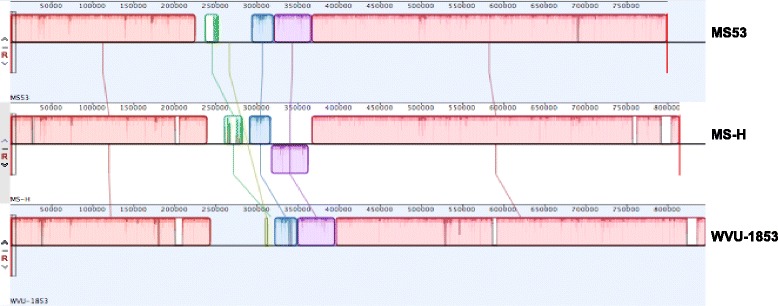
MAUVE alignment analysis of M. synoviae genomes, using MS-H as a reference. Similarly colored blocks with connecting lines represent homologous regions. A block below the centre line indicates a region that aligned in reverse complement (inverse) orientation
Fig. 2.
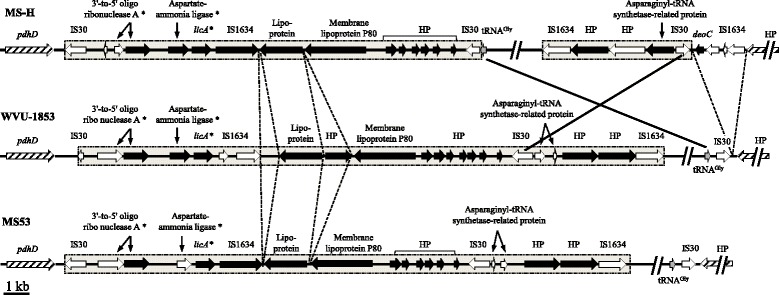
Comparative structural analysis of the genomic island (GI) in MS-H, WVU-1853 and MS53. The grey boxes with dotted lines indicate genes identified as part of the genomic island. Hatched arrows indicate genes flanking the GI. The inverted region in MS-H relative to MS53 and WVU-1853 is indicated by crossed solid lines. Inserted/deleted sequences are indicated by dashed lines. Genes with a complete predicted CDS are depicted in black; pseudogenes are depicted in white; tRNA genes are depicted in grey. HP: hypothetical protein. The asterisks indicate sequences duplicated elsewhere in the host genome
Large mobile genetic elements
Several large mobile genetic elements were identified in all three genomes. Two distinct clusters of phage-related sequences were found in MS-H, MS53 and WVU-1853. These two clusters were similarly organised across the strains and were located at nucleotide positions 446,358–459,743 and 126,056–134,674 in MS-H, 442,958–456,345 and 123,762–132,823 in MS53, and 472,072–485,459 and 124,910–133,971 in WVU-1853. Their GC contents were similar (28.72 ± 0.11% for the first cluster and 29.24 ± 0.05% for the second cluster) and slightly higher than the average GC content of the host genomes. In contrast, a third region, with significantly lower GC content (22.2%–24.2%) was predicted by IslandViewer3 analysis to be a putative genomic island (GI) in MS53 (nucleotide positions 302,828–327,410) and WVU-1853 (nucleotide positions 329,954–356,584). In MS-H, a related region was identified by sequence alignment with the two other strains; this region was adjacent to, and partially affected by, the 55 kb [tRNAGly - IS30] genomic inversion (Fig. 2) on its right side. Specifically, the left portion of the GI was located at nucleotide positions 303,839–321,538 while the right portion was found at 360049–367358 as a result of the genomic inversion. The GI encoded lipoproteins, hypothetical proteins and transposases of the IS30 and IS1634 families. Three coding sequences (CDSs) adjacent to the GI, encoding an oligoribonuclease A, an aspartate-ammonia ligase and a lichenan-specific IIA (LicA) component of a phosphotransferase system (PTS), were also found at another chromosomal locus. In the GI, these CDSs were flanked by an IS30 and an IS1634 (Fig. 2). In MS53, the GI-encoded aspartate-ammonia ligase was present as a pseudogene, while the other chromosomal copy was intact. In all 3 strains, the much lower GC content of the GI suggests that it was horizontally acquired by M. synoviae from another organism.
