

Abstract
Mutations in fibrillin-1 cause Marfan syndrome (MFS), the most common heritable disorder of connective tissue. Fibrillin-1 assemblies (microfibrils and elastic fibers) represent a unique dual-function component of the architectural matrix. The first role is structural for they endow tissues with tensile strength and elasticity, transmit forces across them and demarcate functionally discrete areas within them. The second role is instructive in that these macroaggregates modulate a large variety of sub-cellular processes by interacting with mechanosensors, and integrin and syndecan receptors, and by modulating the bioavailability of local TGFβ signals. The multifunctional, tissue-specific nature of fibrillin-1 assemblies is reflected in the variety of clinical manifestations and disease mechanisms associated with the MFS phenotype. Characterization of mice with ubiquitous or cell type-restricted fibrillin-1 deficiency has unraveled some pathophysiological mechanisms associated with the MFS phenotype, such as altered mechanotransduction in the heart, dysregulated TGFβ signaling in the ascending aorta and perturbed stem cell fate in the bone marrow. In each case, potential druggable targets have also be identified. However, the finding that distinct disease mechanisms underlie different organ abnormalities strongly argues for developing multi-drug strategies to mitigate or even prevent both life-threatening and morbid manifestations in pediatric and adult MFS patients.
Keywords: Cardiomyopathy, Fibrillin, Marfan syndrome, Mechanotransduction, Osteopenia, Stem cell niche, TGFβ, Thoracic aortic aneurysm
1. Introduction
Elastic assemblies (microfibrils and elastic fibers) are heterogeneous extracellular macroaggregates that, together with collagens, constitute the architectural scaffold of connective tissues [1]. Elastic assemblies are widely distributed in virtually every organ system where they support critical physiological functions, such as breathing and maintaining cardiovascular tone. Elastic fibers consist of insoluble elastin complexed with non-collagenous microfibrils 10–12 nm in diameter, which can also exist as individual structures in both pericellular and interstitial matrices of elastic and non-elastic tissues. Microfibrils are predominantly made of fibrillin molecules associated with other extracellular matrix (ECM) proteins that modify microfibril biogenesis and function. Human mutations in different components of elastic assemblies give rise to clinically distinct syndromes [2]. This review focuses on recent advances in our understanding of how mutations in fibrillin-1 translate into organ-specific manifestations in Marfan syndrome (MFS), the most common among heritable disorders of elastic assemblies – in point of fact, the most common of all heritable disorders of connective tissue [3]. While other diseases of elastic assemblies are discussed in this same issue of Matrix Biology, the reader is referred to several excellent reviews for additional details about fibrillin biology and MFS pathophysiology [4–9].
2. MFS; clinical features
MFS is an autosomal-dominant inherited disease with an incidence of approximately 1 in 5,000 live births and cardinal manifestations in the cardiovascular, skeletal and ocular systems [3]. Additional abnormalities of variable penetrance can involve the lung, integument and skeletal muscle. In broad terms, the disease can have either an often-fatal cardiovascular presentation during neonatal life or a progressively severe cardiovascular morbidity during adolescence and adulthood. In spite of significant progress in characterizing key genetic and molecular aspects of the disease, MFS diagnosis remains strictly clinical and according to the criteria codified in the recently revised Ghent nosology [10].
2.1 Cardiovascular system
Major cardiovascular manifestations of MFS include thoracic aortic aneurysm (TAA), mitral valve prolapse (MVP) and cardiac dysfunction [4]. Progressive dilatation of the proximal ascending aorta is associated with alterations in smooth muscle cell (SMC) phenotype, localized inflammatory infiltrates and maladaptive matrix remodeling that, together, predispose the vessel wall to dissection and rupture [4]. Major histopathological findings include elastic lamellae fragmentation, disorganized aortic tissue architecture with excessive collagen and mucopolysaccharide accumulation, and significantly fewer SMCs than normal. Prevention of untimely death from TAA complications currently relies on early detection by routine imaging, chronic administration of anti-hypertensive drugs and prophylactic repair by surgical procedures [4].
MVP associated with atrioventricular and semilunar valve elongation and myxomatous thickening is a frequent finding in MFS [4]. In contrast to adult patients, MVP in MFS neonates can often cause severe regurgitation and cardiac dysfunction leading to congestive heart failure. Although MVP can be surgically treated in adult patients, these individuals are at a greater risk of developing an acute TAA with dissection secondary to the rapid increase in cardiac output. Left ventricle (LV) dysfunction, including both systolic and diastolic function, has traditionally been considered a secondary phenomenon due to valvular insufficiency causing LV volume overload [4]. Increased arterial stiffness is also believed to contribute to cardiomyopathy by altering hemodynamic load on the LV. However, occasional reports of MFS patients with cardiomyopathy out of proportion to valvular problems have suggested a primary defect in cardiac muscle function [4, 6].
