

Abstract
The reactions of three model compounds (1−3) for cyclic acyl disulfides and cyclic acyl selenylsulfides are studied. These compounds were found to be effective precursors for persulfides (RSSH) and selenylsulfides (RSeSH) upon reacting with nucleophilic species. They could also act as H2S donors when interacting with cellular thiols. The most interesting discovery was the generation of RSeSH from compound 3 under mild conditions. Selenylsulfides (RSeSH) are expected to be important regulating molecules involved in Sec-related redox signaling. The method of producing RSeSH should allow researchers to better understand the chemical biology of RSeSH.
Graphical abstract
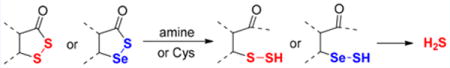
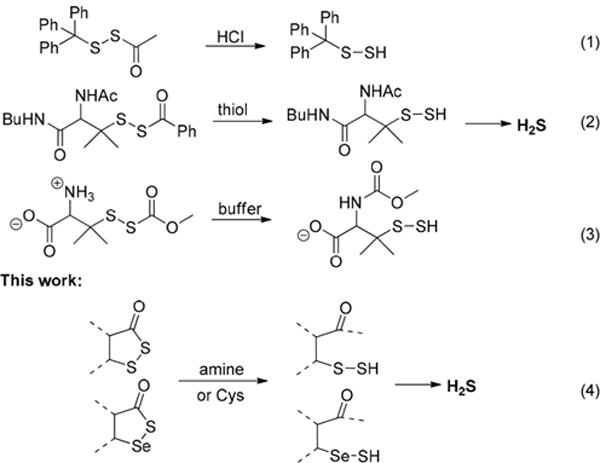
Persulfides (RSSH) belong to the family of reactive sulfur species, which play regulatory roles in redox biology.1 The importance of persulfides was recently recognized due to their links to H2S signaling.1 Some studies have revealed that significant levels of persulfides naturally exist. For example, cysteine persulfide (CysSSH, ∼1−4 μM) and glutathione persulfide (GSSH, up to 150 μM) exist in human and mouse tissues and cells.2 The formation of persulfides under physiological conditions can be attributed to several known reactions.3 For example, enzymes involved in sulfur metabolism (including cystathionine β-synthase CBS, cystathionine γ-lyase CSE, and 3-mercaptopyruvate sulfur-transferase 3MST) can promote sulfur-transfer reactions or cystine elimination to form persulfides. H2S can react with disulfides or S-modified cysteine residues (like nitrosothiols SNO and sulfenic acids SOH) to form disulfides. Cysteines may also react with reactive sulfane sulfurs, such as hydrogen polysulfides (H2Sn), to form persulfides. From a chemistry point-of-view, persulfides are highly reactive and unstable molecules, and understanding their chemical biology can be challenging due to their instability. In the past several years, exploring methods to chemically generate persulfides and study their fundamental chemistry have attracted considerable attention. In particular, acyclic acyl disulfides are commonly used as persulfide precursors (Scheme 1).4 Acid-mediated hydrolysis (Scheme 1, eq 1) or acyl transfer reactions (inter- or intramolecular, Scheme 1, eqs 2 and 3) can convert acyclic acyl disulfides to the corresponding persulfides, and further produce H2S in some cases.4 In contrast, cyclic acyl disulfides as persulfide or H2S precursors have not been reported. In this study, we tested the conversion of cyclic acyl disulfides to persulfides and the mechanisms. Their activity as H2S donors was also studied. Moreover, a cyclic acyl selenylsulfide was found to be a unique precursor for selenylsulfide (RSeSH), which is a selenol-based counterpart of persulfides. Selenylsulfides are expected to be important intermediates in sulfur redox processes involving selenocysteine or selenocysteine-containing proteins, but their chemistry and properties are still largely unknown. The studies reported here provide a facile way to generate persulfide and selenylsulfide model molecules under mild conditions, which should allow us to better study their chemical biology.
Scheme 1.
