

Abstract
Genetic composition and major histocompatibility complex polymorphisms unequivocally predispose to autoimmune disease, but environmental factors also play a critical role in precipitating disease in susceptible individuals. Notorious among these has been microbial infection. Older studies describing associations between microbial infection and autoimmune disease are now followed by new studies demonstrating correlations between susceptibility to autoimmune disease and commensal colonization of the intestinal tract. T helper 17 (TH17) cells have gained a prominent role in autoimmune disease, and notably, their development within the intestine has been linked to colonization with specific commensal bacteria. Here, we consider current views on how microbes, TH17 cells, and autoimmunity are connected. We speculate on how the intricate relationships among commensal, pathogen, and the host might ultimately determine susceptibility to autoimmune disease.
Keywords: Autoimmune disease, Multiple sclerosis, Type 1 diabetes mellitus, Rheumatoid arthritis, Inflammatory bowel disease, Pathogen, Commensal, Host-pathogen interaction, Host-commensal interaction, T helper 17, Autoreactivity, Autoimmunity, Central tolerance, Peripheral tolerance, Infection, Antigen presentation, Major histocompatibility complex, MHC, T cell, CD4 T cell, B cell, Lymphocyte, Germ free, Specific pathogen free, Toll-like receptor, Innate immunity, Nod-like receptor
Introduction
Genetic polymorphisms in the major histocompatibility complex (MHC) constitute the strongest genetic association with the major human autoimmune diseases: multiple sclerosis (MS), type 1 diabetes mellitus (T1D), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and inflammatory bowel diseases (IBD) (reviewed in [1]). Genome-wide association studies, where variations in hundreds of thousands of genes are concomitantly revealed, have tied more than 200 genetic loci with one or more autoimmune disease [2]. These studies have implicated a critical role for discrete functional pathways in innate immunity and host defense, lymphocyte activation and differentiation, and cytokine/cytokine receptor signaling [2]. While high concordance rates among monozygotic twins support the predominant involvement of genetic factors, disease discordance has also been observed, instigating the search for environmental triggers of autoimmune disease [3, 4]. Popular contenders are microbes, both viruses and bacteria, as strong epidemiological links are present between infections with certain pathogens and the development of autoimmune disease in genetically susceptible individuals [5]. Here, we examine the associations of microbial pathogens and commensal microorganisms with different autoimmune diseases focusing primarily on the role of T cells, particularly TH17 cells.
Avoiding and triggering autoimmunity
Central tolerance
The first step in prevention of autoimmunity is the elimination of self-reactive T cells during thymic development in a process called central tolerance. Its aim is to achieve a T cell repertoire that is tolerant to self-antigens (reviewed in [6]). Immature lymphocytes arriving in the cortex of the thymus from the bone marrow initiate expression of their T cell receptor (TCR) via assembly of three separate gene segments, the variable (V), diversity (D), and joining (J) genes, in a process called V(D)J recombination. New TCRs are then tested for reactivity with MHC molecules bearing peptides derived from various self-proteins (pMHC complexes) displayed on the surface of cortical thymic epithelial cells. The first goal is to achieve MHC restriction, that is, the ability to bind pMHC with a minimal threshold of reactivity that signals survival and thus positive selection. To survive, a minority of thymocytes undergo TCRα chain editing by V(D)J recombination replacing their TCRα chain with one that might deliver just the right signals for positive selection. These signals also entail shut down of V(D)J recombination, TCR downregulation, and chemokine receptor upregulation to guide thymocytes toward the thymic medulla where another round of testing eliminates, via negative selection, those thymocytes bearing TCRs with high avidity to self-pMHC complexes, which are now displayed on the surfaces of medullary thymic epithelial cells (MTECs) and dendritic cells (DCs). Throughout these processes, the threshold of TCR signaling critically determines thymocyte survival such that mutations in the signal transducing ζ-chain (TCR)-associated protein kinase of 70 kDa (ZAP-70), which reduce accurate perception of this threshold, result in impaired positive and negative selection of autoreactive thymocytes causing autoimmune arthritis in mice [7].
A fascinating aspect of negative selection relates to how self-pMHC complexes in the thymus, upon which thymocyte TCR avidity is tested, could possibly represent the variety of self-proteins that a T cell might encounter in tissues, such as the pancreas, liver, or intestine, once it exits to the periphery. Here, the autoimmune regulator (AIRE) plays a critical role (reviewed in [8]). AIRE is a transcription factor predominantly expressed by MTECs and functions in mediating ectopic expression of peripheral tissue restricted proteins within these cells by releasing stalled RNA polymerases [9]. MTECs isolated from Aire−/− mice show lost or reduced expression of an estimated 1,200 genes. These include pancreatic preproinsulin II, salivary gland proteins 1 and 2, liver α-1-microglobulin/bikunin precursor, Paneth cell cryptidin-related sequence 2, and multiple tissue-specific proteins such as cytochrome P450, lactotransferrin, prostaglandin D, and insulin-like growth factor II. But even with AIRE, a number of genes, like the pancreatic protein GAD65 encoding gene, are not expressed in the thymus, or expressed at lower levels that preclude their detection. Interestingly, proteins encoded by these genes are often targets for autoimmunity.
An immune function for AIRE was first recognized when genetic AIRE mutations were linked to a rare and severe autosomal recessive autoimmune disease, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), also known as autoimmune polyendocrine syndrome type I (APS-1). More than 60 mutations of AIRE have now been identified, and phenotype varies depending on the mutation. Diagnosis relies on presentation of at least two of the most common manifestations: Addison disease, hypoparathyroidism, and chronic cutaneous candidiasis, while other disease components can also occur such as diabetes mellitus, autoimmune hepatitis, chronic diarrhea, type A autoimmune gastritis, keratitis, alopecia, and vitiligo [10]. Besides the presence of autoantibodies against IFN-α and IFN-ω in the plasma of APS-I patients, these patients also develop autoantibodies to IL-17A, IL-17F, and IL-22 but not to other cytokines like IL-6, IL-23, IL-21, IL-1β, or TGF-β. These autoantibodies (specifically to IL-17A) neutralize TH17-cell function, which predisposes to mucocutaneous candidiasis, a common ailment in APS-1 patients [11]. Despite the importance of TH17 cells in immunity to a number of pathogens in mice [12], APS-I patients are uniquely susceptible to infections with Candida albicans, defense against which requires the TH17 effector cytokine IL-17 in mouse models [13], but surprisingly not to infections with other pathogens.
Peripheral tolerance
A dominant role for central tolerance ensures that all developing lymphocytes bearing TCRs with high affinity to self-pMHC are deleted from the T cell repertoire. However, autoreactive T cells do escape central tolerance [14] and have been detected in the peripheral blood of not only patients with autoimmune disease (multiple sclerosis and type 1 diabetes), but also healthy individuals [15–17]. Self-reactive T cells that escape central tolerance are generally capable of only low-affinity interactions with their cognate ligands, but as mentioned above, there is the potential for T cells bearing high-affinity TCRs, specific to tissue-restricted antigens, to escape central tolerance when this antigen is poorly expressed by MTECs. Therefore, peripheral tolerance is required to prevent activation of such autoreactive T cells [18].
The mechanisms of peripheral tolerance generally fall under several categories including T cell ignorance, where restricted trafficking of naïve T cells between the lymph and blood limits their encounters with self-antigens expressed in peripheral tissues, and T cell anergy, where recognition of cognate self-pMHC at steady state, in the absence of infection or inflammation induced costimulatory molecule expression, aborts proliferation, and prevents sustained clonal expansion. For example, distinct populations of splenic CD8α+ [19] and tissue CD103+ DC (for example, in the lung [20]) are specialized in cross-presentation of apoptotic cell-derived antigens at steady state, thereby enforcing peripheral tolerance of potentially self-reactive CD8+ T cells. Multiple E3 ubiquitin ligases such as Cbl/b and Itch regulate T cell activation by ubiquitinating target proteins downstream of TCR signaling [21], and mice deficient in these molecules are susceptible to autoimmune disease as exemplified by rapid disease development as a result of combined deficiency for AIRE and Cbl-b [22]. Molecules such as CTLA-4 and PD-1, expressed by T cells following their activation and critical for limiting T cell proliferation, have also been recognized as important mediators of peripheral tolerance based on the observations that mice deficient for CTLA-4 or PD-1 (or its two ligands PDL-1 and PDL-2) develop spontaneous autoimmunity [23]. Genome-wide association scans have discovered autoimmunity risk alleles in genes encoding CTLA-4 and PD-1 along with other genes encoding costimulatory molecules such as inducible T cell costimulator (ICOS) ligand, CD58, CD40, CD244, CD226, and tumor necrosis factor superfamily members 4 and 15 (TNFSF4 and TNFSF15, respectively) [24].
T cell deletion can also occur in the periphery where autoreactive T cells die by apoptosis dependent on the pro-survival BCL-2 antagonist BIM and triggered by the death receptor FAS. Faslpr MRL mice express a mutant allele of Fas that fails to transmit a death-inducing signal, and these mice suffer from severe T cell lymphoproliferative and autoimmune disease [25]. Deficiency in pro-apoptotic BIM leads to progressive lymphadenopathy and a systemic autoimmune disease with older mice developing plasmacytosis and autoimmune kidney disease. Many of these symptoms were reproduced by overexpression of BCL-2 [26].
Finally, the most important mechanism of peripheral tolerance is mediated through the function of suppressor FOXP3+ T regulatory (Treg) cells (reviewed in [27, 28]). Originally described merely as CD4+CD25+ T cells, their depletion led to autoimmunity and IBD in otherwise healthy mice, and deletion of FOXP3 in mice or loss of function mutations in FOXP3 (Scurfy mice) leads to fatal autoimmune-like disease. Mutations in human FOXP3 lead to IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome manifesting in aggressive multisystem autoimmunity where presentation with diarrhea, insulin-dependent diabetes mellitus, thyroid disorders, and eczema occurs early in life. These observations demonstrate that FOXP3+ Treg cells function to inhibit autoimmune responses. A large body of work has revealed that FOXP3+ Treg cells mediate their suppressive functions via multiple mechanisms including the production of immunosuppressive cytokines like IL-10, IL-35, and TGF-β, Granzyme- and perforin-dependent cytolysis of effector T cells, as well as the controversial deprivation of IL-2 needed by those cells. Treg cells can also downmodulate antigen-presenting cell (APC) function via interaction of CTLA4 on FOXP3+ Treg cells with CD80/CD86 on APC. This interaction initiates an immunosuppressive program within APC characterized by decreased expression of T cell costimulatory molecules and activation of the indoleamine-2,3-dioxygenase (IDO) and FOXO3 pathways that restrain APC inflammatory cytokine production [29].