2.2 Skeleton
The most striking and immediately evident abnormality in MFS patients is a dysregulated linear bone growth causing serious malformations of limbs, anterior chest wall and spine, which are often associated with severe chronic pain [4]. Substantial chest deformities, particularly pectus excavatum, can be problematic during surgical interventions to restore cardiovascular function. Joint laxity is another hallmark of the disease that can cause dislocation of the hip with severe pain and stiffness. Reduced bone mass (osteopenia) is a controversial finding in MFS, especially in pediatric patients, due to the inherent difficulty of assessing bone mineral density (BMD) in affected vs. healthy individuals. As a result, the contribution of osteopenia to increased long-term risk for fractures remains uncertain [4].
2.3 Eye and other organ systems
Displacement of the lens (ectopia lentis) usually occurs before the age of 20 and can cause nearsightedness and blurred vision [4]. MFS patients are also at a higher risk of developing early cataracts, retinal detachment and glaucoma. Additional abnormalities that can be associated with MFS include decreased skeletal muscle mass and tone, spontaneous pneumothorax, recurrent incisional hernia and stretch marks in areas of the skin subjected to flexural stress [4].
3. MFS; molecular pathophysiology
Fibrillin-1 assemblies represent a unique dual-function component of the architectural matrix [1]. The first role is “structural” in that they endow tissues with tensile strength and elasticity, transmit mechanical forces across them and demarcate functionally discrete areas of the matrix. The second role of fibrillin-1 assemblies is “instructive” for they regulate the activities of resident cells by interacting with mechanosensors\ and integrin and syndecan receptors, and by modulating endogenous (local) TGFβ activity. The latter function is exerted via fibrillin-1 interaction with latent TGFβ-binding proteins (LTBPs) that are part of large latent complexes (LLCs) containing bioactive TGFβ dimers non-covalently bound to the processed pro-peptides (aka, latency associated peptide or LAP) [11]. Intracellular association of TGFβ with LTBPs 1, 3 or 4 facilitates secretion and tethering of the LLC to the ECM from which the signaling molecule is released by proteolytic and/or non-proteolytic disruption of the LAP-TGFβ interaction. LLC incorporation into the ECM is believed to promote spatial distribution and proper concentration of TGFβ molecules for either immediate presentation to cells or subsequent release during tissue remodeling/repair [11].
Mutations in fibrillin-1 are therefore expected to negatively impact both ECM integrity and local TGFβ activity. Just one year before the discovery of the genetic lesion in MFS, Hollister et al. [12] made the seminal observation that tissues and cell cultures from MFS patients contained less immunoreactive fibrillin-1 than healthy specimens. This observation implicitly predicted a loss-of-function phenotype at the tissue level -i.e., deficiency of fibrillin-1 microfibrils-irrespective of whether FBN1 mutations perturb ECM assembly and/or stability through haploinsufficient, dominant-negative or gain-of-function mechanisms. Characterization of mice with genetically engineered Fbn1 mutations have corroborated this early prediction, in addition to yielding invaluable insights into molecular processes secondary to fibrillin-1 deficiency that are associated with onset and progression of cardiovascular and bone abnormalities.
3.1 Arterial disease
The pathophysiological mechanism responsible for TAA development in MFS mice remains unresolved due to conflicting results regarding TGFβ’s role in this process. Early studies of MFS mice with non-dissecting TAA (Fbn1C1039G/+ mice) concluded that aneurysm formation is principally the result of over-stimulation of TGFβ production and signaling by improper activity of the angiotensin II (AngII) type I receptor (AT1r), a disease-causing process further amplified by uncontrolled LLC release from the fibrillin-1-deficient matrix (Fig. 1A) [13]. This disease model is largely based on the finding that either AT1r antagonism (by losartan) or TGFβ inhibition (by a neutralizing antibody) prevented TAA formation in Fbn1C1039G/+ mice. Losartan administration to MFS mice also normalized expression and activation of Mmp2 and Mmp9, downstream targets of TGFβ signals promoting media degeneration [14]. Additional genetic evidence correlated the scaffold protein β-arrestin2 in mediating AT1r stimulation of Mmp2 and Mmp9 through Erk activation independently of TGFβ [15]. As expected, the non-specific MMP inhibitor doxycycline mitigated TAA progression in MFS mice [16, 17]. Additional analyses of Fbn1C1039G/+ mice suggested that TGFβ signaling through the Erk pathway is a prominent driver of TAA formation that is inhibited by AT2r-mediated AngII signaling (Fig. 1A) [18]. Losartan therapy therefore emerged as a promising new strategy that could blunt AT1r stimulation of non-canonical TGFβ signaling and downstream targets, while concomitantly inhibiting Erk activation by shunting AngII signaling through AT2r. However, losartan treatment of MFS patients was subsequently found to mitigate the rate of aneurysm growth to the same extent as the traditionally prescribed anti-hypertensive drug and without modifying clinical endpoints of the disease (i.e., aortic dissection, elective aortic surgery, cardiovascular death) [19]. While losartan dosage could have accounted for the disappointing outcome, an alternative explanation is that additional signaling pathways may contribute to arterial disease progression in MFS [19].
