

Abstract
Hematological malignancies manifest as lymphoma, leukemia, and myeloma, and remain a burden on society. From initial therapy to endless relapse-related treatment, societal burden is felt not only in the context of healthcare cost, but also in the compromised quality of life of patients. Long-term therapeutic strategies have become the standard in keeping hematological malignancies at bay as these cancers develop resistance to each round of therapy with time. As a result, there is a continual need for the development of new drugs to combat resistant disease in order to prolong patient life, if not to produce a cure. This review aims to summarize advances in targeting lymphoma, leukemia, and myeloma in both cutting-edge and well established platforms. Current standard of treatment will be reviewed for these malignancies and emphasis will be made on new therapy development in the areas of antibody engineering, epigenetic small molecule inhibiting drugs, vaccine development, and chimeric antigen receptor cell engineering. In addition, platforms for the delivery of these and other drugs will be reviewed including antibody-drug conjugates, micro- and nanoparticles, and multimodal hydrogels. Lastly, we propose that tissue engineered constructs for hematological malignancies are the missing link in targeted drug discovery alongside mouse and patient-derived xenograft models.
Keywords: lymphoma, leukemia, multiple myeloma, B cell receptor, biomaterial, vaccine, epigenetic inhibitors, nanoparticles, EZH2
Graphical abstract
1. Introduction to B and T cell tumors and need for drug discovery and delivery technologies
Hematological malignancies are lymphoproliferative disorders of B and T cells that comprise of lymphomas, leukemias, and myelomas. Occurrences of these cancers can be attributed to a variety of factors including but not limited to aging, exposure to toxins, radiations, and chemicals, repeated infections and a weakened immune system, as well as side effects of several biologics (e.g. Humira™). Chronic use of immunosuppressive medications for diseases such as inflammatory bowel disease, weakens the host immune response allowing development of lymphoid malignancies1. Most of the hematological malignancies represent a heterogeneous disease associated with a variety of clinical presentations and genetic diversity2. The heterogeneity of these hematological malignancies present unique challenges to conventional cancer treatment. Owing to the inherent heterogeneity of B and T cell tumors3, there are very different responses to treatments. Many patients with these tumors do not show a durable response to treatments, necessitating salvage therapies such as autologous stem cell transplant that often have poor patient outcomes. A myriad of independently predictive biomarkers for resistance have been identified, including gene expression signature, stromal signatures, epigenetic silencing of specific genes, and specific mutations4, 5. But none are sufficient to predict resistance in a given patient and few are helpful in guiding selection of targeted therapies. Therefore, new treatments are needed to improve clinical outcome of tumors of immune cells. Moreover, many of the first-line immunochemotherapy regimens are too toxic to be tolerated by the elderly or by people in the developing world, where infectious diseases are more difficult to manage following treatment. Clearly, novel approaches to tumor treatment are needed across drug discoveries and delivery of therapeutics.
Lymphoma represents an umbrella term applied to over 30 distinct clinical malignancies of B and T cells. While T cell lymphomas are highly prevalent, most lymphomas originate from B lymphocytes, and their diversity can often be traced to the normal precursor B cell6. The bulk of these B cells are antigen-experienced germinal center or post-germinal center B cells which undergo massive proliferation, somatic hypermutation, and antibody class switching6. Lymphoma originates in the lymph nodes and the lymphatics and comprises of two categories of diagnosis: Hodgkins and Non-Hodgkins. Hodgkins lymphoma is identified by the presence of Reed Sternberg cells, a form of malignant mature B lymphocytes7. Non-Hodgkin lymphoma encompasses all other forms of lymphocytic and myeloid cancers with origins in the lymphatics, and includes several subtypes including hairy cell lymphoma, mantle cell lymphoma, diffuse large B cell lymphoma (DLBCL), Burkitt lymphoma8, and peripheral T cell lymphomas. DLBCL is the most common lymphoma representing ~30% of all B cell Non-Hodgkin lymphomas, and gene expression profiling has allowed sub-classification into germinal center B cell-like (GCB) DLBCL and activated B cell-like (ABC) DLBCL subtypes. Improved therapies are needed for all DLBCLs but most urgently for ABC-DLBCLs, which are the most chemo-resistant (5-year overall survival ~ 45%2, 4). Peripheral T cell lymphomas are a molecular and clinical heterogeneous group of non-Hodgkin lymphomas that include, as the most prevalent sub-types, the nodal type peripheral T cell lymphoma not otherwise specified, anaplastic large cell lymphoma, and angioimmunoblastic T cell lymphoma. Although some subtypes may follow a more benign prolonged course, the vast majority of T cell lymphoma patients have poor prognoses due to the combination of an aggressive clinical course and the lack of specific treatments9, 10. Most T cell lymphomas have been commonly treated with therapeutic regimens originally designed for B cell lymphomas, resulting in poor efficacy and frequent relapses10, 11. 2016 incident estimates for the United States predicts 136,960 new lymphoid neoplasms with the most significant forms of lymphoma, with regards to prevalence, remain to be small lymphocytic lymphoma (chronic lymphocytic leukemia), mantle cell lymphoma, diffuse large B-cell lymphoma, follicular lymphoma, and multiple myeloma12.
On the other hand, leukemia are cancers of B cells and other leukocytes with origins in the bone. There are four subgroups of leukemia, defined by the age at onset (acute or chronic) and the category of cell type that is afflicted (myeloid or lymphoid): acute myeloid leukemia (AML), acute lymphoid leukemia (ALL), chronic myeloid leukemia (CML), and chronic lymphocytic leukemia (CLL). Estimates for 2016 predict over 60,000 new cases and over 24,000 leukemia related deaths in the United States alone13. While the death rates for acute lymphocytic and chronic myeloid leukemia have significantly dropped since the introduction of Bruton’s tyrosine kinase inhibitors in leukemia treatment13, there remains room for drug discovery and development for tyrosine kinase inhibitor resistant leukemias. Finally, a third type of hematological malignancy is Multiple myeloma, which is a cancer of plasma cells. Plasma cells, in the healthy state, mass produce antibodies in the body. Estimates from 2015 predict that the number of myeloma cases in the United States in 2020 will increase to 11,581 cases from the 9,083 cases in 201014. In addition, the median survival for multiple myeloma is only 6 years post-diagnosis and there has yet to be a cure for the disease15. Treatment options vary by patient age, as the median age at diagnosis is 69 years old15.
With incredible diversity in cancerous cell types and behaviors within lymphoma, leukemia, and myeloma, diverse therapeutic strategies are required for disease control and achieving remission. Current standards of care for common lymphomas, leukemias, and myeloma are summarized in Table 1 by disease progression stage. It should be noted though, that disease treatment strategy also largely depends on other factors like age and comorbidities rather and the stage of cancer progression and these elements are not represented in this table. From therapeutic delivery standpoint, major challenges within the cancer therapeutics lie with innate drug toxicity and barriers against intracellular drug delivery. The scope of this review is to highlight the advancements in lymphoma, leukemia, and myeloma therapies from drug discovery and drug delivery standpoint. We will review biomolecule engineering including antibody based therapies, targeted enzymes, and small molecule inhibitors. In addition, new cell, tissue, and material engineering techniques will be covered with respect to their impact on potential treatments for the hematological malignances highlighted here.
Table 1.
Gold standards of treatment for most common lymphoma, leukemia and myeloma subtypes.
| Malignancy | Early stage therapy | Late stage therapy | Therapy after primary refractory | Post remission therapy | Therapy after relapse |
|---|---|---|---|---|---|
| Hodgkin lymphoma | ABVD combination chemo with IFRT to lymph nodes223’224 | ABVD or BEACOPP225 | HDC with ASCT224 | – | Treatment with initial chemo- 80% CR rate226; vinorelbine227 or gemcitabine228 |
| DLBCL | R-CHOP229 | – | HDC with ASCT in <60 years old230 | – | Rituximab, Gemcitabine, and oxiplatin231 HDC with ASCT230 |
| Follicular lymphoma | Radiation therapy232 | (when symptomatic) R-CHOP or BR233 | R-CHOP234 | – | – |
| AML | Induction therapy of cytarabine and and doxorubicin235; followed by consolidation chemotherapy or allogeneic stem cell transplant236 | – | – | Cytarabine consolidation therapy235; Allogeneic HSCT237 |
Allogeneic stem cell transplant237, 238 |
| ALL | Hyper-CVAD239; Hyper-CVAD and Imatinib in Philadelphia chromosome translocation240 | – | – | Allogeneic stem cell transplant239 | Hyper CVAD241 high dose cytarabine and anthracycline242; methotrexate and asparaginase243; VAD244; methotrexate and asparaginase245 |
| CML | TKIs: imatinib mesylate246, dasatinib247, or nilotinib248 | After resistance to initial TKIs: Bosutinib249, ponatinib,250 | Switch to unused TKI251 | – | After failure of 2 or more TKIs: allogeneic stem cell transplant252 |
| CLL | FCR253 or BR254 in fit patients; Rituximab255 or obinutuzumab256 plus chlorambucil in unfit patients | – | – | – | Kinase inhibitors: Idelalisib257 or Ibrutinib258; Allogeneic stem cell transplant259; or Alemtuzumab260, 261 |
| Multiple Myeloma | Thalidomide and dexamethasone262, lenalidomide263, bortezomib264 | – | ASCT265 | – | Bortezomib, thalidomide, or lenalidomide266; ASCT267 |
Acronyms used: doxorubicin, bleomycin, vinblastine, dacarbazine (ABVD combination chemo); Involved-field radiation therapy (IFRT); bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone (BEACOPP); High-dose chemotherapy (HDC); autologous stem cell transplant (ASCT); complete response (CR); rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone (R-CHOP); Bendamustine plus rituximab (BR); hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (hyper-CVAD); vincristine, doxorubicin, and dexamethasone (VAD); tyrosine kinase inhibitors (TKIs), fludarabine, cyclophosphamide, rituximab (FCR)
2. Engineered Antibodies, Enzymes, and Antibody-Drug Conjugates to Improve Therapies against B and T Cell Tumors
Engineered therapeutic antibodies as drugs have resulted in substantial benefits to public health, in particular to the area of cancer. To date, more than 20 recombinant therapeutic antibodies have received approval for the treatment of various diseases. These antibodies are mostly of the immunoglobulin G (IgG) class 1. In order to achieve clinical efficacy, two hallmark functionalities have been identified for engineered therapeutic antibodies: target-specific binding by the antigen binding Fab fragment and Fc domain mediated effector functions. The Fc function involves antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity16, 17. Generally, engineered antibodies eliminate target cancer cells through four separate pathways: antibody-dependent cellular cytotoxicity, antibody-dependent cellular phagocytosis, complement-dependent cytotoxicity, and through the cancer specific delivery of conjugated drug18. Fc receptors (FcRs) are a group of glycoproteins organized into classes, such as FcαR, FcγR and FcεR; FcγRs are expressed mostly on leukocytes, and are the class of receptors most relevant for the function of therapeutic antibodies. FcRs are the link between antigen-bound antibody and immune effector cell elimination. FcγRs have been categorized according to their affinity for specific IgG subclasses and the type of signaling pathway that they trigger; that is, whether it is inhibitory or activating19. Fc glycosylation is necessary for therapeutic antibodies to elicit effector functions20. Complement activation by IgG is made possible by asparagine 297 with its associated glycans in the CH2 domain of the antibody21. Such glycans are protected within the secondary and tertiary structure, supporting the fact that glycosylation of this area is critical for proper binding to FcγR despite minimal contact between the glycan and its respective receptor22. Along similar lines, manipulation of another saccharide, fucose, has shown to enhance binding of IgG1 to FcγRIIIa leading to greater efficiency in binding and antibody-dependent cellular phagocytosis downstream23–26
Antibody engineering has been implemented for the treatment of cutaneous T cell lymphoma27, 28, follicular lymphoma29, 30, diffuse large B cell lymphoma30, Hodgkins lymphoma31, 32, and Burkitt’s lymphoma33. Antibody engineering for hematological malignancies gained popularity after the landmark success of rituximab (sold under the brand name Rituxan), a CD20 targeting, chimeric anti-human monoclonal IgG1 antibody that effectively targets several cell types within the B cell lineage34,35. Rituximab has achieved clinical success in B cell malignancies including follicular lymphoma29, 36 diffuse large B cell lymphoma35, 37, and relapsed CLL38, and has served as a model for the clinical potential of antibody engineering in hematological malignancies. Antibody engineering for lymphoma therapeutics targets CD2029, 30, 35, 37, 39–41, killer cell immunogloblulin-like receptors41, CD2230, 33, C-C motif chemokine receptor 4 (CCR4)27, 28, 42, 43, CD8029, CD2744, CD4032, CD1945, CD3031, CD7046 and even soluble interleukin (IL)-647, IL-231, and IL-1231. In the case of B cell malignancies, additional effector mechanisms are present in which direct cytotoxicity occurs from antibody internalization and the triggering of apoptotic or non-apoptotic events48. Likewise, Mogamulizumab (KW-0761), an defucosylated humanized monoclonal antibody was found to successfully target C-C motif chemokine receptor 4 (CCR4) in both peripheral and cutaneous T cell lymphomas in CCR4 positive patients in a phase II Japanese study42. It is important to consider genetic diversity and polymorphism in designing therapeutic antibodies. Genetic analyses have revealed that lymphoma patients carrying the FcγRIIIa-v158 allotype responded better to treatment with the chimeric CD20-specific IgG1 antibody rituximab than those patients carrying the FcγRIIIa-F158 allotype49, 50. In 2014, Obinutuzumab, also known as GA101 or Gazyva51, a glycoengineered type II humanized anti-CD20 monoclonal antibody was named a successor to its own long-time blockbuster drug Rituxan (rituximab). Gazyva, a fully humanized antibody, has a dual mechanism of action where the antibody binds and directly kills B cells by introducing caspase-independent programmed cell death, and through antibody dependent cytotoxicity it recruits the immune system to attack B cells. Gazyva is a type II antibody, which binds to a different epitope on CD20, rather than a type I antibody like Rituxan, which engages complement dependent cytotoxicity and antibody dependent cytotoxicity but does not potently induce programmed cell death upon binding to CD20. Finally, the discovery of high expression levels of programmed cell death 1 Ligand 1(PD-L1) in classical Hodgkin’s lymphoma by programmed cell death protein 1 (PD-1), a key immune-inhibitory molecule expressed on T cells has led to the revolutionary field of immunotherapy. It was discovered that PD-L1 expressing cells can escape T-cell-mediated cellular cytotoxicity by exploiting the inhibitory PD-1 immune checkpoint. Therefore, blocking the PD-L1 with therapeutic antibodies that block the PD-1–PD-L1 axis induce durable clinical responses against a growing list of solid tumors as well as against both Hodgkin’s’ and Non-Hodgkin’s lymphomas, including Diffuse large B Cell Lymphomas52–54.