Microbial triggers of autoimmunity
Alongside the above-described genetic defects in central or peripheral tolerance mechanisms, which can confer increased susceptibility to autoimmune diseases, autoimmunity has also been epidemiologically linked to infections. Many theories have been proposed to explain how microbes could initiate or predispose to autoimmunity (Fig. 1a), and these include bystander activation of autoreactive T cells by pathogen encoded superantigens or inflammatory cytokines produced by APC responding to infection. The dominant theory is “molecular mimicry”, or more specifically “epitope mimicry”, where self-reactive B or T cells are activated inappropriately upon recognition of peptides derived from pathogens that share sequence or structural homology with peptides derived from self-antigens (reviewed in [30]). However, molecular mimicry, especially in the case of T cell epitope mimicry, has been controversial due to varying views regarding the experimental evidence in animal models, and the lack of strong evidence in human disease [5, 31–34]. The case may be stronger for B cell epitope mimicry with recent evidence in human SLE (more on this below) [35]. More recently, in a dramatically different manner of associating microbes with autoimmune disease, colonization with certain commensals appears to determine susceptibility to autoimmune disease. In the next two sections, we examine some of the associations that have been made between pathogens and autoimmune disease, and how some have been explained by molecular mimicry. We also review newer studies looking at the role of commensals in modulating autoimmune disease. The precise molecular basis underlying these associations, for either pathogen or commensal, remains poorly understood.
Fig. 1.
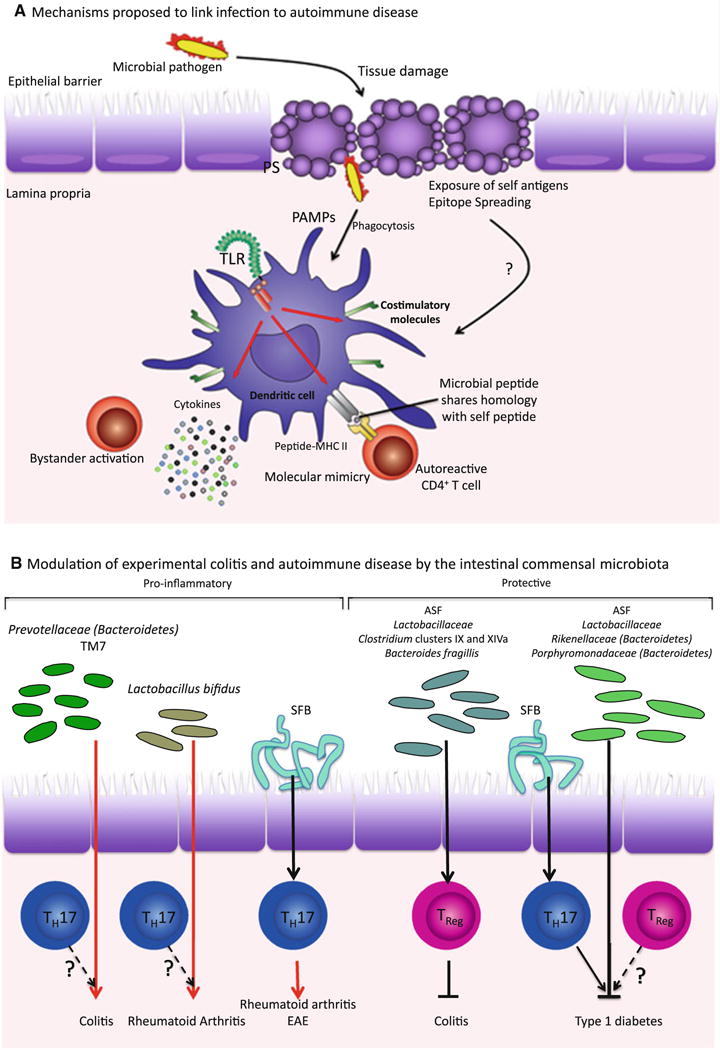
Both pathogenic and commensal microbes can influence development of autoimmunity. a In molecular mimicry, or T cell epitope mimicry, peptides derived from a pathogen share homology with some self-peptides. During infection, autoreactive T cells that have escaped clonal deletion in the thymus, can become activated by antigen-presenting cells (APC) presenting these “mimic” antigens. Pathogens may also contribute to autoimmunity by causing tissue damage and allowing abnormal exposure of self-antigens. Self-antigens presented by APC activated by PAMPs present during infection can then stimulate autoreactive T cells by a process known as epitope spreading. Bystander activation occurs when “bystander” T cells are activated via the pro-inflammatory cytokine milieu surrounding an infection. b In addition to pathogenic bacteria, commensals also influence the development of autoinflammatory conditions, by promoting inflammation in certain cases and preventing it in others. However, the mechanism by which commensals can be pro-inflammatory or protective remains unclear—although TH17 and TReg and the balance in their numbers clearly seem to be involved in autoimmunity, whether protective commensals such as Lactobacillus promote tolerance via Treg activity or otherwise is not known. Some pro-inflammatory commensals such as SFB have been shown to induce TH17 cells, which exacerbate the development of rheumatoid arthritis. However, for the most part, these links and the antigen specificity of the T cells involved, have yet to be fully elucidated. Specific links that have not been tested are indicated with question marks. ASF, Altered Schaedler Flora, SFB, segmented filamentous bacteria
Infections with pathogenic microbes
There are several autoimmune disease states that have been associated with infection with specific pathogens [36, 37]. Perhaps the best known example is Guillain-barré syndrome (GBS) [38]. GBS is characterized by immune mediated destruction of nerves in the peripheral nervous system. Despite recent speculation as to whether GBS should continue to be classified as an autoimmune disease due to its acute onset and transient nature [39], it is thought to occur via an infection which then triggers autoimmune pathology. Two-thirds of GBS patients report a recent gastrointestinal or respiratory infection. While Epstein-Barr virus (EBV), cytomegalovirus (CMV) and Mycoplasma pneumoniae have all been reported to precede GBS, Campylobacter jejuni, a gram-negative enteric bacillus, has emerged as the principal infectious agent implicated in the induction of GBS, although of note only 1/1,000 patients with C. jejuni enteritis present with GBS [38, 40]. The lipopolysaccharides of C. jejuni isolated from patients with GBS were found to have outer core oligosaccharides identical to those of GM1 and GD1a gangliosides expressed by peripheral nerve axons [41]. Immunization of rabbits with the LPS oligosaccharide fraction obtained from a clinical isolate of C. jejuni, containing homology to GM1 or GD1a, led to development of anti-GM1 IgG antibodies along with infiltration of macrophages into the periaxonal space and subsequent flaccid limb paralysis [40]. Anti-GM1 or anti-GD1a specific IgG autoantibodies are seen in GBS patients suggesting an involvement of CD4+ T cell help, likely from CD1-restricted lipid-specific T cells, although no evidence for this is reported [38].
Another frequently cited example of microbial infection leading to autoimmune pathology is infection with the spirochete Borrelia burgdorferi as a result of a deer tick bite, which causes Lyme disease (reviewed in [42]). A chronic inflammatory joint disease, Lyme arthritis, can be a complication of infection with B. burgdorferi when infection is not treated or is refractory to treatment with antibiotics [43]. However, even with successful treatment and subsequent confirmation of the absence of B. burgdorferi DNA in synovial fluid, arthritis persists in a small percentage of patients, pointing to an autoreactive etiology to the inflammation. Patients mount both a T cell and antibody response to the B. burgdorferi outer-surface protein A (OspA), which correlates with the severity of arthritis. Epitope mimicry was invoked with the identification of partial sequence homology to a peptide derived from human lymphocyte function–associated antigen-1 (hLFA-1α), and CD4+ T cells from patients with antibiotic refractory arthritis produced IFN-γ to both OspA and hLFA-1. However, later studies using OspA-specific T cell clones could confirm these results with only 10 % of the clones, and a search for other self-derived epitopes that might cross-react with OspA did not yield results that could be applicable to the large majority of patients. On the other hand, strong evidence demonstrates that HLA-DR4 presentation of OspA165–173 confers increased susceptibility and severity of chronic Lyme arthritis [42].
Another well-known example of a post-infection autoimmune disease is rheumatic fever following pharyngeal infection with β-hemolytic Streptococci. The autoimmune pathology following infection with β-hemolytic Streptococci presents as an acutely febrile illness that can involve multiple organs, including kidneys, joints, and the heart, and can develop even with proper treatment or prophylaxis (reviewed in [31]). Disease pathogenesis is thought to be caused largely by T cells in the myocardium, and a pre-dominance of infiltrating CD4+ T cells are found in rheumatic lesions of affected individuals. Several studies have demonstrated cross-reactivity of these infiltrating T cells with streptococcal M proteins, the major virulence factors of Streptococci, and cardiac myosin, laminin or tropomyosin [31]. Several streptococcal and human cross-reactive antibodies, particularly to cardiac myosin, have also been described in the sera of rheumatic fever patients [44].
A gram-negative bacterium causing chronic infection in gastric mucosa, Helicobacter pylori, has also been associated with some autoimmune conditions, particularly autoimmune gastritis. Autoimmune gastritis in the context of H. pylori infection is thought to be T cell mediated via molecular mimicry [45], and described autoantibodies have been found to be produced by B cells after activation by components of H. pylori, including urease [46]. A potential role for bacterial heat-shock proteins (HSP) has been postulated because of the high level homology between bacterial and human HSP sequences. However, H. pylori has also been associated with protection from certain autoimmune diseases, including IBD, possibly because of its propensity for chronic low level infection and tolerance induction (reviewed in [47]).
Besides associations with bacterial pathogens, infections with viruses, particularly enteroviruses such as Coxsackie virus B4, which have tropisms for pancreatic β islet cells, have been associated with the development of T1D [48–50]. These viruses can be detected in pancreatic but also intestinal biopsies from patients with T1D, especially after recent onset [50]. Innate recognition of enteroviral nucleic acids through Toll-like receptor (TLR) 3 and/or RIG-I and MDA-5, pattern recognition receptors that mediate viral nucleic acid recognition and subsequent type I interferon (IFN) production [51], are thought to be critical in initiating a sequence of events including recruitment of inflammatory cells, apoptosis of infected β islet cells, presentation of apoptotic β cell-derived antigens by activated DCs to autoreactive T cells, and increased levels of MHC class I on remaining β islet cells leading to their targeting by activated autoreactive T cells [52]. Consistent with the importance of type I IFNs in these events, four rare polymorphisms in the gene encoding MDA-5, Ifih1 (interferon induced with helicase C domain 1), which confers recognition of RNA from picornaviruses (the class to which enteroviruses belong), were predicted to reduce MDA-5 function and were associated with lowered risk of developing T1D [53]. Besides T1D, several other human autoimmune diseases are thought to arise from viral infection, including cardiomyopathy (myocarditis), also from infection with group B (B3) Coxsackie viruses and group A streptococci [54], as well as acute disseminating encephalomyelitis and MS, which have been associated with viral pathogens such as EBV, measles virus, and HHV-6 [30]. In MS, clinical studies have shown that infection with EBV is a consistent and strong risk factor. In fact, it has been reported that in individuals infected with EBV in early childhood, the risk of MS is about tenfold higher compared to EBV-negative individuals, and at least 20-fold greater if they also have developed mononucleosis [55, 56]. SLE is also associated with prior viral infection. Prior to clinical onset of SLE, the initial autoantibodies that develop in patients to the 60KDa protein Ro, a common target in SLE, were found to cross-react with a peptide from the latent EBV viral protein Esptein-Barr virus nuclear antigen-1 (EBNA-1) [35]. Rabbits immunized with either Ro or the cross-reactive EBNA-1 peptide developed autoantibodies that bound to multiple epitopes of Ro, and the animals progressively developed symptoms of lupus [35]. These data support a role for B cell epitope mimicry in the initiation of SLE.