Strain-variable sequences in M. synoviae genomes and vaccine-specific genetic markers
Comparison of the gene repertoires of the three strains (excluding the vlhA locus, which has complex patterns of variability within M. synoviae) revealed that 18 predicted gene products were variably distributed amongst the 3 strains (Table 3). These genes were either present in only one of the strains or present in two strains, but absent from the third. Strain-variable sequences are of particular interest because they could be used as molecular markers for differentiating the MS-H vaccine from field strains and understanding the recent evolution of M. synoviae and other avian mollicutes. Remarkably, most of the loci containing strain-variable genes were associated with, and often flanked by, IS elements. The comparative organisation of the major strain-variable loci is illustrated in Fig. 3. Only a few of the putative gene products from these loci had a predicted function at the time of analysis. They include a deoxyribose-phosphate aldolase (deoC) fragment (MSH_03520) (Additional file 2: Table S2), an ATP-dependent DNA helicase (MSH_07190), an N-acetylneuraminate lyase (nanA) variant (MSH_00300), an integrase (MSH_07560, VY93_03735), a DNA methylase (MSH_02150, VY93_01015/VY93_02585), and several type I, II and III related restriction-modification proteins. The deoC fragment and the ATP-dependent DNA helicase sequences were located at two distinct chromosomal sites in the MS-H genome, and were absent in WVU-1853 and MS53 (Fig. 3a and b). While a complete copy of nanA was found in MS-H, WVU-1853 and MS53 at a conserved locus, a second truncated sequence variant lacking the first 134 nt and adjacent to an IS1634 family transposase gene, was located between the conserved glpQ and rpiR genes in MS-H only (Fig. 3c). NanA is involved in sialic acid scavenging and degradation and is proposed to be associated to virulence in M. synoviae [30, 31]. Whether the nanA variant found in MS-H influences the sialic acid metabolism of the strain remains to be explored. A locus containing an integrase gene, several of hypothetical protein CDSs and sequences related to type I and type III restriction-modification systems were found in MS-H and WVU-1853 but not in MS53 (Fig. 3d). The type I and type III restriction-modification related sequences were located at both ends of the locus, in opposite orientations. These sequences were pseudogenes (see below) and had similarities with CDSs for two restriction S subunits, one restriction R subunit and one DNA-methyltransferase M subunit for the type I system, and two restriction-modification system methylation subunits for the type III system. Genes encoding type I restriction modification (RM) systems are known to be unstable and to display allelic variability [32]. Accordingly, no type I RM system was found in MS53 and significant sequence variations were seen between the specificity subunits; MS-H and WVU-1853 had four type III RM system methylation subunits, while only one was seen in MS53. Moreover, a locus encoding a type II restriction enzyme homologous to MjaIII (prototype MboI) and a methyl-directed repair DNA adenine methylase, flanked by IS1634 copies was detected in MS-H and WVU-1853, but not in MS53 (Fig. 3e). Overall, the analysis of the strain-variable gene repertoires indicated that strains MS-H and WVU-1853, which have distinct geographical and historical origins, were more closely related to each other than strain MS53, which was isolated from the same continent as WVU-1853. Whether these findings are representative of strain diversity in North and South America compared to Australia remains to be explored.
Table 3.
Summary of variable genes across M. synoviae strains MS-H, MS53 and WVU-1853
| Gene product | ORF (locus taga) | ||
|---|---|---|---|
| MS-H | MS53 | WVU-1853 | |
| N-acetylneuraminate lyase variant | MSH_00300 | – | – |
| Transcriptional regulator | MSH_00310b MSH_00330b |
MS53_0024 | VY93_00145 |
| Translation elongation factor G | MSH_00500 | MS53_0039b | VY93_00220b |
| Multiple sugar ABC transporter, ATP-binding protein | MSH_01200c | MS53_0102 | VY93_00560 |
| Hypothetical protein | MSH_01840c | – | VY93_00865 |
| Hypothetical protein | MSH_01960 | – | VY93_00910 |
| Hypothetical protein | MSH_01930 MSH_01970 |
– | VY93_00915 |
| Type II restriction enzyme MjaIII | MSH_02140 | – | VY93_01010 VY93_02580 |
| Methyl-directed repair DNA adenine methylase | MSH_02150 | – | VY93_01015 VY93_02585 |
| 3′-to-5′ oligoribonuclease A | MSH_02940b MSH_02950b MSH_02960b |
MS53_0278b MS53_0279b |
VY93_01505b VY93_01510b |
| Aspartate-ammonia ligase | MSH_02990 | MS53_0281b | VY93_01515 |
| Hypothetical protein | MSH_03480b MSH_03490b |
MS53_0293 | VY93_01595b |
| Asparaginyl-tRNA synthetase-related protein | MSH_03500 | MS53_0291b MS53_0700b |
VY93_01590b |
| Deoxyribose-phosphate aldolase | MSH_03520 | – | – |
| Oligopeptide transport ATP-binding protein OppF | MSH_03740b MSH_03750b |
MS53_0345 | VY93_01875 |
| Lipoprotein | MSH_03790 | MS53_0349 | VY93_01895b VY93_01900b |
| Hypothetical protein | MSH_04620 | MS53_0430b | VY93_02305 |
| Hypothetical protein | MSH_04790b | MS53_0442 | |
| Hypothetical protein | MSH_04880 | VY93_02420 | |
| Hypothetical protein | MSH_04960 | MS53_0458b MS53_0459b |
VY93_02465 |
| Hypothetical protein | MSH_05850b MSH_05860b |
MS53_0713b MS53_0540b |
VY93_02935b |
| CRISPR-associated protein, Csn1 | MSH_06430b MSH_06440b |
MS53_0582 | VY93_03200b VY93_03205b |
| IS30 family transposase | MSH_07180 MSH_07210 |
– | VY93_00200 VY93_00900 VY93_01540 VY93_02375 |
| ATP-dependent DNA helicase | MSH_07190 | – | – |
| Hypothetical protein | MSH_07360b | MS53_0668 | VY93_03635 |
| Hypothetical protein | MSH_07370b | MS53_0669b | VY93_03640 |
| ABC transporter ATP-binding and permease protein | MSH_07380 | MS53_0670b | VY93_03645b |
| Hypothetical protein | MSH_07610c | MS53_0680 | VY93_03760b |
| PTS system enzyme IIB component | MSH_07640b | MS53_0682 | VY93_03770 |
| Ascorbate-specific PTS system, EIIC component | MSH_07650b MSH_07660b MSH_07670b MSH_07680b |
MS53_0683 | VY93_03775b |
| Type III restriction-modification system methylation subunit | MSH_07490b MSH_07500b |
– | VY93_03700b VY93_03705b |
| Hypothetical protein | MSH_07510 | – | VY93_03710b |
| Hypothetical protein | MSH_07520 | – | VY93_03720b |
| Hypothetical protein | MSH_07530 | – | VY93_03725b |
| Hypothetical protein | MSH_07540 b | – | VY93_03730 |
| Integrase | MSH_07560 | – | VY93_03735 |
| Type I restriction-modification system, specificity subunit S | MSH_07570b MSH_07580b |
– | VY93_03740b VY93_03745b |
| Type I restriction-modification system, restriction subunit R | MSH_07590 | – | VY93_03750 |
| Type I restriction-modification system, DNA-methyltransferase subunit M | MSH_07600 | – | VY93_03755 |
aPseudogenes that have lost more than 30% of their full length; nucleotide positions corresponding to the locus tags of MS-H are listed in Additional file 1: Table S1. IS elements and the vlhA locus were not included in pseudogenisation analysis. bPseudogenes that result from a frameshift mutation. c Pseudogenes that result from acquisition of an internal stop codon. “−” indicates the absence of the gene in a strain. Genes without a specified locus tag are not indicated in this table
Fig. 3.