Figure 1.
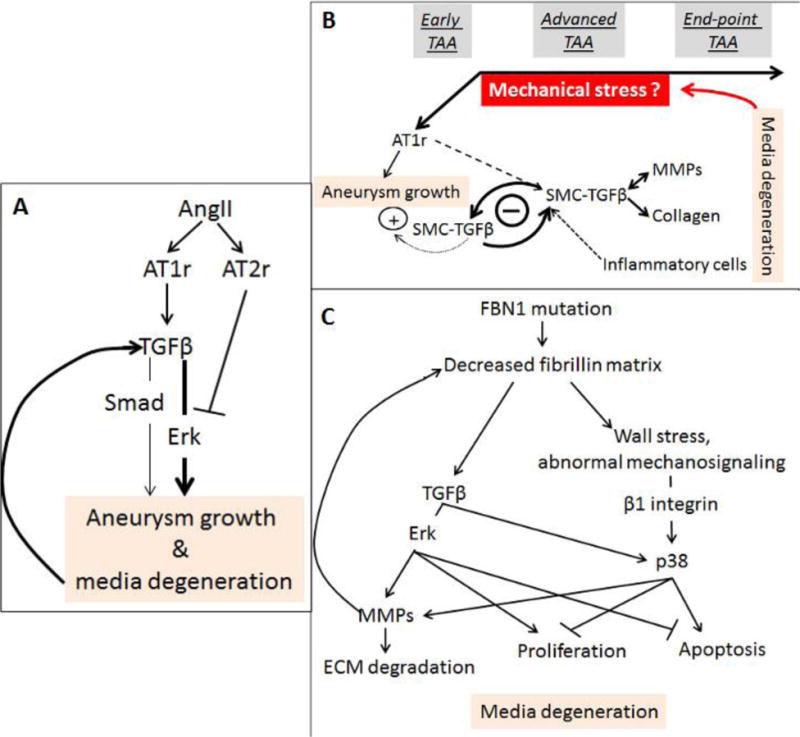
(A) Original disease model of a linear relationship between AT1r activation and TGFβ signaling derived from the characterization of TAA in Fbn1C1039G/+ mice (modified from Habashi et al, 2006 and Holm et al, 2011). (B) Revised model of TAA progression derived from studies of Fbn1mgR/mgR mice (modified from Cook et al, 2015). (C) Model of medial degeneration during TAA progression derived from the analysis of MFS-iPSCs differentiated into vascular SMCs (modified from Granata et al, 2017).
Several findings have corroborated and expanded the above postulate. The milder TAA phenotype of Fbn1C1039G/+ mice indicates that aneurysm growth and media degeneration are not by themselves sufficient to precipitate dissection and rupture of the vessel wall [20]. Similarly, media degeneration throughout the entire aortic tree of Fbn1mgR/mgR mice implies that segment-specific factors (e.g., hemodynamic load and/or SMC’s embryonic origin) are major contributors to the dilatation and subsequent dissection and rupture of a structurally compromised ascending aorta [21]. With regard to the TGFβ controversy, two research teams have independently reported that genetic disruption of TGFβ signaling in post-natal SMCs of Fbn1C1039G/+ mice exacerbates (as opposed to mitigate) TAA pathology [22, 23]. The implication of these studies that TGFβ plays a protective role in TAA development contrasts the earlier postulate of a prominent (AT1r-dependent) pathogenic role of TGFβ signaling [13, 18]. The reason of this discrepancy is unclear since all aforementioned studies have employed the same mouse model of MFS [13, 18, 22, 23].
Accelerated TAA progression was also the unexpected finding when MFS mice with dissecting (lethal) TAA (Fbn1mgR/mgR mice) were subjected to systemic TGFβ neutralization starting at post-natal day 16 (P16) [24]. By documenting an absolute requirement for basal TGFβ signaling in supporting post-natal vessel growth, these pharmacological studies independently supported the aforementioned genetic experiments excluding a prominent role of TGFβ in TAA onset. The additional finding of attenuated arterial disease in Fbn1mgR/mgR mice subjected to systemic TGFβ neutralization from P45 onward revealed a dimorphic, stage-specific role of TGFβ during TAA development [24]. Accordingly, it was argued that the primary consequence of fibrillin-1 deficiency is loss of TGFβ’s protective activity on the growing vessel combined with persistent AT1r over-activation throughout postnatal life; and that the secondary consequence of the ECM defect is to promote TGFβ-driven maladaptive tissue remodeling/repair at later stages of TAA progression (Fig. 1B) [24]. Consistent with this new disease model, losartan administration from P16 onward combined with TGFβ neutralization from P45 onward prevented TAA formation and thus, premature death of Fbn1mgR/mgR mice from vascular complications [24]. Additional studies of MFS mice have further increased the molecular complexity of arterial disease by identifying several pro-inflammatory determinants of TAA pathology, including IL-6, NOS2 and COX-2 [25–27], in addition to documenting mitigation of aneurysm growth by mild aerobic exercise, statins or long-term miR29 suppression [28–30].