Release of H2S from Acyl Disulfides
In previous studies of acyclic acyl disulfides, tertiary thiol derived acyl disulfides (Scheme 1) were often used as the substrates.4a This is because tertiary thiol derived persulfides are believed to have greater stability due to steric hindrance.4d Cyclic acyl disulfides, however, are expected to have different reactivity due to their cyclic structures. In our study, two five-member ring substrates (dithiolane 1 and benzodithiolone 2) were selected as the substrates. 1 was prepared from penicillamine in three steps (Scheme S1, in Supporting Information (SI)). 2 was obtained using the protocol reported in our previous work.5 As amines and thiols are common nucleophiles in biological environments, we first tested the reactions of 1 and 2 with a model amine (n-BuNH2) and a model thiol (n-BuSH). As shown in Scheme 2, the reaction between dithiolane 1 and n-BuNH2 was found to be fast (<20 min) and clean in most organic solvents such as CH2Cl2, CHCl3, THF, and dioxane, as well as in aqueous solutions such as THF/H2O (1:5) or CH3CN/H2O (1:5). A mixture of trisulfide and tetrasulfide 4 was obtained in good yield. Clearly, in this process the amine directly attacked the carbonyl group and a persulfide intermediate 5 was formed, which could be trapped by thiol blocking reagents such as iodoacetamide (Scheme S2, SI). As an unstable species, 5 easily decomposed to form the polysulfide mixture 4.
Scheme 2.
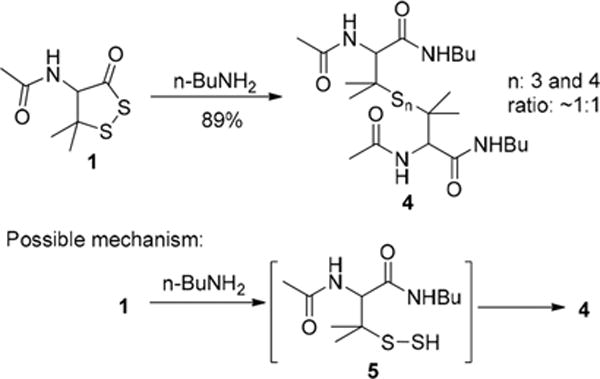
Reaction of 1 and nBuNH2
The reaction between 1 and n-BuSH was more complicated. When pure organic solvents were used as the reaction media, no reaction was observed and only the starting materials were recovered. However, when the reaction was carried out in aqueous buffer solutions, we obtained two products 6 and 7 in good combined yields (Scheme 3). Presumably the thiol was at least partially converted into a more nucleophilic thiolate in buffers and therefore facilitated the reaction. The formation of 6 and 7 suggested two possible mechanisms: (a) BuSH attacked the sulfur atom next to the carbonyl to form the intermediate 8, which then cyclized to form thiolactone 10 and BuSSH 9. 10 could further react with BuSH to provide thioester 6, whose −SH residue could also react with 9 to provide disulfide 7. (b) BuSH directly attacked the carbonyl group to form persulfide intermediate 11. Then, BuSH could react with either a sulfur atom of 11 to furnish 6 or 7.
Scheme 3.
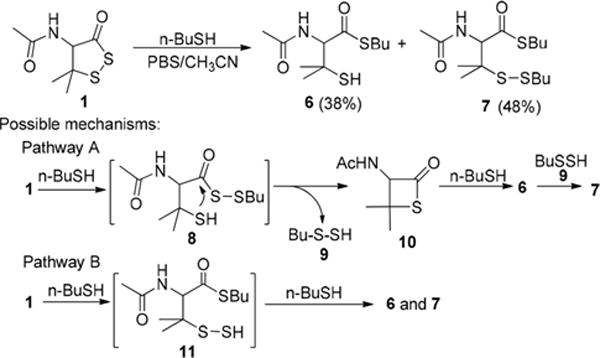
Reaction of 1 and nBuSH
To figure out which pathway is more favorable, theoretical calculation was carried out using Gaussian packages.6 The calculated relative energies indicated that the first step in both pathways A and B is slightly exothermic (−0.88 and −1.54 kcal/mol; see Table S1 in SI). However, the step forming 10 from 8 is quite endothermic with an ∼9.8 kcal/mol difference. One of the possible explanations is that the formation of a four-membered ring in 10 is energetically less favorable. Therefore, pathway B should be more favorable than pathway A.