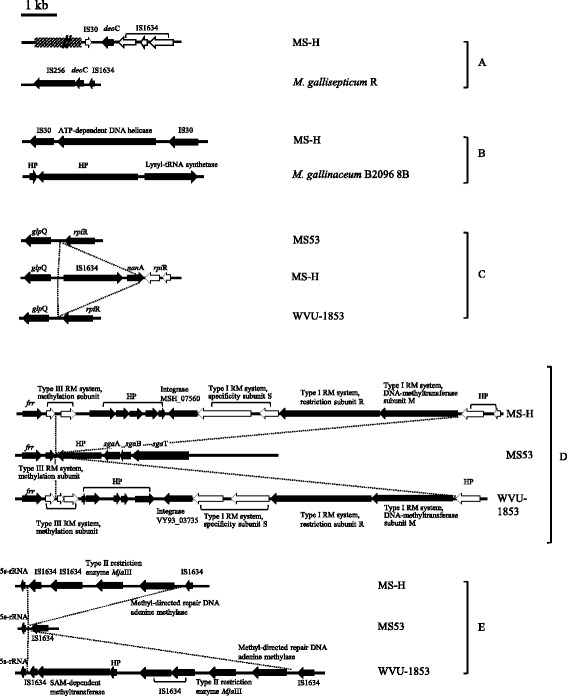
a Location of the deoC gene in M. gallisepticum corresponding to the deoC fragment found in MS-H. b Locations of the ATP-dependent DNA helicase genes in MS-H and M. gallinaceum strain B2096 8B. c, d, e Comparison of unique genomic loci identified in Mycoplasma synoviae strains MS-H, WVU-1853 and MS53. The inserted/deleted sequences are indicated by dotted lines. Genes with complete predicted CDSs are depicted in black; pseudogenes are depicted in white. HP: hypothetical protein
Pseudogenisation of transport systems and their potential impact on attenuation of virulence
Excluding the IS elements and the vlhA locus, a total of 27 pseudogenes were identified in MS-H, WVU-1853 and MS53, with some differences in pseudogene repertoires between strains (Table 3). MS-H, WVU-1853 and MS53 had 17, 15 and 9 pseudogenes, respectively. Of these pseudogenes, 8 were unique to MS-H, 4 to WVU-1853 and 3 to MS53. Only 3 of the 27 were caused by a point mutation creating a TAA stop codon within the same frame. The majority of the pseudogenes were the result of single nucleotide insertions or deletions resulting in frameshifts. Overall, the numbers of pseudogenes were relatively low and similar across all strains, albeit slightly higher in MS-H. Active pseudogenisation in a live vaccine could lower its immunogenic capacity and efficacy by disrupting the expression of protective antigens. Based on our analysis, pseudogenisation does not seem to be significantly more prevalent in MS-H, and is unlikely to affect its repertoire of expressed antigens. The potential impact of pseudogenisation on the attenuation of virulence in MS-H was also considered. A remarkable example of pseudogenisation was found in the oligopeptide permease (Opp) system. Multiple copies of opp operons are commonly seen in both Gram-negative and Gram-positive bacteria, including M. gallisepticum, and can play a role in virulence [33–36]. The Opp systems comprise an extracellular substrate binding protein OppA, two transmembrane proteins OppB and OppC, which form the pore, and two cytoplasmic ATPases OppD and OppF, which provide the energy for peptide translocation [37, 38]. As in most mycoplasmas, all 3 M. synoviae strains possessed two Opp systems, hereafter named opp-I and opp-II. The phylogenic relations between opp operons within the same species are complex. It has been suggested that in the Hominis group the opp operons underwent duplication and divergence. In M. gallisepticum, one of the opp operons was proposed to be horizontally acquired from a member of the Hominis group [36]. The correct annotation of oppA sequences in Mycoplasma spp. genomes is problematic because the gene is often identified only as a lipoprotein [39]. In this study, we found a putative oppA gene in the MS-H opp-I cluster, based on the 28% similarity of the product to M. canadense oppA. Unlike M. gallisepticum, where the two operons are found in tandem, the M. synoviae opp-I and opp-II systems were distant from each other within the chromosome. Moreover, the order of the opp genes differed between the two operons in MS-H and WVU-1853, as has been previously noted for MS53 [39]. Homologous proteins encoded by the two gene clusters share low sequence similarity, possibly indicating that they are involved in the acquisition of different nutrients. The M. synoviae opp-I operon (Fig. 4a) is similar to the opp-1 subtype of Staphylococcus aureus [40] but contains 2 pseudogenes, oppF in MS-H and oppA in WVU-1853. In WVU-1853, a premature stop codon in oppA was close to 3′ end of the gene while the frameshift mutation in MS-H oppF allows only the translation of 157 of 797 amino acids, most likely resulting in loss of function of the protein. In contrast, the opp-II operon (Fig. 4b) is intact and identically organised in all three strains, but differs from other avian mycoplasma species with respect to its gene arrangement [39]. Co-existing opp systems with different, but partially redundant, substrate specificity have been described in Bacillus subtilis, in which the inactivation of one system may be compensated for by the presence of an active second operon [41, 42]. It is tempting to speculate that in WVU-1853 the oppA pseudogene from opp-I is at least partially complemented by the full oppA copy from the opp-II operon, while in MS-H the pseudogenisation of oppF in opp-I is not compensated by the opp-II copy, which has resulted in attenuation of virulence. Experimental verification of this hypothesis is required to establish whether the two OppA transporters can co-operate or display broad substrate specificity, whereas OppF and other components of the Opp system have more exacting interactions in M. synoviae. Alternatively, as has been hypothesized for OppD1 in M. gallisepticum [36], it is possible that the two OppF proteins form dimers in the transport complex and the product of the partial copy of oppF-I in MS-H may retain the capacity to form a heterodimer with the product of full-length copy of oppF-II, resulting in a functional reduction thus a decrease in virulence.