Zilberberg et al. [31] have investigated a different aspect of TAA development by focusing on LTBP-3 contribution to arterial disease in mice with progressively severe MFS. The premise of the study was based on the investigators’ prior demonstration that fibrillin-1 microfibrils dictate proper association of LTBP-3 and -4 (but not LTBP-1) with the vascular ECM [32]. Contrary to prior studies predicting that lowering TGFβ activity during post-natal vessel growth would lead to premature death [24], genetic disruption of LTBP-3 synthesis in Fbn1mgR/mgR mice prevented TAA development and death of the double mutant animals by normalizing the transcriptome of aortic cells, including transcription of genes associated with TGFβ hyperactivity [31]. To reconcile the apparent discrepancy between the pharmacological findings of Cook et al [24] and the genetic findings of Zilberberg et al. [31] it was argued that distinct TGFβ isotypes might subsume protective and pathological roles during TAA onset and progression, respectively, and that LTBP-3 binds the disease-driving isotype(s) [31]. As TGFβ-2 haploinsufficiency in Fbn1C1039G/+ mice had previously been shown to exacerbate TAA pathology [33], it was further argued that selective association of LTBP-3 with TGFβ-1 and/or TGFβ-3 might specify the LLC complex driving degeneration of the fibrillin-1-deficient tunica media later in TAA progression [31]. In this view, disruption of spatiotemporally distinct interactions between LTBP and TGFβ molecules could represent another key determinant of TAA progression.
To gain additional mechanistic insights into medial degeneration in the MFS aorta, Granata et al. [34] have recently employed a clinically relevant cell-based approach that lends itself to more extensive biochemical analyses than studies in mice (Fig. 1C). MFS patient-derived induced pluripotent stem cells (MFS-iPSCs) differentiated into vascular SMCs were found to phenocopy molecular and cellular abnormalities previously noted in aortic tissue and cells isolated from MFS patients, including TGFβ hyperactivity, MMP up-regulation, ECM degradation, increased SMC apoptosis, reduced muscle contractility and perturbed Ca2+ flux. TGFβ hyperactivity in MFS-iPSC cultures was associated with increased phosphorylation of Smad, Erk and p38 proteins, which were in turn correlated with regulating distinct cellular phenotypes at different stages of in vitro differentiation. For example, Erk1/2 activation augmented MMP-driven ECM degradation late in MFS-iPSC differentiation, but inhibited apoptosis and stimulated proliferation of SMCs throughout the entire process. The study also implicated p38-mediated TGFβ signaling in promoting MMP up-regulation and ECM degradation. Unlike p-Erk1/2, however, p38 activation remained abnormally high at more mature stages of cell differentiation. Furthermore, p38 activation antagonized p-Erk1/2 protective action on SMC survival by stimulating apoptosis and inhibiting proliferation. Cyclic stretch further increased p38 activation, conceivably through the action of β1 integrin, thus suggesting a potential connection with dysregulated mechanosignaling (Fig. 1C). This possibility is in line with recent reports indicating that the aortic wall of Fbn1mgR/mgR mice is mechanically compromised, as evidenced by the loss of elastic energy storage capability and mechanical degradation of the media [35, 36]. Hence, the emerging evidence-based hypothesis that progressive hemodynamic load on a structurally vulnerable aorta may represent the primary trigger and driver of TAA development in MFS [5, 9].
3.2 Cardiac manifestations
Cardiac valve disease and stiffening of the dilating aortic wall have traditionally been thought to cause heart dysfunction in MFS by imposing volume overload on the LV [3, 6]. However, recent analyses of mice with tissue-specific Fbn1 gene inactivation have refuted this notion by demonstrating that fibrillin-1 deficiency in the pericellular matrix of the myocardium is both necessary and sufficient to trigger dilated cardiomyopathy (DCM) [37]. Characterization of subclinical cardiomyopathy in Fbn1C1039G/+ mice has independently reached the same conclusion [38]. These findings suggest that fibrillin-1 deficiency significantly weakens the physical properties of the myocardium and this in turn translates into abnormal myocyte mechanosignaling and impaired muscle contractility. In support of this argument, fibrillin-1 deficient cardiomyocytes display dysregulated activity of cardiac mechanosensors AT1r and β1 integrin, as evidenced by increased β-arrestin2-mediated Erk1/2 signaling and abated FAK signaling respectively (Fig. 2A). Additional evidence suggests a probable cross talk between the two receptors stimulated by mechanical unloading of the entire fibrillin-1-deficient ECM (Fig. 2A) [37]. In contrast to TAA, TGFβ hyperactivity was not found to be a prominent determinant of DCM development. Consistent with these findings, losartan treatment prevented DCM formation in Fbn1mgR/mgR mice and mitigated ventricular dysfunction in a small cohort of MFS patients [37, 39].