In the context of Crohn’s disease (CD), an IBD that affects mainly the ileum but can also occur anywhere in the intestinal tract, recent work in mice has shown that viral infection can also contribute to the development of IBD in a susceptible genetic background [57]. Since the autophagy gene Atg16l1 has been linked to CD in humans, a mouse model was generated where the expression of the ATG16L1 protein is rendered hypomorphic (HM) by inhibiting the expression of intact mRNA after gene trap-mediated disruption of the gene [58]. ATG16L1HM mice are viable and survive to adulthood. While the morphology of both ileum and colon is intact in these mice, evident abnormalities in granule exocytosis were detected as well as in the transcriptional profile of Paneth cells derived from these mice, consistent with similar observations in patients homozygous for the Atg16l1 risk allele. Strikingly, these abnormalities were absent if the same line of mice were raised in an enhanced barrier facility, and chronic infection by the murine norovirus was required for the observed phenotype [57]. Treatment with dextran sulfate sodium (DSS), which induces colitis in mice via toxicity toward intestinal epithelial cells leading to disruption of intestinal barrier integrity and consequent inflammation, generates an altered response to injury in ATG16L1HM mice and recapitulates the main characteristics of IBD. It was concluded that both gene susceptibility and virus infection are likely responsible for triggering Crohn’s-like disease in these mice.
Infection and intestinal homeostasis seem to also be related to peripheral autoimmune diseases such as RA, which causes chronic inflammation of the joints and of the tissues around the joints, as well as in other organs in the body [59, 60]. Several epidemiological studies in patients affected with arthritis have reported a correlation between the onset of the disease and preceding gut infections [59]. It has been estimated that the annual incidence of enteric infection-related arthritis within a population approaches 1/1,000 [61, 62]. These observations suggest that the outcome of an enteric infection can have significant impact on the development of arthritis in humans.
Colonization with commensal microbes
The commensal microbial community populating the mammalian gut has recently emerged as a critical player that contributes to both health and disease, affecting a range of systemic diseases. Skewing of the commensal community or “dysbiosis” can result in autoimmune pathology in the intestine as well as at distant sites. While host genetics underlies the susceptibility to autoimmunity, accumulating evidence emerging from both clinical and experimental studies shows that the intestinal microbiota can also modulate the susceptibility to autoimmune disease (Conclusions from studies in animal models are schematized in Fig. 1b).
The first piece of evidence comes from studies in the context of IBD defined as chronic inflammatory disorders affecting the intestinal tract. The most common types of IBD are CD described above, and ulcerative colitis (UC)—an inflammation of the colonic mucosa. The main cause of these diseases is still unclear and despite an essential role for the genetic background in determining susceptibility to disease, several observations in humans and mice suggest an important contribution of the intestinal microbiota (reviewed in [63]). We mention only a handful of studies here. First, abnormal composition and activity of the microbiota have been observed in IBD patients as compared to healthy individuals. An increased frequency of mucosa-associated bacteria and the emergence of adherent/invasive strains of Escherichia coli has been reported in patients with CD. Also, a serological response against commensal-derived antigens such as E. coli outer membrane protein C (OmpC), the Crohn’s related bacterial sequence I2 from Pseudomonas fluorescence, and bacterial flagellin have been detected in patients with CD. Approximately 70 % of patients with UC have anti-neutrophil cytoplasmic antibody (pANCA), which curiously cross-reacts with select species of commensal bacteria suggesting that pANCA may be directed toward a phylogenetically conserved sequence within histone-H1 [64]. Analysis of the C3H/HeJBir strain of mice, which spontaneously develop colitis, has shown the presence of CD4+ TH1 cells proliferating in response to enteric bacterial flora, and these cells are able to transfer the disease in scid/scid recipients after antigen-specific activation. More recently, TReg cells were shown to promote production of intestinal IgA specific to the immunodominant flagellin antigen CBir1 [65]. In addition, several polymorphisms in genes predisposing to IBD are involved in innate immune recognition of bacterial ligands and their processed byproducts. These include polymorphisms in the nucleotide-binding oligomerization domain containing 2 (NOD2) and TLRs 2, 4 and 5. In humans, clinical experience suggests that antibiotic treatment or administration of probiotics and prebiotics can treat symptoms of IBD although their benefits in carefully designed clinical trials have not been formally established. In genetic mouse models of IBD, the development of colitis and ileitis is often blocked after antibiotic treatment or housing in germ-free conditions. Some examples include Il10−/− mice and Tg(ε26) mice, a transgenic line that expresses high copy number of the CD3ε human gene leading to an arrest in T cell development (reviewed in [66]). In the Samp-1/Yit strain of mice, which develop a spontaneous ileitis similar in many features to human CD, as well as Il2−/− mice, manifestations of IBD are also attenuated in germ-free conditions [66]. Similarly, ATG16L1HM mice infected with norovirus are protected against DSS-induced colitis after antibiotic treatment [57]. Colonization of mice with an experimentally standardized microbiota called the Altered Schaedler Flora (ASF) or with commensal bacterial species, such as Clostridium, protects against DSS-induced colitis by promoting colonic generation of Treg cells [67, 68].
An altered fecal microbiota has been shown to be responsible for an increased susceptibility to DSS-induced colitis in mice deficient for the inflammasome components NLRP6, ASC and caspase-1 [69]. These proteins form a large multi-protein complex called the inflammasome, assembly and activation of which is initiated by members of the Nod-like receptor (NLR) family. Assembly of the inflammasome leads to the activation of inflammatory caspases, cysteine proteases, that are produced as zymogens and which induce cell death or inflammation upon activation. Caspase-1 interacts with some members of the NLR family via a CARD domain-containing protein called PYCARD (or ASC), and when activated upon cleavage, leads to the cleavage of pro-IL-1β and pro-IL-18 to generate the biologically active forms of these cytokines [70]. In this context it has been shown that NLRP6 inflammasome expression in epithelial cells and consequent IL-18 production limits the host colonization by bacterial phyla Bacteroidetes (Prevotellaceae) and TM7, which are highly colitogenic and increase susceptibility to DSS-induced colitis [69]. Consistently, antibiotic treatment of NLRP6 deficient mice decreases the abundance of these species and susceptibility of the mice to colitis. In contrast to these findings, mice deficient for activation-induced cytidine deaminase (AID), which is expressed by germinal center B cells and acts as a central regulator of somatic hypermutation and class switch recombination, are often used as a spontaneous model of gastritis. Interestingly, no change in the development of autoimmunity has been observed in AID deficient mice during germ-free housing [71].
As has been shown for IBD, alterations in the gut microbiota have also been reported in patients with RA [72]. The development of RA in mice deficient for the IL-1 receptor antagonist (Il1rn−/−) is associated with spontaneous autoimmune T cell-mediated arthritis because of excessive IL-1 signaling. Notably, development of arthritis in this model is dependent on commensals because germ-free mice do not develop the disease [73]. Interestingly, monocolonization with Lactobacillus bifidus induces rapid onset of arthritis in germ-free Il1rn−/−animals. In a more recent study, the onset of autoimmune arthritis in a different model was also shown to be attenuated under germ-free conditions [74]. The authors used the K/BxN mouse model of inflammatory arthritis where mice express both the NOD strain MHC class II molecule Ag7 and the transgenic TCR KRN specific to a self-peptide derived from glucose-6-phosphate isomerase (GPI) presented within the context of Ag7 [75]. The autoreactive KRN T cell response induces high levels of anti-GPI antibodies, which recognize cationic GPI associated specifically with the cartilage surface, leading to inflammatory deposition of immune complexes at the cartilage surface. Development of arthritis is significantly reduced in K/BxN mice housed in germ-free conditions, but these mice develop arthritis within two weeks of being transferred into a specific pathogen free (SPF) facility [74]. The titer of anti-GPI autoantibodies was reduced in germ-free mice, and gene expression analysis of their splenic CD4+ T cells showed an impaired TH17 and TH1 signature compared to their SPF counterparts [74].
Contrary to these observations, T1D has long been known to be fully penetrant in germ-free NOD mice, while housing under SPF conditions reduces disease incidence [76, 77]. Surprisingly, SPF housed NOD mice are completely protected from T1D when bred onto a background deficient for the TLR signaling adaptor MyD88 [78]. Compared to Myd88+/− NOD mice, the intestinal microbiota in Myd88−/− NOD mice was altered in composition (for example, significantly lower ratio of Firmicutes to Bacteroidetes), and exposure of germ-free NOD mice to this altered microbiota led to severe attenuation of pancreatic islet immune cell infiltration [78]. Therefore, in an experimental model of diabetes, the lack of microbiota increases disease penetrance, although some commensal species, such as Bacteroidetes, can be protective. It has been suggested that production of anti-inflammatory short chain fatty acids from dietary fiber, which Bacteroidetes have been described to do, induces systemic anti-inflammatory responses that protect against diabetes [79]. Furthermore, based on the increased susceptibility of germ-free mice to infections with Coxsackie B virus, member of the aforementioned enteroviruses that have been linked to diabetes, colonization with specific commensals is speculated to prevent infection with Coxsackie virus, and thereby protect from development of subsequent diabetes [80].