Fig. 4.
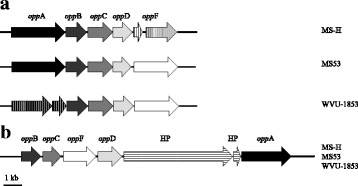
Organisation of the opp operons in MS-H, MS53 and WVU-1853. a Comparison of operon I in strains MS-H, MS53 and WVU-1853; (b) organisation of operon II in all three strains. Solid arrows indicate functional genes, while vertically hatched arrows indicate pseudogenes. Horizontally hatched arrows indicate hypothetical proteins (HP)
Other transport systems operons were also found to contain pseudogenes. Advanced pseudogenisation was noted for the sgaABT gene cluster, which is predicted to encode an ascorbate-specific PTS and which is located next to a strain-variable region containing Type I and Type III RM systems, as well as an integrase gene (see Fig. 3d). Sequences encoding the subunits EIIC (sgaT-2), IIB (sgaB) and IIA (sgaA) appeared to be apparently intact in MS53, but various degrees of pseudogenisation were observed in the 2 other strains. In MS-H and WVU-1853, sgaT-2 was split into four fragments. In MS-H, sgaB contained a frameshift very close to the start codon, but this gene was intact in MS53 and WVU-1853. The integrity of the PTS transporter locus in MS53 might be correlated with the absence of a RM system and associated sequences in its vicinity (see Fig. 3d). As with the opp-I and opp-II operons, a second ascorbate-specific PTS locus containing intact sequences for the transport subunits IIA, IIB and EIIC was identified in the genome of all 3 M. synoviae strains. This second locus may compensate for the pseudogenisation seen in MS-H and WVU-1853. Multiple copies of the ascorbate-specific PTS are often seen in bacteria [43], and are involved in acquisition of L-ascorbate by the organism under anaerobic conditions [44, 45]. However, orphan IIA and/or IIB and/or IIC homologues of these systems are also often found, and may be residues of genomic minimisation [46]. In MS-H, the rpiR-like transcriptional regulator gene, lying downstream of the nanA variant and the IS1634, was also split into two pseudogenes, but was intact in MS53 and WVU-1853 (Fig. 3c). It is known that NanA is involved in the breakdown and utilisation of sialic acid [47], and RpiR belongs to a family of transcriptional regulators, some of which are able to repress or activate the expression of nan genes [48–51]. As transcription regulatory systems in mycoplasmas are not well understood, it is not possible to predict whether the pseudogenisation of rpiR gene might modify sialic acid metabolism in M. synoviae. In addition, restriction modification sequences are often present as pseudogenes in bacterial genomes. In MS-H, these pseudogenes affect a type I restriction-modification specificity subunit S and a type III restriction-modification system methylation subunit.