A more advanced and rapidly emerging use of antibody engineering is in targeted drug delivery of drugs and other molecules. Over the years, antibody engineering has evolved from monoclonal antibody development to the engineering of antibody drug conjugates (ADCs) for targeted, site-specific cancer drug delivery. ADCs are engineered by chemically conjugating single or multiple cytotoxic small-molecule drugs to a surface antigen receptor-targeted antibody55, 56, and function as targeted drug delivery system to guide the toxic conjugated drugs specifically to malignant cells while minimizing potential systemic toxicity. Multiple mechanisms have been exploited for cytosolic delivery. For example, the reducing nature of the cytosol has been used extensively in protein-conjugate chemistry to trigger release of the payload56, 57. Anti-cancer drugs like dimeric pyrrolobenzodiazepine have been conjugated onto monoclonal antibodies to target cancerous cell types46. Pyrrolobenzodiazepine combats uncontrolled cell growth by crosslinking double stranded DNA, effectively damaging beyond the ability of cellular DNA repair mechanisms therefore triggering target cell death46. Similarly, calicheamicin has also been incorporated into ADCs as the therapeutic component of inotuzumab ozogamicin for follicular lymphoma, DLBCL, and refractory Non-Hodgkin’s lymphomas30. This anti-tumor antibiotic cleaves double stranded DNA with remarkable specificity58, and ADCs with this drug have proven to be effective in refractory Non-Hodgkin’s lymphomas in combined treatment with rituximab. Other ADC drugs implemented for the treatment of lymphoma include DM4, a maytan-sinoid that blocks cell division at the microtubule level45. For T cell lymphomas, such as the relapsed anaplastic large cell lymphomas, SGN-35 (Brentuximab vedotin, Adcetris®) has emerged as a new anti-CD30 ADC combination with the potent synthetic drug monomethyl auristatin E. The antibody is conjugated through an enzyme cleavable linker, which makes it stable in the bloodstream but enzymatically cleavable upon internalization into CD30-expressing T cell tumors, releasing monomethyl auristatin E and leading to cell death. For better understanding of new targets of therapy in T-cell lymphomas, readers are recommended to consult the review by Erter et al.59
In addition to Non-Hodgkin’s lymphomas, CD20 (rituximab), CD33(gemtuzumab ozogamicin), CD22 (inotzumab ozogamacin and epratuzumab), CD19 (blinatumomab), and CD52(Alemtuzumab/Campath 1-H) have served as targets of antibody therapy for the treatment of acute lymphoblastic leukemia60. Recent research efforts have focused on new cellular surface targets like CD2744 and EphA361, as well as improving cancer eradication though development of antibody drug conjugates for CD2262. Anti-CD22 antibody therapies have arguably been the most successful, and clinically translatable in treating ALL and other leukemias and lymphomas. Notable innovations to antibody-drug conjugates have evolved from the success of anti-CD22 monoclonal antibody therapies. Since upon binding with CD22, the antibody therapy is endocytosed, CD22 antibodies are an intriguing drug delivery platform for drugs with intracellular actions. Satake et al. used monoclonal anti-CD22 antibodies to traffic antisense oligonucleotides for inhibiting translation of MYC-associated factor X (MAX) dimerization protein 3 (MXD3), an important transcription factor involved in preB-cell acute lymphoblastic leukemia62. Results of their preclinical study showed that the conjugate successfully trafficked the oligonucleotides to the leukemia cells without off target effects and at one-twentieth of the dose required for similar effects from anti-CD22 monoclonal antibody therapy alone62,63.
Anti-CD33 ADCs have also been developed for the targeting of leukemia cells and multiple generations of CD33 targeted ADCs have evolved, as summarized in Fig. 1 and reviewed in detail elsewhere56. Gemtuzumab ozogamicin (GO) is a calichaemicin linked to a CD33-specific human antibody and functions by cleavage of the reductively labile disulfide-based linker57. GO was the first antibody-drug conjugate to be approved by the FDA for acute promyelocytic leukemia63. The treatment of other AML subtypes has proved to be less efficacious with antibody-based therapies as gemtuzumab ozogamicin (GO) with induction and post-consolidation chemotherapeutic regimens did not improve final outcomes for younger patients64. With a lack of success and mounting safety concerns, GO was removed from the market in 2010, and is no longer available commercially64. Nevertheless, clinical trials are still underway for the discovery of safe applications of GO including maintenance of the remission state following anti-cancer treatment with trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia65. Recent positive results with minimized toxicity and safety concerns in the latest GO phase III trial have suggested that there are applications for GO where benefits outweigh risks that manifested in prior applications65. Further work in this area has led to second and third generations of ADCs, where components of third generation vadastuxumab talirine include anti-CD33 humanized monoclonal antibody with two drug warheads of pyrrolobenzodiaezpine connected by protease cleavable linker56 (Fig. 1). For greater depth on drug discovery in AML, see the review article by Sallman et al.66.
Figure 1. Components of antibody-drug conjugates for targeting acute myeloid leukemia.
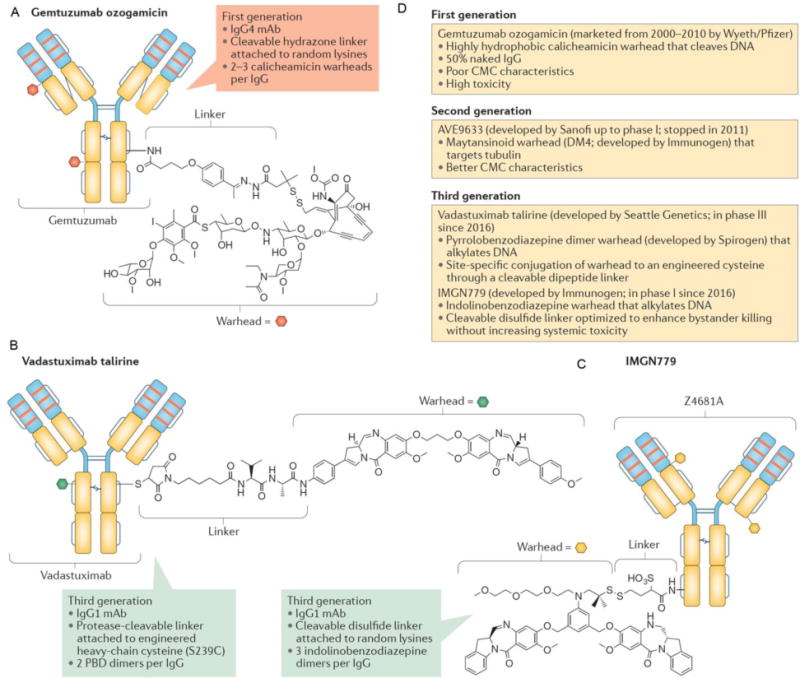
(A) Structure of first generation antibody-drug conjugate, gemtuzumab ozogamacin, including humanized anti-CD33 IgG4 monoclonal antibody, hydrazine linker at lysine residues, and calicheamicin drug. (B) Components of third generation vadastuxumab talirine including anti- CD33 humanized monoclonal antibody with two drug warheads of pyrrolobenzodiaezpine connected by protease cleavable linker. (C) Components of third generation antibody-drug conjugate IMGN779 including humanized IgG1 monoclonal antibody with three drug units consisting of a cleavable disulfide linker and indolinobenzodiazepine. (D) Summary of benefits and trade-offs for utilizing first, second, or third generation antibody-drug conjugates for treatment of acute myeloid leukemia. Adapted with permission from56.
Treatment of CLL has shown success in phase I study of Moxetumomab Pasudotox, an immunoconjugate with specificity for CD2267. When compared to rituximab and cladribine – the current standards of care for CLL– fewer side effects were observed, and healthy lymphocyte populations showed better rebounding post-treatment. Despite promising results, several patients treated with Moxetumomab pasudotox were forced to end treatment before complete response after developing antibodies against the immunotoxin67. As such, Moxetumomab pasudotox was recently redesigned into a new immunotoxin, LMB11, with less antigenicity68. Reduced immunogenicity in LMB11 was achieved by removing excess regions and modifications to surface amino acids of the Pseudomonas exotoxin68. Because the designs for reduced immunogenicity were made with humans in mind, mouse studies could not be performed to demonstrate reduced immunogenicity in humans68, although antigenicity studies suggest that LMB11 has a longer half-life in the blood while being tolerated at high doses in mice68.
Antibody engineering for CML has taken a different approach than engineering for other leukemias. Monoclonal antibodies have been designed to block the interleukin-1 receptor accessory protein (ILRAP/IL1R3) which is vital for IL-1 signaling pathways, and is expressed more consistently than IL-1 in CML69. IL-1 signaling has been shown to promote strong proliferation in CML stem cells and is exclusively expressed on primitive CML cells and not healthy hematopoietic stem cells69. Specific expression of ILRAP therefore allows for antibody-dependent cellular cytotoxicity and phagocytosis by NK cells and macrophages, respectively69.
Several antibody-based therapeutics have also been developed to combat multiple myeloma. In recent research history, monoclonal antibody therapies for multiple myeloma target CD4870, signaling lymphocytic activation molecule family 7 protein (SLAMF)71, and IL-647. CD48 has been proposed as a target form monoclonal antibody therapies because it is known to specifically be expressed by cells within the hematopoietic lineage70. This protects the patient from off target binding that is unrelated to their myeloma. CD48 was found to be expressed on over 90% of multiple myeloma cells in 22 patients out of 24 in a pilot study by Hosen et al, proving that it could be a powerful target for antibody therapy70. That same research group went on to develop a murine anti-CD48 monoclonal antibody to drive myeloma specific cytotoxicity70. Impressive results were shown in a subcutaneous tumor mouse models despite limited evidence of antibody dependent cell cytotoxicity in in vitro studies70. Due to the murine origin of the pilot antibody, human trials were unable to be performed to show the full potential of anti-CD48 monoclonal antibodies in treating multiple myeloma, although the potential targeting of healthy lymphocytes and monocytes may cause the risks of the such a therapy to outweigh the benefits in such a trial.
As a result, other attempts at monoclonal antibody therapy have been made for the treatment of multiple myeloma. SLAMF has been proposed as an alternate target to CD48, because it is more widely expressed in myeloma cells while being independent of cytogenic abnormalities inherent to the myeloma71. The humanized IgG1 monoclonal anti-SLAMF antibody (elotuzumab) was approved by the FDA in 2015 after a successful phase 3 clinical trial in relapsed myeloma patients71. Although elotuzumab resulted more frequently in adverse reactions of all severities, the clinical trial showed it increased the response rate and extended the period for additional treatment a full year when compared to lenaliodomide plus dexamethasone71. In addition to CD48 and SLAMF, IL-6 was assessed as a target for antibody therapy for the treatment of myeloma. Slituximab is an chimeric anti-IL6 monoclonal antibody that showed promising applications for myeloma and Castleman’s disease47. Despite successful phase one trials, further exploration in a phase two study failed to show that outcomes in relapsed myeloma patients were improved by inclusion of Slituximab with bortezomib when compared to bortexomib in combination with a placebo72.
Modest and insignificant success with monoclonal antibodies for myeloma has triggered recent development of antibody-drug conjugates for its treatment. SGN-CD352A, an anti- SLAMF (CD352) cysteine antibody conjugated to pyrrolobenzodiazepine has been shown to interact with myeloma cells through clathrin-dependent endocytosis73. Endocytosis of SGN-CD325A was followed by caspase 3 and 7 dependent apoptosis had minimal affect to off target cell types73. Preclinical in vitro and in vivo studies in mouse xenograft models suggest that SGN-CD325A could prove to be a viable option for treating multiple myeloma in clinical trials73. Antibody-drug conjugates for treating multiple myeloma have also been recently developed targeting B-cell maturation antigen (BCMA) with monomethyl auristatin F (MMAF)(GSK2857916)74, CD46 with MMAF75, and CD56 with the maytansinoid DM176. While the anti-CD56 ADC, lorvotuzumab mertansine, has moved onto clinical trials (NCT00991562), the specificity towards CD46 (CD46-ADC) is the most novel myeloma antibody-drug conjugate developed recently. This approach is notable because of the previously underappreciated targeting of CD46. Although, CD46 is not a surface marker required, by myeloma cells or other B-cell lineage cells, it has been shown to be upregulated in myeloma cells because of the copy number duplications on chromosome 1q, but remains unexpressed outside placenta and prostate tissues75. Chromosome 1q amplifications are a known marker for poor prognosis in multiple myeloma75, so targeting this subset of myeloma is a relatively untapped niche of myeloma research and should, therefore, be developed further.
While antibody production and engineering has been investigated for decades now, antibody and human enzyme based-research is still evolving. Some of the most promising results for harnessing the host’s own immune cells have been through the administration of bispecific T-cell engager (BiTE) antibodies. BitE antibodies bind to both the target cell as well as a cytotoxic T cell to drive cell-mediated cytotoxicity. BitE antibodies have been produced with anti-CD1977–79 for B-cell lineage leukemia and lymphoma cell types paired with anti-CD378, 79 or anti-CD577 regions to join T cells to the cancerous targets. The resulting connection between target cells and the T cell promotes immune synapse formation and target cell lysis79. In addition, recombinant human engineered enzymes have also been proposed as a potential therapy in AML. Pegylated human recombinant arginase I cobalt (HuArgI(Co)-PEG5000, AEB1102) was developed to combat arginase accumulation in AML cells80. Depletion of arginase stores in AML cells was shown to hinder growth in all nine AML cell lines tested and without off-target effects on healthy hematopoietic cells80. Varying levels of arginine dependence were observed, but the dependence was present in all cells tested. Translation of AEB1102 into mice and monkeys proved successful81, and clinical trials are currently underway to show efficacy in treating relapsed or refractory acute myeloid leukemia as well as myelodysplastic syndrome (clinicaltrials.gov, NCT02732184).
3. Novel small molecule inhibitor targets of signaling molecules and epigenetic mediators in hematological malignancies
The standard combination chemotherapy, CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) was the frontline therapy for DLBCL for >30 years. Rituximab (R-CHOP) was the only major improvement in spite of tens of millions of dollars in expenditure on clinical trials, and still a significant percentage of DLBCL patients are not cured82. Therefore, alternative drug treatments are needed to target this particularly difficult malignancy. The initiation and progression of cancer is controlled by both genetic and epigenetic alterations, where the former is almost impossible to reverse. In contrast, epigenetic aberrations are potentially reversible, allowing the malignant cell population to revert to a more normal state. Therefore, in addition to the antibody and emerging protein approaches, identification of signaling pathways and small molecule inhibitors of these targets have emerged as promising highly specific alternatives with low toxicity that potentially reprogram the cancer cells to make them more susceptible to death. Such is the case of enhancer of zeste homologue 2 (EZH2), a histone methyltransferase83epigenetic target, which is required for B cells to form germinal, centers (GCs). Expression of EZH2 is linked to a highly proliferative state of cells not only found in cancer, but in healthy stem cells as well83. Prostate cancer, DLBCL, and breast cancer are just a few types of cancers where EZH2 over expression is a driving forces for tumorigenesis83, 84. Somatic mutation in histone modifying enzymes is the genetic hallmark of GCB-DLBCL. Gain of function mutations of H3K27 methyltransferase EZH2, and loss of function mutations of H3K27 histone acetyltransferases CREBBP, EP300 and H3K4 methyltransferase KMT2D are present in >80% of patients and are early hits during disease pathogenesis85–92. Recent work has shown that each of these mutations causes lymphomas through aberrant epigenetic programming of their respective histone marks85, 87, 93. Beyond the effects of its mutations, EZH2 is also required for the growth of DLBCLs with wild-type EZH285, 87, 93. In addition, mutations in the SET domain of EZH2 has been identified in DLBCL94–96. Since the discovery that EZH2 inhibitors can halt the progression DLBCL, several unique drugs have been under development to specifically reduce EZH2 function including Tazemetostat (EPZ-6438) for NHL97–99, El1 for DLBCL83, GSK126 for DLBCL90, CPI-360 for NHL100, Compound 3 in EZH2 mutant GCB-DLBCL84, and EPZ011989 for DLBCL101. As more cancer types are discovered to be dependent on EZH2 mutations and drug specific resistance emerges in the treatment of those cancers, there will always be a need for the development98 of a plethora of EZH2 inhibitors especially in DLBCL102.