Two recent studies have also highlighted the role of commensal flora in the context of experimental autoimmune encephalomyelitis (EAE), a mouse model of MS. Disease is caused by damage to the myelin sheath of neurons, and activation of autoreactive T and B cells is involved in disease development. One study has shown that the induction of EAE upon injecting animals with a peptide derived from myelin oligodendrocyte glycoprotein (MOG) in Complete Freund’s Adjuvant (CFA) was attenuated if animals were housed in germ-free conditions compared to their SPF housed counterparts [81]. In a second study, a spontaneous model of EAE was used where TCR transgenic mice termed relapsing-remitting (RR) mice express a TCR specific for MOG peptide92–106 in the context of I–As [82]. Backcrossed to the EAE susceptible SJL/J background, most RR mice spontaneously develop EAE sharing many features of human MS. The authors demonstrated that commensal bacteria were required for the development of spontaneous EAE since RR germ-free mice were fully protected while SPF animals developed disease within 3–8 months [82]. In parallel, reduced titers of anti-MOG antibodies in the serum and impaired germinal center formation in the cervical lymph nodes of germ-free RR mice were also observed. MOG-specific antibody titers were promptly increased upon recolonization with conventional commensal microbiota [82].
The role of T helper 17 cells (TH17) cells
TH17 cells occupy center stage in autoimmune disease
Interest in the last several years has centered on the association of autoimmune inflammation with TH17 cells, a T helper cell subset known for its role in pathogenesis of organ-specific autoimmunity in both animal models as well as human disease. TH17 cells are also important for host defense against certain microbial infections, and a variety of pathogenic bacteria induce TH17 cells in the intestine or at other mucosal surfaces.
Even before the definition of TH17 cells, IL-17 cytokine produced by CD4+ T cells has been associated with host defense against infectious pathogens as well as with autoimmune diseases. At the turn of the century, it became clear that CD4+ T cells that express IL-17 do not co-express interferon-γ (IFN-γ) and vice versa [83]. Subsequent studies of mice deficient for IL-23 in autoimmune disease models showed that although this cytokine was not important for expression of IFN-γ, it was required for expression of IL-17, indicating that IL-17 and IFN-γ are differentially regulated [84, 85]. IL-23 was subsequently shown to selectively induce proliferation of in vivo primed IL-17-expressing CD4+ T cells [86]. T cells induced to proliferate with IL-23 express a distinctive set of genes: they do not produce IFN-γ or IL-4 but instead express IL-23 receptor (IL-23R) and IL-17. IL-23R expression was later found to be essential for successful completion of the differentiation process in TH17 but not TH1 cells in vivo [87]. In investigating the requirements for TH17-cell differentiation, two groups independently showed that naïve CD4+ T cells uniquely differentiate into IL-17-expressing T cells distinct from TH1 or TH2 under the influence of TGF-β and IL-6, with IL-23 driving the expansion of these cells [88, 89]. TH17 cells have since been recognized as a separate lineage of T-helper cells that plays a crucial role in T cell-mediated adaptive immunity. This idea has been supported in the last several years with many studies describing their unique cytokine profile and transcriptional regulation, both in human and mouse systems (reviewed in [90]). TH17 cells are not only distinct from other T-helper cell subsets in terms of gene expression and regulation, but also in their biological function. TH17 cells are generally thought to be pro-inflammatory, especially through the production of IL-17 (reviewed in [12]). They play a role in recruiting neutrophils and macrophages to infected tissues, and participate in other functions such as abscess formation and induction of antimicrobial peptides.
The importance of TH17 cells in the pathogenesis of organ-specific autoimmune inflammation has now been well established and demonstrated in many different animal models (reviewed in [91]). As IL-17 being the signature cytokine produced by TH17 cells, looking for its presence both at the transcript and protein levels or disrupting IL-17 signaling has predominated earlier studies. EAE was found to be severely attenuated in mice lacking IL-17, IL-17 receptors A (IL-17RA) or IL-17RC [92–94], while treatment with antibody to IL-17 inhibited chemokine expression, and thereby inflammation, in the brain [89]. Using various models of arthritis such as collagen-induced arthritis or spontaneous arthritis in K/BxN or Il1rn−/− mice, deficiency for IL-17 or IL-17R demonstrated a critical role for IL-17A in arthritis (reviewed in [95]). IL-17 was found to increase joint inflammation by inducing synovial pro-inflammatory cytokine and chemokine production, and aggravate bone and cartilage erosion in murine collagen-induced arthritis, also acting synergistically with IL-1 and TNF. Its neutralization before the onset of arthritis prevented the development of arthritis in both collagen-induced and spontaneous models of the disease. Consistent with these studies, loss of IL-23 is protective in autoimmune arthritis in mouse models, while loss of IL-12 correlates with increased severity of disease as well as increased numbers of IL-17-expressing lymphocytes [85]. IL-17 is also implicated in mouse models of IBD. Blocking IL-23, but not IL-12, is sufficient to prevent onset of colitis in Il10−/− mice, by a mechanism involving inhibition of IL-17, and administration of recombinant IL-23 accelerated colitis induced by CD4+CD45RBhigh T cell transfer into Rag−/− mice [96]. Also, in a different acute colitis model of IBD employing the use of 2,4,6 trinitrobenzenesulfonic acid (TNBS), IL-17R deficient mice showed lower levels of colonic IL-6 and MIP-2, with a resultant reduction in colonic neutrophil infiltration and reduced severity of colonic inflammation [97]. Conversely, treatment of control C57BL/6 J mice with an adenoviral vector encoding a soluble IL-17R-IgG fusion protein significantly reduced IL-6 and MIP-2 levels. Finally, via secretion of IL-22, TH17 cells also play a pathogenic role in dermal inflammation and psoriasis, an autoimmune disease characterized by faster multiplication of skin cells and recruitment of leukocytes in both dermis and epidermis (reviewed in [98]). Increased IL-22 mRNA and protein levels have been found both in the skin and blood of psoriatic patients. In a CD4+ T cell-dependent mouse model of psoriasis where CD4+CD45RBhi T cells depleted of CD25+ Treg cells were transferred into lymphocyte deficient mice, IL-22 neutralization prevented disease development.
High levels of IL-17A in the synovial fluid and systemic levels of IL-23 have been detected in patients with RA [99–102]. Increased IL-17 transcripts along with IL-6 and IFN-γ transcripts were found in central nervous system autopsy lesions as well as CSF collected from patients with MS [103, 104]. Also, IL-17-expressing perivascular lymphocytes have been described in brain lesions of patients with active MS, while these IL-17+ cells were reduced in patients with quiescent MS [105]. IL-23 expression by monocyte-derived DCs was found to be significantly higher in MS patients than healthy subjects [106]. This observation correlated with more production of IL-17 by CD4+ T cells isolated from patients with MS. In patients with UC and CD, elevated IL-17 mRNA levels were found in the colonic mucosa as compared to corresponding samples from either normal controls or patients with infectious or ischemic colitis [107]. IL-17-producing TH17 cells have also been observed in gut mucosa of CD patients. Along with IL-17, these cells also produced IFN-γ and expressed IL-23R and CCR6 [108]. Increased numbers of lamina propria CD14+ IL-23-producing macrophages were noted in patients with CD [109]. Recently, CD3+ lamina propria mononuclear cells from both patients with CD and UC were shown to produce increased levels of IL-17 and IFN-γ compared to healthy controls [110]. Similarly, IL-17 mRNA can be detected in psoriatic skin lesions, and IL-17-producing cells have also been isolated from dermis of psoriatic lesions [111, 112].
Despite all the evidence linking IL-17 and TH17 cells to autoimmune disease, none of the genome-wide association studies looking for autoimmunity risk alleles have identified the Il17 locus as a susceptibility gene for human autoimmune disease. On the other hand, Il23R polymorphisms have been linked to a range of autoimmune diseases including MS, IBD, RA, psoriasis and psoriatic arthritis [113–118]. Similarly, polymorphisms in the genes encoding Janus kinase 2 and STAT3, downstream of IL-23R signaling, have also been identified as risk alleles [119, 120]. How does the IL-23R contribute to the pathogenesis of autoimmune disease? Although the connection between IL-23 (the ligand for IL-23R) and IL-17 has been solidified by experimental evidence in mouse models of autoimmune disease [86, 121, 122], IL-23 rather than IL-17 has emerged as the master regulator of disease [123]. A previously unsuspected function for IL-23 relates to its role in mediating a conversion in the cytokine expression profile of TH17 cells into one that is more TH1-like in nature. Adding IL-23 to TH17 cells that had previously been generated with TGF-β and IL-6 led to a decrease in the frequency of IL-17-expressing cells and a concomitant increase in the frequency of IFN-γ-producing T cells in a STAT4- and T-bet-dependent manner [124]. Generation of Il17a-CreR26ReYFP fate reporter mice, which allowed tracing of IL-17A-expressing cells in inflammatory responses in vivo, showed that after immunization with MOG and CFA to induce EAE, IL-17-producing TH17 cells proceeded from expressing IL-17A, to co-expressing IL-17A with IFN-γ, to expressing IFN-γ alone [125]. In fact, the main source of the IFN-γ was MOG-specific CD4+ T cells that derived mostly from ex-TH17 cells (eYFP+). Ex-TH17 cells expressed the TH1 transcription factor T-bet as well as IL-12Rβ2, which was co-expressed with IL-12Rβ1 imparting high affinity binding to IL-12. Ex-TH17 had also downregulated the TH17 transcription factor ROR-γt and IL-23R. Unlike bona fide TH1 cells, ex-TH17 IFN-γ-producing cells retained expression of the aryl hydrocarbon receptor, previously shown to be expressed by TH17 cells [126], and also expressed IL-1R1. Most importantly, when Il17a-CreR26ReYFP reporter mice were crossed to IL-23p19 deficient mice, IL-17A+IFN-γ+ cells as well as single IFN-γ+ cells were no longer found suggesting that IL-23 was critical for the switch in TH17 cells toward IFN-γ production.