Next we turned to benzodithiolone 2, and its reactions with BuNH2 and BuSH were studied (Scheme 4). The fused benzene ring significantly enhanced the stability of the thiolone functional group. Compound 2 was found to be completely inert to BuSH. No reaction was observed when 2 was treated with BuSH in both organic and aqueous solutions (with or without bases). 2 did react with BuNH2, however, at a slower rate. In this process, polysulfides 12 could be observed initially. Since the reaction was slow, polysulfides 12 were further converted into more stable disulfide 13 while the starting material 2 was consumed. Overall, disulfide 13 was obtained in an excellent yield with elemental sulfur as the byproduct. These results again demonstrate that the five-membered thiolone group can effectively react with effective nucleophiles (such as amines) to produce persulfides.
Scheme 4.
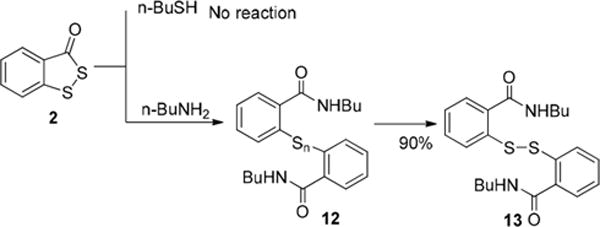
Reactions of 2 and nBuSH/nBuNH2
Having demonstrated that cyclic acyl disulfides 1 and 2 can be effective persulfide precursors, we turned our attention to their selenyl analogs. Selenocysteine (Sec) is the 21st proteinogenic amino acid and found in selenoproteins, many of which are redox enzymes. Cys and Sec share many properties, as there are only minor differences between sulfur and selenium in terms of electronegativity, ionic radius, and available oxidation states.7 However, the pKa of the selenol group (∼5.3) in Sec is much lower than the thiol in Cys (∼8.3). Sec also has a lower redox potential than Cys (−381 mV vs −180 mV). This means Sec is mostly deprotonated at physiological pH and very sensitive to redox regulation. The involvement of Sec in sulfur-related redox signaling is an interesting research topic but still poorly studied. We envisioned that −SeH could react similarly to −SH with reactive sulfur species to form selenylsulfides (−SeSH). We also expected that selenylsulfides could have a unique property/function in redox biology. It should be noted that the chemistry and properties of RSeSH are largely unknown in literature, except for a few theoretical studies.8 The prerequisite of studying selenylsulfides is to have a method that can easily generate RSeSH under mild conditions. As such, we expected acyl selenylsulfides, such as benzothiaselenol-3-one 3, to be the precursors.
Compound 3 was obtained from our previous work on diselenide-based H2S sensors.9 We then tested its reactions with BuSH and BuNH2 (Scheme 5). Compound 3 appeared to be even more inert than 2 showing no reactivity toward BuSH. Reactions of compound 3 with BuNH2 were also found to be slower than in the case of 2. Diselenide 15 and a mixture of diselenylsulfides 16 were isolated as the products. The reaction times and ratios of 15/16 varied, depending on the solvents used (Table 1). Diselenylsulfides 16 belong to the sulfane sulfur family, and their identity was further confirmed by a specific sulfane sulfur probe SSP4 (Figure S1 in the SI). We also found diselenylsulfides 16 to be unstable. They slowly decomposed to form more stable diselenide 15. Nevertheless, the formation of 15 and 16 indicated that selenylsulfide 14 was generated in the process. Diselenide 15 could be generated directly from 14, or from the decomposition of 16.
Scheme 5.
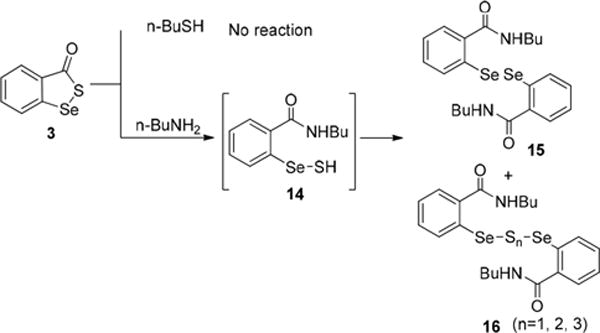
Reactions of 3 and nBuSH/nBuNH2
Table 1.