Similar to EZH2 inhibitors, other methylation effectors have been successfully inhibited for targeting DLBCL. DNA methyltransferase (DNMT) 1, DNMT2, and DNMT3 are proteins that methylate at CpG dinucleotides in the DNA sequence and the effector sites of epigenetic regulator drugs103 as indicated in Fig. 2. While DNMT2 and DNMT3 have larger role in initiating methylation at new sites, the function of DNMT1 is more closely aligned with the maintenance of preexisting methylation sites. Targeting DNMTs has potential to halt methylation, thereby reversing gene silencing by allowing for initiation of gene transcription104. 5-aza-2′-deoxycytidine and azacitidine are drugs that inactivate DNMTs by an irreversible formation of a covalent bond between the two upon association with DNA as pyrimidine nucleoside analogs104. As a result, methylation groups are steadily reduced at cytosine residues in daughter strands of DNA upon cell division effectively turning gene transcription back on105. Because of the heavy requirement of successive cell division on the successful function of DNMT-targeting drugs, recent efforts to promote their application focused on combination therapy with DLBCL as the target malignancy because of its highly proliferative nature106. Another way to understand the role of epigenetic modulators is their involvement in immune response. Treatment with DNMT, histone deacetylase (HDAC), and EZH2 inhibitors, lead to enhanced antigen production, antigen processing, CD40 expression, PDL-1 checkpoint target expression in tumor cells, major histocompatibility complex (MHC) class 1 expression, and production of chemokine, cytokines, and interferons107–109 (Fig. 3A). At the same time, HDAC inhibitors can increase the expression of receptors on natural killer (NK) cells and ligands on AML cells, promoting AML blast killing110. Similarly, HDAC inhibitors have been shown to enhance CD8+ T cell killing and regulation of regulatory T cells in tumors111–113.
Figure 2. Effector sites of epigenetic regulator drugs.
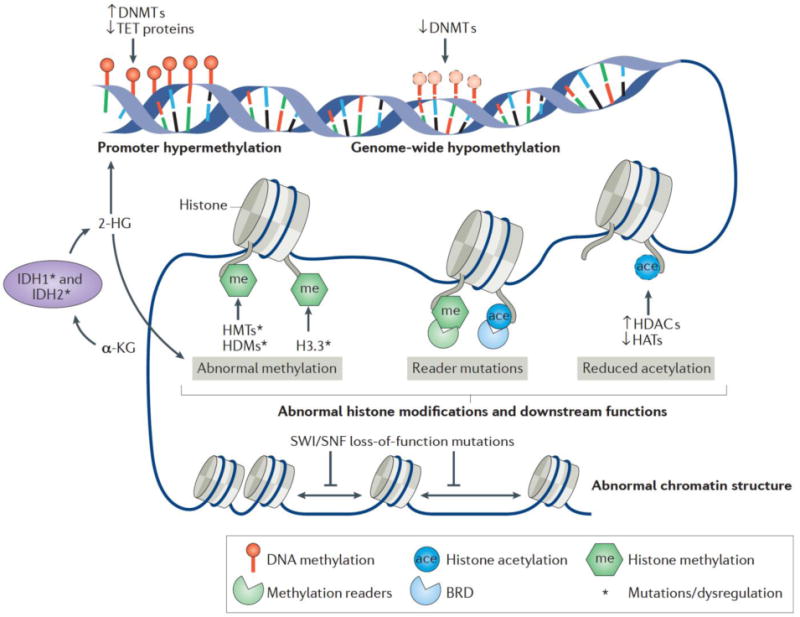
Upregulation of DNMTs and HDACS alter gene transcription leading to human cancers including hematological malignancies. Similarly, loss of TET supporting proteins leads to decreased gene transcription. DNMT, IDH1, and IDH2 inhibitors inhibit DNA methylation pathways to mitigate loss of function, gene down regulation from methylation. Other epigenetic regulators function through histone modifications that can lead to abnormal chromatin compaction. These include HDACs which reduce acetylation and histone methyl transferases (HMTs) which promote histone methylation. EZH2 inhibitors target the EZH2 HMT to reduce histone methylation to correct gene dysregulation. Adapted with permission from103.
Figure 3. Effect of epigenetic inhibitors on immune cells and modes of epigenetic drug delivery.
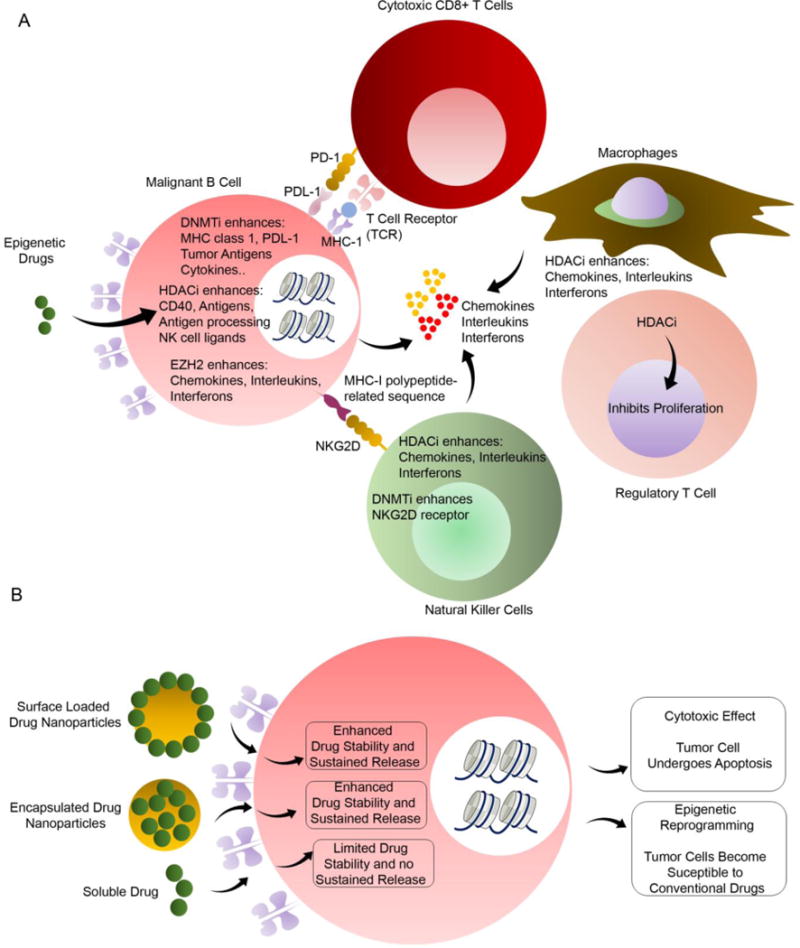
(A) Epigenetic drugs offer multi-modal approach to targeting malignant B cells. While EZH2 inhibitors exclusively affect malignant B cells, HDAC inhibitors (HDACi) also enhance chemokines, interleukins, and interferons while inhibiting the proliferation of regulatory T cells. Similarly, DNMT inhibitors (DNMTi) enhances nKGG2D receptors on natural killer cells while enhancing MHC class I, PDL-1, tumor antigens, and cytokines from malignant B cells. (B) Targeted drug delivery to cancer cells function through two pathways: cytotoxicity of cancer cells and epigenetic reprogramming through various epigenetic regulating drugs. Nanoparticle drug delivery platforms improve outcomes through enhanced drug stability and sustained drug release over soluble drug delivery paradigms.
In addition to epigenetic reprogramming drugs, recent discoveries have looked into B and T cell signaling pathways as potential target for suppressing tumors. One prominent target is B cell receptor (BCR) pathway. Biological dependence on the BCR signaling pathway is a major hallmark of ABC DLBCLs2, 114–119. The BCR is a transmembrane protein located on the outer surface of healthy and malignant B cells120. It is a heterodimer composed of heavy-chain and light-chain immunoglobulins, CD79A/Igα and CD79B/Igβ. BCR phosphorylation facilitates recruitment of proteins including Bruton’s tyrosine kinase (BTK), phospholipase C (PLC)-2, and phosphatidylinositol (PI)-3 kinases. The BCR pathway is chronically activated in ABC-DLBCLs in part due to somatic mutations of CD79A/B (~20% of ABC-DLBCLs)114, CARD11 (~10%)121, and several others. Given this scenario, a number of BCR pathway targeted therapies are in development – including kinase inhibitors targeting SYK, BTK, PI3K, PKC, MAPKs and AKT117. Among these, ibrutinib, an irreversible BTK inhibitor, has shown activity in clinical trials for ABC-DLBCL patients who have CD79 mutations, although patients with mutations affecting proteins downstream of BTK, such as CARD11 are resistant115, 122. Moreover resistance can develop due to mutations in BTK and other downstream BCR components that are directly or indirectly dependent on tumor microenvironment119, 123, 124.
Another key pathway target for BCR in ABC-DLBCLs is mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1). MALT1 is an attractive target for ABC-DLBCLs and other BCR-dependent lymphoma subtypes. Many of the activating mutations of the BCR pathway cause constitutive MALT1 paracaspase activity, which in turn explains, at least in part how lymphoma cells maintain NFκB activation. Interestingly, translocations of MALT1 lead to: 1) overexpression of the protein or 2) formation of the fusion protein API2-MALT1 in MALT lymphoma125–127. Amplification of MALT1 locus has been linked to increased MALT1 mRNA expression in ABC-DLBCL128. Moreover, a loss-of-function shRNA screening, determined that MALT1 was necessary for ABC but not GCB-DLBCL survival129. MALT1 is the only paracaspase in the human genome and hence its enzymatic pocket is structurally unique compared to other caspase family members, offering the possibility of developing highly specific inhibitors. Loss of MALT1 paracaspase activity is less toxic than targeting NFκB signaling more broadly as demonstrated by the fact that MALT1 knockout mice are born at the expected Mendelian rate, are fertile, and present only immune response activation defects130, 131, 132. Accordingly, studies show that ABC-DLBCLs are biologically dependent on and addicted to MALT1132–136. Hence targeting MALT1 has the potential to impact a broad cross section of ABC-DLBCL patients without causing toxicity to other organs and with reduced likelihood of off-target effects. Along these lines, Melnick and colleagues have reported a small molecule compound MI-2 as a potent and irreversible MALT1 enzymatic inhibitor132 while others have reported phenothiazines as reversible allosteric MALT1 inhibitors (e.g. Mepazine, an anti-psychotic drug)135.
Similarly, small molecule inhibitors have been developed for the treatment of leukemia. Isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) inhibitors (Fig. 2) have emerged as powerful treatments for AML. Isocitrate dehydrogenase is an enzyme that is responsible for catalysis of isocitrate to alpha-ketoglutarate137. It manifests in two locations with IDH1 in the cytoplasm and IDH2 in the mitochondria of the cell137. IDH1 and IDH2 are linked with progression through differentiation and upregulation or mutation of these enzymes are found in some AML patients137. Mutations at R172 and R140 of IDH2 are present in approximately 15–20% of AML cases137. Inhibition of function has been shown to be a viable treatment for IDH-related cancers, and as a result several small molecule inhibitors have emerged to inhibit IDH1137–144. While many IDH inhibitory drugs have been discovered over the years, few drugs have gone beyond in vitro validation. The IDH2 inhibitor AG-221 was one of the few to even progress to phase I clinical trials (trial number NCT 01915498 additional also found online at ClinicalTrials.gov)137, 138.
Despite increased numbers of small molecule inhibitors being discovered and engineered for the treatment of hematological malignancies, one major hurdle to their successful application is the method of delivery. Systemic administration of these drugs may result in unwanted side effects or at least inefficient treatment. As a result, drug delivery platforms have been developed to solve this issue by targeting healthy and malignant immune cell populations and effectively usher the use of small molecule inhibitors into modern cancer treatment. Nanoparticle and microparticle engineering has been proposed to solve this problem by acting as drug vehicles for targetted toxicity to cancer cells and sustained drug release in lymphoma145–147, leukemia148–150, and myeloma151–154. Nanoparticle research has built on basic principles of nanomedicine to design silver and gold nanoparticles to specifically target tumors of interest149, 150, 154. For example, a simian immunodeficiency virus accessory protein, Vpx delivered through virus-like particles inhibited the function of deoxynucleoside triphosphate (dNTP) triphosphohydrolase SAM domain and HD domain 1 (SAMHD1), which has been shown to be critical in turning over ara-CTP pools within cancerous cells, and thereby enhanced AML susceptibility to ara-C therapy155.
While many drugs can have devastating effects on cancer cells, they often come with undesirable side effects and general toxicity resulting from systemic rather than targeted delivery. While some particles engineered for AML actually lack AML cell specificity150, nanoparticle cancer specificity has been achieved by aptamer modification of gold nanoparticles for the targeted delivery of daunorubicin to the ALL cell line Molt-4154. In this application, Danesh et al. used Sgc8c, an aptamer specific for tyrosine kinase 7 in T cell leukemias, to drive nanoparticle contact with ALL cell lines154. Aptamer-cell connections, as used here, deliver chemotherapeutics directly to targeted cells while minimizing off target drug delivery and potentially the undesirable side effects that come with systemic chemotherapy. Similarly, alternative targeting techniques use monoclonal antibodies to drive nanoparticle delivery to hematological malignancies. Significant strides to this field were achieved through Fe3O4 nanoparticles with conjugated ACBG2 monoclonal antibody and paclitaxel (McAb-PTX-NPs)151. In the comparative in vitro and in vivo study, the engineered nanoparticles outperformed all other standards of care tested including melphalan and prednisone in combination151. In addition to the McAb-PTX-NPs, simpler drug conjugated nanoparticle systems have also been proposed for treatment of myeloma using the chemotherapeutics carilzomib153, and daunorubicin154.