Figure 2.
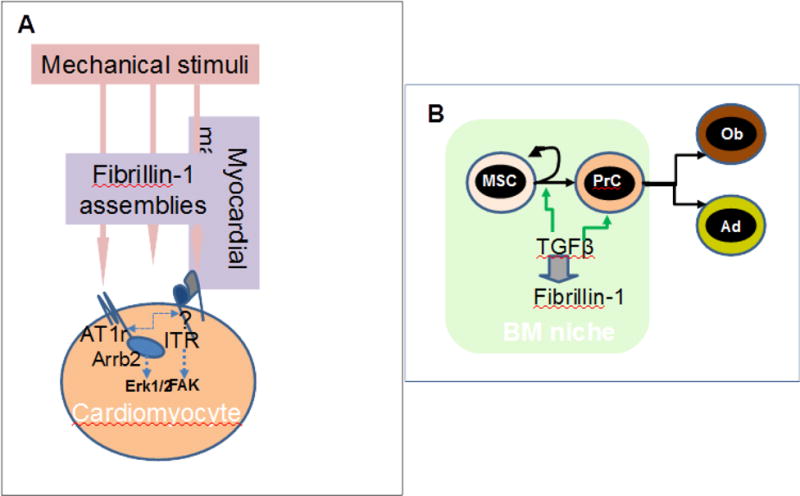
(A) Schematic representation of the mechanotransducing role of fibrillin-1 assemblies in the myocardium as inferred from DMC characterization in Fbn1mgR/mgR mice. In this model, fibrillin-1 assemblies influence cross talk between AT1r and integrin by participating in ECM loading on the mechanosensors (B) Proposed role of fibrillin-1 in stem cell fate regulation by modulating TGFβ bioavailability within the bone marrow niche, as implied from the characterization of progressive bone loss in Fbn1Prx−/− mice. Arrows points to cellular processes of self-renewal, commitment and differentiation, while abbreviations signify mesenchyme stem cell (MSC), progenitor cell (PrC), osteoblast (Ob) and adipocyte (Ad).
MVP in Fbn1C1039G/+ mice has been correlated with increased cellular proliferation and decreased apoptosis consistent with changes associated with myxomatous degeneration, as well as increased expression of TGFβ-induced stimulators of ECM remodeling [40]. Treatment of mutant mice with a TGFβ-neutralizing antibody normalized both the length and thickness of MV leaflets, implying participation of TGFβ hyperactivity in valve disease [40]. It remains to be determined if also in this case, mechanical stress might be the primary trigger of MVP and TGFβ hyperactivity a secondary driver of maladaptive tissue remodeling/repair [41].
3.3 Osteopenia
Characterization of the natural history of progressive bone loss in MFS mice has revealed that fibrillin-1 is a structural component of the bone marrow niche that supports self-renewal and commitment of mesenchyme stem cells (MSCs), and lineage determination of progenitor cells [42]. While other ECM proteins have been associated with the structural microenvironment of a functional stem cell niche [42], fibrillin-1 is the first element of the architectural matrix to be implicated in regulating stemness. By advancing our primitive understating of ECM role in defining the niche microenvironment, this finding may also impact future development of more effective stem cell-based therapies in regenerative medicine.
Reduced BMD in Fbn1mgR/mgR mice was originally correlated with accelerated in vitro maturation of calvarial osteoblasts and increased osteoblast-dependent osteoclast activity [43]. Whereas TGFβ-dependent stimulation of RANKL expression by osteoblasts accounted for augmented osteoclastogenesis, accelerated osteoblast differentiation was associated with both TGFβ and BMP hyperactivity. Subsequent analyses of mice with restricted Fbn1 inactivation in limb mesenchyme cells (Fbn1Prx1−/− mice) revealed that progressive bone loss is driven by constitutively enhanced bone resorption combined with premature depletion of MSCs and osteoprogenitor cells [44]. Fibrillin-1 deficiency also perturbed MSC commitment to adipogenesis resulting in osteopenic bones unusually depleted of marrow fat. All these cellular abnormalities were associated with improper over-activation of latent TGFβ complexes. By normalizing the number of MSCs, osteoprogenitor cells, osteoblasts, osteoclasts and adipocytes, systemic treatment of Fbn1Pr1x−/− mice with a TGFβ neutralizing antibody improved bone mass and trabecular microarchitecture, and restored marrow adipogenesis [44]. In contrast to cardiovascular manifestations, losartan treatment did not mitigate bone loss in MFS mice [43]. Consistent with these in vivo findings, Quarto et al. [45] have independently reported TGFβ-dependent inhibition of the osteoblastogenic potential of MFS-iPSC cultures. Both in vitro and in vivo findings therefore demonstrate that fibrillin-1 regulates MSC and progenitor cell fate by modulating TGFβ bioavailability within the functional microenvironment of marrow niches (Fig. 2B). With regard to enhanced bone resorption, dysregulated marrow hematopoiesis in Fbn1Pr1x−/− mice might also contribute to increasing the number of osteoclasts [46]. A decreased amount of proteolytic fragments of fibrillin-1 that normally restrict osteoclast differentiation through RANKL sequestration and NFATc inhibition might potentially be another stimulator of bone catabolism in Fbn1Pr1x−/− mice [47].