Clustered regularly interspersed palindromic repeats and evidence of horizontal gene transfer
All 3 M. synoviae strains harbor a typical CRISPR-associated Cas system, with csn1 sequentially followed by cas1, cas2 and a putative csn2 in a contiguous operon (Fig. 5). Homologs of these proteins are also encoded in the genomes of other Mycoplasma species, including M. ovipneumoniae, M. arthritidis, M. hyosynoviae and M. gallisepticum [52, 53]. The operon structure was consistent with a type II (Nmeni subtype) CRISPR/Cas system [52, 54, 55]. However, MS53 was the only M. synoviae strain found to possess a CRISPR array, formed by 11 spacers separated by 36 -bp repeat units, suggesting a record of past foreign DNA invasions in this strain. Small duplicated fragments of csn1 and cas1 were positioned downstream of the CRISPR array (Fig. 5). Among the CRISPR- associated genes, csn1 appeared intact in MS53, but was present as a pseudogene in WVU-1853 and MS-H. In WVU-1853, csn1 was disrupted by a single nucleotide deletion resulting in a frameshift [11]. In MS-H, csn1 was disrupted by an 11 nucleotide insertion. In both strains these mutations were positioned within the 5′ region of the coding sequence and were therefore likely to result in the loss of CRISPR function, as has been observed previously in Streptococcus thermophilus [56]. An IS element was found downstream of the CRISPR -associated genes in all three strains. Genome alignments of all 3 M. synoviae strains with M. gallisepticum, another mycoplasma species infecting chickens, detected 11 regions sharing similarity of more than 90%, suggesting that some horizontal gene transfer has occurred between these two species. The majority of these regions have been previously reported as putatively transferred [10], and notably include the vlhA haemagglutinin genes [57, 58]. In addition, the deoxyribose-phosphate aldolase (deoC) sequence fragment (MSH_03520), which was located upstream of an IS1634 in MS-H (Fig. 3a) and was not found in WVU-1853 and MS53, was very similar to a sequence in Mycoplasma gallisepticum strain R, sharing 94% amino acid similarity with it. In Mycoplasma gallisepticum strain R this sequence is also flanked by IS-related sequences. Moreover, while MS53 and WVU-1853 have only one ATP-binding helicase CDS, a second helicase gene was found in MS-H (MSH_07190) (Table 3). This second copy, flanked by two IS30 family transposase genes (Fig. 3b), is also detected in other Mycoplasma species, including M. gallinaceum strain B2096-8B, with which it had 93% amino acid similarity. M. gallinaceum is a mycoplasma of low pathogenicity that shares the same habitat [59, 60] and phylogenic group [61] as M. synoviae. Additionally, two siderophore-mediated iron transport proteins were also identified as putatively horizontally transferred in a single M. synoviae genome region (536617–551,599 in MS-H). Siderophores are specific Fe (III)-binding agents produced by many microorganisms that mediate iron scavenging from the environment of the host [62]. Siderophore-mediated iron transport system related genes are considered as virulence factors for many bacterial pathogens because of their critical role in adaption to iron-limited conditions within the host [63, 64]. Although the iron-acquisition mechanisms in mycoplasmas remain largely unknown [65], this putative siderophore-associated iron transport system raises the possibilities that siderophores may contribute to the pathogenesis of infection with M. synoviae.
Fig. 5.
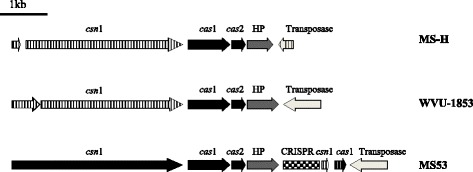
Organisation of the CRISPR/Cas system genes in the 3 M. synoviae genomes. Solid arrows represent genes. Vertically hatched arrows represent pseudogenes. The checked rectangle indicates the CRISPR repeats
Conclusion
The MS-H live vaccine has been used to prevent infection with virulent M. synoviae in poultry industry for more than two decades and is now used in many countries around the world. As a result it is possible that MS-H is the most prevalent strain of M. synoviae globally. Comparative genome analyses revealed a number of features unique to MS-H, WVU-1853 or MS53, but the genomes of these three strains were largely similar to each other. In particular, striking similarities were found between strains MS-H and WVU-1853, despite their distinct geographical origins and dates of isolation. This apparently low genome variability within the species supports the evidence from the field that MS-H protects against M. synoviae in a wide range of countries. Differentiation of vaccine strains from field strains is always challenging because of the limited number of genetic markers available for development of routine tests. In this study, we identified genetic features unique to the MS-H vaccine strain, that may be able to be targeted in the future for diagnostic purpose. Strain-variable sequences, putatively acquired by horizontal transfer, and pseudogenes play a critical role in the genomic plasticity of M. synoviae. This is exemplified by the discovery of a large inversion in the vaccine strain MS-H adjacent to a short region potentially transferred from M. gallisepticum, and an ATP-binding helicase gene putatively acquired from M. gallinaceum. Although the 3 M. synoviae strains had similar numbers of pseudogenes, the slightly more advanced pseudogenisation of MS-H could be explained by the chemical mutagenesis used to produce the vaccine. It is not yet clear whether the pseudogenisation of some of these genes is associated with attenuation of virulence that is characteristic of the MS-H vaccine. The complete genomic sequencing of MS-H is the first step towards a thorough comparison with its parental strain 86,079/7NS, currently underway in our laboratory, that will help address this question.
Additional files
Composition of the inverted region in MS-H. (DOCX 23 kb)
Nucleotide positions of the unique gene locus tags in MS-H genome. (DOCX 21 kb)
Acknowledgements
The authors would like to acknowledge the assistance from the staff of Asia-Pacific Centre for Animal Health (APCAH), Faculty of Veterinary and Agricultural Sciences, The University of Melbourne.
Funding
Funding for this study was provided by APCAH.
Availability of data and materials
The whole-genome sequence of MS-H has been deposited in GenBank under accession number CP021129. The genome sequences of MS53 and WVU-1853 were retrieved from GenBank (accession numbers AE017245 and CP011096).
Abbreviations
- CRISPR
Clustered Regularly Interspersed Short Palindromic Repeats
- DeoC
Deoxyribose-phosphate aldolase
- GI
Genomic Island CDS: Coding Sequence
- IS
Insertion Sequences
- LicA
Lichenan-specific IIA component
- NanA
Acetylneuraminate lyase
- NCBI
National Centre for Biotechnology Information
- NGS
Next Generation Sequencing
- ORF
Open Reading Frame
- PHAST
PHAge Search Tool
- PTS
Phosphotransferase system
- RM system
Restriction Modification system
- Opp
Oligopeptide permease
Authors’ contributions
Conception and design: LZ, MAS, AHN, MSM; Acquisition of data: LZ, MAS, JM, MSM; Analysis and interpretation of data: LZ, MAS, MSM, AHN, GFB; Manuscript: LZ, MSM, AHN, GFB, MAS. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4501-8) contains supplementary material, which is available to authorized users.