Epigenetic effector drugs present unique problems with drug delivery because of their quick degradation and excretion rates in vivo. Enzymes like cytidine deaminase in the liver and other tissues have been shown to cause increased degradation of the epigenetic drug decitabine156, 157, thereby minimizing therapeutic effects of treatment. Effectively similar, due to the relatively small size of these drugs and inability to bind to other proteins in the body, excretion and clearance rates are also fast, therefore requiring continuous infusion for therapeutically relevant levels of drug function158. Nanoscale delivery systems can offer protection from degradation and cancer-targeted delivery (Fig. 3B), and have been a natural solution for epigenetic drug delivery. These delivery methods include dendrimers, nanogels, nanoparticles, and liposomes, and the diversity among them provide tailor made properties for different malignancy types. Liposomal carriers with encapsulated anacardic acid (epigenetic drug) in the liposomal bilayer with a vitamin C gradient, loaded with mitoxantrone has been developed159. The efficacy of N-isopropylacrylamide (NIPAM)-based biodegradable nanogels with decitabine was shown in drug resistant solid tumors and leukemia cells. Decitabine sustained release from nanogels prolonged cell arrest in the G2/M cell-cycle phase, and enhanced efficacy of the drug160. Most hematological malignancies allow for increased access for drug delivery, but in the case of solid tumors, nanoparticles and liposomes have been shown to aggregate at tumor sites due to leaky lymphatics and vasculature to overcome general drug inaccessibility161, 162. As more of the epigenetic and signaling targets as well as nanoscale delivery methods are being developed, the diversity of their therapeutic potential in treating the various types of hematological malignancies is increasingly being understood.
4. Targeting the immune cell populations using vaccines and drug delivery systems
For some time now, vaccination has been proposed as a possible tool for the elimination of cancer by the patient’s own immune system. With the proper antigen presentation to cytotoxic T cells, cancer cells can be targeted similarly to old or virally infected cells. Cyclin D1, a frequently upregulated cyclin in mantle cell lymphoma has been singled out as an effective antigen for vaccination therapy163. The frequent over expression of cyclin D1 in mantle cell lymphoma and its potency in T cell activation upon cross presentation contribute to why cyclin D1 makes an excellent target for vaccines163. Along other lines, Mattarollo et al. showed that a cancer antigen cocktail with an α–galactosylceramide (α-GalCer) adjuvant could successfully activate natural killer T cells against B cell lymphoma and subsequently inhibit its growth in mice lymphoma models164. Similar strategies have been translated to the targeting of leukemia. An α-GalCer adjuvant vaccine with irradiated acute leukemia cells was tested in mice as a possibility for targeting leukemia165. Unfortunately, the vaccine proved to only be effective at preventing onset and relapse rather than mitigating an already established leukemia165. Other approaches utilized a multi-epitope vaccine for CML by delivering the fusion protein BCR-ABL and Wilms’ tumor 1 (WT1) epitopes166. Lentiviral delivery of these antigens into primary human dendritic cells successfully stimulated autologous CD3+ T cells in vitro166. While validation of this system has yet to be done in vivo, lentiviral delivered vaccines contain significant promise for the future of leukemia treatment. Cancer vaccines have also been developed for the treatment of myeloma when first line proteasome inhibitors and immunomodulatory agents fail. An mucin 1 (MUC1) targeted vaccine (ImMucin) has been developed for myeloma patients who have not seen responses though traditional treatment pathways167. MUC1 is a myeloma associated surface marker that can be recognized through a 21-mer peptide loaded vaccine167. The mechanism of action for the MUC1 vaccine relies on already present MUC1 specific MHC complexes that, after binding, mount a T cell response against the MUC1 expressing myeloma cells167. Initial phase I/II studies have shown that ImMucin can initially stabilize myeloma levels although there is no indication that it improves patient outcome in the long-term167.
Recruitment of the adaptive immune response through antigen recognition, processing, and presentation followed by T cell activation is critical for cancer vaccine function. Vaccine efficiency has been known to be hindered due to early antigen degradation prior to the recognition stage. Both DNA- and protein-based vaccines are susceptible to both extracellular and intracellular degradation, the latter which prevents antigen translocation in the cytoplasm or to the nucleus. Encapsulating drug delivery platforms such as micro- or nanoparticles offer protective properties from in vivo degradation, and as such, have been proposed as cancer vaccine carriers168–170. Polyesters171, 172, polyanhydrides173, and liposomes174 have served as effective base biomaterials for micro- and nanoparticles in vaccine delivery. Upon delivery to sites of malignancy, immunostimulatory and immunomodulatory components such as pathogen-associated molecular patterns are used to target immune cells such as dendritic, tumor, or T cells. For example, cationic microparticles with polyethyleneimine attached to the surface, improve cell transfection, and activation of APC by the idiotype pDNA antigen (A20 idiotype-chemokine (MCP-3) fusion plasmid), yet they survive harsh endosomal environments175, 176. Altogether, this vaccine platform led to anti-tumor immunity in a prophylactic model of B cell lymphoma importantly without adjuvant support.
Another major strategy in particle engineering, is the utilization of small interfering RNAs (siRNAs)147, 148, 152, CpG oligodeoxynucleotides (CpG ODN)145, or both147. siRNA block expression of target genes in tumor cells. Both oncogenes and signaling cytokines have been targeted using the technique to reduce proliferation and alter the immune response, respectively147, 152. CpG ODN on the other hand, act in an adjuvant fashion to drive maturation of dendritic cells though binding to toll-like receptor 9 to allow for tumor antigen presentation to cytotoxic T cells and an increased Th1 immune response145.
In addition to micro- and nanoparticle delivery systems, multi-modal biomaterial delivery systems have expanded on the importance of porosity, degradability, and general physical structure for vaccine and drug delivery177–179. For example, dendritic cell migration through delivery materials for antigen discovery was promoted by increasing the size of material pores, faster in vivo degradation rates, cytokines, and danger signal presentation178, 179. All the while, this system allowed dendritic cells to migrate out of the material to transition to draining lymph nodes and initiate the immune response. This feat was achieved by an injectable in situ cross linking dextran vinyl sulfone and tetra-functional polyethylene-glycol thiol hydrogel that degrades quickly in vivo for effective delivery of plasmid DNA antigen, siRNA for inhibition of IL-10, and MIP-3α for dendritic cell recruitment178, 179. This work considers several factors that affect multi-modal delivery systems including matrix gelation time (which can be affected by reactive group substitution), the effects of physiological temperature, the dependence of swelling ratio on polymer combinations, and the promotion of dendritic cell invasion by material class and porosity178, 179. Ultimately, all of these factors came together to allow for a multimodal fast-degrading in situ crosslinking hydrogel system that can effectively shift immune response towards Th1 responses, despite poor immunogenicity of B cell lymphoma in a mouse model, leading to notably increased survivorship when challenged with a tumor cells in lethal quantities147, 179.
Since siRNAs require intracellular access to properly perform their function of blocking gene expression, additional approaches have been developed using lipid nanoparticles that fuse with the cell membrane for transport into the cell148. This drug delivery platform, SLP-301R, has been shown to prefer transfection with suspension leukemia cell lines with low expression of the Cav1 and Cav2, cell membrane surface lipid rafts, that have historically be difficult to transfect with standard lipid nanoparticle systems148. The transfection interaction was correlated to the decreased expression of the Cav1 and Cav2 genes, that are also under-expressed in neuronal cells, which are also difficult to transfect148. This may suggest that while siRNA delivery was only presented in the bone marrow and spleen, there could be potential off target siRNA delivery in the nervous system, and that further research into lipid nanoparticles should address misdirected drug delivery to healthy tissues.
5. Cell Engineering Approaches to Target B and T cell Tumors
In recent years, the engineering of immune cells has emerged to the forefront of hematological cancer treatment. Through lentiviral delivery, chimerical antigen receptors (CAR) are introduced into natural killer and T cells for a cancer cell targeted cytotoxic cellular response. First generation CARs link the antigen receptor of choice to a CD3ζ domain for intracellular signal transduction, while second generation CARs also include either CD28 or 4–1BB (CD137) as costimulatory membrane proteins180, 181. Engineered CAR cells have been implemented in both T cells180, 182–187, natural killer cells (NK)188, 189, and cytokine induced killer cells (CIK)190, 191 to channel their cytotoxic functions at CAR-complemented cancer cells. They have been developed for AML, CLL, multiple myeloma, B cell lymphoma, and ALL. AML targeting CAR cells typically express anti-CD33182, 191 or anti-123184, 190 CARs. While both showed success in targeting AML blasts, comparison of these two target surface proteins showed that targeting CD123 had less off target cytotoxicity to hematopoietic progenitor cells than anti-CD33 CAR T CIKs190, 191. Engineered CAR cells for AML all used primary human cell lines either collected through apheresis from peripheral blood draws or from umbilical cord blood. Emphasis was placed on autologous cell sources for T cells to maintain HLA restriction for in vivo applications (NCT02623582 NCT01864902 NCT02799680).
Major efforts towards fighting hematological malignancies have used CD19 targeted CAR T cells (NCT03029338, NCT02822326, NCT02975687, NCT02842138, NCT02546739, NCT02656147, NCT02081937). Anti-CD19 CAR T cells specifically target healthy and malignant B cells, and have implemented for CLL192, B cell ALL183, 186, 193, and follicular lymphoma194. In all applications, autologous T cells were used from with mixed ratios of CD4 and CD8 positive T cells that were highly patient dependent187. Despite the differences in cytotoxicity profile of the harvested patient T cells, no correlation was found between the ratio of cell type and patient outcome187. Patient outcomes were improved though through at least 5 rounds of oral ibrutinib (a Bruton’s tyrosine kinase inhibitor of the BCR pathways) prior to anti-CD19 CAR T cell infusions192. Ibrutinib not only targeted the malignant B cells, but reduced expression of programmed cell death 1 protein, an immunosuppressive membrane protein in T cells192. In addition to attacking malignant cells, engineered CAR cells have been proposed to tackle other issues with cancer treatment. In B cell ALL, Epstein Bar Virus- and cytomegalovirus-specific autologous memory T cells transfected with anti-CD19 CARs have been suggested to protect immunosuppressed patients from these viruses after hematopoietic stem cell transplant, and through specific effector T cell selection, reduce the risk of graft versus host disease triggered by the CAR T cell therapy183. Anti-CD19 CAR T cells have also been applied to follicular lymphoma185, 194. Recent research in this area has shown that by programming CAR T cells to also produce soluble herpes virus entry mediator (HVEM), the effectiveness of engineered cell therapy can be enhanced194. This could provide the framework for future directions in engineered CAR T cells through the expression of soluble effector molecules to enhance cancer targeted cytotoxicity.
Multiple myeloma targeted CAR therapies have also been developed targeting CD138 (syndecan 1)189, B-cell maturation antigen (BCMA)195, 196, and CS1180, 188. CD138, BCMA, and CS1 are highly expressed on multiple myeloma cells but are more importantly lowly expressed on hematopoietic stem cells180, 189. CS1 has proved a successful target, as shown by the success of the CS1 targeted therapy Elotuzumab188. While engineering of CS1 cells relied on primary T180 and NK188 cells, Jiang et al. took a different approach and used the NK-92MI cell line, a derivative of large granular lymphoma cells189. Cell lines can be used in this case because NK cells are not MHC/HLA restricted and cell lines offer greater ease for in vitro expansion prior to therapy. To mitigate risks associated with cell therapy using immortalized cell lines, the anti-CD138 CAR NK cells were irradiated by 10 Gy to stop cell division without a reduction in cytotoxic effect when tested in a myeloma xenograft mouse model189. Even with these interesting outcomes with CD138 targeted therapy, CS1 has been shown to be more widely and specifically expressed in patient myeloma cells than CD40, CD56, CD138, and CD74188. Although CS1 may be a better target for CAR T cell therapy conceptually, clinical trials for anti-CD138 CAR T cells (NCT01886976) and anti-BCMA CAR T cells are currently underway196 (NCT02658929, NCT02215967, NCT02954445, NCT02786511).
6. Engineered ex vivo models of hematological malignancies and their role in drug discovery
While many cancer studies begin with experiments on cancer cell lines, they all must be studied in an animal model before clinical translation. Mouse models tend to be the animals of choice because of their small size, relative cost-effectiveness, and mammalian similarities to humans. All hematological malignancies reviewed here possess mouse models to mimic disease within the complexity of a complete organism. Many animal models for hematological malignancies begin with NOD/Scid mice to which cancerous cell lines or patient explants can be introduced and allowed to flourish. Introduction of cancer cell lines intravenously, intramuscularly, or subcutaneously has been the standard platform for hematological malignancy mouse models for some time. Examples of this technique are found in AML140, 141, 155, 184, 191, ALL197, DLBCL101, 164, 198–200, and multiple myeloma180, 188, 201. For liquid tumors, this alone is sufficient for a working cancer model, but in palpable tumors like lymphoma, cells are often injected with 50% Matrigel as the scaffold for lymphoma cells to inhabit97, 200. These models have been the standard for preclinical research because they recapitulate spontaneous tumorigenesis in human cancers and allow for the use of human cancer cell lines over mouse cell lines that may not accurately represent human disease. Immortalized human cancer cell lines though, have their drawbacks. Through countless passages in vitro, cell phenotype can be altered. Older, more established cell lines then may behave differently when reintroduced in vivo than newer cells. In fact, when gene expression of 90 cell lines were compared to primary tumor cell samples, the cell lines tended to cluster with greater similarity to other cells lines regardless of tumor origin than the primary cancer cells202. In addition, through the cell culturing process, established cell lines have indirectly been selecting for rapidly dividing cells over cells with slower proliferation202. This selection process reduces cell line heterogeneity and could account for discrepancies in cell line based and clinical trials.
For this reason, patient derived xenografts (PDX) have been proposed to better recapitulate human cancers. In a PDX, patient malignant cells or tumorous tissue are engrafted into immunodeficient mice as a model for the patient’s specific cancer. This allows for exact matching of oncogenic mutations and minimizes culture time ex vivo that can lead to genotypic and phenotypic changes in cancer cell lines. Mouse PDX models provide the patient cells with organismal context to better recapitulate the systematic complexities of cancer. Solid patient tumors are often passaged three or more times between animals, and this process has shown to alter the integrity of the tumor stroma203. While typical patient tumor explants contain human tumor and stroma, through prolonged implantation, stromal cells, matrix components, nutrient supplying vessels drift to overwhelmingly murine in origin203.
Within the many rotating parts of xenograft models of, it can be difficult to discern which elements of the xenograft are eliciting the observed response in the context of pharmacological studies. Because many of the elements of xenografts are out of the researcher’s control, three dimensional in vitro models of hematological malignancies have been developed for AML204, 205, follicular lymphoma206, 207, B cell lymphoma208, 209, and multiple myeloma210–214. With the modularity of tissue engineering, these 3D models are highly adaptable to be disease specific. While culturing techniques like using the hanging drop method for the formation of aggregates207, 215 required little engineering for microenvironment development, microenvironment tailoring has been achieved through manipulation of biomaterial stiffness209, strength212, integrin ligand presentation208, 209, inclusion of stromal cell support204, 213, and morphology205. Even though tailored tissue engineered constructs are over-simplifications of the tumor microenvironment, the presence of these 3D structures in cancer drug trials has been shown to produce different responses when compared to traditional 2D cell culture204, 206–209, 212. For example, our group has developed specific integrin ligand presenting designer 4-arm polyethylene glycol hydrogel organoids of B and T cell lymphomas208. The biological findings in the study showed heterogeneity in the expression of various integrin receptors across subclasses of B cell lymphomas and T cell lymphomas. The presentation of integrin ligand-specific amino acid polymer segments, in a designer and tailored fashion, provide a biological connection between cancer cells and their supporting extracellular structure. Transmission of these survival cues through integrins, has been shown to alter lymphoma cell phenotype within the context of chemotherapy resistance and promotion of overall survival208, 216, 217. While the mechanisms of the promotion of survival vary between cancer types and some remain unclear, B cell lymphomas survival was correlated to the presence of stromal support cells and the upregulation of IgM response the 3D engineered microenvironment208, 217. As these mechanisms become more fully understood, it is becoming increasingly clear that the extracellular environment, including biochemical and mechanical cues, significantly alters cancer cell response to chemotherapy in vitro218, 219. As such, greater effort should be made to improve in vitro models of B and T cell tumors in the hopes to surpass animal models in their predictive power while improving statistical power from high throughput capabilities all the while reducing study costs for drug discovery218, 220.