4. Concluding remarks and perspectives
Findings discussed in this review demonstrate that fibrillin-1 assemblies play a critical role in post-natal organ growth and homeostasis by integrating both structural and instructive properties of connective tissue. As noted fifteen years ago in a Matrix Biology review [48], these architectural matrix components “may function in the extracellular milieu in a manner similar to the way scaffold proteins act in the intracellular environment by focusing diverse molecules (both structural and signaling molecules) to promote their interactions.” The reviewers also argued that “the unique organization of matrix in individual tissues (may) define which (extrinsic signals and sub-cellular process) will be modulated (and how).” Characterization of cardiovascular and bone disease in MFS mice have validated these earlier predictions by causally linking disruption of fibrillin-1-centered networks of extracellular protein interactions with tissue-specific perturbations in mechanotransduction, TGFβ signaling and stem cell fate. An important corollary to these findings is the notion that multi-drug treatments will be required to manage both life-threatening and morbid manifestations of MFS.
In light of the above considerations, it is safe to predict that much research effort will be devoted in the future to develop unbiased study designs that can more effectively unravel the complex dynamics of fibrillin-1-centered regulatory networks associated with distinct MFS pathologies. For example, the progressive nature of skeletal complications constitutes a significant morbidity factor in MFS patients, particularly in older individuals and severely affected children. Unfortunately, effective treatment is hampered by the lack of an evidence-based mechanistic understanding of how a loss-of-function tissue defect (fibrillin-1 deficiency) translates into a gain-of-function organ phenotype (linear bone overgrowth). Resolving this fascinating scientific mystery will not only advance fundamental knowledge of molecular determinants of postnatal bone growth, but also improve the clinical management and life quality of MFS patients.
Highlights.
Mutations in the architectural matrix component fibrillin-1 cause Marfan syndrome (MFS)
Consistent with the multiple functions of fibrillin-1 assemblies, molecular abnormalities in MFS mice include dysregulated mechanotransduction, TGFβ signaling and stem cell fate.
Association of organ-specific manifestations with distinct pathophysiological mechanisms argues for combinatorial drug treatments in MFS
Acknowledgments
We apologize to our colleagues whose primary publications were not referenced in this review. We also thank Ms. Karen Johnson for organizing the manuscript. Studies performed in the authors’ laboratory were supported by grants from the National Institute of Arthritis, Musculoskeletal and Skin Diseases (NIAMS), the National Heart, Blood and Lung Institute (NHLBI), the National Marfan Foundation (NMF) and the Elster family’s research endowment.
Abbreviations used
- AngII
Angiotensin II
- AT1r
AngII type I receptor
- BMD
Bone mineral density
- DCM
Dilated cardiomyopathy
- ECM
Extracellular matrix
Human (FBN1) and mouse (Fbn1) fibrillin-1 genes
- iPSC
Induced pluripotent stem cells
- LAP
Latency-associated peptide
- LTBP
Latent TGFβ-binding protein
- LLC
Large latent complex
- LV
Left ventricle
- MFS
Marfan syndrome
- MSC
Mesenchyme stem cell
- MVP
Mitral valve prolapse
- SMC
Smooth muscle cell
- TAA
Thoracic aortic aneurysm
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ramirez F, Sakai LY. Biogenesis and function of fibrillin assemblies. Cell Tiss Res. 2010;339:71–82. doi: 10.1007/s00441-009-0822-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramirez F. Introduction to the mini-review series “Extracellular determinants of cell signaling”. Matrix Biol. 2015;47:1–2. doi: 10.1016/j.matbio.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Pyeritz PE. The Marfan syndrome. Annu Rev Med. 2000;51:481–510. doi: 10.1146/annurev.med.51.1.481. [DOI] [PubMed] [Google Scholar]
- 4.Doyle JJ, Gerber EE, Dietz HC. Matrix-dependent perturbation of TGFβ signaling and disease. FEBS Lett. 2012;586:2003–2015. doi: 10.1016/j.febslet.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. 2015;116:1448–1461. doi: 10.1161/CIRCRESAHA.114.304936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pyeritz RE. Recent progress in understanding the natural and clinical histories of the Marfan syndrome. Trends Cardiovasc Med. 2016;26:423–428. doi: 10.1016/j.tcm.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Sakai LY, Keene DR, Renard M, DeBacker J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene. 2016;591:279–291. doi: 10.1016/j.gene.2016.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen SA, Handford PA. New insights into the structure, assembly and biological roles of 10–12 nm connective tissue microfibrils from fibrillin-1 studies. Biochem J. 2016;473:827–838. doi: 10.1042/BJ20151108. [DOI] [PubMed] [Google Scholar]