Collectively, the findings from these studies were notable not only because they revealed a fundamental aspect of T-helper cell biology demonstrating surprising plasticity in the differentiation fates of these cells (reviewed in [127]), but also explained previous data that seemed at odds with a center stage for TH17 cells in autoimmune disease. These included reports describing (1) the presence of double-producing IL-17A+IFNγ+ cells in inflamed tissues [128, 129], which in light of the new studies are perhaps transitional TH17 cells on the way to becoming IFN-γ producers, (2) that deficiency for STAT4 or T-bet prevented development of EAE [130, 131], again perhaps because these transcription factors that act downstream of IL-23 were shown to be important in mediating the conversion to IFN-γ-producing cells, (3) how transfer of TH17 polarized cells induced IFN-γ-dependent disease as in the case of islet-specific TCR transgenic TH17 cells inducing diabetes and islet injury dependent on IFN-γ [132, 133], and induction of colitis upon transfer of ex vivo polarized TH17 cells into Rag−/− mice associated with a decrease in IL-17A and IL-17F production and an induction of IFN-γ in the transferred T cells [124], (4) how in the absence of IL-23, TH17 cells demonstrated reduced production of inflammatory cytokines and increased secretion of IL-10, which correlated with an impaired ability to transfer EAE [134], and (5) how compared to IL-17A, IL-17RA or IL-17RC deficient mice [92, 93, 135], and even IL-23p19 deficient mice [84], IL-12 p40 deficient mice are completely protected from EAE [136]. The most logical conclusion from these studies is one where adoption of a TH1-like cytokine profile by autoreactive TH17 cells has been speculated to be the main cause of pathology in autoimmune disease [125]. However, some experimental evidence in T1D exists that does not fully support this, and the picture may be even more complex. An attempt to identify IL-17A-producing T cells using IL-17A-enhanced green fluorescent protein (eGFP) bicistronic reporter mice in the CD8+ T cell-driven lymphocytic choriomeningitis virus (LCMV)-induced model of T1D, where LCMV glycoprotein is transgenically expressed as self in the pancreas, failed to locate CD4+ or CD8+ T cells producing IL-17A in either diabetic or pre-diabetic mice [137]. Furthermore, adoptively transferred IL-17A-eGFP reporter, LCMV glycoprotein-specific TCR transgenic CD4+ T cells produced copious amounts of IFN-γ, but no IL-17. While these results appear consistent with those where IFN-γ producers were detected after transfer of TH17 polarized islet-specific TCR transgenic into NOD/SCID mice [132, 133], the status of IL-23 expression in these T1D mouse models remains to be investigated.
One final note: it appears that one more important step needs to take place for TH17 cells to adopt a TH1-like fate, which is that T-bet and IFN-γ can only be expressed in the absence of signals from TGF-β [124, 138]. It has been conjectured that perhaps this could explain how pathogenic TH17 cells arise in the absence of TGF-β [125, 139]. Consistent with this notion, Treg cells and TGF-β have been shown to inhibit the conversion of TH17 into colitogenic IFN-γ-producing TH1-like cells [124, 140].
Commensals, TH17 and autoimmune disease
The studies we discussed in the section regarding commensal microbes demonstrate that colonization with commensals critically impacts development of autoimmune disease in different animals models for autoimmunity. Although the exact mechanisms are far from being fully elucidated, a correlation has emerged between the appearance of intestinal TH17 cells upon colonization with commensals and development of autoimmune disease. A number of studies have focused on the homeostatic balance in the numbers of TH17 and TReg cells within the intestinal lamina propria given the close interface of the intestinal epithelium with the abundant intestinal microbiota. TH17 cells are not detected in the small intestinal lamina propria of germ-free mice while the numbers of FOXP3+ TReg cells are increased, even systemically, in these mice [81, 141–143]. Notably, TReg cells are decreased in the colons of germ-free mice suggesting a dichotomy in the signals inducing their generation in the colon versus the small intestine [67]. Furthermore, antibiotic treatment of mice housed in SPF conditions significantly decreases the number of intestinal TH17 cells over time [142]. Consistent with these observations, the frequencies of such TH17 cells are variable in mice purchased from different vendors: mice from Taconic Farms show elevated numbers of intestinal TH17 cells compared to those from The Jackson Laboratory [141]. Detailed investigations of the composition of the small intestinal microbiota showed that Taconic but not Jackson mice were enriched for commensal segmented filamentous bacteria (SFB), gram-positive anaerobes of the order Clostridiales. Monocolonization of germ-free mice with SFB specifically induces TH17 generation in both the small and large intestine while decreasing Treg cell numbers, providing strong evidence that commensal bacteria influence homeostasis of the intestinal immune system [67, 74, 141, 144]. Certain members of the intestinal microbiota also affect TReg cell generation in the colon. The ASF specifically increases the frequency of CD4+CD25+Foxp3+ cells, which in turn were shown to prevent generation of TH17 and TH1 in an IL-10-dependent manner [68]. Colonization of mice with various commensal species of Lactobacillus, Clostridium clusters IV and XIVa, or Bacteroides fragilis also increased TReg cells in the intestinal lamina propria [67, 145–148]. Consistent with these observations, high-throughput sequencing has revealed that many Treg cells in the colon possess unique TCRs that recognized commensal-derived antigens including those from Clostridium species [149].
What is the correlation between autoimmune disease and TH17 cells in the small intestine? Based on the aforementioned higher incidence of T1D in germ-free NOD mice, the prediction would be that TH17 cells, development of which is impaired under those conditions, would inhibit autoimmune disease. Protection could be mediated via the function of TH17 effector cytokines such as IL-22, which induces synthesis of anti-microbial peptides by intestinal epithelial cells [98]. The action of these anti-microbial peptides would be expected to shape the composition of the intestinal microbiota, which perhaps like the microbiota in Myd88−/− NOD mice, would attenuate T1D. IL-22 is also important for maintaining integrity of the intestinal epithelial barrier [98], compromise of which by enteric pathogens has been linked to T1D [150]. Consistent with the prediction, when female NOD mice were housed in SPF facilities that were positive for SFB, which induced intestinal TH17 cells, the incidence of T1D was significantly lower than those housed in SFB-negative SPF facilities [151]. Notably, these results, along with those in Myd88−/− NOD mice [78], highlighted the importance of composition of the microbiota rather than general germ-free versus SPF conditions per se in determining the incidence of T1D. Therefore, under SPF conditions, it is the increased representation of SFB and/or Bacteroidetes (mentioned above) that appears to critically dictate the attenuation of T1D.
However, contrary to the prediction and results based on T1D in the NOD model, the incidence of autoimmune disease is strongly reduced under germ-free conditions in other mouse models of autoimmune disease. Indeed, colonization of germ-free K/BxN mice with SFB induced the appearance of small intestinal TH17 cells, but provoked rapid onset of arthritis within 3 days of oral gavage (compared to 2 week onset in SPF conditions) [74]. Time of disease onset correlated with the appearance of splenic TH17 cells that expressed α4β7, a receptor expressed by gut-imprinted T cells, suggesting that intestinal TH17 cells had migrated to the spleen. In fact, arthritis could successfully be blocked by neutralization of IL-17, and IL-17R deficient B cells transferred along with splenocytes from arthritic K/BxN mice into BxN Rag−/−recipients could not support germinal center formation [74]. Presumably, TH17 cells specific to the aforementioned GPI might differentiate within the small intestine upon SFB colonization, accumulate in the spleen during their recirculation, and via the production of IL-17, provide help to IL-17R+ B cells to promote germinal center formation and GPI autoantibody production. In light of the ubiquitous nature of GPI and the increased frequency of autoreactive GPI-specific T cells in the K/BxN model, the authors argued that the need for molecular mimicry, i.e. cross-reactivity with an epitope derived from an intestinal microbe, was not necessary [74].
Other studies have also supported a link between TH17 cells and autoimmune disease in the context of commensals. Splenocytes isolated from germ-free Il1rn−/− mice which, unlike their SPF housed counterparts fail to develop RA, were less sensitive to anti-CD3 and TLR stimulation and showed decreased production of IL-17 [73]. In the context of EAE induced by injection of MOG peptide with CFA, attenuated disease in germ-free mice was linked to a reduction in both IL-17- and IFN-γ-producing cells harvested from cervical draining lymph nodes or spinal cords, while CD4+CD25+Foxp3+ Treg cells were increased [81]. In vitro experiments showed that DCs from germ-free mice were less efficient in inducing TH17 and TH1 differentiation of TCR transgenic MOG-specific CD4+ T cells, suggesting a role for the microbiota in modulating the capacity of DCs to induce T cell differentiation. Monocolonization of germ-free mice with SFB restored the susceptibility to EAE consistent with a considerable increase in IL-17- and IFN-γ-producing cells in the intestines as well as the spinal cords [81]. In another report, a requirement of commensal bacteria for developing EAE was observed in the aforementioned relapsing-remitting (RR) model of spontaneous disease [82]. Again, less TH17 cells were found specifically in the intestinal lamina propria and Peyer’s patches of germ-free mice, and both IL-17 and IFN-γ levels produced by splenocytes after restimulation with anti-CD3 or MOG peptide were lower than those made by splenocytes from SPF housed mice. Given the direct correlation between EAE incidence, commensal colonization and TH17 generation, it was possible that TH17 cells generated in response to a commensal antigen were cross-reactive with MOG. However, MOG-specific molecular mimicry was excluded based on the observation that both polyclonal CD4+ T cells derived from wild type (WT) SJL/J mice and transgenic MOG-specific CD4+ T cells derived from RR mice pro-liferated to the same extent, and only within the lamina propria upon adoptive transfer into WT SPF mice, but remarkably, not mice treated with antibiotics [82]. This suggested that a general inflammatory milieu in the gut, rather than a specific epitope, drove proliferation of MOG-specific CD4+ T cells. More convincingly, SPF housed RR mice deficient for the MOG protein failed to develop anti-MOG autoantibody despite a normal microbiota, and conversely, injection of MOG with CFA into RRxMOG−/−mice readily induced the production of MOG-specific autoantibody [82]. Furthermore, it was demonstrated that specificity of B cells to MOG and presence of the MOG protein were both required to form cervical lymph node germinal centers. Based on these data, the authors proposed a model where increased frequencies of pathogenic MOG-specific autoreactive IL-17-producing T cells (TH17 cells) in RR mice proliferate in the intestine under inflammatory signals initiated upon commensal colonization, and provide help to MOG-specific B cells in cervical lymph node germinal centers.
Collectively, with the notable exception of T1D in NOD mice, the studies above have established a connection between commensal colonization in the gut, generation of intestinal TH17 cells, and initiation of a non-gut autoimmune disease. An important point to raise here is that despite this connection, the antigen specificities of intestinal TH17 cells at homeostasis is currently not known. It is likely that these cells harbor self-reactive T cells, especially in light of recent observations showing that like TReg cells, TH17 cells can be selected on self-antigens within the thymus, express α4β1 and CCR6, and migrate to the lung, liver and gut [152]. It is also possible that these TH17 cells are specific to commensals like SFB similar to the specificities found in intestinal TReg cells to Clostridium, for example [149]. On the other hand, there is evidence from TCR transgenic mice that intestinal lamina propria TH17 cells can be generated at relatively normal numbers in the absence of the transgenic TCR’s cognate antigen [68, 153].