7. Conclusion and Perspective
As cancers continue to evolve in the presence of chemotherapies, there will likely always be a need for new anti-cancer drugs in the treatment of the majority of hematological malignancies. As such, efforts should remain not only in drug discovery, but in discovery platforms as well. A simple schematic of future drug research is summarized in Figure 4. While many of the reviewed methods have shown efficacy in targeting hematological malignancies, multi-faceted or novel approaches have come to be seen as the future of these fields of cancer research. One example of such a therapeutic would be the anti-CD20 nanodisks developed by Crosby and colleagues. This approach utilizes apolipoprotein A-I nanoparticle chemotherapy delivery systems that are typically protected by a protein ring, and replaced typical ring structures with a single chain variable antibody fragment specific to CD20 for B-cell malignancy targeting221. The resulting fusion protein, termed nanodisks, were shown to specifically deliver curcumin, an anti-oxidant polyphenol, to CD20 positive lymphoma cells in vitro with successful intracellular release upon binding221. The result of this process was decreased viability in Ramos, a CD20 positive B cell lymphoma cell, further showing the effectiveness of this nanodisk drug delivery system and the promising therapeutic potential it holds. Similar lipid nanoparticle targeting techniques have also been developed for multiple myeloma by conjugating very-late antigen 4-specific ligand to lipid nanoparticles loaded with carfilzomib153. Additional novel approaches to targeted hematological malignancy therapy include the gene therapy drug delivery platform engineered by Dong et al.222. In this approach, lentiviral delivery of suicide genes was directed by an anti-MUC1 antibody for gene therapy targeted to leukemia cells222. The suicide genes included herpes simplex virus thymidine kinase and Escherichia coli cytosine deaminase followed by treatment with the prodrugs ganciclovir and 5-fluorocytosine, respectively. Lentiviral delivery of suicide genes allowed for intra-leukemia cellular drug activation leading to leukemia cell death by direct and bystander mechanisms that are yet to be fully understood222. This leukemia therapy represents a targeted therapy by the anti-MUC1 antibody that minimizes off-target cytotoxicity. In addition, they targeted leukemia cells with two drugs for a multifaceted approach and greater leukemia cell death. Moving forward, smart and intelligent drug delivery system that could function in an “on-demand” fashion is warranted to tackle relapsed tumors. The authors imagine these systems to circulate in the body for very long period and prevent any relapse tumor buildup. Finally, we proposed the translation of cancer drug discovery and initial drug trials from two dimensional cultures to tissue engineered constructs at least in the case of solid hematological tumors. While they may not provide identical scenarios to human disease, they continue to provide more patient representative features of growing tumors when compared to traditional cancer monocultures. For drug discovery to shift away from traditional cell cultures, greater efforts should be made to develop tissue engineered constructs that are simple, replicable, scalable, and representative of both the healthy and diseased tissues they aim to recapitulate. In essence, many such new platforms for drug discovery and drug delivery will have to consider the genetic and epigenetic heterogeneity of hematological malignancies, which continues to remain a major roadblock in the field.
Figure 4. Proposed pathway for drug discovery and translation of therapeutics in hematological malignancies.
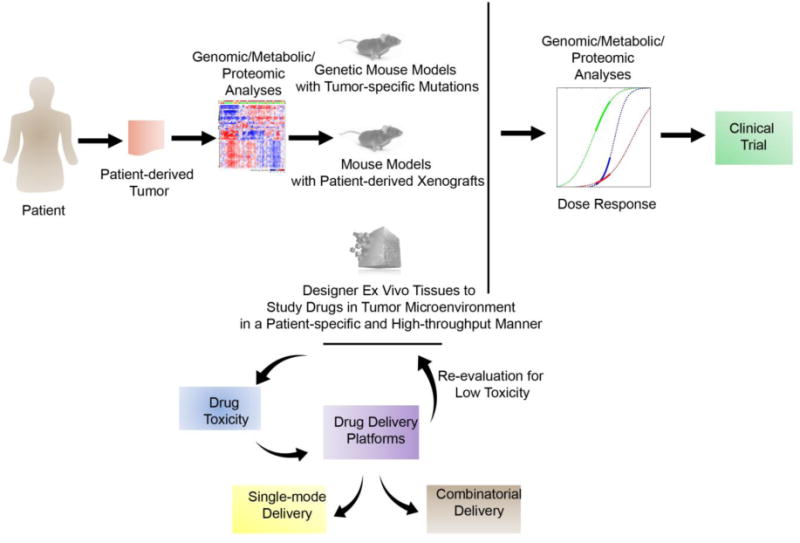
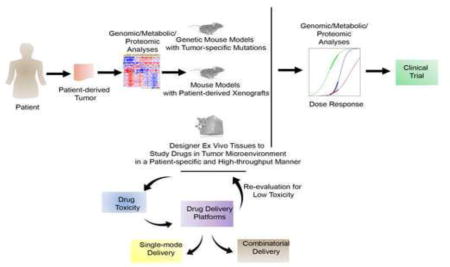
Drug discovery is often required on a special patient- and mutation-specific basis. Understanding the oncogenic profile of patients can be achieved through genomic, metabolic, and proteomic analyses. With greater understanding of the mutation types from the patient, tumor specific cytotoxicity can be evaluated in genetic mouse models, patient derived xenograft models, or in ex vivo engineered tissues, each with distinct advantages and disadvantages. Each of these models can be used to evaluate efficacy and toxicity of drugs, followed by formulation development using biomaterials with reduced toxicity effects. Drug delivery platforms could be engineered for both single-mode and combinatorial delivery of drugs to establish improved treatment protocols for patient disease. Upon discovery of tumor-targeting drug and suitable delivery platform, dose response studies evaluating anti-tumor effect, genomic, metabolic, and proteomic changes in tumor cell response can initiate new clinical trials to treat human hematological malignancies.
Acknowledgments
The authors acknowledge financial support from the National Cancer institute of the National Institute of Health (1R21CA185236–01, A.S; 1 R33 CA212968–01, A.S.). The authors would also like to acknowledge the Dean’s Excellence Fellowship to R.S. from the Graduate School of Cornell University.
Glossary of Acronyms and Abbreviations
- ABC
Activated B cell-like
- ADC
Antibody-drug conjugates
- ALL
Acute lymphoid leukemia
- AML
Acute myeloid leukemia
- BCMA
B-cell maturation antigen
- BiTE
Bispecific T-cell engager antibodies
- BTK
Bruton’s tyrosine kinase
- CAR
Chimerical antigen receptor
- CCR4
CC motif chemokine receptor 4
- CHOP
Cyclophosphamide, doxorubicin, vincristine, and prednisone
- CIK
Cytokine inducer killer cells
- CLL
Chronic lymphocytic leukemia
- CML
Chronic myeloid leukemia
- CpG ODN
CpG oligodeoxynucleotides
- DLBCL
Diffuse large B cell lymphoma
- DNMT
DNA methyltransferase
- dNTP
Deoxynucleoside triphosphate
- EZH2
Enhancer of zeste homologue 2
- FcR
Fc receptors
- GC
Germinal center
- GCB
Germinal center
- GO
Gemtuzumab ozogamacin
- HDAC
Histone deacetylase
- HVEM
Herpes Virus Entry Mediator
- IDH1
Isocitrate dehydrogenase 1
- IDH2
Isocitrate dehydrogenase 2
- IgG
Immunoglobulin G
- IL
Interleukin
- ILRAP/IL1R3
Interleukin-1 receptor accessory protein
- MALT1
Mucosa-associated lymphoid tissue lymphoma translocation protein 1
- MAX
MYC-associated factor X
- MHC
Major histocompatibility complex
- MMAF
Monomethyl auristatin F
- MUC1
Mucin 1
- MXD3
MYC-associated factor X dimerization protein 3
- NIPAM
N-isopropylacrylamide
- NK
Natural killer
- PD-1
Programmed cell death protein 1
- PD-L1
Programmed cell death ligand 1
- PDX
Patient derived xenografts
- PI
Phosphatidylinositol
- PLC
Phospholipase C
- R-CHOP
Rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone
- SAMHD1
Triphosphohydrolase SAM domain and HD domain 1
- siRNA
Small interfering RNA
- SLAMF
Signaling lymphocytic activation molecule family 7 protein
- WT1
Wilms’ tumor 1
- α-GalCer
α–galactosylceramide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bewtra M, Lewis JD. Update on the risk of lymphoma following immunosuppressive therapy for inflammatory bowel disease. Expert Rev Clin Immunol. 2010;6:621–631. doi: 10.1586/eci.10.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014;11:12–23. doi: 10.1038/nrclinonc.2013.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:1398–1403. doi: 10.1073/pnas.1205299110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lenz G, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125:22–32. doi: 10.1182/blood-2014-05-577189. [DOI] [PubMed] [Google Scholar]
- 6.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12:229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ansell SM, et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. The New England Journal of Medicine. 2015;372:311–319. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campo E, et al. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117:5019–5032. doi: 10.1182/blood-2011-01-293050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gazzola A, et al. CDKN1B/p27 expression in peripheral T cell lymphoma not otherwise specified. J Clin Pathol. 2011;64:83–87. doi: 10.1136/jcp.2010.083832. [DOI] [PubMed] [Google Scholar]
- 10.Foss FM, et al. Peripheral T-cell lymphoma. Blood. 2011;117:6756–6767. doi: 10.1182/blood-2010-05-231548. [DOI] [PubMed] [Google Scholar]
- 11.Reddy NM, Savani BN. Management of T-cell lymphomas: overcoming challenges and choosing the best treatment. Semin Hematol. 2014;51:1–4. doi: 10.1053/j.seminhematol.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Teras LR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA: A Cancer Journal for Clinicians. 2016;66:443–459. doi: 10.3322/caac.21357. [DOI] [PubMed] [Google Scholar]
- 13.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: A Cancer Journal for Clinicians. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 14.Weir HK, Thompson TD, Soman A, Møller B, Leadbetter S. The past, present, and future of cancer incidence in the United States: 1975 through 2020. Cancer. 2015;121:1827–1837. doi: 10.1002/cncr.29258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rollig C, Knop S, Bornhauser M. Multiple myeloma. LANCET. 2015;385:2197–2208. doi: 10.1016/S0140-6736(14)60493-1. [DOI] [PubMed] [Google Scholar]
- 16.Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Current Opinion in Immunology. 2008;20:471–478. doi: 10.1016/j.coi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 17.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10:301–316. doi: 10.1038/nri2761. [DOI] [PubMed] [Google Scholar]
- 18.Vedi A, Ziegler DS. Antibody therapy for pediatric leukemia. Frontiers in oncology. 2014;4:82. doi: 10.3389/fonc.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nimmerjahn F, Ravetch JV. Fc[gamma] receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 20.Jiang XR, et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. 2011;10:101–111. doi: 10.1038/nrd3365. [DOI] [PubMed] [Google Scholar]
- 21.Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther. 2007;7:1401–1413. doi: 10.1517/14712598.7.9.1401. [DOI] [PubMed] [Google Scholar]
- 22.Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. 2000;406:267–273. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- 23.Shields RL, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 24.Shinkawa T, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–3473. doi: 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- 25.Niwa R, et al. IgG subclass-independent improvement of antibody-dependent cellular cytotoxicity by fucose removal from Asn297-linked oligosaccharides. J Immunol Methods. 2005;306:151–160. doi: 10.1016/j.jim.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Ferrara C, et al. Modulation of therapeutic antibody effector functions by glycosylation engineering: influence of Golgi enzyme localization domain and co-expression of heterologous beta1, 4-N-acetylglucosaminyltransferase III and Golgi alpha-mannosidase II. Biotechnol Bioeng. 2006;93:851–861. doi: 10.1002/bit.20777. [DOI] [PubMed] [Google Scholar]
- 27.Duvic M, et al. Phase 1/2 study of mogamulizumab, a defucosylated anti-CCR4 antibody, in previously treated patients with cutaneous T-cell lymphoma. (blood-2014-2009-600924).Blood. 2015 doi: 10.1182/blood-2014-09-600924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang D-K, et al. Humanization of an Anti-CCR4 Antibody That Kills Cutaneous T-Cell Lymphoma Cells and Abrogates Suppression by T-Regulatory Cells. Molecular Cancer Therapeutics. 2012;11:2451–2461. doi: 10.1158/1535-7163.MCT-12-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Czuczman MS, et al. Phase II trial of galiximab (anti-CD80 monoclonal antibody) plus rituximab (CALGB 50402): Follicular Lymphoma International Prognostic Index (FLIPI) score is predictive of upfront immunotherapy responsiveness. Annals of Oncology. 2012;23:2356–2362. doi: 10.1093/annonc/mdr620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fayad L, et al. Safety and Clinical Activity of a Combination Therapy Comprising Two Antibody-Based Targeting Agents for the Treatment of Non-Hodgkin Lymphoma: Results of a Phase I/II Study Evaluating the Immunoconjugate Inotuzumab Ozogamicin With Rituximab. Journal of Clinical Oncology. 2013;31:573–583. doi: 10.1200/JCO.2012.42.7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jahn T, et al. An Il12-Il2-Antibody Fusion Protein Targeting Hodgkin’s Lymphoma Cells Potentiates Activation Of Nk And T Cells For An Anti-Tumor Attack. PLOS ONE. 2012;7:e44482. doi: 10.1371/journal.pone.0044482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fanale M, et al. Phase IA/II, multicentre, open-label study of the CD40 antagonistic monoclonal antibody lucatumumab in adult patients with advanced non-Hodgkin or Hodgkin lymphoma. Br J Haematol. 2014;164:258–265. doi: 10.1111/bjh.12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berguig GY, et al. Intracellular Delivery and Trafficking Dynamics of a Lymphoma-Targeting Antibody–Polymer Conjugate. Mol Pharm. 2012;9:3506–3514. doi: 10.1021/mp300338s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cang S, Mukhi N, Wang K, Liu D. Novel CD20 monoclonal antibodies for lymphoma therapy. Journal of Hematology & Oncology. 2012;5:64. doi: 10.1186/1756-8722-5-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Habermann TM, et al. Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. Journal of Clinical Oncology. 2006;24:3121–3127. doi: 10.1200/JCO.2005.05.1003. [DOI] [PubMed] [Google Scholar]
- 36.McLaughlin P, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. Journal of clinical oncology. 1998;16:2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 37.Coiffier B, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346 doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- 38.Badoux XC, et al. Fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy is highly effective treatment for relapsed patients with CLL. Blood. 2011;117:3016–3024. doi: 10.1182/blood-2010-08-304683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salles G, et al. Phase 1 study results of the type II glycoengineered humanized anti-CD20 monoclonal antibody obinutuzumab (GA101) in B-cell lymphoma patients. Blood. 2012;119:5126–5132. doi: 10.1182/blood-2012-01-404368. [DOI] [PubMed] [Google Scholar]
- 40.Forero-Torres A, et al. Results of a phase 1 study of AME-133v (LY2469298), an Fc-engineered humanized monoclonal anti-CD20 antibody, in FcgammaRIIIa-genotyped patients with previously treated follicular lymphoma. Clin Cancer Res. 2012;18 doi: 10.1158/1078-0432.CCR-11-0850. [DOI] [PubMed] [Google Scholar]
- 41.Kohrt HE, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. 2014;123:678–686. doi: 10.1182/blood-2013-08-519199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogura M, et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. Journal of Clinical Oncology. 2014;32:1157–1163. doi: 10.1200/JCO.2013.52.0924. [DOI] [PubMed] [Google Scholar]
- 43.Kato K, et al. Diffuse panbronchiolitis after humanized anti-CCR4 monoclonal antibody therapy for relapsed adult T-cell leukemia/lymphoma. International journal of hematology. 2013;97:430–432. doi: 10.1007/s12185-013-1278-z. [DOI] [PubMed] [Google Scholar]