Formation of 15 and 16 from the Reactions of 3 with BuNH2
| entry | solvent | time | yield of 15/16 (%) |
|---|---|---|---|
| 1 | CH2Cl2 | overnight | 64/33 |
| 2 | CHCl3 | overnight | 53/42 |
| 3 | EtOAc | 4 h | 53/38 |
| 4 | THF | 4 h | 49/42 |
| 5 | CH3CN | 3 h | 95/0 |
| 6 | THF/PBS | overnight | 88/0 |
| 7 | CH3CN/PBS | 7 h | 88/0 |
To probe the reaction mechanism and further demonstrate the generation of the reactive selenylsulfide 14 in this reaction, we attempted to trap 14 using a thiol blocking reagent MSBT.10 Interestingly, we obtained 2-mercaptobenzthiazole 18, as well as diselenylsulfides 16 and diselenide 15, as the major product (Scheme 6). The isolation of 18 indicates that MSBT indeed trapped the selenylsulfide intermediate. However, since this reaction is a slow process, the trapped product 17 further reacted with 14 to form 16 and its desulfurated product 15.
Scheme 6.
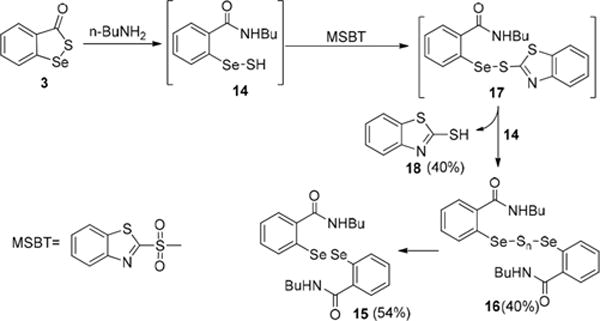
Trapping the Reaction Intermediate by MSBT
The reaction between 3 and nucleophiles such as amines provides an efficient way to generate selenylsulfides (RSeSH). It is also interesting to observe the formation of diselenylsulfides (RSeSnSeR) from RSeSH. If Sec-derived selenylsulfide (Sec-SeSH) is formed endogenously, the corresponding diselenylsulfides (Sec-Se-Sn-Se-Sec) might be the main end products. We suspected that such sulfur-containing species could serve as a reserved H2S pool. We then decided to explore the generation of H2S from 16 in the presence of endogenous reductants. NADH/NAD+ is a dominant redox regulation system in biology.11 As a potent electron donor, NADH participates in many electron-transfer processes and functions as a powerful reductant. As NADH is ubiquitous in cells, we wondered if NADH could directly react with 16 to produce H2S. Hantzsch ester (HEH) is a commonly used model compound of NADH. We then tested the reaction between 16 and HEH (Scheme 7). The formation of diselenide 15 and oxidized HEH 19 in almost quantitative yields was observed. Moreover, we also carried out a H2S gas trapping experiment using the protocol established in our lab. As shown in Figure 1, the reaction between HEH and 16 produced obvious H2S gas signals by the standard methylene blue (MB) assay. These results suggest selenylsulfides and diselenylsulfides could serve as a reserved H2S pool and produce H2S not by enzymatic, but by redox regulation.
Scheme 7.
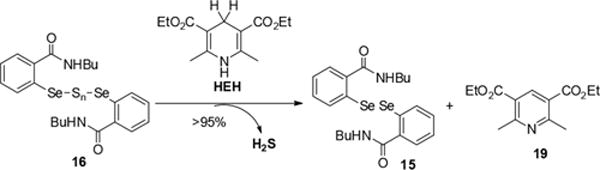
Reaction of 16 and HEH
Figure 1.