Concluding remarks and perspective
Importance of the intestinal commensal microbiota for determining susceptibility to autoimmune disease has added an exciting new dimension to the complexity of autoimmune diseases. Much work remains in understanding how commensals within the intestine can affect the course of autoimmune disease in distant organs. On the other hand, the link between infections and autoimmune diseases has been known for a long time, and as discussed in this review, many mechanisms have been presented, foremost of which has been “epitope mimicry”. However, an accurate understanding of the course of events during microbial infection that set the stage for later autoimmune disease is still elusive. We became interested in the link between microbe and autoimmune diseases during the course of our studies on phagocytosis of dying cells and impact of this process on T cell activation. We had found that innate recognition of apoptotic cells carrying ligands for TLRs drives differentiation of naïve CD4+ T cells into TH17 cells [154]. We had reported that blocking apoptosis during infections with Citrobacter rodentium impairs the ensuing TH17 response [154]. We have subsequently been interested in understanding the outcome of antigen presentation within the context of phagocytosis of infected apoptotic cells, specifically whether self-antigens derived from infected apoptotic cells could be presented within an inflammatory/T cell costimulatory context. Phagosomes carrying infected apoptotic cells should contain peptides derived from both self and non-self. Based on our previous work [155], the prediction would be that compartmentalized TLR signals from a phagosome carrying infected apoptotic cells would optimally tailor that phagosome for antigen presentation, providing equal opportunity for both self and non-self-peptides to be loaded onto MHC class II molecules. This is very different than the simultaneous phagocytosis of microbe and apoptotic cell into distinct phaogosomes, where only the phagosome carrying microbe is favored for antigen presentation [155]. Our working hypothesis is that presentation of self-antigens within the context of phagocytosis of infected apoptotic cells would, under conditions where T cell tolerance has been compromised, lead to activation of self-reactive T cells (schematized in Fig. 2). Could these cells differentiate into TH17 cells? Would they be pathogenic or protective? Could this event at the forefront of innate immunity lead to autoimmunity and eventually, within the appropriate genetic susceptibility background, culminate in autoimmune disease?
Fig. 2.
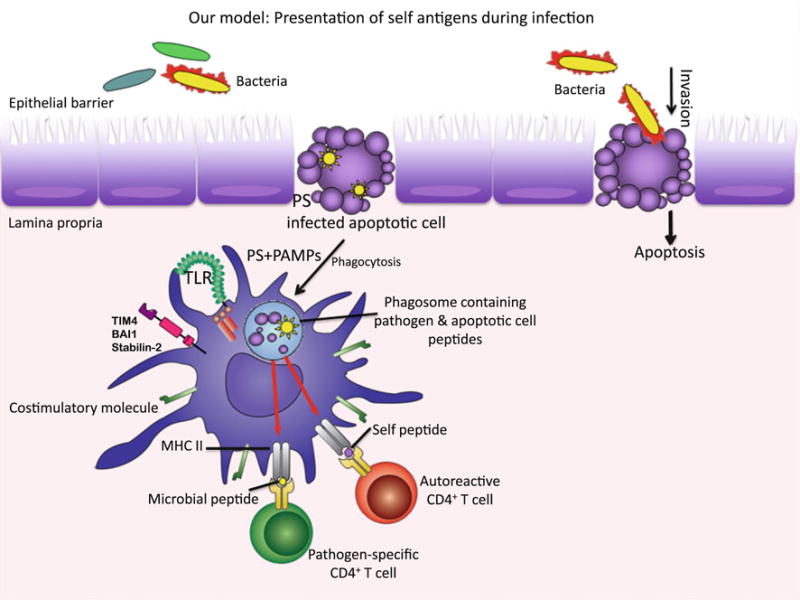
Model proposing the potential MHC II presentation of self-peptides after phagocytosis of infected apoptotic cells. During infection that causes apoptosis of host cells, APCs recognize “eat me” signals such as phosphatidylserine (PS) and engage receptors such as Stabilin-2, BAI-1 and TIM-4 along with TLRs [156]. Following phagocytosis of infected apoptotic cells, apoptotic cell (self) and microbial (non-self) peptides contained simultaneously within TLR ligand carrying phagosomes, can become presented by the APC, leading to stimulation of both pathogen-specific and self-reactive CD4+ T cells
Finally, rather than viewing the associations of pathogens or commensals with autoimmune disease as separate entities (Table 1), could it be that these associations are all interconnected, and that the outcome of the relationships among host, commensal and pathogen is what ultimately shapes susceptibility to autoimmune disease? For example, colonization with a certain commensal (an attenuator of risk for autoimmune disease) might protect the host from an infection with a certain pathogen (a precipitator of autoimmune disease). On the other hand, infection with a pathogen that may have no relation to autoimmune disease might alter the composition of the host microbiota to one that predisposes to autoimmune disease in a genetically susceptible host.
Table 1.
The associations between pathogen or commensal and autoimmune disease
| Disease type or experimental model | Role of infection in onset of the disease and associated pathogens | Role of microbiota in onset of the disease and associated commensal species | Refs |
|---|---|---|---|
| GBS (peripheral nervous system) | Induction of autoimmunity (EBV, CMV, Mycoplasma pneumoniae, Campylobacter jejuni) | Not reported | [38, 40] |
| Myocarditis | Induction of autoimmunity (B3 coxsackievirus and group A streptococci) | Not reported | [54] |
| MS, enchephalomyelitis and EAE (MOG + CFA, RR mice) (central nervous system) | Induction of autoimmunity (EBV, measles virus, HHV-6) | Induction of autoimmunity (SFB, MOG + CFA), Germ-free housing (MOG + CFA, RR mice) and antibiotic treatment (RR mice) are protective | [30, 35, 56, 81, 82] |
| Lyme arthritis | Induction of autoimmunity (Borrelia burgdorferi) | Not reported | [42, 43] |
| Rheumatic fever | Induction of autoimmunity (β-hemolytic Streptococci) | Not reported | [31, 44] |
| Clinical and experimental rheumatoid arthritis (Il1rn−/−, K/BxN) | Induction of autoimmunity (Enteropathogenic bacteria, EBV, CMV and parvovirus in humans) | Induction of autoimmunity (Lactobacillus bifidus, Il1rn−/−), SFB (K/BxN) Germ-free housing is protective (Il1rn−/−, K/BxN) |
[59–62, 72–75] |
| Autoimmune gastritis | Induction of autoimmunity (Helicobacter pylori) Aicda−/− mice | No change in autoimmunity in germ-free conditions | [45, 46, 71] |
| Crohn’s disease, DSS in ATG16L1HM mice, Samp-I/Yit mice | Induction of Crohn’s like symptoms in ATG16L1HM mice after DSS treatment (murine norovirus) | Humans: Altered composition and activity of microbiota and response against commensal-derived antigens (invasive escherichia coli, Pseudomonas fluorescence) Mice: Germ-free housing (Samp-I/Yit) and antibiotic treatment (ATG16L1HM mice) are protective |
[57, 63, 64, 66] |
| Ulcerative colitis, genetic models. Not reported of colitis (C3H/HeJBir, Il10−/−, Tg(ε26), Il2−/−) and DSS treatment | Not reported | Humans: Composition and activity microbiota of and response against commensal-derived antigens Mice: Induction of colitis (Bacteroidetes (Prevotellaceae) and TM7) Protection of colitis (ASF, Clostridium, Lactobacillus, Bacteroides fragilis) Germ-free housing (Il10−/−, Tg(ε26), Il2−/−) is protective |
[63, 65, 66–69] |
| Type I diabetes, NOD mice | Induction of autoimmunity (Coxsackie virus B4 in humans) | Commensal Bacteroidetes are protective SPF housing is protective correlating with presence of commensal SFB |
[48–50, 76–78, 80] |
Biography

J. Magarian Blander
References
- 1.Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD. Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet. 2008;4:e1000024. doi: 10.1371/journal.pgen.1000024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho JH, Gregersen PK. Genomics and the multifactorial nature of human autoimmune disease. N Engl J Med. 2011;365:1612–23. doi: 10.1056/NEJMra1100030. [DOI] [PubMed] [Google Scholar]
- 3.Bogdanos DP, Smyk DS, Rigopoulou EI, Mytilinaiou MG, Heneghan MA, Selmi C, Eric Gershwin M. Twin studies in autoimmune disease: Genetics, gender and environment. J Autoimmun. 2011 doi: 10.1016/j.jaut.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Javierre BM, Hernando H, Ballestar E. Environmental triggers and epigenetic deregulation in autoimmune disease. Discov Med. 2011;12:535–45. [PubMed] [Google Scholar]
- 5.Munz C, Lunemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9:246–58. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–76. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 7.Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S, Sakaguchi S. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003;426:454–60. doi: 10.1038/nature02119. [DOI] [PubMed] [Google Scholar]
- 8.Akirav EM, Ruddle NH, Herold KC. The role of AIRE in human autoimmune disease. Nat Rev Endocrinol. 2011;7:25–33. doi: 10.1038/nrendo.2010.200. [DOI] [PubMed] [Google Scholar]
- 9.Giraud M, Yoshida H, Abramson J, Rahl PB, Young RA, Mathis D, Benoist C. Aire unleashes stalled RNA polymerase to induce ectopic gene expression in thymic epithelial cells. Proc Natl Acad Sci USA. 2012;10:535–40. doi: 10.1073/pnas.1119351109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Husebye ES, Perheentupa J, Rautemaa R, Kampe O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J Intern Med. 2009;265:514–29. doi: 10.1111/j.1365-2796.2009.02090.x. [DOI] [PubMed] [Google Scholar]
- 11.Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachee-Chardin M, Toulon A, Bustamante J, Al-Muhsen S, Al-Owain M, Arkwright PD, Costigan C, McConnell V, Cant AJ, Abinun M, Polak M, Bougneres PF, Kumararatne D, Marodi L, Nahum A, Roifman C, Blanche S, Fischer A, Bodemer C, Abel L, Lilic D, Casanova JL. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. 2010;207:291–7. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, Filler SG, Masso-Welch P, Edgerton M, Gaffen SL. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouneaud C, Kourilsky P, Bousso P. Impact of negative selection on the T cell repertoire reactive to a self-peptide: a large fraction of T cell clones escapes clonal deletion. Immunity. 2000;13:829–40. doi: 10.1016/s1074-7613(00)00080-7. [DOI] [PubMed] [Google Scholar]
- 15.Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. autoreactive T cells in healthy individuals. J Immunol. 2004;172:5967–72. doi: 10.4049/jimmunol.172.10.5967. [DOI] [PubMed] [Google Scholar]
- 16.Raddassi K, Kent SC, Yang J, Bourcier K, Bradshaw EM, Seyfert-Margolis V, Nepom GT, Kwok WW, Hafler DA. Increased frequencies of myelin oligodendrocyte glycoprotein/MHC class II-binding CD4 cells in patients with multiple sclerosis. J Immunol. 2011;187:1039–46. doi: 10.4049/jimmunol.1001543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang J, Danke N, Roti M, Huston L, Greenbaum C, Pihoker C, James E, Kwok WW. CD4+ T cells from type 1 diabetic and healthy subjects exhibit different thresholds of activation to a naturally processed proinsulin epitope. J Autoimmun. 2008;31:30–41. doi: 10.1016/j.jaut.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 18.van Noort JM, van Sechel A, Boon J, Boersma WJ, Polman CH, Lucas CJ. Minor myelin proteins can be major targets for peripheral blood T cells from both multiple sclerosis patients and healthy subjects. J Neuroimmunol. 1993;46:67–72. doi: 10.1016/0165-5728(93)90234-p. [DOI] [PubMed] [Google Scholar]