- 44.Vitale LA, et al. Development of a Human Monoclonal Antibody for Potential Therapy of CD27-Expressing Lymphoma and Leukemia. Clinical Cancer Research. 2012;18:3812–3821. doi: 10.1158/1078-0432.CCR-11-3308. [DOI] [PubMed] [Google Scholar]
- 45.Ribrag V, et al. A Dose-Escalation Study of SAR3419, an Anti-CD19 Antibody Maytansinoid Conjugate, Administered by Intravenous Infusion Once Weekly in Patients with Relapsed/Refractory B-cell Non-Hodgkin Lymphoma. Clinical Cancer Research. 2014;20:213–220. doi: 10.1158/1078-0432.CCR-13-0580. [DOI] [PubMed] [Google Scholar]
- 46.Jeffrey SC, et al. A potent anti-CD70 antibody–drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjugate chemistry. 2013;24:1256–1263. doi: 10.1021/bc400217g. [DOI] [PubMed] [Google Scholar]
- 47.Kurzrock R, et al. A Phase I, Open-Label Study of Siltuximab, an Anti–IL-6 Monoclonal Antibody, in Patients with B-cell Non-Hodgkin Lymphoma, Multiple Myeloma, or Castleman Disease. Clinical Cancer Research. 2013;19:3659–3670. doi: 10.1158/1078-0432.CCR-12-3349. [DOI] [PubMed] [Google Scholar]
- 48.Honeychurch J, et al. Antibody-induced nonapoptotic cell death in human lymphoma and leukemia cells is mediated through a novel reactive oxygen species-dependent pathway. Blood. 2012;119:3523–3533. doi: 10.1182/blood-2011-12-395541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cartron G, Watier H, Golay J, Solal-Celigny P. From the bench to the bedside: ways to improve rituximab efficacy. Blood. 2004;104:2635–2642. doi: 10.1182/blood-2004-03-1110. [DOI] [PubMed] [Google Scholar]
- 50.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–3947. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 51.Ratner M. Genentech’s glyco-engineered antibody to succeed Rituxan. Nat Biotechnol. 2014;32:6–7. doi: 10.1038/nbt0114-6b. [DOI] [PubMed] [Google Scholar]
- 52.Georgiou K, et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood. 2016;127:3026–3034. doi: 10.1182/blood-2015-12-686550. [DOI] [PubMed] [Google Scholar]
- 53.Goodman A, Patel SP, Kurzrock R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol. 2017;14:203–220. doi: 10.1038/nrclinonc.2016.168. [DOI] [PubMed] [Google Scholar]
- 54.He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Sci Rep. 2015;5:13110. doi: 10.1038/srep13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang X-R, et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat Rev Drug Discov. 2011;10:101–111. doi: 10.1038/nrd3365. [DOI] [PubMed] [Google Scholar]
- 56.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017 doi: 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- 57.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 58.Zein N, Sinha AM, McGahren WJ, Ellestad GA. Calicheamicin gamma 1I: An Antitumor Antibiotic That Cleaves Double-Stranded DNA Site Specifically. Science. 1988;240:1198. doi: 10.1126/science.3240341. [DOI] [PubMed] [Google Scholar]
- 59.Erter J, et al. New targets of therapy in T-cell lymphomas. Curr Drug Targets. 2010;11:482–493. doi: 10.2174/138945010790980376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoelzer D. Anti-CD22 therapy in acute lymphoblastic leukaemia. The lancet oncology. 2012;13 doi: 10.1016/S1470-2045(12)70010-4. [DOI] [PubMed] [Google Scholar]
- 61.Charmsaz S, et al. EphA3 as a target for antibody immunotherapy in acute lymphoblastic leukemia. Leukemia. 2017 doi: 10.1038/leu.2016.371. [DOI] [PubMed] [Google Scholar]
- 62.Satake N, et al. Novel targeted therapy for Precursor B-Cell acute Lymphoblastic Leukemia: anti-CD22 antibody-MXD3 antisense Oligonucleotide Conjugate. Molecular Medicine. 2016;22:632. doi: 10.2119/molmed.2015.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cowan AJ, Laszlo GS, Estey EH, Walter RB. Antibody-based therapy of acute myeloid leukemia with gemtuzumab ozogamicin. Frontiers in bioscience (Landmark edition) 2013;18:1311. doi: 10.2741/4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petersdorf SH, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121:4854–4860. doi: 10.1182/blood-2013-01-466706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lancet JE, et al. ATRA, Arsenic Trioxide (ATO), and Gemtuzumab Ozogamicin (GO) Is Safe and Highly Effective in Patients with Previously Untreated High-Risk Acute Promyelocytic Leukemia (APL): Final Results of the SWOG/Alliance/ECOG S0535 Trial. Blood. 2016;128:896–896. [Google Scholar]
- 66.Sallman DA, Lancet JE. What are the most promising new agents in acute myeloid leukemia? Current opinion in hematology. 2016 doi: 10.1097/MOH.0000000000000319. [DOI] [PubMed] [Google Scholar]
- 67.Kreitman RJ, et al. Phase I Trial of Anti-CD22 Recombinant Immunotoxin Moxetumomab Pasudotox (CAT-8015 or HA22) in Patients With Hairy Cell Leukemia. Journal of Clinical Oncology. 2012;30:1822–1828. doi: 10.1200/JCO.2011.38.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bera TK, Onda M, Kreitman RJ, Pastan I. An improved recombinant Fab-immunotoxin targeting CD22 expressing malignancies. Leuk Res. 2014;38:1224–1229. doi: 10.1016/j.leukres.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ågerstam H, et al. IL1RAP antibodies block IL-1–induced expansion of candidate CML stem cells and mediate cell killing in xenograft models. Blood. 2016;128:2683–2693. doi: 10.1182/blood-2015-11-679985. [DOI] [PubMed] [Google Scholar]
- 70.Hosen N, et al. CD48 as a novel molecular target for antibody therapy in multiple myeloma. Br J Haematol. 2012;156:213–224. doi: 10.1111/j.1365-2141.2011.08941.x. [DOI] [PubMed] [Google Scholar]
- 71.Raedler LA. Empliciti (Elotuzumab): First SLAMF7 Antibody Therapy Approved for the Treatment of Patients with Previously Treated Multiple Myeloma. American health & drug benefits. 2016;9:74. [PMC free article] [PubMed] [Google Scholar]
- 72.Orlowski RZ, et al. A phase 2, randomized, double‐blind, placebo‐controlled study of siltuximab (anti‐IL‐6 mAb) and bortezomib versus bortezomib alone in patients with relapsed or refractory multiple myeloma. American journal of hematology. 2015;90:42–49. doi: 10.1002/ajh.23868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lewis T, et al. Abstract 1195: SGN-CD352A: A novel humanized anti-CD352 antibody-drug conjugate for the treatment of multiple myeloma. Cancer Research. 2016;76:1195–1195. [Google Scholar]
- 74.Tai Y-T, et al. Novel anti–B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123:3128–3138. doi: 10.1182/blood-2013-10-535088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sherbenou DW, et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. Journal of Clinical Investigation. 2016;126:4640+. doi: 10.1172/JCI85856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berdeja JG, et al. Phase I Study of Lorvotuzumab Mertansine (LM, IMGN901) in Combination with Lenalidomide (Len) and Dexamethasone (Dex) in Patients with CD56-Positive Relapsed or Relapsed/Refractory Multiple Myeloma (MM) Blood. 2012;120:728–728. [Google Scholar]
- 77.Lüttgau S, et al. Immunotherapy of B-Cell Lymphoma with an Engineered Bispecific Antibody Targeting CD19 and CD5. Antibodies. 2013;2:338–352. [Google Scholar]
- 78.Klinger M, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell–engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119:6226–6233. doi: 10.1182/blood-2012-01-400515. [DOI] [PubMed] [Google Scholar]
- 79.Przepiorka D, et al. FDA Approval: Blinatumomab. CLINICAL CANCER RESEARCH. 2015;21:4035–4039. doi: 10.1158/1078-0432.CCR-15-0612. [DOI] [PubMed] [Google Scholar]
- 80.Tanios R, et al. Human recombinant arginase I (Co)-PEG5000 [HuArgI (Co)-PEG5000]-induced arginine depletion is selectively cytotoxic to human acute myeloid leukemia cells. Leuk Res. 2013;37:1565–1571. doi: 10.1016/j.leukres.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 81.Rowlinson SW, et al. Abstract 1042: Development of AEB1102, an engineered human arginase 1 for patients with solid tumors. Cancer Research. 2016;76:1042–1042. [Google Scholar]
- 82.Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematology Am Soc Hematol Educ Program. 2011;2011:498–505. doi: 10.1182/asheducation-2011.1.498. [DOI] [PubMed] [Google Scholar]
- 83.Qi W, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. P Natl Acad Sci USA. 2012;109:21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garapaty-Rao S, et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. doi: 10.1016/j.chembiol.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 85.Beguelin W, et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell. 2016;30:197–213. doi: 10.1016/j.ccell.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Beguelin W, et al. EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell. 2013;23:677–692. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Beguelin W, et al. EZH2 and BCL6 cooperate to create the germinal center B-cell phenotype and induce lymphomas through formation and repression of bivalent chromatin domains. Blood. 2013 ASH Abstract 2013 Annual Meeting. [Google Scholar]
- 88.Caganova M, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. The Journal of clinical investigation. 2013;123:5009–5022. doi: 10.1172/JCI70626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lue JK, SA P, Liu Y, O’Conner OA, Amengual JE. Epigenetic Targeting with EZH2 and HDAC Inhibitors Is Synergistic in EZH2 Deregulated Lymphomas; 58th ASH Annual Meeting & Exposition; December 3–6, 2016; San Diego, CA. 2016. Session: 802. Chemical Biology and Experimental Therapeutics: Drug Combinations and Synthetic Lethality. [Google Scholar]
- 90.McCabe MT, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 91.Melnick A. Epigenetic therapy leaps ahead with specific targeting of EZH2. Cancer Cell. 2012;22:569–570. doi: 10.1016/j.ccr.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jiang Y, et al. CREBBP Inactivation Promotes the Development of HDAC3 Dependent Lymphomas. Cancer Discov. 2016 doi: 10.1158/2159-8290.CD-16-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Béguelin W, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–692. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McCabe MT, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27) Proceedings of the National Academy of Sciences. 2012;109:2989–2994. doi: 10.1073/pnas.1116418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ryan RJ, et al. EZH2 codon 641 mutations are common in BCL2-rearranged germinal center B cell lymphomas. PloS one. 2011;6:e28585. doi: 10.1371/journal.pone.0028585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Morin RD, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nature genetics. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Knutson SK, et al. Selective Inhibition of EZH2 by EPZ-6438 Leads to Potent Antitumor Activity in EZH2 -Mutant Non-Hodgkin Lymphoma. Molecular Cancer Therapeutics. 2014;13:842–854. doi: 10.1158/1535-7163.MCT-13-0773. [DOI] [PubMed] [Google Scholar]
- 98.Knutson SK, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. P Natl Acad Sci USA. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ribrag V, et al. Phase 1 Study of Tazemetostat (EPZ-6438), an Inhibitor of Enhancer of Zeste-Homolog 2 (EZH2): Preliminary Safety and Activity in Relapsed or Refractory Non-Hodgkin Lymphoma (NHL) Patients. Blood. 2015;126:473–473. [Google Scholar]
- 100.Bradley William D, et al. EZH2 Inhibitor Efficacy in Non-Hodgkin’s Lymphoma Does Not Require Suppression of H3K27 Monomethylation. Chemistry & Biology. 2014;21:1463–1475. doi: 10.1016/j.chembiol.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 101.Campbell JE, et al. EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust in Vivo Activity. ACS Med Chem Lett. 2015;6:491–495. doi: 10.1021/acsmedchemlett.5b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gibaja V, et al. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene. 2016;35:558–566. doi: 10.1038/onc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pfister SX, Ashworth A. Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discov. 2017 doi: 10.1038/nrd.2016.256. [DOI] [PubMed] [Google Scholar]
- 104.Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell. 2010;19:698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 105.Momparler RL. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine) Semin Hematol. 2005;42:S9–16. doi: 10.1053/j.seminhematol.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 106.Clozel T, et al. Mechanism-Based Epigenetic Chemosensitization Therapy of Diffuse Large B Cell Lymphoma. Cancer discovery. 2013;3:1002–1019. doi: 10.1158/2159-8290.CD-13-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Maeda T, Towatari M, Kosugi H, Saito H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood. 2000;96:3847–3856. [PubMed] [Google Scholar]
- 108.Magner WJ, et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165:7017–7024. doi: 10.4049/jimmunol.165.12.7017. [DOI] [PubMed] [Google Scholar]
- 109.Peng D, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–253. doi: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kopp LM, et al. Decitabine has a biphasic effect on natural killer cell viability, phenotype, and function under proliferative conditions. Mol Immunol. 2013;54:296–301. doi: 10.1016/j.molimm.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 111.West AC, Smyth MJ, Johnstone RW. The anticancer effects of HDAC inhibitors require the immune system. Oncoimmunology. 2014;3:e27414. doi: 10.4161/onci.27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gameiro SR, Malamas AS, Tsang KY, Ferrone S, Hodge JW. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget. 2016;7:7390–7402. doi: 10.18632/oncotarget.7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zheng H, et al. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res. 2016;22:4119–4132. doi: 10.1158/1078-0432.CCR-15-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Davis RE, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wilson WH, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nature medicine. 2015;21:922–926. doi: 10.1038/nm.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nature reviews Drug discovery. 2013;12:229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fontan L, Melnick A. Molecular pathways: targeting MALT1 paracaspase activity in lymphoma. Clin Cancer Res. 2013;19:6662–6668. doi: 10.1158/1078-0432.CCR-12-3869. [DOI] [PubMed] [Google Scholar]
- 118.Chen L, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood. 2008;111:2230–2237. doi: 10.1182/blood-2007-07-100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer. 2014;14:517–534. doi: 10.1038/nrc3774. [DOI] [PubMed] [Google Scholar]
- 120.Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5:251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- 121.Lenz G, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 122.Brower V. Ibrutinib promising in subtype of DLBCL. Lancet Oncol. 2015;16:e428. doi: 10.1016/S1470-2045(15)00192-8. [DOI] [PubMed] [Google Scholar]
- 123.Woyach JA, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. The New England journal of medicine. 2014;370:2286–2294. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nagel D, et al. Combinatorial BTK and MALT1 inhibition augments killing of CD79 mutant diffuse large B cell lymphoma. Oncotarget. 2015;6:42232–42242. doi: 10.18632/oncotarget.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dierlamm J, et al. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood. 1999;93:3601–3609. [PubMed] [Google Scholar]