- 9.Milewicz DM, Prakash SK, Ramirez F. Therapeutics targeting drivers of thoracic aortic aneurysm and acute aortic dissections; insights from predisposing genes and mouse models. Annu Rev Med. 2017;68:51–67. doi: 10.1146/annurev-med-100415-022956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loeys BL, Dietz HC, Braverman AC, Callewaert BL, Devereux RB, Hilhorst-Hofstee Y, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 11.Ramirez F, Rifkin DB. Extracellular microfibrils: contextual platforms for TGFβ and BMP signaling. Curr Opin Cell Biol. 2009;21:616–622. doi: 10.1016/j.ceb.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollister DW, Godfrey M, Sakai LY, Pyeritz RE. Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. N Engl J Med. 1990;323:152–159. doi: 10.1056/NEJM199007193230303. [DOI] [PubMed] [Google Scholar]
- 13.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang HH, Kim JM, Chum E, van Breemen C, Chung AWY. Long-term effects of losartan on structure and function of the thoracic aorta in a mouse model of Marfan syndrome. British J Pharmcol. 2009;158:1503–1512. doi: 10.1111/j.1476-5381.2009.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wisler JW, Harris EM, Raisch M, Mao L, Kim J, Rockman HA, et al. The role of β-arrestin2-dependent signaling in thoracic aortic aneurysm formation in a murine model of Marfan syndrome. Am J Physiol Heart Circ Physiol. 2015;309:H1516–H1527. doi: 10.1152/ajpheart.00291.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung AW, Yang HH, Radomski MW, van Breeman C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in Marfan syndrome through the inhibition of matrix metalloproteinase −2 and −9. Circ Res. 2008;102:e73–85. doi: 10.1161/CIRCRESAHA.108.174367. [DOI] [PubMed] [Google Scholar]
- 17.Xiong W, Knipsel RA, Dietz HC, Ramirez F, Baxter BT. Doxycycline delays aneurysm rupture in a mouse model of Marfan syndrome. J Vasc Surg. 2008;47:166–172. doi: 10.1016/j.jvs.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Backer J. Marfan and Sartans: time to wake up! Eur Heart J. 2015;36:2131–2133. doi: 10.1093/eurheartj/ehv228. [DOI] [PubMed] [Google Scholar]
- 20.Chung AW, Yang HH, van Breemen C. Imbalanced synthesis of cyclooxygenase-derived thromboxane A2 and prostacyclin compromises vasomotor function of the thoracic aorta in Marfan syndrome. Br J Pharmacol. 2007;152:305–312. doi: 10.1038/sj.bjp.0707391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwill S, Seppelt P, Grunhagen J, Ott CE, Jugold M, Rugparwar A, et al. The fibrillin-1 hypomorphic mgR/mgR murine model of Marfan syndrome shows severe elastolysis in all segments of the aorta. J Vasc Surg. 2013;57:1628–1636. doi: 10.1016/j.jvs.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Li W, Li Q, Qin L, Ali R, Zhou J, Ferruzzi J, et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124:755–767. doi: 10.1172/JCI69942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei H, Hu JH, Angelov SN, Fox K, Yan J, Enstrom R, et al. Aortopathy in a mouse model of Marfan syndrome is not mediated by altered transforming growth factor β signaling. J Am Heart Assoc. 2017;6:e004968. doi: 10.1161/JAHA.116.004968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S. Dimorphic effects of TGFβ signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35:911–917. doi: 10.1161/ATVBAHA.114.305150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo G, Ott CE, Grunhagen J, Munoz-Garcia B, Pletschacher A, Kallenback K, et al. Indomethacin prevents the progression of thoracic aortic aneurysm in Marfan syndrome mice. Aorta. 2013;1:5–12. doi: 10.12945/j.aorta.2013.13.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ju X, Ijaz T, Sun H, Lejeune W, Vargas G, Shilagard T, et al. IL-6 regulates extracellular matrix remodeling associated with aortic dilation in a fibrillin-1 hypomorphic mgR/mgR mouse model of severe Marfan syndrome. J Am Heart Assoc. 2014;3:e000476. doi: 10.1161/JAHA.113.000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oller J, Mendez-Barbero N, Ruiz EJ, Villahoz S, Renard M, Canelas LI, et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat Med. 2017;23:200–212. doi: 10.1038/nm.4266. [DOI] [PubMed] [Google Scholar]
- 28.McLoughlin D, McGuinness J, Byrne J, Terzo E, Huuskonen V, McAllister H, et al. Pravastatin reduces Marfan aortic dilation. Circulation. 2011;124:S168–S173. doi: 10.1161/CIRCULATIONAHA.110.012187. [DOI] [PubMed] [Google Scholar]