- 19.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 20.Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH, Caminschi I, Shortman K, Henson PM, Jakubzick CV. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J Exp Med. 2011;208:1789–97. doi: 10.1084/jem.20110538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoyne GF. Mechanisms that regulate peripheral immune responses to control organ-specific autoimmunity. Clin Dev Immunol. 2011;2011:294968. doi: 10.1155/2011/294968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teh CE, Daley SR, Enders A, Goodnow CC. T-cell regulation by casitas B-lineage lymphoma (Cblb) is a critical failsafe against autoimmune disease due to autoimmune regulator (Aire) deficiency. Proc Natl Acad Sci USA. 2010;107:14709–14. doi: 10.1073/pnas.1009209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–82. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- 24.Maier LM, Hafler DA. Autoimmunity risk alleles in costimulation pathways. Immunol Rev. 2009;229:322–36. doi: 10.1111/j.1600-065X.2009.00777.x. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–7. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 26.Strasser A, Whittingham S, Vaux DL, Bath ML, Adams JM, Cory S, Harris AW. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc Natl Acad Sci USA. 1991;88:8661–5. doi: 10.1073/pnas.88.19.8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 28.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- 29.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A. 2011;108:5354–9. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cusick MF, Libbey JE, Fujinami RS. Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol. 2012;42:102–11. doi: 10.1007/s12016-011-8294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Azevedo PM, Pereira RR, Guilherme L. Understanding rheumatic fever. Rheumatol Int. 2011 doi: 10.1007/s00296-011-2152-z. [DOI] [PubMed] [Google Scholar]
- 32.Benoist C, Mathis D. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat Immunol. 2001;2:797–801. doi: 10.1038/ni0901-797. [DOI] [PubMed] [Google Scholar]
- 33.Christen U, Hintermann E, Holdener M, von Herrath MG. Viral triggers for autoimmunity: is the ‘glass of molecular mimicry’ half full or half empty? J Autoimmun. 2010;34:38–44. doi: 10.1016/j.jaut.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rose NR, Mackay IR. Molecular mimicry: a critical look at exemplary instances in human diseases. Cell Mol Life Sci. 2000;57:542–51. doi: 10.1007/PL00000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med. 2005;11:85–9. doi: 10.1038/nm1167. [DOI] [PubMed] [Google Scholar]
- 36.Fairweather D, Kaya Z, Shellam GR, Lawson CM, Rose NR. From infection to autoimmunity. J Autoimmun. 2001;16:175–86. doi: 10.1006/jaut.2000.0492. [DOI] [PubMed] [Google Scholar]
- 37.Regner M, Lambert PH. Autoimmunity through infection or immunization? Nat Immunol. 2001;2:185–8. doi: 10.1038/85228. [DOI] [PubMed] [Google Scholar]
- 38.Hardy TA, Blum S, McCombe PA, Reddel SW. Guillain-barre syndrome: modern theories of etiology. Curr Allergy Asthma Rep. 2011;11:197–204. doi: 10.1007/s11882-011-0190-y. [DOI] [PubMed] [Google Scholar]
- 39.Steiner I, Rosenberg G, Wirguin I. Transient immunosuppression: a bridge between infection and the atypical autoimmunity of Guillain-Barre syndrome? Clin Exp Immunol. 2010;162:32–40. doi: 10.1111/j.1365-2249.2010.04223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuki N, Kuwabara S. Axonal Guillain-Barre syndrome: carbohydrate mimicry and pathophysiology. J Peripher Nerv Syst. 2007;12:238–49. doi: 10.1111/j.1529-8027.2007.00153.x. [DOI] [PubMed] [Google Scholar]
- 41.Moran AP. Structure and conserved characteristics of Campylobacter jejuni lipopolysaccharides. J Infect Dis. 1997;176(Suppl 2):S115–21. doi: 10.1086/513781. [DOI] [PubMed] [Google Scholar]
- 42.Steere AC, Drouin EE, Glickstein LJ. Relationship between immunity to Borrelia burgdorferi outer-surface protein A (OspA) and Lyme arthritis. Clin Infect Dis. 2011;52(Suppl 3):s259–65. doi: 10.1093/cid/ciq117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh SK, Girschick HJ. Lyme borreliosis: from infection to autoimmunity. Clin Microbiol Infect. 2004;10:598–614. doi: 10.1111/j.1469-0691.2004.00895.x. [DOI] [PubMed] [Google Scholar]
- 44.Guilherme L, Fae K, Oshiro SE, Kalil J. Molecular pathogenesis of rheumatic fever and rheumatic heart disease. Expert Rev Mol Med. 2005;7:1–15. doi: 10.1017/S146239940501015X. [DOI] [PubMed] [Google Scholar]
- 45.Amedei A, Bergman MP, Appelmelk BJ, Azzurri A, Benagiano M, Tamburini C, van der Zee R, Telford JL, Vandenbroucke-Grauls CM, D’Elios MM, Del Prete G. Molecular mimicry between Helicobacter pylori antigens and H+, K+–adenosine triphosphatase in human gastric autoimmunity. J Exp Med. 2003;198:1147–56. doi: 10.1084/jem.20030530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamanishi S, Iizumi T, Watanabe E, Shimizu M, Kamiya S, Nagata K, Kumagai Y, Fukunaga Y, Takahashi H. Implications for induction of autoimmunity via activation of B-1 cells by Helicobacter pylori urease. Infect Immun. 2006;74:248–56. doi: 10.1128/IAI.74.1.248-256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller A, Oertli M, Arnold IC. H. pylori exploits and manipulates innate and adaptive immune cell signaling pathways to establish persistent infection. Cell Commun Signal. 2011;9:25. doi: 10.1186/1478-811X-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drescher KM, Tracy SM. The CVB and etiology of type 1 diabetes. Curr Top Microbiol Immunol. 2008;323:259–74. doi: 10.1007/978-3-540-75546-3_12. [DOI] [PubMed] [Google Scholar]
- 49.Fairweather D, Rose NR. Type 1 diabetes: virus infection or autoimmune disease? Nat Immunol. 2002;3:338–40. doi: 10.1038/ni0402-338. [DOI] [PubMed] [Google Scholar]
- 50.Filippi CM, von Herrath MG. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: viruses, autoimmunity and immunoregulation. Clin Exp Immunol. 2010;160:113–9. doi: 10.1111/j.1365-2249.2010.04128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawasaki T, Kawai T, Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev. 2011;243:61–73. doi: 10.1111/j.1600-065X.2011.01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5:219–26. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- 53.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Root-Bernstein R, Vonck J, Podufaly A. Antigenic complementarity between coxsackie virus and streptococcus in the induction of rheumatic heart disease and autoimmune myocarditis. Autoimmunity. 2009;42:1–16. doi: 10.1080/08916930802208540. [DOI] [PubMed] [Google Scholar]
- 55.Alotaibi S, Kennedy J, Tellier R, Stephens D, Banwell B. Epstein-Barr virus in pediatric multiple sclerosis. JAMA. 2004;291:1875–9. doi: 10.1001/jama.291.15.1875. [DOI] [PubMed] [Google Scholar]
- 56.Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: the role of infection. Ann Neurol. 2007;61:288–99. doi: 10.1002/ana.21117. [DOI] [PubMed] [Google Scholar]
- 57.Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–45. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS, Virgin HWt. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hill Gaston JS, Lillicrap MS. Arthritis associated with enteric infection. Best Pract Res Clin Rheumatol. 2003;17:219–39. doi: 10.1016/s1521-6942(02)00104-3. [DOI] [PubMed] [Google Scholar]
- 60.Scher JU, Abramson SB. The microbiome and rheumatoid arthritis. Nat Rev Rheumatol. 2011;7:569–78. doi: 10.1038/nrrheum.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Townes JM. Reactive arthritis after enteric infections in the United States: the problem of definition. Clin Infect Dis. 2010;50:247–54. doi: 10.1086/649540. [DOI] [PubMed] [Google Scholar]
- 62.Townes JM, Deodhar AA, Laine ES, Smith K, Krug HE, Barkhuizen A, Thompson ME, Cieslak PR, Sobel J. Reactive arthritis following culture-confirmed infections with bacterial enteric pathogens in Minnesota and Oregon: a population-based study. Ann Rheum Dis. 2008;67:1689–96. doi: 10.1136/ard.2007.083451. [DOI] [PubMed] [Google Scholar]
- 63.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–94. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 64.Targan SR, Karp LC. Defects in mucosal immunity leading to ulcerative colitis. Immunol Rev. 2005;206:296–305. doi: 10.1111/j.0105-2896.2005.00286.x. [DOI] [PubMed] [Google Scholar]
- 65.Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci USA. 2009;106:19256–61. doi: 10.1073/pnas.0812681106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 67.Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–41. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, McCoy KD, Macpherson AJ. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806. doi: 10.1016/j.immuni.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 69.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–57. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 71.Hase K, Takahashi D, Ebisawa M, Kawano S, Itoh K, Ohno H. Activation-induced cytidine deaminase deficiency causes organ-specific autoimmune disease. PLoS One. 2008;3:e3033. doi: 10.1371/journal.pone.0003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vaahtovuo J, Munukka E, Korkeamaki M, Luukkainen R, Toivanen P. Fecal microbiota in early rheumatoid arthritis. J Rheumatol. 2008;35:1500–5. [PubMed] [Google Scholar]
- 73.Abdollahi-Roodsaz S, Joosten LA, Koenders MI, Devesa I, Roelofs MF, Radstake TR, Heuvelmans-Jacobs M, Akira S, Nicklin MJ, Ribeiro-Dias F, van den Berg WB. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Investig. 2008;118:205–16. doi: 10.1172/JCI32639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–27. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Monach PA, Mathis D, Benoist C. The K/BxN arthritis model. Curr Protoc Immunol. 2008:22. doi: 10.1002/0471142735.im1522s81. Chapter 15: Unit 15. [DOI] [PubMed] [Google Scholar]
- 76.Pozzilli P, Signore A, Williams AJ, Beales PE. NOD mouse colonies around the world–recent facts and figures. Immunol Today. 1993;14:193–6. doi: 10.1016/0167-5699(93)90160-M. [DOI] [PubMed] [Google Scholar]
- 77.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 78.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–13. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kranich J, Maslowski KM, Mackay CR. Commensal flora and the regulation of inflammatory and autoimmune responses. Semin Immunol. 2011;23:139–45. doi: 10.1016/j.smim.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 80.Pang IK, Iwasaki A. Control of antiviral immunity by pattern recognition and the microbiome. Immunol Rev. 2012;245:209–26. doi: 10.1111/j.1600-065X.2011.01073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Nat Acad Sci USA. 2011;108(Suppl 1):4615–22. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–41. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- 83.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165:6107–15. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- 84.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 85.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–7. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O’Shea JJ, Cua DJ. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–24. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 89.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 91.Ghoreschi K, Laurence A, Yang XP, Hirahara K, O’Shea JJ. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 2011;32:395–401. doi: 10.1016/j.it.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gonzalez-Garcia I, Zhao Y, Ju S, Gu Q, Liu L, Kolls JK, Lu B. IL-17 signaling-independent central nervous system autoimmunity is negatively regulated by TGF-beta. J Immunol. 2009;182:2665–71. doi: 10.4049/jimmunol.0802221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hu Y, Ota N, Peng I, Refino CJ, Danilenko DM, Caplazi P, Ouyang W. IL-17RC is required for IL-17A- and IL-17F-dependent signaling and the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4307–16. doi: 10.4049/jimmunol.0903614. [DOI] [PubMed] [Google Scholar]