- 126.Sanchez-Izquierdo D, et al. MALT1 is deregulated by both chromosomal translocation and amplification in B-cell non-Hodgkin lymphoma. Blood. 2003;101:4539–4546. doi: 10.1182/blood-2002-10-3236. [DOI] [PubMed] [Google Scholar]
- 127.Streubel B, et al. T(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood. 2003;101:2335–2339. doi: 10.1182/blood-2002-09-2963. [DOI] [PubMed] [Google Scholar]
- 128.Dierlamm J, et al. Gain of chromosome region 18q21 including the MALT1 gene is associated with the activated B-cell-like gene expression subtype and increased BCL2 gene dosage and protein expression in diffuse large B-cell lymphoma. Haematologica. 2008;93:688–696. doi: 10.3324/haematol.12057. [DOI] [PubMed] [Google Scholar]
- 129.Ngo VN, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ruefli-Brasse AA, French DM, Dixit VM. Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science. 2003;302:1581–1584. doi: 10.1126/science.1090769. [DOI] [PubMed] [Google Scholar]
- 131.Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity. 2003;19:749–758. doi: 10.1016/s1074-7613(03)00293-0. [DOI] [PubMed] [Google Scholar]
- 132.Fontan L, et al. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell. 2012;22:812–824. doi: 10.1016/j.ccr.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ferch U, et al. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2009;206:2313–2320. doi: 10.1084/jem.20091167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hailfinger S, et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2009;106:19946–19951. doi: 10.1073/pnas.0907511106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nagel D, et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell. 2012;22:825–837. doi: 10.1016/j.ccr.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 136.Ngo VN, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 137.Stein EM. Molecular pathways: IDH2 mutations—co-opting cellular metabolism for malignant transformation. Clinical Cancer Research. 2016;22:16–19. doi: 10.1158/1078-0432.CCR-15-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Deng G, et al. Selective inhibition of mutant isocitrate dehydrogenase 1 (IDH1) via disruption of a metal binding network by an allosteric small molecule. Journal of Biological Chemistry. 2015;290:762–774. doi: 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xie X, et al. Allosteric Mutant IDH1 Inhibitors Reveal Mechanisms for IDH1 Mutant and Isoform Selectivity. Structure. 2017 doi: 10.1016/j.str.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 140.Chaturvedi A, et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia. 2017 doi: 10.1038/leu.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Okoye-Okafor UC, et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol. 2015;11:878–886. doi: 10.1038/nchembio.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kernytsky A, et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood. 2015;125:296–303. doi: 10.1182/blood-2013-10-533604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang F, et al. Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 144.Sarkaria SM, Christopher MJ, Klco JM, Ley TJ. Primary acute myeloid leukemia cells with IDH1 or IDH2 mutations respond to a DOT1L inhibitor in vitro. Leukemia (08876924) 2014;28:2403–2406. doi: 10.1038/leu.2014.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Makkouk A, et al. Biodegradable Microparticles Loaded with Doxorubicin and CpG ODN for In Situ Immunization Against Cancer. The AAPS Journal. 2015;17:184–193. doi: 10.1208/s12248-014-9676-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Demir E, Castranova V. Genotoxic effects of synthetic amorphous silica nanoparticles in the mouse lymphoma assay. Toxicology Reports. 2016;3:807–815. doi: 10.1016/j.toxrep.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pradhan P, et al. The effect of combined IL10 siRNA and CpG ODN as pathogen-mimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials. 2014;35:5491–5504. doi: 10.1016/j.biomaterials.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.He W, et al. Discovery of siRNA lipid nanoparticles to transfect suspension leukemia cells and provide in vivo delivery capability. Molecular Therapy. 2014;22:359–370. doi: 10.1038/mt.2013.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Guo DW, et al. The Cellular Uptake and Cytotoxic Effect of Silver Nanoparticles on Chronic Myeloid Leukemia Cells. JOURNAL OF BIOMEDICAL NANOTECHNOLOGY. 2014;10:669–678. doi: 10.1166/jbn.2014.1625. [DOI] [PubMed] [Google Scholar]
- 150.Guo DW, et al. Anti-leukemia activity of PVP-coated silver nanoparticles via generation of reactive oxygen species and release of silver ions. BIOMATERIALS. 2013;34:7884–7894. doi: 10.1016/j.biomaterials.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 151.Yang C, et al. Target therapy of multiple myeloma by PTX-NPs and ABCG2 antibody in a mouse xenograft model. Oncotarget. 2015;6:27714. doi: 10.18632/oncotarget.4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Taylor CA, et al. Modulation of eIF5A expression using SNS01 nanoparticles inhibits NF-κB activity and tumor growth in murine models of multiple myeloma. Molecular Therapy. 2012;20:1305–1314. doi: 10.1038/mt.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ashley JD, et al. Liposomal carfilzomib nanoparticles effectively target multiple myeloma cells and demonstrate enhanced efficacy in vivo. Journal of Controlled Release. 2014;196:113–121. doi: 10.1016/j.jconrel.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 154.Danesh NM, Lavaee P, Ramezani M, Abnous K, Taghdisi SM. Targeted and controlled release delivery of daunorubicin to T-cell acute lymphoblastic leukemia by aptamer-modified gold nanoparticles. International journal of pharmaceutics. 2015;489:311–317. doi: 10.1016/j.ijpharm.2015.04.072. [DOI] [PubMed] [Google Scholar]
- 155.Herold N, et al. Targeting SAMHD1 with the Vpx protein to improve cytarabine therapy for hematological malignancies. Nature Medicine. 2017 doi: 10.1038/nm.4265. [DOI] [PubMed] [Google Scholar]
- 156.Jabbour E, Issa JP, Garcia-Manero G, Kantarjian H. Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer. 2008;112:2341–2351. doi: 10.1002/cncr.23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yoo CB, et al. Delivery of 5-aza-2′-deoxycytidine to cells using oligodeoxynucleotides. Cancer Res. 2007;67:6400–6408. doi: 10.1158/0008-5472.CAN-07-0251. [DOI] [PubMed] [Google Scholar]
- 158.Karahoca M, Momparler RL. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2′-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clinical epigenetics. 2013;5:3. doi: 10.1186/1868-7083-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Legut M, et al. Anacardic acid enhances the anticancer activity of liposomal mitoxantrone towards melanoma cell lines - in vitro studies. Int J Nanomedicine. 2014;9:653–668. doi: 10.2147/IJN.S54911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vijayaraghavalu S, Labhasetwar V. Efficacy of decitabine-loaded nanogels in overcoming cancer drug resistance is mediated via sustained DNA methyltransferase 1 (DNMT1) depletion. Cancer Lett. 2013;331:122–129. doi: 10.1016/j.canlet.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135–146. doi: 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 162.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 163.Chen J, et al. A novel vaccine for mantle cell lymphoma based on targeting cyclin D1 to dendritic cells via CD40. Journal of hematology & oncology. 2015;8:35. doi: 10.1186/s13045-015-0131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mattarollo SR, et al. NKT cell adjuvant-based tumor vaccine for treatment of myc oncogene-driven mouse B-cell lymphoma. Blood. 2012;120:3019–3029. doi: 10.1182/blood-2012-04-426643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gibbins JD, et al. An autologous leukemia cell vaccine prevents murine acute leukemia relapse after cytarabine treatment. Blood. 2014;124:2953–2963. doi: 10.1182/blood-2014-04-568956. [DOI] [PubMed] [Google Scholar]
- 166.Dong W, et al. Development and immunological evaluation of HLA-specific chronic myeloid leukemia polyepitope vaccine in Chinese population. Vaccine. 2014;32:3501–3508. doi: 10.1016/j.vaccine.2014.04.041. [DOI] [PubMed] [Google Scholar]
- 167.Carmon L, et al. Phase I/II study exploring ImMucin, a pan-major histocompatibility complex, anti-MUC1 signal peptide vaccine, in multiple myeloma patients. Br J Haematol. 2015;169:44–56. doi: 10.1111/bjh.13245. [DOI] [PubMed] [Google Scholar]
- 168.Boussif O, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Neu M, Fischer D, Kissel T. Recent advances in rational gene transfer vector design based on poly (ethylene imine) and its derivatives. The journal of gene medicine. 2005;7:992–1009. doi: 10.1002/jgm.773. [DOI] [PubMed] [Google Scholar]
- 170.Jilek S, Merkle HP, Walter E. DNA-loaded biodegradable microparticles as vaccine delivery systems and their interaction with dendritic cells. Advanced drug delivery reviews. 2005;57:377–390. doi: 10.1016/j.addr.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 171.Hedley ML, Curley J, Urban R. Microspheres containing plasmid-encoded antigens elicit cytotoxic T-cell responses. Nat Med. 1998;4:365–368. doi: 10.1038/nm0398-365. [DOI] [PubMed] [Google Scholar]
- 172.Klencke B, et al. Encapsulated plasmid DNA treatment for human papillomavirus 16-associated anal dysplasia. Clinical Cancer Research. 2002;8:1028–1037. [PubMed] [Google Scholar]
- 173.Mathiowitz E, Jacob JS, Jong YS, Carino GP. Biologically erodable microspheres as potential oral drug delivery systems. Nature. 1997;386:410. doi: 10.1038/386410a0. [DOI] [PubMed] [Google Scholar]
- 174.Wang D, et al. Intranasal immunization with liposome-encapsulated plasmid DNA encoding influenza virus hemagglutinin elicits mucosal, cellular and humoral immune responses. Journal of Clinical Virology. 2004;31:99–106. doi: 10.1016/j.jcv.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 175.Kasturi SP, Sachaphibulkij K, Roy K. Covalent conjugation of polyethyleneimine on biodegradable microparticles for delivery of plasmid DNA vaccines. Biomaterials. 2005;26:6375–6385. doi: 10.1016/j.biomaterials.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 176.Kasturi SP, et al. Prophylactic anti-tumor effects in a B cell lymphoma model with DNA vaccines delivered on polyethylenimine (PEI) functionalized PLGA microparticles. Journal of Controlled Release. 2006;113:261–270. doi: 10.1016/j.jconrel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 177.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection-mimicking materials to program dendritic cells in situ. Nature materials. 2009;8:151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Singh A, Suri S, Roy K. In-situ crosslinking hydrogels for combinatorial delivery of chemokines and siRNA–DNA carrying microparticles to dendritic cells. Biomaterials. 2009;30:5187–5200. doi: 10.1016/j.biomaterials.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Singh A, et al. An injectable synthetic immune-priming center mediates efficient T-cell class switching and T-helper 1 response against B cell lymphoma. Journal of controlled release. 2011;155:184–192. doi: 10.1016/j.jconrel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 180.Chu J, et al. Genetic modification of T cells redirected toward CS1 enhances eradication of myeloma cells. Clinical Cancer Research. 2014;20:3989–4000. doi: 10.1158/1078-0432.CCR-13-2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hoyos V, Borrello I. The immunotherapy era of myeloma: monoclonal antibodies, vaccines, and adoptive T-cell therapies. Blood. 2016;128:1679–1687. doi: 10.1182/blood-2016-05-636357. [DOI] [PubMed] [Google Scholar]
- 182.Minagawa K, et al. In Vitro Pre-Clinical Validation of Suicide Gene Modified Anti-CD33 Redirected Chimeric Antigen Receptor T-Cells for Acute Myeloid Leukemia. PLoS One. 2016;11 doi: 10.1371/journal.pone.0166891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Terakura S, et al. Generation of CD19-chimeric antigen receptor modified CD8(+) T cells derived from virus-specific central memory T cells. Blood. 2012;119:72–82. doi: 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mardiros A, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122:3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kochenderfer JN, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Park JH, et al. CD19-Targeted 19–28z CAR Modified Autologous T Cells Induce High Rates of Complete Remission and Durable Responses in Adult Patients with Relapsed, Refractory B-Cell ALL. Blood. 2014;124:382–382. [Google Scholar]
- 187.Langer T, et al. Abstract 2305: Comparative evaluation of peripheral blood T cells and resultant engineered anti-CD19 CAR T cell products from relapsed/refractory non-Hodgkin’s lymphoma (NHL) patients. Cancer Research. 2016;76:2305–2305. [Google Scholar]
- 188.Chu J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–927. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jiang H, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Molecular Oncology. 2014;8:297–310. doi: 10.1016/j.molonc.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tettamanti S, et al. Targeting of acute myeloid leukaemia by cytokine‐induced killer cells redirected with a novel CD123‐specific chimeric antigen receptor. Br J Haematol. 2013;161:389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 191.Pizzitola I, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28:1596–1605. doi: 10.1038/leu.2014.62. [DOI] [PubMed] [Google Scholar]
- 192.Fraietta JA, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127:1117–1127. doi: 10.1182/blood-2015-11-679134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Grupp SA, et al. T Cells Engineered with a Chimeric Antigen Receptor (CAR) Targeting CD19 (CTL019) Have Long Term Persistence and Induce Durable Remissions in Children with Relapsed, Refractory ALL. Blood. 2014;124:380–380. [Google Scholar]
- 194.Boice M, et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell. 2016;167:405–418.e413. doi: 10.1016/j.cell.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Carpenter RO, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clinical Cancer Research. 2013;19:2048–2060. doi: 10.1158/1078-0432.CCR-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ali SA, et al. T cells expressing an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128:1688–1700. doi: 10.1182/blood-2016-04-711903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Conrad DP, et al. Leukemia cell-rhabdovirus vaccine: personalized immunotherapy for acute lymphoblastic leukemia. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:3832–3843. doi: 10.1158/1078-0432.CCR-12-3199. [DOI] [PubMed] [Google Scholar]