Contributor Information
Ling Zhu, Email: ling.zhu@unimelb.edu.au.
Muhammad A. Shahid, Email: drakbarshahid@gmail.com
John Markham, Email: jmarkham@unimelb.edu.au.
Glenn F. Browning, Email: glenfb@unimelb.edu.au
Amir H. Noormohammadi, Email: amirh@unimelb.edu.au
Marc S. Marenda, Email: mmarenda@unimelb.edu.au
References
- 1.Kleven SH. Mycoplasmas in the etiology of multifactorial respiratory disease. Poult Sci. 1998;77:1146–1149. doi: 10.1093/ps/77.8.1146. [DOI] [PubMed] [Google Scholar]
- 2.Lockaby SB, Hoerr FJ, Lauerman LH, Kleven SH. Pathogenicity of Mycoplasma synoviae in broiler chickens. Vet Pathol. 1998;35:178–190. doi: 10.1177/030098589803500303. [DOI] [PubMed] [Google Scholar]
- 3.Olson NO, Bletner JK, Shelton DC, Munro DA, Anderson GC. Enlarged joint condition in poultry caused by infectious agent. Poult Sci. 1954;33:1075. [Google Scholar]
- 4.Feberwee A, Morrow CJ, Ghorashi SA, Noormohammadi AH, WJM L. Effect of a live Mycoplasma synoviae vaccine on the production of eggshell apex abnormalities induced by a M. Synoviae infection preceded by an infection with infectious bronchitis virus D1466. Avian Pathol. 2009;38:333–340. doi: 10.1080/03079450903183652. [DOI] [PubMed] [Google Scholar]
- 5.WJM L. Is Mycoplasma synoviae outrunning Mycoplasma gallisepticum? A viewpoint from the Netherlands. Avian Pathol. 2014;43:2–8. doi: 10.1080/03079457.2014.881049. [DOI] [PubMed] [Google Scholar]
- 6.Markham JF, Morrow CJ, Whithear KG. Efficacy of a temperature-sensitive Mycoplasma synoviae live vaccine. Avian Dis. 1998;42:671–676. doi: 10.2307/1592701. [DOI] [PubMed] [Google Scholar]
- 7.Markham JF, Scott PC, Whithear KG. Field evaluation of the safety and efficacy of a temperature-sensitive Mycoplasma synoviae live vaccine. Avian Dis. 1998;42:682–689. doi: 10.2307/1592703. [DOI] [PubMed] [Google Scholar]
- 8.Ogino S, Munakata Y, Ohashi S, Fukui M, Sakamoto H, Sekiya Y, Noormohammadi AH, Morrow CJ. Genotyping of Japanese field isolates of Mycoplasma synoviae and rapid molecular differenciation from the MS-H vaccine strain. Avian Dis. 2011;55:187–194. doi: 10.1637/9461-071310-Reg.1. [DOI] [PubMed] [Google Scholar]
- 9.Noormohammadi AH, Jones JF, Harrigan KE, Whithear KG. Evaluation of the non-temperature-sensitive field clonal isolates of the Mycoplasma synoviae vaccine strain MS-H. Avian Dis. 2003;47:355–360. doi: 10.1637/0005-2086(2003)047[0355:EOTNFC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 10.Vasconcelos ATR, Ferreira HB, Bizarro CV, Bonatto SL, Carvalho MO, Pinto PM, Almeida DF, Almeida LG, Almeida R, Alves-Filho L, Assunção EN, et al. Swine and poultry pathogens: the complete genome sequences of two strains of Mycoplasma hyopneumoniae and a strain of Mycoplasma synoviae. J Bacteriol. 2005;187:5568–5577. doi: 10.1128/JB.187.16.5568-5577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.May MA, Kutish GF, Barbet AF, Michaels DL, Brown DR. Complete genome sequence of Mycoplasma synoviae strain WVU 1853T. Genome Announc. 2015;3:e00563–e00515. doi: 10.1128/genomeA.00563-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whithear KG. Avian mycoplasmosis. In: Corner LA, Bagust TJ, editors. Australian standard diagnostic techniques for animal diseases. East Melbourne: CSIRO for the Standing Committee on Agriculture and Resource Management; 1993. pp. 1–12. [Google Scholar]
- 13.Green MR, Sambrook J. Isolation of high-molecular-weight DNA using organic solvents to purify DNA. Cold Spring Harb Protoc. 2017; 10.1101/pdb.prot093450. [DOI] [PubMed]
- 14.Green MR, Sambrook J. Precipitation of DNA with ethanol. Cold Spring Harb Protoc. 2016; 10.1101/pdb.prot093377. [DOI] [PubMed]
- 15.Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010. [Google Scholar]
- 16.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loman NJ, Quinian AR. Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics. 2014;30:3399–3401. doi: 10.1093/bioinformatics/btu555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berlin K, Koren S, Chin CS, Drake PJ, Landolin JM, Phillippy AM. Assembled large genomes with single-molecule sequencing and locality-sensitive hashing. Nat Biotechnol. 2015;33:623–630. doi: 10.1038/nbt.3238. [DOI] [PubMed] [Google Scholar]
- 19.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 21.Langille M, Brinkman F. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics. 2009;25:664–665. doi: 10.1093/bioinformatics/btp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucl Acids Res. 2011;39:1–6. doi: 10.1093/nar/gkr595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucl Acids Res. 2007;35:53–57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucl Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shahid MA, Markham PF, Markham JF, Marenda MS, Noormohammadi AH. Mutations in GTP binding protein Obg of Mycoplasma synoviae vaccine strain MS-H: implications in temperature-sensitivity phenotype. PLoS One. 2013;8:e73954. doi: 10.1371/journal.pone.0073954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Zheng H, Liu Y, Jiang Y, Xin J, Chen W, Song Z. The complete genome sequence of Mycoplasma bovis strain Hubei-1. PLoS One. 2011;6:e20999. doi: 10.1371/journal.pone.0020999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suyama M, Bork P. Evolution of prokaryotic gene order: genome rearrangements in closely related species. Trends Genet. 2001;17:10–13. doi: 10.1016/S0168-9525(00)02159-4. [DOI] [PubMed] [Google Scholar]