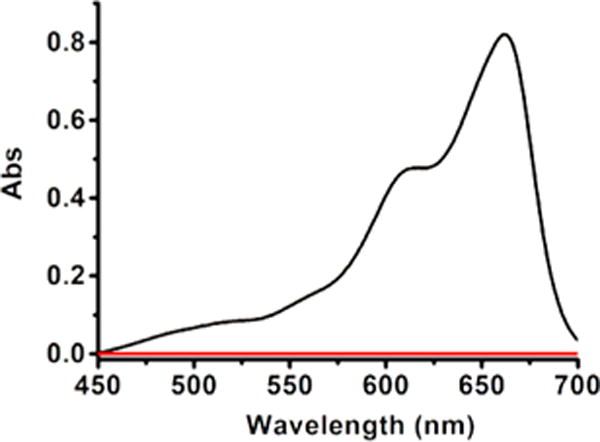
UV absorption spectra of methylene blue assay. A 0.5 mL aliquot of trapping solution is mixed with 0.5 mL of water, 0.5 mL of FeCl3, and 0.5 mL of DMPD and then incubated overnight before UV measurements. Black line: the reaction between HEH and 16; red line: control (trapping solution only).
Having studied the reactions of compounds 1−3 with nucleophiles such as amines and thiols, we wondered if these compounds could function as H2S donors in the presence of cellular thiols such as cysteine and glutathione (GSH), as those thiols contain both amine and thiol groups in their structures. We then tested their H2S-releasing ability in PBS buffers with the treatment of cysteine or GSH. The produced H2S was measured and quantified by the MB assay. The results were shown in Figure 2 (for cysteine) and Figure S2 (for GSH). Time-dependent H2S release from all of these compounds was observed. Compound 1 exhibited the strongest H2S release with both cysteine and GSH, which correlated well with its strong reactivity toward both BuSH and BuNH2. Compounds 2 and 3 showed much slower and weaker H2S release, which could also be attributed to their decreased reactivity toward amines and thiols. We also verified the H2S production of 2/3 (in the presence of cysteine and GSH) using the H2S gas trapping experiments (Figure S3). Overall these results suggest cyclic acyl disulfides such as 1 can be effective thiol-triggered H2S donors while benzene-fused cyclic acyl disulfides or selenylsulfides such as 2/3 are very weak donors. The slow or weak release of H2S from 2 and 3 could be useful for some biological applications when such release profiles are needed.
Figure 2.
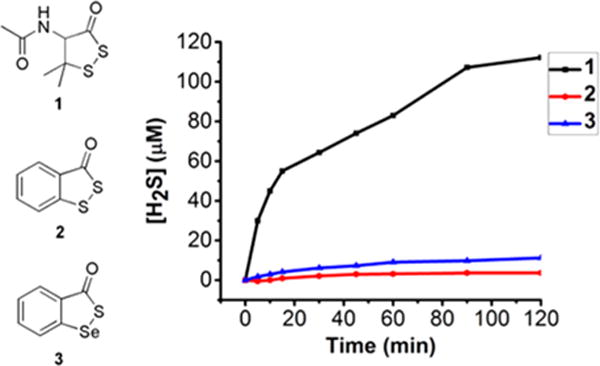
Time-dependent H2S release profiles of 1−3 (200 μM) in the presence of cysteine (5 equiv) in PBS buffer (pH 7.4, 50 mM).
In summary, we studied the reactions of three model compounds (1−3) of cyclic acyl disulfides and cyclic acyl selenylsulfides. These compounds were found to be effective precursors for persulfides (RSSH) and selenylsulfides (RSeSH) upon reacting with nucleophilic species. They could also act as H2S donors when interacting with cellular thiols. The most interesting discovery was the generation of RSeSH from compound 3 under mild conditions. Selenylsulfides (RSeSH) are expected to be important regulating molecules involved in Sec-related redox signaling. The method of producing RSeSH under mild conditions should allow researchers to better understand the chemical biology of RSeSH. The studies of other acyl selenylsulfide substrates are currently ongoing in our laboratory.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (R01GM125968). This work was also supported in part by funds provided for medical and biological research by the State of Washington Initiative Measure No. 171.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.7b04005.
Experimental and characterization of each compound (PDF)
ORCID Ming Xian: 0000-0002-7902-2987
Notes The authors declare no competing financial interest.