- 198.Sakamaki I, et al. Lenalidomide enhances the protective effect of a therapeutic vaccine and reverses immune suppression in mice bearing established lymphomas. Leukemia (08876924) 2014;28:329–337. doi: 10.1038/leu.2013.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cardenas MG, et al. Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. The Journal of Clinical Investigation. 2016;126:3351–3362. doi: 10.1172/JCI85795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fontan L, et al. MALT1 Small Molecule Inhibitors Specifically Suppress ABC-DLBCL In Vitro and In Vivo. Cancer Cell. 2012;22:812–824. doi: 10.1016/j.ccr.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Qian J, et al. Active vaccination with Dickkopf-1 induces protective and therapeutic antitumor immunity in murine multiple myeloma. Blood. 2012;119:161–169. doi: 10.1182/blood-2011-07-368472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gillet J-P, Varma S, Gottesman MM. The clinical relevance of cancer cell lines. J Natl Cancer Inst. 2013;105:452–458. doi: 10.1093/jnci/djt007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hidalgo M, et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discovery. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Houshmand M, et al. Mimicking the Acute Myeloid Leukemia Niche for Molecular Study and Drug Screening. Tissue Engineering Part C: Methods. 2016;23:72–85. doi: 10.1089/ten.TEC.2016.0404. [DOI] [PubMed] [Google Scholar]
- 205.Nair MS, et al. Development and molecular characterization of polymeric micro-nanofibrous scaffold of a defined 3-D niche for in vitro chemosensitivity analysis against acute myeloid leukemia cells. Int J Nanomedicine. 2015;10:3603. doi: 10.2147/IJN.S80397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Decaup E, et al. Anti-tumor activity of obinutuzumab and rituximab in a follicular lymphoma 3D model. Blood Cancer J. 2013;3:e131. doi: 10.1038/bcj.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Gravelle P, et al. Cell growth in aggregates determines gene expression, proliferation, survival, chemoresistance, and sensitivity to immune effectors in follicular lymphoma. The American journal of pathology. 2014;184:282–295. doi: 10.1016/j.ajpath.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 208.Tian YF, et al. Integrin-specific hydrogels as adaptable tumor organoids for malignant B and T cells. Biomaterials. 2015;73:110–119. doi: 10.1016/j.biomaterials.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Apoorva F, et al. Lymph Node Stiffness Mimicking Hydrogels Regulate Human B Cell Lymphoma Growth and Cell Surface Receptor Expression in a Molecular Subtype‐ Specific Manner. Journal of Biomedical Materials Research Part A. 2017 doi: 10.1002/jbm.a.36031. [DOI] [PubMed] [Google Scholar]
- 210.Zhang W, Lee WY, Siegel DS, Tolias P, Zilberberg J. Patient-specific 3D microfluidic tissue model for multiple myeloma. Tissue Engineering Part C: Methods. 2014;20:663–670. doi: 10.1089/ten.TEC.2013.0490. [DOI] [PubMed] [Google Scholar]
- 211.de la Puente P, et al. AACR. 2015 [Google Scholar]
- 212.Reagan MR, et al. Investigating osteogenic differentiation in multiple myeloma using a novel 3D bone marrow niche model. Blood. 2014;124:3250–3259. doi: 10.1182/blood-2014-02-558007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Jakubikova J, et al. A novel 3D mesenchymal stem cell model of the multiple myeloma bone marrow niche: biologic and clinical applications. Oncotarget. 2016;7 doi: 10.18632/oncotarget.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Narayanan NK, Duan B, Butcher JT, Mazumder A, Narayanan BA. Characterization of multiple myeloma clonal cell expansion and stromal Wnt/β-catenin signaling in hyaluronic acid-based 3D hydrogel. In Vivo. 2014;28:67–73. [PubMed] [Google Scholar]
- 215.Giese C, et al. Immunological substance testing on human lymphatic micro-organoids in vitro. J Biotechnol. 2010;148:38–45. doi: 10.1016/j.jbiotec.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 216.Cayrol F, et al. Integrin alphavbeta3 acting as membrane receptor for thyroid hormones mediates angiogenesis in malignant T cells. Blood. 2015;125:841–851. doi: 10.1182/blood-2014-07-587337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Apoorva F, et al. Lymph Node Stiffness Mimicking Hydrogels Regulate Human B Cell Lymphoma Growth and Cell Surface Receptor Expression in a Molecular Subtype-Specific Manner. J Biomed Mater Res A. 2017 doi: 10.1002/jbm.a.36031. [DOI] [PubMed] [Google Scholar]
- 218.Shah SB, Singh A. Creating artificial lymphoid tissues to study immunity and hematological malignancies. Current opinion in hematology. 2017 doi: 10.1097/MOH.0000000000000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Shah SB, Singh A. Cellular self-assembly and biomaterials-based organoid models of development and diseases. Acta Biomater. 2017;53:29–45. doi: 10.1016/j.actbio.2017.01.075. [DOI] [PubMed] [Google Scholar]
- 220.Singh A. Biomaterials innovation for next generation ex vivo immune tissue engineering. Biomaterials. 2017;130:104–110. doi: 10.1016/j.biomaterials.2017.03.015. [DOI] [PubMed] [Google Scholar]
- 221.Crosby NM, et al. Anti-CD20 single chain variable antibody fragment–apolipoprotein A-I chimera containing nanodisks promote targeted bioactive agent delivery to CD20-positive lymphomas. Biochemistry and Cell Biology. 2015;93:343–350. doi: 10.1139/bcb-2015-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Dong X-Y, et al. Antibody-directed double suicide gene therapy targeting of MUC1-positive leukemia cells in vitro and in vivo. Current gene therapy. 2013;13:346–357. doi: 10.2174/15665232113136660029. [DOI] [PubMed] [Google Scholar]
- 223.Bonadonna G, et al. ABVD plus subtotal nodal versus involved-field radiotherapy in early-stage Hodgkin’s disease: long-term results. Journal of Clinical Oncology. 2004;22:2835–2841. doi: 10.1200/JCO.2004.12.170. [DOI] [PubMed] [Google Scholar]
- 224.Ansell SM. Hodgkin lymphoma: 2016 update on diagnosis, risk‐stratification, and management. American journal of hematology. 2016;91:434–442. doi: 10.1002/ajh.24272. [DOI] [PubMed] [Google Scholar]
- 225.Eich HT, et al. Intensified chemotherapy and dose-reduced involved-field radiotherapy in patients with early unfavorable Hodgkin’s lymphoma: final analysis of the German Hodgkin Study Group HD11 trial. Journal of Clinical Oncology. 2010;28:4199–4206. doi: 10.1200/JCO.2010.29.8018. [DOI] [PubMed] [Google Scholar]
- 226.Longo DL, et al. Conventional-dose salvage combination chemotherapy in patients relapsing with Hodgkin’s disease after combination chemotherapy: the low probability for cure. Journal of Clinical Oncology. 1992;10:210–218. doi: 10.1200/JCO.1992.10.2.210. [DOI] [PubMed] [Google Scholar]
- 227.Devizzi L, Santoro A, Bonfante V, Viviani S, Bonadonna G. Vinorelbine: a new promising drug in Hodgkin’s disease. Leuk Lymphoma. 1996;22:409–414. doi: 10.3109/10428199609054778. [DOI] [PubMed] [Google Scholar]
- 228.Santoro A, et al. Gemcitabine in the treatment of refractory Hodgkin’s disease: results of a multicenter phase II study. Journal of Clinical Oncology. 2000;18:2615–2619. doi: 10.1200/JCO.2000.18.13.2615. [DOI] [PubMed] [Google Scholar]
- 229.Sehn LH, et al. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. Journal of Clinical Oncology. 2005;23:5027–5033. doi: 10.1200/JCO.2005.09.137. [DOI] [PubMed] [Google Scholar]
- 230.Martelli M, et al. Diffuse large B-cell lymphoma. Critical reviews in oncology/hematology. 2013;87:146–171. doi: 10.1016/j.critrevonc.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 231.El Gnaoui T, et al. Rituximab, gemcitabine and oxaliplatin (R-GEMOX): An effective regimen for relapsed and refractory B-cell lymphoma. ASCO Meeting Abstracts. 2006;24 [Google Scholar]
- 232.Guadagnolo BA, et al. Long-term outcome and mortality trends in early-stage, Grade 1–2 follicular lymphoma treated with radiation therapy. International Journal of Radiation Oncology* Biology* Physics. 2006;64:928–934. doi: 10.1016/j.ijrobp.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 233.Flinn IW, et al. Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood. 2014;123:2944–2952. doi: 10.1182/blood-2013-11-531327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Freedman A. Follicular lymphoma: 2015 update on diagnosis and management. American journal of hematology. 2015;90:1171–1178. doi: 10.1002/ajh.24200. [DOI] [PubMed] [Google Scholar]
- 235.Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2016;127:53–61. doi: 10.1182/blood-2015-08-604520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Roboz GJ. Current treatment of acute myeloid leukemia. Current opinion in oncology. 2012;24:711–719. doi: 10.1097/CCO.0b013e328358f62d. [DOI] [PubMed] [Google Scholar]
- 237.Koreth J, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. Jama. 2009;301:2349–2361. doi: 10.1001/jama.2009.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Cornelissen JJ, et al. Results of a HOVON/SAKK donor versus no-donor analysis of myeloablative HLA-identical sibling stem cell transplantation in first remission acute myeloid leukemia in young and middle-aged adults: benefits for whom? Blood. 2007;109:3658–3666. doi: 10.1182/blood-2006-06-025627. [DOI] [PubMed] [Google Scholar]
- 239.Narayanan S, Shami PJ. Treatment of acute lymphoblastic leukemia in adults. Critical reviews in oncology/hematology. 2012;81:94–102. doi: 10.1016/j.critrevonc.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 240.Thomas DA, et al. Outcome after frontline therapy with the hyper-CVAD and imatinib mesylate regimen for adults with de novo or minimally treated Philadelphia (Ph) positive acute lymphoblastic leukemia (ALL) Journal of Clinical Oncology. 2008;26:7019–7019. [Google Scholar]
- 241.Koller CA, et al. The hyper-CVAD regimen improves outcome in relapsed acute lymphoblastic leukemia. Leukemia (08876924) 1997;11:2039. doi: 10.1038/sj.leu.2400861. [DOI] [PubMed] [Google Scholar]
- 242.Hiddemann W, et al. Treatment of refractory acute lymphoblastic leukemia in adults with high dose cytosine arabinoside and mitoxantrone (HAM) Leukemia. 1990;4:637–640. [PubMed] [Google Scholar]
- 243.Yap B, McCredie K, Keating M, Bodey G, Freireich E. Asparaginase and methotrexate combination chemotherapy in relapsed acute lymphoblastic leukemia in adults. Cancer treatment reports. 1980;65:83–87. [PubMed] [Google Scholar]
- 244.Kantarjian HM, et al. Experience with vincristine, doxorubicin, and dexamethasone (VAD) chemotherapy in adults with refractory acute lymphocytic leukemia. Cancer. 1989;64:16–22. doi: 10.1002/1097-0142(19890701)64:1<16::aid-cncr2820640104>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 245.Yap BS, McCredie KB, Keating MJ, Bodey GP, Freireich EJ. Asparaginase and methotrexate combination chemotherapy in relapsed acute lymphoblastic leukemia in adults. Cancer Treat Rep. 1981;65(Suppl 1):83–87. [PubMed] [Google Scholar]
- 246.Deininger M, et al. International Randomized Study of Interferon vs STI571 (IRIS) 8-Year Follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood. 2009;114:1126–1126. [Google Scholar]
- 247.Kantarjian H, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. New England Journal of Medicine. 2010;362:2260–2270. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- 248.Larson RA, et al. American Society of Clinical Oncology. 2014 [Google Scholar]
- 249.Cortes JE, et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome–positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011;118:4567–4576. doi: 10.1182/blood-2011-05-355594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Cortes JE, et al. A phase 2 trial of ponatinib in Philadelphia chromosome–positive leukemias. New England Journal of Medicine. 2013;369:1783–1796. doi: 10.1056/NEJMoa1306494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2016 update on diagnosis, therapy, and monitoring. American journal of hematology. 2016;91:252–265. doi: 10.1002/ajh.24275. [DOI] [PubMed] [Google Scholar]
- 252.Jabbour E, et al. Results of allogeneic hematopoietic stem cell transplantation for chronic myelogenous leukemia patients who failed tyrosine kinase inhibitors after developing BCR-ABL1 kinase domain mutations. Blood. 2011;117:3641–3647. doi: 10.1182/blood-2010-08-302679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hallek M, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. The Lancet. 2010;376:1164–1174. doi: 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- 254.Fischer K, et al. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. Journal of Clinical Oncology. 2012;30:3209–3216. doi: 10.1200/JCO.2011.39.2688. [DOI] [PubMed] [Google Scholar]
- 255.Hillmen P, et al. Rituximab plus chlorambucil in patients with CD20-positive B-cell chronic lymphocytic leukemia (CLL): final response analysis of an open-label phase II study. Blood. 2010;116:697–697. [Google Scholar]
- 256.Goede V, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. New England Journal of Medicine. 2014;370:1101–1110. doi: 10.1056/NEJMoa1313984. [DOI] [PubMed] [Google Scholar]
- 257.Brown JR, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123:3390–3397. doi: 10.1182/blood-2013-11-535047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Byrd JC, et al. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. New England Journal of Medicine. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Dreger P, et al. Managing high-risk CLL during transition to a new treatment era: stem cell transplantation or novel agents? Blood. 2014;124:3841–3849. doi: 10.1182/blood-2014-07-586826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Elter T, et al. Fludarabine in combination with alemtuzumab is effective and feasible in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: results of a phase II trial. Journal of Clinical Oncology. 2005;23:7024–7031. doi: 10.1200/JCO.2005.01.9950. [DOI] [PubMed] [Google Scholar]
- 261.Rai K, et al. Alemtuzumab in previously treated chronic lymphocytic leukemia patients who also had received fludarabine. Journal of Clinical Oncology. 2002;20:3891–3897. doi: 10.1200/JCO.2002.06.119. [DOI] [PubMed] [Google Scholar]
- 262.Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. Journal of clinical oncology. 2006;24:431–436. doi: 10.1200/JCO.2005.03.0221. [DOI] [PubMed] [Google Scholar]
- 263.Anderson KC. in Seminars in Hematology. Vol. 42. Elsevier; 2005. pp. S3–S8. [DOI] [PubMed] [Google Scholar]
- 264.Harousseau JL, et al. VELCADE/Dexamethasone (Vel/D) Versus VAD as Induction Treatment Prior to Autologous Stem Cell Transplantion (ASCT) in Newly Diagnosed Multiple Myeloma (MM): Updated Results of the IFM 2005/01 Trial. Blood. 2007;110:450–450. [Google Scholar]
- 265.Alexanian R, Dimopoulos MA, Hester J, Delasalle K, Champlin R. Early myeloablative therapy for multiple myeloma. Blood. 1994;84:4278–4282. [PubMed] [Google Scholar]
- 266.Richardson PG, Hideshima T, Mitsiades C, Anderson KC. The emerging role of novel therapies for the treatment of relapsed myeloma. Journal of the National Comprehensive Cancer Network. 2007;5:149–162. doi: 10.6004/jnccn.2007.0015. [DOI] [PubMed] [Google Scholar]
- 267.Barlogie B, et al. High-dose chemoradiotherapy and autologous bone marrow transplantation for resistant multiple myeloma. Blood. 1987;70:869–872. [PubMed] [Google Scholar]