- 28.Ohtsubo F, Sekine Y. Bacterial insertion sequences. Curr Top Microbiol Immunol. 1996;204:1–26. doi: 10.1007/978-3-642-79795-8_1. [DOI] [PubMed] [Google Scholar]
- 29.Rocha EP, Blanchard A. Genomic repeats, genome plasticity and the dynamics of Mycoplasma evolution. Nucl Acids Res. 2002;30:2031–2042. doi: 10.1093/nar/30.9.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.May M, Kleven SH, Brown DR. Sialidase activity in Mycoplasma synoviae. Avian Dis. 2007;51:829–833. doi: 10.1637/7806-120106-REGR.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.May M, Brown DR. Genetic variation in sialidase and linkage to N-acetylneuraminate catabolism in Mycoplasma synoviae. Microb Pathog. 2008;45:38–44. doi: 10.1016/j.micpath.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’ Neill M, Chen A, Murray N. The restriction-modification genes of Escherichia coli K-12 may not be selfish: they do not resist loss and are readily replaced by alleles conferring different specificities. Proc Natl Acad Sci. 1997;94:14596–14601. doi: 10.1073/pnas.94.26.14596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eitinger T, Rodionov DA, Grote M, Schneider E. Canonical and ECF-type ATP-binding cassette importers in prokaryotes: diversity in modular organization and cellular functions. FEMS Microbiol Rev. 2011;35:3–67. doi: 10.1111/j.1574-6976.2010.00230.x. [DOI] [PubMed] [Google Scholar]
- 34.Jones MM, Johnson A, Koszelak-Rosenblum M, Kirkham C, Brauer AL, Malkowski MG, Murphy TF. Role of the oligopeptide permease ABC transporter of Moraxella catarrhalis in nutrient acquisition and persistence in the respiratory tract. Infect Immun. 2014;82:4758–4766. doi: 10.1128/IAI.02185-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moraes PM, Seyffert N, Silva WM, Castro TL, Silva RF, Lima DD, Hirata R, Silva A, Miyoshi A, Azevedo V. Characterization of the Opp peptide transporter of Corynebacterium pseudotuberculosis and its role in virulence and pathogenicity. Biomed Res Int. 2014;2014:7. doi: 10.1155/2014/489782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tseng CW, Chiu CJ, Kanci A, Citti C, Rosengarten R, Browning GF, Markham PF. The oppD gene and putative peptidase genes may be required for virulence in Mycoplasma gallisepticum. Infect Immun. 2017;85:e00023–e00017. doi: 10.1128/IAI.00023-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monnet V. Bacterial oligopeptide-binding proteins. Cell Mol Life Sci. 2003;60:2100–2114. doi: 10.1007/s00018-003-3054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardan R, Besset C, Guillot A, Gitton C, Monnet V. The oligopeptide transport system is essential for the development of natural competence in Streptococcus thermophilus strain LMD-9. J Bacteriol. 2009;19:4647–4655. doi: 10.1128/JB.00257-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wium M, Botes A, Bellstedt DU. The identification of oppA gene homologues as part of the oligopeptide transport system in mycoplasmas. Gene. 2015;558:31–40. doi: 10.1016/j.gene.2014.12.036. [DOI] [PubMed] [Google Scholar]
- 40.Yu D, Pi B, Yu M, Wang Y, Ruan Z, Feng Y, Yu Y. Diversity and evolution of oligopeptide permease systems in staphylococcal species. Genomics. 2014;104:8–13. doi: 10.1016/j.ygeno.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 41.Koide A, Perego M, Hoch JA. ScoC regulates peptide transport and sporulation initiation in Bacillus Subtilis. J Bacteriol. 1999;181:4114–4117. doi: 10.1128/jb.181.13.4114-4117.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koide A, Hoch JA. Identification of a second oligopeptide transport system in Bacillus Subtilis and determination of its role in sporulation. Mol Microbiol. 1994;13:417–426. doi: 10.1111/j.1365-2958.1994.tb00436.x. [DOI] [PubMed] [Google Scholar]
- 43.Barabote RD, Saier MH. Comparative genomic analyses of the bacterial phosphotransferase system. Microbiol Mol Biol Rev. 2005;69:608–634. doi: 10.1128/MMBR.69.4.608-634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Z, Aboulwafa M, Smith MH, Saier Jr MH. The ascorbate transporter of Escherichia coli. J Bacteriol. 2003;185:2243–2250. doi: 10.1128/JB.185.7.2243-2250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hvorup R, Chang AB, Saier MH., Jr Bioinformatic analyses of the bacterial L-ascorbate phosphotransferase system permease family. J Mol Microbiol Biotechnol. 2003;6:191–205. doi: 10.1159/000077250. [DOI] [PubMed] [Google Scholar]
- 46.Moran NA. Microbial minimalism: genome reduction in bacterial pathogens. Cell. 2002;108:583–586. doi: 10.1016/S0092-8674(02)00665-7. [DOI] [PubMed] [Google Scholar]