- 29.Gibson C, Nielsen C, Alex R, Cooper K, Farney M, Gaufin D, et al. Mild aerobic exercise blocks elastin fiber fragmentation and aortic dilatation in a mouse model of Marfan syndrome associated aortic aneurysm. J Appl Physiol. 2017;123:147–160. doi: 10.1152/japplphysiol.00132.2017. [DOI] [PubMed] [Google Scholar]
- 30.Okamura H, Emrich F, Trojan J, Chiu P, Dalal AR, Arakawa M, et al. Long-term miR-29b suppression reduces aneurysm formation in a Marfan mouse model. Physiol Rep. 2017;5:e13257. doi: 10.14814/phy2.13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zilberberg L, Phoon CK, Robertson I, Dabovic B, Ramirez F, Rifkin DB. Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc Natl Acad Sci USA. 2015;112:14012–14017. doi: 10.1073/pnas.1507652112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zilberberg L, Todorovic V, Dabovic B, Horiguchi M, Courousse T, Sakai LY, et al. Specificity of latent TGF-β binding protein (LTBP) incorporation into matrix: role of fibrillins and fibronectin. J Cell Physiolo. 2012;227:3828–3836. doi: 10.1002/jcp.24094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44:922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, et al. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet. 2017;49:97–109. doi: 10.1038/ng.3723. [DOI] [PubMed] [Google Scholar]
- 35.Bellini C, Korneva A, Zilberberg L, Ramirez F, Rifkin DB, Humphrey JD. Differential ascending and descending aortic mechanics parallel aneurysmal propensity in a mouse model of Marfan syndrome. J Biomech. 2016;49:2383–2389. doi: 10.1016/j.jbiomech.2015.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JJ, Galatioto J, Rao S, Ramirez F, Costa KD. Losartan attenuates degradation of aorta and lung tissue micromechanics in a mouse model of severe Marfan syndrome. Ann Biomed Eng. 2016;44:2994–3006. doi: 10.1007/s10439-016-1616-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cook JR, Carta L, Benard L, Chemaly ER, Chiu E, Rao SK, et al. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest. 2014;124:1329–1339. doi: 10.1172/JCI71059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campens L, Renard M, Trachet B, Segers P, Muino Mosquera L, DeSutter J, et al. Intrinsic cardiomyopathy in Marfan syndrome: results from in-vivo and ex-vivo studies of the Fbn1C1039G/+ model and longitudinal findings in humans. Pediatr Res. 2015;78:256–263. doi: 10.1038/pr.2015.110. [DOI] [PubMed] [Google Scholar]
- 39.den Hartog AW, Franken R, van den Berg MP, Zwinderman AH, Timmermans J, Scholte AJ, et al. The effect of losartan therapy on ventricular function in Marfan patients with haploinsufficient or dominant negative FBN1 mutations. Neth Heart J. 2016;24:675–681. doi: 10.1007/s12471-016-0905-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, et al. TGF-β-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gould RA, Sinha R, Aziz H, Rouf R, Dietz HC, 3rd, Judge DP, et al. Multi-scale biomechanical remodeling in aging and genetic mutant murine mitral valve leaflets: insights into Marfan syndrome. PLoS One. 2012;7:e44639. doi: 10.1371/journal.pone.0044639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smaldone S, Ramirez F. Fibrillin microfibrils in bone physiology. Matrix Biol. 2016;52–54:191–197. doi: 10.1016/j.matbio.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nistala H, Lee-Arteaga S, Carta L, Cook JR, Smaldone S, Siciliano G, et al. Differential effects of alendronate and losartan therapy on osteopenia and aortic aneurysm in mice with severe Marfan syndrome. Hum Mol Genet. 2010;19:4790–4798. doi: 10.1093/hmg/ddq409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smaldone S, Clayton N, del Solar M, Pasqual-Gonzales G, Cheng S, Wentworth B, et al. Fibrillin-1 regulates skeletal stem cell differentiation by modulating TGFβ activity within the marrow niche. J Bone Miner Res. 2016;31:86–97. doi: 10.1002/jbmr.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quarto N, Leonard B, Marchand M, Anderson E, Behr B, Francke U, R, et al. Skeletogenic phenotype of human Marfan embryonic stem cells faithfully phenocopied by patient-specific induced pluripotent stem cells. Proc Natl Acad Sci USA. 2012;109:215–220. doi: 10.1073/pnas.1113442109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smaldone S, Bigarella CL, del Solar M, Ghaffari S, Ramirez F. Fibrillin-1 microfibrils influence adult bone marrow hematopoiesis. Matrix Biol. 2016;52–54:88–94. doi: 10.1016/j.matbio.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tiedemann K, Boraschi-Diaz I, Rajakumar I, Kaur J, Roughley P, Reinhardt DP, et al. Fibrillin-1 directly regulates osteoclast formation and function by a dual mechanism. J Cell Sci. 2013;126:4187–4189. doi: 10.1242/jcs.127571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramirez F, Rifkin D. Cell signaling events: a view from the matrix. Matrix Biol. 2003;22:101–107. doi: 10.1016/s0945-053x(03)00002-7. [DOI] [PubMed] [Google Scholar]