- 94.Ishigame H, Nakajima A, Saijo S, Komiyama Y, Nambu A, Matsuki T, Nakae S, Horai R, Kakuta S, Iwakura Y. The role of TNFalpha and IL-17 in the development of excess IL-1 signaling-induced inflammatory diseases in IL-1 receptor antagonist-deficient mice. Ernst Schering Research Foundation workshop. 2006:129–53. doi: 10.1007/3-540-37673-9_8. [DOI] [PubMed] [Google Scholar]
- 95.Lubberts E. IL-17/Th17 targeting: on the road to prevent chronic destructive arthritis? Cytokine. 2008;41:84–91. doi: 10.1016/j.cyto.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 96.Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–6. doi: 10.1172/JCI21404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm Bowel Dis. 2006;12:382–8. doi: 10.1097/01.MIB.0000218764.06959.91. [DOI] [PubMed] [Google Scholar]
- 98.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–90. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 99.Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–70. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 100.Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000;164:2832–8. doi: 10.4049/jimmunol.164.5.2832. [DOI] [PubMed] [Google Scholar]
- 101.Rasmussen TK, Andersen T, Hvid M, Hetland ML, Horslev-Petersen K, Stengaard-Pedersen K, Holm CK, Deleuran B. Increased interleukin 21 (IL-21) and IL-23 are associated with increased disease activity and with radiographic status in patients with early rheumatoid arthritis. J Rheumatol. 2010;37:2014–20. doi: 10.3899/jrheum.100259. [DOI] [PubMed] [Google Scholar]
- 102.Melis L, Vandooren B, Kruithof E, Jacques P, De Vos M, Mielants H, Verbruggen G, De Keyser F, Elewaut D. Systemic levels of IL-23 are strongly associated with disease activity in rheumatoid arthritis but not spondyloarthritis. Ann Rheum Dis. 2010;69:618–23. doi: 10.1136/ard.2009.107649. [DOI] [PubMed] [Google Scholar]
- 103.Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 104.Ishizu T, Osoegawa M, Mei FJ, Kikuchi H, Tanaka M, Takakura Y, Minohara M, Murai H, Mihara F, Taniwaki T, Kira J. Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain J Neurol. 2005;128:988–1002. doi: 10.1093/brain/awh453. [DOI] [PubMed] [Google Scholar]
- 105.Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–55. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vaknin-Dembinsky A, Balashov K, Weiner HL. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol. 2006;176:7768–74. doi: 10.4049/jimmunol.176.12.7768. [DOI] [PubMed] [Google Scholar]
- 107.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–61. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, Sakuraba A, Kitazume MT, Sugita A, Koganei K, Akagawa KS, Hibi T. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Investig. 2008;118:2269–80. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rovedatti L, Kudo T, Biancheri P, Sarra M, Knowles CH, Rampton DS, Corazza GR, Monteleone G, Di Sabatino A, Macdonald TT. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut. 2009;58:1629–36. doi: 10.1136/gut.2009.182170. [DOI] [PubMed] [Google Scholar]
- 111.Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645–9. doi: 10.1046/j.1523-1747.1998.00347.x. [DOI] [PubMed] [Google Scholar]
- 112.Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, Bowman EP, Krueger JG. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128:1207–11. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 113.Liu Y, Helms C, Liao W, Zaba LC, Duan S, Gardner J, Wise C, Miner A, Malloy MJ, Pullinger CR, Kane JP, Saccone S, Worthington J, Bruce I, Kwok PY, Menter A, Krueger J, Barton A, Saccone NL, Bowcock AM. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008;4:e1000041. doi: 10.1371/journal.pgen.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nunez C, Dema B, Cenit MC, Polanco I, Maluenda C, Arroyo R, de las Heras V, Bartolome M, de la Concha EG, Urcelay E, Martinez A. IL23R: a susceptibility locus for celiac disease and multiple sclerosis? Genes Immun. 2008;9:289–93. doi: 10.1038/gene.2008.16. [DOI] [PubMed] [Google Scholar]
- 116.Hazlett J, Stamp LK, Merriman T, Highton J, Hessian PA. IL-23R rs11209026 polymorphism modulates IL-17A expression in patients with rheumatoid arthritis. Genes Immun. 2011 doi: 10.1038/gene.2011.80. [DOI] [PubMed] [Google Scholar]
- 117.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, Matsunami N, Ardlie KG, Civello D, Catanese JJ, Leong DU, Panko JM, McAllister LB, Hansen CB, Papenfuss J, Prescott SM, White TJ, Leppert MF, Krueger GG, Begovich AB. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–90. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–52. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–4. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 122.Vanden Eijnden S, Goriely S, De Wit D, Willems F, Goldman M. IL-23 up-regulates IL-10 and induces IL-17 synthesis by polyclonally activated naive T cells in human. Eur J Immunol. 2005;35:469–75. doi: 10.1002/eji.200425677. [DOI] [PubMed] [Google Scholar]
- 123.Neurath MF. IL-23: a master regulator in Crohn disease. Nat Med. 2007;13:26–8. doi: 10.1038/nm0107-26. [DOI] [PubMed] [Google Scholar]
- 124.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12:255–63. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hirota K, Martin B, Veldhoen M. Development, regulation and functional capacities of Th17 cells. Semin Immunopathol. 2010;32:3–16. doi: 10.1007/s00281-009-0187-y. [DOI] [PubMed] [Google Scholar]
- 127.O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4 + T cells. Science. 2010;327:1098–102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17 + T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 129.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, Bowman EP, Sgambellone NM, Chan CC, Caspi RR. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205:799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chitnis T, Najafian N, Benou C, Salama AD, Grusby MJ, Sayegh MH, Khoury SJ. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108:739–47. doi: 10.1172/JCI12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bending D, De la Pena H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B, Cooke A. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Investig. 2009;119:565–72. doi: 10.1172/JCI37865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol. 2009;39:216–24. doi: 10.1002/eji.200838475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–7. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 135.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–73. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 136.Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, Kamoun M, Rostami A. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–10. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- 137.Van Belle TL, Esplugues E, Liao J, Juntti T, Flavell RA, von Herrath MG. Development of autoimmune diabetes in the absence of detectable IL-17A in a CD8-driven virally induced model. J Immunol. 2011;187:2915–22. doi: 10.4049/jimmunol.1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol. 2006;7:1151–6. doi: 10.1038/ni1391. [DOI] [PubMed] [Google Scholar]
- 139.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O’Shea JJ. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. 2010;467:967–71. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sujino T, Kanai T, Ono Y, Mikami Y, Hayashi A, Doi T, Matsuoka K, Hisamatsu T, Takaishi H, Ogata H, Yoshimura A, Littman DR, Hibi T. Regulatory T cells suppress development of colitis, blocking differentiation of T-helper 17 into alternative T-helper 1 cells. Gastroenterology. 2011;141:1014–23. doi: 10.1053/j.gastro.2011.05.052. [DOI] [PubMed] [Google Scholar]
- 141.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–98. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–49. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K, Takeda K. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455:808–12. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 144.Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, Eberl G, Snel J, Kelly D, Cerf-Bensussan N. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31:677–89. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 145.Kruis W, Fric P, Pokrotnieks J, Lukas M, Fixa B, Kascak M, Kamm MA, Weismueller J, Beglinger C, Stolte M, Wolff C, Schulze J. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut. 2004;53:1617–23. doi: 10.1136/gut.2003.037747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kwon HK, Lee CG, So JS, Chae CS, Hwang JS, Sahoo A, Nam JH, Rhee JH, Hwang KC, Im SH. Generation of regulatory dendritic cells and CD4+ Foxp3+ T cells by probiotics administration suppresses immune disorders. Proc Natl Acad Sci U S A. 2010;107:2159–64. doi: 10.1073/pnas.0904055107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Livingston M, Loach D, Wilson M, Tannock GW, Baird M. Gut commensal Lactobacillus reuteri 100–23 stimulates an immunoregulatory response. Immunol Cell Biol. 2010;88:99–102. doi: 10.1038/icb.2009.71. [DOI] [PubMed] [Google Scholar]
- 148.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–7. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh CS. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–4. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee AS, Gibson DL, Zhang Y, Sham HP, Vallance BA, Dutz JP. Gut barrier disruption by an enteric bacterial pathogen accelerates insulitis in NOD mice. Diabetologia. 2010;53:741–8. doi: 10.1007/s00125-009-1626-y. [DOI] [PubMed] [Google Scholar]
- 151.Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci USA. 2011;108:11548–53. doi: 10.1073/pnas.1108924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Marks BR, Nowyhed HN, Choi JY, Poholek AC, Odegard JM, Flavell RA, Craft J. Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulate peripheral inflammation. Nat Immunol. 2009;10:1125–32. doi: 10.1038/ni.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lochner M, Berard M, Sawa S, Hauer S, Gaboriau-Routhiau V, Fernandez TD, Snel J, Bousso P, Cerf-Bensussan N, Eberl G. Restricted microbiota and absence of cognate TCR antigen leads to an unbalanced generation of Th17 cells. J Immunol. 2011;186:1531–7. doi: 10.4049/jimmunol.1001723. [DOI] [PubMed] [Google Scholar]
- 154.Torchinsky MB, Garaude J, Martin AP, Blander JM. Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature. 2009;458:78–82. doi: 10.1038/nature07781. [DOI] [PubMed] [Google Scholar]
- 155.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–12. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 156.Torchinsky MB, Garaude J, Blander JM. Infection and apoptosis as a combined inflammatory trigger. Curr Opin Immunol. 2010;22:55–62. doi: 10.1016/j.coi.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]