References
- 1.(a) Park CM, Weerasinghe L, Day JJ, Fukuto JM, Xian M. Mol BioSyst. 2015;11:1775–1785. doi: 10.1039/c5mb00216h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ono K, Akaike T, Sawa T, Kumagai Y, Wink D, Tantillo DJ, Hobbs AJ, Nagy P, Xian M, Lin J, Fukuto JM. Free Radical Biol Med. 2014;77:82–94. doi: 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mishanina TV, Libiad M, Banerjee R. Nat Chem Biol. 2015;11:457–464. doi: 10.1038/nchembio.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T. Proc Natl Acad Sci U S A. 2014;111:7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Filipovic MR, Zivanovic J, Alvarez B, Banerjee R. Chem Rev. 2017 doi: 10.1021/acs.chemrev.7b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yadav PK, Martinov M, Vitvitsky V, Seravalli J, Wedmann R, Filipovic MR, Banerjee R. J Am Chem Soc. 2016;138:289–299. doi: 10.1021/jacs.5b10494. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cuevasanta E, Moller MN, Alvarez B. Arch Biochem Biophys. 2017;617:9–25. doi: 10.1016/j.abb.2016.09.018. [DOI] [PubMed] [Google Scholar]; (d) Kasamatsu S, Nishimura A, Morita M, Matsunaga T, Abdul Hamid H, Akaike T. Molecules. 2016;21:1721. doi: 10.3390/molecules21121721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Bailey TS, Zakharov LN, Pluth MD. J Am Chem Soc. 2014;136:10573–10576. doi: 10.1021/ja505371z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Artaud I, Galardon E. ChemBioChem. 2014;15:2361–2364. doi: 10.1002/cbic.201402312. [DOI] [PubMed] [Google Scholar]; (c) Zhao Y, Bhushan S, Yang C, Otsuka H, Stein JD, Pacheco A, Peng B, Devarie NO, Aguilar HC, Lefer DJ, Xian M. ACS Chem Biol. 2013;8:1283–1290. doi: 10.1021/cb400090d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bailey TS, Pluth MD. Free Radical Biol Med. 2015;89:662. doi: 10.1016/j.freeradbiomed.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen W, Liu C, Peng B, Zhao Y, Pacheco A, Xian M. Chem Sci. 2013;4:2892–2896. doi: 10.1039/C3SC50754H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, revision E.01. Gaussian, Inc.; Wallingford, CT: 2013. [Google Scholar]
- 7.(a) Mousa R, Reddy PS, Metanis N. Synlett. 2017;28:1389–1393. [Google Scholar]; (b) Metanis N, Hilvert D. Curr Opin Chem Biol. 2014;22:27–37. doi: 10.1016/j.cbpa.2014.09.010. [DOI] [PubMed] [Google Scholar]; (c) Steinbrenner H, Speckmann B, Klotz LO. Arch Biochem Biophys. 2016;595:113–119. doi: 10.1016/j.abb.2015.06.024. [DOI] [PubMed] [Google Scholar]; (d) Huber RE, Criddle RS. Arch Biochem Biophys. 1967;122:164–173. doi: 10.1016/0003-9861(67)90136-1. [DOI] [PubMed] [Google Scholar]
- 8.(a) Bachrach SM, Demoin DW, Luk M, Miller JV., Jr J Phys Chem A. 2004;108:4040–4046. [Google Scholar]; (b) Bachrach SM, Walker CJ, Lee F, Royce S. J Org Chem. 2007;72:5174–5182. doi: 10.1021/jo070578s. [DOI] [PubMed] [Google Scholar]; (c) Benkova Z, Kona J, Gann G, Fabian WF. Int J Quantum Chem. 2002;90:555–565. [Google Scholar]
- 9.Peng B, Zhang C, Marutani E, Pacheco A, Chen W, Ichinose F, Xian M. Org Lett. 2015;17:1541–1544. doi: 10.1021/acs.orglett.5b00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Zhang D, Devarie-Baez NO, Li Q, Lancaster JR, Jr, Xian M. Org Lett. 2012;14:3396–3399. doi: 10.1021/ol301370s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen X, Wu H, Park CM, Poole T, Keceli G, Devarie-Baez NO, Tsang AW, Lowther WT, Poole LB, King SB, Xian M, Furdui CM. ACS Chem Biol. 2017;12:2201–2208. doi: 10.1021/acschembio.7b00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagel ZD, Klinman JP. Chem Rev. 2006;106:3095–3118. doi: 10.1021/cr050301x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.