- 47.Walters DM, Stirewalt VL, Melville SB. Cloning, sequence, and transcriptional regulation of the operon encoding a putative N-acetylmannosamine-6-phosphate epimerase (nanE) and sialic acid lyase (nanA) in Clostridium perfringens. J Bacteriol. 1999;181:4526–4532. doi: 10.1128/jb.181.15.4526-4532.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnston JW, Zaleski A, Allen S, Mootz JM, Armbruster D, Gibson BW, Apicella MA, Munson RS. Regulation of sialic acid transport and catabolism in Haemophilus influenzae. Mol Microbiol. 2007;66:26–39. doi: 10.1111/j.1365-2958.2007.05890.x. [DOI] [PubMed] [Google Scholar]
- 49.Kim BS, Hwang J, Kim MH, Choi SH. Cooperative regulation of the Vibrio Vulnificus nan gene cluster by NanR protein, cAMP receptor protein, and N-acetylmannosamine 6-phosphate. J Biol Chem. 2011;286:40889–40899. doi: 10.1074/jbc.M111.300988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Afzal M, Shafeeq S, Ahmed H, Kuipers OP. Sialic acid-mediated gene expression in Streptococcus Pneumoniae and role of NanR as a transcriptional activator of the nan gene cluster. Appl Environ Microbiol. 2015;81:3121–3131. doi: 10.1128/AEM.00499-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Therit B, Cheung JK, Rood JI, Melville SB. NanR, a transcriptional regulator that binds to the promoters of genes involved in sialic acid metabolism in the anaerobic pathogen Clostridium perfringens. PLoS One. 2015;10:e0133217. doi: 10.1371/journal.pone.0133217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bumgardner EA, Kittichotichotirat W, Bumgarner R, Lawrence PK. Comparative genomic analysis of seven Mycoplasma hyosynoviae strains. Microbiology. 2015;4:343–359. doi: 10.1002/mbo3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leclerq S, Dittermer J, Bouchon O, Cordaux R. Phylogenomics of “Candidatus Hepatoplasma crinochetorum,” a lineage of mollicutes associated with noninsect arthropods. Genome Biol Evol. 2014;6:407–415. doi: 10.1093/gbe/evu020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haft DH, Selengut J, Mongodin EF, Nelson KE. A guide of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, Van Der Oost J, Koonin EV. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. Phage response to CRISPR-encoded resistance in Streptococcus Thermophilus. J Bacteriol. 2008;190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Noormohammadi AH, Markham PF, Duffy MF, Whithear KG, Browning GF. Multigene families encoding the major hemagglutinins in phylogenetically distinct mycoplasmas. Infect Immun. 1998;66:3470–3475. doi: 10.1128/iai.66.7.3470-3475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benčina D. Haemagglutinins of pathogenic avain mycoplasmas. Avian Pathol. 2002;31:535–547. doi: 10.1080/0307945021000024526. [DOI] [PubMed] [Google Scholar]
- 59.Jordan FTW, Ernø H, Cottew GS, Hinz KH, Stipkovits L. Characterization and taxonomic description of five mycoplasma serovars (serotypes) of avian origin and their elevation to species rank and further evaluation of the taxonomic status of Mycoplasrna synoviae. Int J Syst Evol Microbiol. 1982;32:108–115. [Google Scholar]
- 60.Abolnik C, Beylefeld A. Complete genome sequence of Mycoplasma gallinaceum. Genome Announc. 2015;3:e00712–e00715. doi: 10.1128/genomeA.00712-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramírez AS, Naylor CJ, Pitcher DG, Bradbury JM. High inter-species and low intra-species variation in 16S–23S rDNA spacer sequences of pathogenic avian mycoplasmas offers potential use as a diagnostic tool. Vet Microbiol. 2008;128:279–287. doi: 10.1016/j.vetmic.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 62.Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu Rev Microbiol. 2000;54:881–941. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- 63.Richardson PT, Park SF. Enterochelin acquisition in campylobacter coli: characterizationof components of a binding-protein-dependenttransport system. Microbiology. 1995;141:3181–3191. doi: 10.1099/13500872-141-12-3181. [DOI] [PubMed] [Google Scholar]
- 64.Payne SM, Lawlor KM. Molecular studies on iron acquisition by non-Escherichia coli species. In: Iglewski BH, Clark VL, editors. The molecular basis of bacterial pathogenesis. New York: Academic Press; 1990. pp. 225–248. [Google Scholar]
- 65.Madsen ML, Nettleton D, Thacker EL, Minion FC. Transcriptional profiling of Mycoplasma hyopneumoniae during iron depletion using microarrays. Microbiology. 2006;152:937–944. doi: 10.1099/mic.0.28674-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Composition of the inverted region in MS-H. (DOCX 23 kb)
Nucleotide positions of the unique gene locus tags in MS-H genome. (DOCX 21 kb)
Data Availability Statement
The whole-genome sequence of MS-H has been deposited in GenBank under accession number CP021129. The genome sequences of MS53 and WVU-1853 were retrieved from GenBank (accession numbers AE017245 and CP011096).